

Summary
Enveloped viruses that rely on a low pH-dependent step for entry initiate infection by fusing with acidic endosomes, whereas the entry sites for pH-independent viruses, such as HIV-1, have not been defined. These viruses have long been assumed to fuse directly with the plasma membrane. Here we used population-based measurements of the viral content delivery into the cytosol and time-resolved imaging of single viruses to demonstrate that complete HIV-1 fusion occurred in endosomes. In contrast, viral fusion with the plasma membrane did not progress beyond the lipid mixing step. HIV-1 underwent receptor-mediated internalization long before endosomal fusion, thus minimizing the surface exposure of conserved viral epitopes during fusion and reducing the efficacy of inhibitors targeting these epitopes. We also show that, strikingly, endosomal fusion is sensitive to a dynamin inhibitor. These findings imply that HIV-1 infects cells via envelope glycoprotein- and dynamin-dependent fusion with intracellular compartments.
Introduction
Endocytosis is an obligatory entry step for enveloped viruses whose fusion proteins are activated by acidic pH (Marsh and Helenius, 2006). In contrast, viruses that undergo fusion upon interacting with cognate cellular receptors irrespective of the pH are thought to fuse directly with a plasma membrane. For instance, HIV-cell fusion initiated upon sequential interactions of the envelope (Env) glycoprotein with CD4 and coreceptors, CCR5 or CXCR4 (e.g., (Doms and Trono, 2000)), has long been assumed to occur at the cell surface, whereas internalized virions were thought to be degraded by cells (Maddon et al., 1988; McClure et al., 1988; Pelchen-Matthews et al., 1995; Stein et al., 1987). This notion is supported by the fact that HIV can mediate fusion between adjacent target cells (“fusion from without”) and that HIV Env expressed on effector cells promotes fusion with target cells at neutral pH. In addition, mutations in CD4 or coreceptors (CR) that impair their ligand-induced internalization do not block HIV-1 infection (Brandt et al., 2002; Maddon et al., 1988). The fact that pseudotyping the HIV core with the low pH-dependent G glycoprotein of Vesicular Stomatitis Virus (VSV) eliminates the Nef requirement for optimal infectivity (Aiken, 1997) is indicative of different entry routes for these and HIV Env-bearing viruses. In addition, the restriction on HIV-1 infection in resting T-cells imposed by the cortical actin is consistent with fusion at the cell surface (Yoder et al., 2008).
On the other hand, several lines of evidence support the existence of an alternative endocytic pathway for HIV-1 entry. First, HIV fusion with endosomes and micropinosomes has been observed by electron microscopy (Marechal et al., 2001; Pauza and Price, 1988). Second, blocking the acidification of endosomal compartments can augment HIV infection, apparently by sparing the virus from degradation in lysosomes (Fredericksen et al., 2002; Schaeffer et al., 2004; Wei et al., 2005). Third, efficient infection by HIV particles pseudotyped with VSV G (Aiken, 1997) shows that there are no apparent restrictions associated with the endocytic entry pathway. Finally, inhibition of clathrin-mediated endocytosis reduces the efficacy of HIV-cell fusion and infection in HeLa-derived cells (Daecke et al., 2005). However, this intervention perturbs important cellular functions and may thus alter the sites of virus entry.
Here, we applied time-resolved single virus imaging and a virus population-based fusion assay to delineate the cellular entry sites of HIV-1. These approaches have revealed that, surprisingly, complete HIV-1 fusion occurred in endosomal compartments, but not at the plasma membrane of epithelial and lymphoid cells. We found that endosomal fusion was delayed relative to HIV-1 uptake via CD4/CR-dependent endocytosis and that the fusion step was enhanced by the large GTPase dynamin. Methodologies developed in this work should help define the entry pathways of other pH-independent viruses.
Results
To elucidate the sites of virus entry, we first compared the effects of fusion inhibitors blocking surface-accessible viruses and universal inhibitors that block all viruses irrespective of their location. If fusion is limited to the cell surface, these interventions should yield identical results. However, the ability to enter into and fuse with endosomes would result in the transient appearance of viruses resistant to external inhibitors but sensitive to inhibitors blocking endosomal fusion. The difference in virus sensitivity to site-specific and universal inhibitors can thus be used to deduce the entry sites of pH-independent viruses.
HIV-1 fusion is delayed relative to its escape from a membrane-impermeant fusion inhibitor
Virus-cell fusion was directly quantified by measuring the cytosolic activity of viral core-associated β-lactamase (BlaM) (Cavrois et al., 2002). HIV-1 cores carrying a BlaM-Vpr chimera were pseudotyped with Env from JRFL (CCR5-tropic) or HXB2 (CXCR4-tropic) HIV-1 strains. We first examined the HIV-1 entry sites in HeLa-derived indicator cells expressing CD4, CCR5 and CXCR4 (designated TZM-bl cells (Wei et al., 2002)). Viruses were allowed to bind to cells in the cold, and fusion was initiated by shifting to 37°C and measured as the extent of cleavage of a fluorogenic substrate by the cytosolic BlaM-Vpr. To determine the kinetics of virus-cell fusion, we stopped the reaction after varied times of incubation at 37°C by adding a recombinant peptide derived from the C-terminal heptad repeat region of HIV-1 gp41 (hereafter referred to as C52L (Deng et al., 2007)). C52L and other gp41-derived peptides inhibit fusion by binding to intermediate gp41 conformations formed upon Env interactions with CD4 and CR and preventing the formation of the final 6-helix bundle structure (reviewed in (Eckert and Kim, 2001)). The time of C52L addition experiments revealed that the kinetics of the JRFL and HXB2 escape from this membrane-impermeant inhibitor was relatively fast, showing little or no lag and reaching completion within ∼2 hrs (Fig. 1A).
Figure 1.

Dissection of surface and endosomal HIV-1 fusion. (A) Virus fusion with TZM-bl cells was stopped by adding C52L after indicated times of incubation at 37°C, and incubation was continued up to 90 min, at which point the cells were briefly placed on ice and loaded with the BlaM substrate. Alternatively, fusion was stopped by placing cells on ice after varied times of incubation at 37°C (TB). After loading the substrate, cells were incubated overnight at 13.5°C regardless of the fusion protocol to allow the substrate cleavage. The red and blue dashed lines were obtained by subtracting the TB plot from the C52L escape plot for JRFL and HXB2, respectively. Unless stated otherwise, data points are means +/-SEM from triplicate measurements. (B) Fusion of VSV G pseudotypes with TZM-bl cells was blocked at indicated times either by treating cells with 2 mg/ml pronase on ice (10 min), adding 50 mM NH4Cl, or chilling the samples (TB). Cells were then loaded with CCF2 and incubated overnight at 12°C. (C, D) After 20 min at 37°C, viruses remaining at the surface of TZM-bl cells were rendered non-fusogenic by adding C52L (arrow), and the extent of fusion over time at 37°C was determined by chilling cells either immediately or at indicated time points (red triangles). (E) HXB2 virus escape from C52L and from the TB in CEMss cells was measured as described above. The dashed blue line represents the difference between the C52L and TB curves. Error bars are SEM (n=4).
To block HIV-1 fusion irrespective of its cellular location, we took advantage of the steep temperature dependence of HIV-1 fusion (Frey et al., 1995; Mkrtchyan et al., 2005). JRFL and HXB2 fusion with TZM-bl cells exhibited a well-defined threshold at ∼22°C (Fig. S1A). By contrast, the cytosolic BlaM was active at temperatures that were not permissive for fusion (data not shown). This allowed kinetic measurements of virus-cell fusion by quickly reducing the temperature after varied times of incubation at 37°C, followed by an overnight incubation at sub-threshold temperature to permit substrate cleavage.
The temperature block (TB) protocol showed that, surprisingly, the cytosolic BlaM delivery was greatly delayed compared to the virus escape from C52L (Fig. 1A). This delayed kinetics can result from two principal mechanisms. Low temperature can either block Env-mediated fusion or inhibit post-fusion steps that may be required for the optimal activity of the viral core-associated BlaM-Vpr. However, previous work (Cavrois et al., 2004) and our data (Fig. S1B) show that the post-fusion uncoating step does not enhance the BlaM activity. We found that this activity was not affected by inhibition of cellular proteases or proteasomes and, importantly, was observed in vitro in the absence of any cytosolic factors (Fig. S1C-E). Thus, the cleavage of BlaM substrate faithfully reports the extent of virus-cell fusion.
Our experimental strategy to elucidate the sites of virus entry was further validated using HIV particles pseudotyped with the low pH-dependent VSV G. As expected for an endocytic entry pathway, escape from the TB was delayed relative to the virus uptake measured by the emergence of the BlaM signal resistant to pronase (Fig. 1B). The temperature-dependent steps of VSV G fusion were completed soon after the completion of low pH-dependent steps, as evidenced by the virus escape from the block imposed by NH4Cl. The quick appearance of the TB-resistant BlaM signal after the low pH-induced fusion further implies that temperature-dependent post-fusion steps are not required to render BlaM-Vpr active.
HIV-1 likely enters lymphoid cells through an endocytic pathway
To define the sites of HIV entry in more natural target cells, we measured the rates of virus escape from C52L and from the TB in lymphoid CEMss cells expressing CD4 and CXCR4. In these cells, both rates were considerably faster than in TZM-bl cells (Fig. 1E vs. 1A). However, the loss of sensitivity to C52L occurred much earlier than the progression beyond temperature-dependent steps, suggesting that endosomal fusion is the major HIV-1 entry route in T-cells.
HIV-1 associated with CD4 and coreceptors spends considerable time in endosomes prior to fusion
The divergent rates of HIV-1 escape from C52L and the TB demonstrate that the actual fusion is much slower than the loss of sensitivity to the membrane-impermeant fusion inhibitor, which has been customarily interpreted as fusion with the plasma membrane. The difference between the C52L- and TB-resistant BlaM signals should reflect the fraction of internalized viruses that have not fused at that time point (Fig. 1A, dashed lines). Within the first 20 min of incubation, ∼40% of viruses appeared in C52L-inaccessible compartments, while only a small fraction acquired resistance to the TB. The high level of internalized viruses was maintained during the next 20 min due to the similar rates of escape from C52L (endocytosis) and the TB (fusion). Likewise, nearly half of the viruses were protected from C52L but did not fuse with CEMss cells within the first 10 min at 37°C (Fig. 1E, dashed line). Collectively, these findings show that HIV-1 fuses primarily, if not exclusively, with endosomes.
In order to separate plasma membrane entry from endosomal entry, viruses were pre-bound to cells in the cold and incubated at 37°C for 20 min, at which point surface-accessible unfused viruses were blocked by C52L. The BlaM signal was then chased by dropping the temperature either immediately or after varied times of incubation at 37°C in the presence of the inhibitor. Under these conditions, any increase in the BlaM signal over time should be exclusively due to viral content release from endosomes. The chase experiments revealed that endosomal fusion progressed slowly (Fig. 1C and D), reaching completion within ∼1 hr at 37°C. As expected, the regular TB protocol yielded much greater extents of fusion compared to the chase protocol (red triangles vs. green squares), which was most likely due to the continued uptake and fusion of surface-accessible viruses in the absence of the inhibitor. Thus, on average, HIV-1 spent about 30 min in C52L-inaccessible compartments prior to releasing its content.
In the absence of surface fusion, protection from C52L should correspond to productive, CD4/CR-mediated HIV endocytosis. This notion is supported by the ability of C52L to block fusion when added at the beginning of incubation, demonstrating that the gp41 coiled coils are exposed prior to virus uptake; this exposure is known to occur upon Env binding to CD4 alone or to CD4 and CR ((Eckert and Kim, 2001) and references therein). Accordingly, HIV-1 acquired resistance to inhibitors blocking CD4 and CR binding before it escaped from C52L (Fig. S1F). Thus, HIV-1 particles internalized by pathways other than CD4/CR-mediated endocytosis do not contribute to fusion. These results and virus imaging data (see below) show that, surprisingly, the major rate-limiting step of HIV-1 fusion occurs after CR binding and virus endocytosis.
Single virus imaging distinguishes between surface and endosomal fusion
To unambiguously identify the sites of HIV-1 entry, we visualized the fusion of viruses co-labeled with the relatively small, diffusible content marker (NC-GFP, Fig. S2B, D, E) and the lipophilic dye DiD ((Markosyan et al., 2005) and Experimental Procedures). Fusion with the plasma membrane should lead to the disappearance of the viral membrane and content markers due to their virtually infinite dilution within the plasma membrane and the cytosol, respectively (Fig. 2A). In contrast, virus fusion with a small intracellular organelle that is not continuous with the plasma membrane should lead to the loss of viral content without the disappearance of a membrane marker. Hence, the fusion sites can be identified based on the dilution of viral markers.
Figure 2.

Identification of HIV-1 fusion sites by single virus imaging. (A) Schematic presentation of redistribution of viral lipid and content markers upon fusion with a plasma membrane (left) and with an endosome (right). Viruses co-labeled with membrane (red) and content (green) markers are pseudocolored yellow. (B, C) Partial fusion of JRFL with the plasma membrane of TZM-bl cells. The time from the beginning of imaging is shown. The two-dimensional projection of the particle's trajectory (cyan) is overlaid on the last image. Changes in fluorescence intensities (in arbitrary units) of membrane (red) and content (green) markers, as well as the instantaneous velocity (blue trace) of the particle, are shown. (D-G) Complete fusion of JRFL (D, E) and HXB2 (F, G) viruses following the fast retrograde movement from the cell periphery (cyan traces on last images). Fusion is evident from the disappearance of GFP signal. Graphs (E and G) show changes in fluorescence of membrane and content markers (smoothed for visual clarity) and 3D trajectories for the viruses marked by arrows in panels D and F. See supplemental movies 3-5.
We validated this strategy by imaging the fusion of pseudoviruses bearing E1/E2 glycoproteins of the low pH-dependent Semliki Forest Virus (SFV). Normally, SFV fuses with acidic endosomes, but it can also be forced to fuse with the plasma membrane by lowering the pH (Marsh and Bron, 1997). As expected, SFV fusion with endosomes resulted in the disappearance of the viral content while the membrane marker remained localized within an endosome (Fig. S3A and movie 1). In contrast, exposure to low pH led to the quick redistribution of viral lipids, but not of viral content (Fig. S3B and movie 2), demonstrating the failure of SFV to undergo full fusion with the plasma membrane.
HIV-1 fusion with the plasma membrane is blocked after the lipid mixing stage
JRFL or HXB2 viruses were pre-bound to TZM-bl cells in the cold and triggered to fuse by quickly shifting to 37°C. We observed three principal outcomes of HIV-cell fusion. First, viruses released their lipid marker, as seen by the disappearance of the red signal, but retained their content for as long as we imaged (Fig. 2B and movie 3). Particles undergoing this type of fusion usually exhibited limited movement before and after the lipid transfer (Fig. 2C). These events were almost assuredly due to the partial fusion at the cell surface that did not result in the cytosolic delivery of viral content. Second, following the transport of viruses towards the cell nucleus, typical for endosomal trafficking (Lakadamyali et al., 2004), the viral content marker disappeared while the lipid marker continued to move as a distinct spot (Figs. 2D-G, movies 4 and 5). These events observed for both JRFL and HXB2 viruses were interpreted as the cytosolic release of viral content through fusion with endosomes. Third, viral markers were released (disappeared) sequentially, often exhibiting a considerable delay between lipid and content transfer (see Fig. 5 below). As discussed below, these events (hereafter dubbed the 2-step fusion) likely reflect the full fusion proceeding in two distinct temporally and, in most cases, spatially separated steps.
Figure 5.

Sequential lipid and content transfer events exhibited by HIV-1. (A, B) A 2-step fusion event exhibiting a short delay between lipid and content transfer. (C, D) A rare 2-step event characterized by stepwise release of viral content. Complete content discharge (double arrow) occurs after the onset of quick movement (see the movie 7). The the predicted dynamics of a fusion pore (panel D) is shown by a thick line above the panel. The initial and final coordinates of particles on 3D-plots are marked by pink crosses and stars, respectively. (E, F) The 2-step fusion of infectious R9 HIV-1 co-labeled with DiD and MA-GFP-CA. Changes in fluorescence intensities of viral lipid and content markers upon incubation with TZM-bl cells at 37°C are smoothed for visual clarity.
JRFL and HXB2 pseudoviruses exhibited distinct fusion phenotypes. The majority of JRFL particles exchanged lipids with the plasma membrane, while the content release from endosomes was less frequent (Fig. 3A). By comparison, most HXB2 particles underwent endosomal fusion, very few released the lipid marker and none exhibited the 2-step phenotype seen for JRFL viruses. The overall low probability of HIV-cell fusion is in agreement with our previous data (Markosyan et al., 2005). In control experiments, viral lipid and content transfer was inhibited by a high concentration of C52L (Fig. 3A), demonstrating that the overwhelming majority of viral lipid and content transfer events were mediated by HIV-1 Env. Viral content release was not detected between JRFL pseudoviruses and HeLa cells expressing CD4 but not CCR5 (Fig. 3A). Thus, under our experimental conditions, the deterioration of the GFP signal caused by low pH in late endosomes/lysosomes was negligible (see also Fig. S2C).
Figure 3.
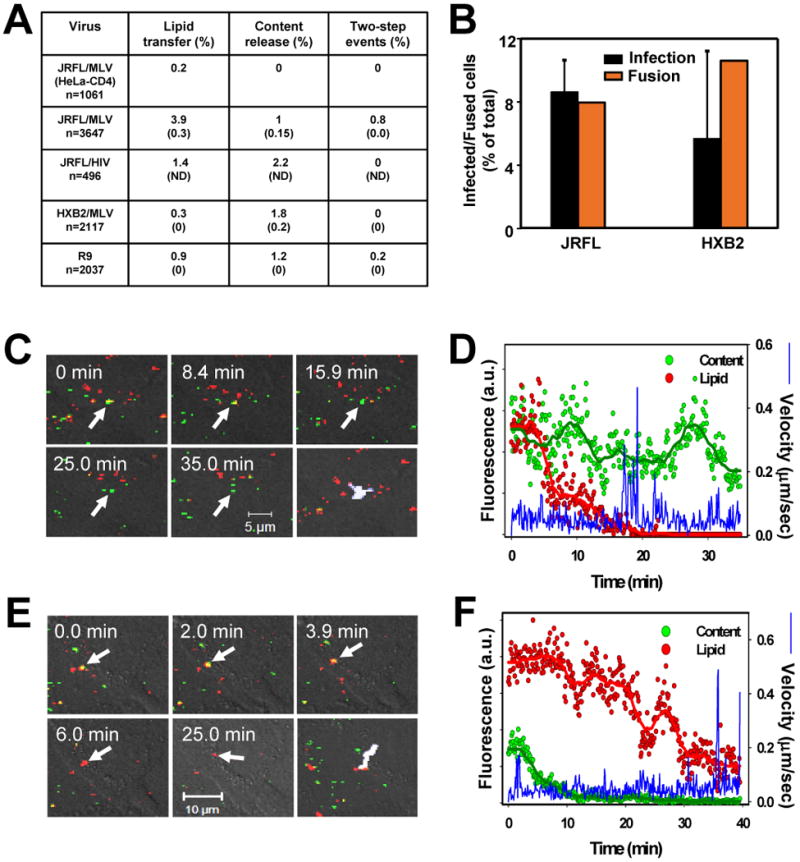
Fusion of HIV core-based pseudoviruses and of infectious HIV-1 with TZM-bl cells. (A) The efficiency of lipid mixing with the plasma membrane, the viral content release from endosomes, and the sequential 2-step fusion events mediated by JRFL Env (pseudotyped with MLV or HIV core), HXB2 Env, and by infectious R9 viruses. The first row is the negative control for JRFL fusion using HeLa-CD4 cells lacking CCR5. The number of respective fusion-related events was normalized to the total number of cell-associated double-labeled virions at the beginning of experiment. The extents of viral lipid and content mixing in the presence of 4 μM C52L are shown in parentheses. The rare false-positive events in the presence of C52L were usually delayed relative to those in the absence of the inhibitor (data not shown) and were minimized by limiting the duration of imaging experiments. ND, not determined. (B) The fraction of cells supporting viral content release was measured by imaging (orange bars), and the fraction of infected cells in the same sample was determined by a β-Gal assay (black bars) after an additional 48 hr-cultivation in the presence of C52L. The somewhat larger fraction of cells supporting HXB2 fusion was due to a more efficient binding of HXB2 to target cells compared to JRFL viruses (data not shown). (C, D) Lipid transfer initiated by the infectious R9 HIV-1 labeled with DiD and MA-GFP-CA at the surface of the TZM-bl cell. (E, F) Complete endosomal fusion of the infectious R9 particle. See the supplementary movie 6.
The restriction on virus fusion at the cell surface was not limited to HIV-1 and SFV pseudoviruses. We found that the Env glycoprotein of pH-independent amphotropic Murine Leukemia Virus (aMLV) also mediated virus fusion with endosomes. Out of 14 detected events, 11 released their content from endosomes (Fig. S3C, D), 2 transferred only the lipid marker at the cell surface, and 1 exhibited sequential (two-step) lipid and content release.
Endosomal fusion can lead to infection
To relate single virus fusion to infectivity, we evaluated the fraction of cells for which at least one content transfer event was detected by imaging. Endosomal fusion was observed for 8.0% and 10.5% of cells incubated with JRFL and HXB2 particles, respectively (Fig. 3B). Under identical conditions, 8.6% of cells were infected by JRFL and 5.7% by HXB2 (i.e., MOI was ∼0.1). The fraction of fusion-supporting cells was clearly underestimated due to the missed events. The relatively low fraction of double-labeled particles produced by the labeling protocol and the relatively short imaging time limited our ability to track all fusion events. Nonetheless, comparable efficacies of viral content delivery and infection indicate that a significant fraction of endosomal fusion established productive infection.
Infectious HIV-1 fuses with an endosome but not with the plasma membrane
To take advantage of the diffusible NC-GFP marker, our initial imaging experiments employed MLV-based pseudoviruses. To ensure adequate incorporation of HIV-1 Env into these particles, we used the gp41 construct lacking the cytoplasmic domain (Fig. S2A, B and (Markosyan et al., 2005)). To rule out the possibility that deletion of the cytoplasmic domain or pseudotyping with the MLV core alters the virus entry sites, we labeled infectious viruses by co-transfecting the cells with the proviral HIV-1 R9 clone encoding a full-length CXCR4-tropic Env (Gallay et al., 1997) and a new vector expressing GFP-tagged HIV Gag. In this construct (referred to as MA-GFP-CA), the EGFP coding sequence (flanked by the viral protease cleavage sites) was inserted between the MA and CA sequences of Gag polyprotein (see Supplementary Experimental Procedures). Upon virus maturation, MA-GFP-CA is cleaved by viral protease, yielding a free GFP (Fig. S2F-I). These viruses were co-labeled with DiD and allowed to fuse with TZM-bl cells.
Similar to pseudovirus fusion, infectious HIV-1 exhibited lipid mixing at the cell surface and content transfer from endosomes (Fig. 3A, 3C-F and movie 6), whereas the sequential lipid and content release (2-step events, see Fig. 5E, F) was less frequent. All fusion-related activities were abrogated in the presence of C52L (n=972). We observed the same fusion phenotype for particles produced by pseudotyping the HIV-1 core with the full-length Env (Figs. 3A and S3E, F). Together, these results imply that, irrespective of the origin of the viral core or the presence of the cytoplasmic domain, HIV-1 Env-mediated content delivery into HeLa-derived target cells occurs through fusion with endosomes. Moreover, in CEMss cells, infectious HIV-1 also underwent partial fusion (lipid transfer) with the plasma membrane and complete fusion with endosomes (Fig. S4).
Endosomal fusion is delayed relative to lipid transfer at the cell surface
The kinetics of single HIV-1 pseudovirus fusion with the plasma membrane and with endosomes was determined by measuring the waiting time from raising the temperature to each lipid or content transfer event, respectively (Fig. 4A). Content release from endosomes started after a considerable delay (∼10 min), whereas lipid transfer proceeded without an apparent lag. The rates of surface and endosomal fusion differed markedly regardless of the virus tropism (JRFL vs. HXB2) and regardless of whether MLV core-based pseudoviruses or infectious HIV-1 viruses (Fig. 4C) were imaged. The delayed release of the viral content marker is consistent with the lag in the cytosolic BlaM delivery measured by the TB protocol (Fig. 1A). In fact, after re-normalization to correct for the shorter imaging time, the rates of viral content delivery measured by single-virus and BlaM assays were indistinguishable (Fig. S5). This finding validates the usage of the TB protocol for measuring the rate of formation of relatively small fusion pores and implies that complete pore dilation is not required for detecting the BlaM signal in the cytosol.
Figure 4.

Lipid mixing at the cell surface precedes the content release from endosomes. (A) The kinetics of JRFL (circles) and HXB2 (triangles) fusion with TZM-bl cells. Waiting times from the shift to 37°C to the point of lipid (red symbols) or content (green symbols) transfer were measured, rank ordered, and plotted as cumulative distributions of the fraction of fused viruses over time. (B) Comparative kinetics of partial fusion at the cell surface (red circles), endosomal fusion (green circles), and of sequential lipid and content transfer exhibited during the 2-step events (red and green triangles, respectively). The time intervals between sequential content (TC) and lipid (TL) transfer were ranked and plotted as cumulative distribution (crosses). (C) The kinetics of lipid and content mixing during fusion of infectious R9 HIV-1 viruses with TZM-bl cells.
HIV-1 fusion may proceed through a stable hemifusion-like intermediate
Even though the loss of a lipid marker during the 2-step fusion precludes unambiguous determination of the site of subsequent content release, the latter step appears to occur in endosomes. First, the rates of sequential lipid and content transfer for the 2-step fusion were statistically indistinguishable (p>0.180 and p>0.594, respectively) from the respective rates of separate surface and endosomal fusion events (Fig. 4B). Hence, by analogy to endosomal fusion, the content release through the 2-step events likely occurs in endosomes. This result also implies that the 2-step events are a subset of “regular” fusion events, in which lipid transfer occurred prior to virus uptake. Second, the pronounced delay between lipid and content transfer during the 2-step events (half-time of about 10 min, Fig. 4B) was sufficiently long to permit virus endocytosis (t1/2=13.5 min, Fig. 1A) prior to content release.
Third, by tracking the 2-step fusion events, we found that viruses tended to accelerate (>0.2 μm/sec) prior to or at the time of content release (Fig. 5A, B). These particles were thus judged to have entered an endocytic pathway and fused with endosomes. Only 1 out of 22 particles released approximately half of its content (Fig. 5C, D, arrow) without a significant prior displacement. However, the incomplete release of viral content shows that, even if this fusion pore was formed at the cell surface, it closed soon after opening (schematically shown by the thick line above 5D). Importantly, viral content release did not resume until after the onset of fast movement (double arrow and movie 7) associated with virus uptake. We also found that the content release during the 2-step events exhibited by infectious HIV-1 usually coincided with the fast particle movement (Fig. 5E, F).
These data imply that, even when HIV-1 establishes lipid continuity with the plasma membrane, it fails to form a fusion pore that permits the transfer of a content marker. This fusion phenotype is operationally defined as membrane hemifusion (Chernomordik and Kozlov, 2005). The temporal separation of lipid and content transfer events suggests that the 2-step fusion proceeds through a remarkably long-lived hemifusion intermediate. We cannot rule out the possibility that the lipid mixing at the cell surface represents a non-productive pathway, in which case, distinct Env trimers would be responsible for subsequent endosomal fusion. However, given the paucity of Env trimers in HIV-1 particles (Zhu et al., 2006), formation of more than one fusion complex per virion appears unlikely, suggesting that hemifusion is a bona fide intermediate of HIV entry.
Clathrin- and dynamin-dependent endocytosis is a prerequisite for HIV-1 fusion
To obtain further evidence that HIV-1 is internalized prior to fusion, we blocked clathrin- and caveolin-mediated endocytosis by pre-treating the TZM-bl cells with dynasore, a small molecule inhibitor of the dynamin GTP-ase activity that prevents the scission of clathrin-coated pits from the plasma membrane (Macia et al., 2006). At a concentration that blocked transferrin uptake (Fig. S7A), dynasore diminished virus internalization and strongly inhibited HIV-1 infection and fusion with TZM-bl and CEMss cells (Fig. 6A and B, respectively). As expected for viruses entering cells via a clathrin-dependent pathway (Sun et al., 2005), fusion and infection by VSV G pseudoviruses were suppressed by the drug. The diminished HIV-1 fusion was not caused by the down regulation of CD4 or CR expression, reduction of virus binding or the compromised ability of Env to promote fusion in the presence of dynasore (Fig. S7B-D).
Figure 6.
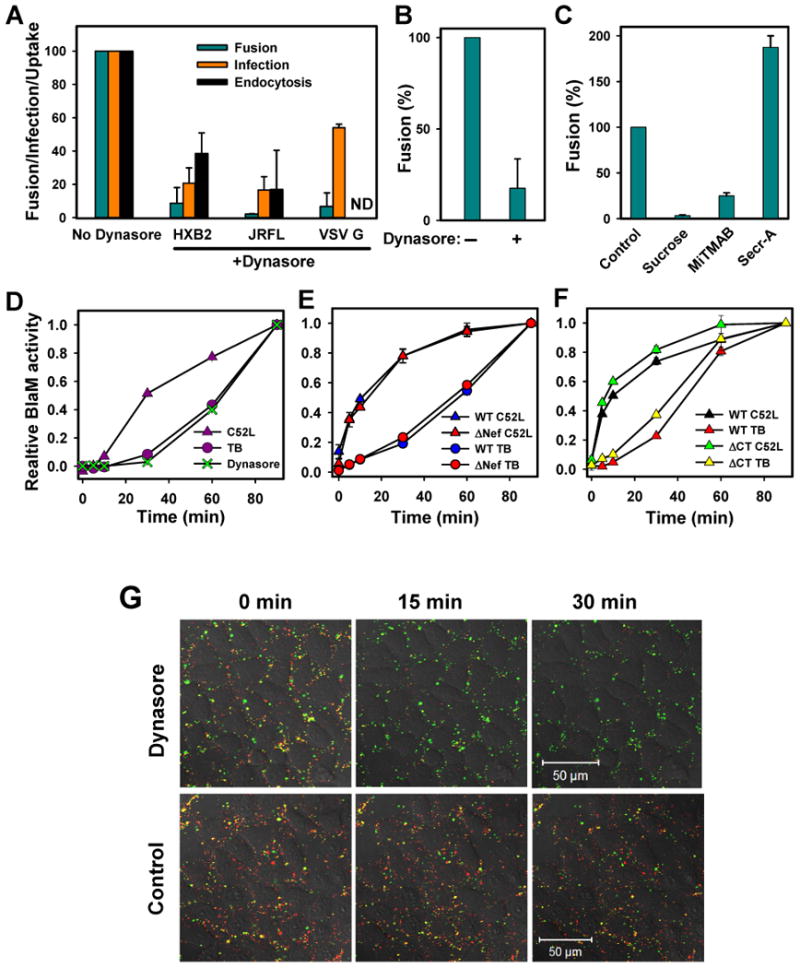
Blocking the dynamin function inhibits HIV-1 uptake, fusion and infection. (A) TZM-bl cells were pre-treated with 80 μM dynasore and allowed to bind viruses in the cold. Virus uptake after a 1 hr-incubation at 37°C was measured by the accumulation of intracellular p24 (black bars, n=6), as described in Supplemental Experimental Procedures. The extent of fusion was quantified by the BlaM assay (dark cyan bars, n=4), and single-cycle infection was measured by a β-Gal assay (orange bars, n=6); (B) Inhibition of HXB2 fusion with CEMss cells pretreated with dynasore. (C) Inhibition of HXB2 fusion in TZM-bl cells pretreated with 0.45 M sucrose, 80 μM MiTMAB, and 15 μM secramine A. (D) The kinetics of HXB2 escape from 80 μM dynasore added at indicated times of incubation at 37°C. The background fusion (∼20% of total) in the presence of dynasore was subtracted from the dynasore curve to ease the comparison with the C52L and TB curves. (E) Fusion of wild-type (WT) and Nef-deficient (ΔNef) viruses bearing HXB2 Env with TZM-bl cells. (F) Kinetics of fusion mediated by the wild-type and cytoplasmic tail-deleted (ΔCT) JRFL Env. (G) Pre-treatment of TZM-bl cells with dynasore blocks the HXB2 content (NC-GFP) transfer while permitting lipid mixing (disappearance of DiD) with the plasma membrane. The reduction in the number of GFP-labeled particles at a later time point is mainly due to virus detachment.
To control against possible adverse effects of dynasore, we assessed the effect of MiTMAB, a surface-active dynamin inhibitor that blocks its interactions with phospholipids (Quan et al., 2007). The diminished HIV-1 fusion in cells pre-treated with MiTMAB (Fig. 6C) supported the essential role of dynamin in virus entry. By comparison, the small molecule inhibitor of Cdc42 GTPase, secramine A (Pelish et al., 2006), augmented HIV-1 fusion. The specificity of small-molecule dynamin inhibitors was further verified by showing that HIV-1 fusion was suppressed in cells over-expressing the dominant-negative K44A mutant of dynamin (Damke et al., 1994) (Fig. S8). These results, along with the inhibition of HIV-1 fusion by a hypertonic medium (Fig. 6C) known to inhibit clathrin-mediated endocytosis, suggest that HIV-1 is internalized via a clathrin-dependent pathway prior to undergoing fusion.
To determine which step of HIV fusion is blocked by dynasore, we performed single virus imaging experiments. Viruses bound to cells pre-treated with the drug exhibited highly restricted mobility compared to control experiments (data not shown), consistent with inhibited virus endocytosis. Most importantly, dynasore abolished the viral content release but permitted the overwhelming majority of viruses to transfer their lipids to the plasma membrane (Fig. 6G). In the absence of virus uptake, partial fusion at the cell surface led to HIV inactivation, as evidenced by the lack of recovery of the BlaM signal after the removal of dynasore (Fig. S7E). These results confirm that HIV-1 is unable to fuse at the cell surface and that the fusion block occurs after the lipid mixing stage.
Endocytosis reduces the window of opportunity for the inhibitory peptide to bind to intermediate conformations of gp41
Clearance from the cell surface prior to the completion of fusion can protect HIV-1 against antibodies and inhibitors targeting Env epitopes that are exposed during the slow fusion reaction. To test this notion, we sought to reversibly slow down virus uptake while permitting its interactions with CD4 and CR. After unsuccessful attempts to reversibly arrest virus uptake using different intervention strategies, we have chosen to create a temperature-arrested stage (TAS) described before (Henderson and Hope, 2006; Mkrtchyan et al., 2005). After binding to cells in the cold, the viruses/cells were incubated for several hours at a temperature that minimized productive endocytosis and virus-cell fusion (for details, see the legend to Fig. S6). This intermediate stage was reversible, as fusion quickly ensued upon raising the temperature (Fig. S6A, C). At this stage, a fraction of fusion events became resistant to CD4- and CR-binding inhibitors, demonstrating the formation of ternary complexes, which in turn resulted in the exposure of the gp41 coiled coil regions targeted by inhibitory peptides, such as T20 and C34 (Eckert and Kim, 2001). We were thus able to control the exposure of pre-triggered gp41 to C34 by varying the interval between adding this peptide at the TAS and inducing endocytosis (and fusion) by shifting to 37°C. The longer exposure to C34 enhanced its inhibitory activity (Fig. S6B, D), consistent with the notion that endocytic entry of HIV-1 might attenuate the potency of this class of fusion inhibitors.
HIV-endosome fusion does not rely on an intact cytoskeleton but depends on dynamin activity
The reliance of HIV-1 fusion on endosomal pathways prompted us to examine the effects of actin- and microtubule-disrupting agents also known to interfere with endosomal trafficking and maturation (Bayer et al., 1998). Pre-treatment of cells with latrunculin A or nocodazole led to a modest reduction of the extent but not the rate of HIV-1 fusion (Fig. S9). This finding indicates that the BlaM signal is not critically affected by actin- and microtubule-dependent processes, in agreement with the previous work (Campbell et al., 2004).
Dynasore's ability to quickly block endocytosis ((Macia et al., 2006) and Fig. S7F) permitted us to perform time of addition experiments and delineate the step(s) of HIV-1 fusion sensitive to this compound. Dynasore was added after varied times of virus-cell incubation, and the drug-resistant fusion was compared to that obtained by the C52L and TB protocols. The loss of sensitivity to inhibitors of virus endocytosis is expected to occur at the time of its escape from C52L. Remarkably, however, HIV-1 escape from dynasore was markedly delayed and was indistinguishable from its escape from the TB (Fig. 6D). This finding indicates that dynamin plays a role both in HIV-1 uptake and in virus-endosome fusion.
Next, we asked whether dynamin-2 could augment Env-mediated fusion with endosomes through its specific binding to the accessory HIV-1 Nef protein (Pizzato et al., 2007) exposed to the cytosol as a result of fusion. However, the identical rates (Fig. 6E) and extents (data not shown) of fusion of wild-type and Nef-deficient viruses did not support this possibility. Thus dynamin appears to promote HIV-1 fusion indirectly, perhaps by interacting with effector protein(s) involved in a variety of cellular processes, such as cytokinesis, membrane trafficking, cell migration, and adhesion (Kim and Chang, 2006; Kruchten and McNiven, 2006; Peters et al., 2004). We also tested whether the unusually long cytoplasmic domain of gp41 is involved in pore formation or dilation either directly or by interacting with its cellular partners. Deletion of the cytoplasmic domain did not considerably alter the kinetics of viral escape from the TB relative to its escape from C52L (Fig. 6F), implying that this domain is not essential for HIV-1 fusion.
Discussion
Time-resolved imaging of single viruses and differential blocking of fusion by site-specific and universal inhibitors revealed that HIV-1 co-opts the endocytic machinery to enter into and fuse with target cells. By contrast, fusion with the plasma membrane did not progress beyond the lipid mixing step, suggesting that endosomal entry is the pathway that leads to productive infection. Endocytic entry offers several advantages, including the sheltering of HIV-1 from antibodies and inhibitors targeting intermediate conformations of Env during the unusually slow fusion reaction. Indeed we found that the delayed virus uptake increased the potency of the inhibitory C34 peptide. Thus, in order to efficiently block intracellular fusion events, the next generation of HIV entry inhibitors must be able to permeate the cell membrane.
The failure of HIV to fuse with the plasma membrane is in stark contrast to cell-cell fusion mediated by Env glycoproteins of this and other pH-independent viruses. While the basis for this discrepancy is unclear, the much larger number of Env involved in cell-cell contact compared to a few Env responsible for virus entry could increase the likelihood of fusion at the cell surface. Another manifestation of differences between these experimental systems is the prolonged lag between CD4/CR-mediated HIV-1 uptake and endosomal fusion. By comparison, the formation of ternary Env-CD4-CR complexes abrogated the lag before cell-cell fusion (Melikyan et al., 2000; Mkrtchyan et al., 2005). The delayed endosomal fusion of HIV-1 is indicative of a rate-limiting step downstream of coreceptor-dependent steps and downstream of a hemifusion-like intermediate. Slow pore enlargement is unlikely to contribute to this delay because both single virus imaging and the BlaM assay appear to detect relatively small pores. It is thus possible that the formation of higher-order Env oligomers and/or gp41 folding into the 6-helix bundle are rate-limiting for HIV-cell fusion. Alternatively, the lag before fusion could reflect the time required for HIV-1 delivery into permissive intracellular compartments, such as late endosomes.
Accumulating evidence suggests that entry of viruses other than HIV-1 by direct fusion with the plasma membrane is also disfavored. For instance, pH-independent MLV and respiratory syncytial virus appear to utilize an endocytic pathway for infection (Beer et al., 2005; Katen et al., 2001; Kolokoltsov et al., 2007). Infection by several low pH-dependent viruses can be hindered when fusion with a plasma membrane is forced by acidic pH (Marsh and Bron, 1997; Matlin et al., 1982; Mothes, 2000). The failure of HIV-1, aMLV and SFV to progress beyond the lipid mixing step at the cell surface (Figs. 2, 3 and S3) shows, for the first time, that the block for pH-dependent and pH-independent infection is at the stage of formation and/or dilation of a fusion pore. The lack of complete fusion at the cell surface could be due to restrictions imposed by the cortical actin or other factors present in or around the plasma membrane. However, the modest effect of actin depolymerization on viral content delivery (Fig. S9) does not support this notion. An alternative possibility discussed below is that viruses rely on yet unidentified endosomal factors to promote complete fusion.
Our data revealed a novel role for dynamins in HIV-1 content release from endosomes. Why would HIV-1 need cellular factors to promote fusion? Several lines of evidence suggest that the formation and enlargement of a fusion pore are the most energy-intensive steps (reviewed in (Melikyan, 2008)) which require the concerted action of several viral proteins. Considering the low number of Env per virion (Zhu et al., 2006), HIV-1 may not be able to sustain a fusion pore on its own without cellular partners. The ability of dynamin to regulate actin remodeling and/or to associate with membrane-bending proteins (Kruchten and McNiven, 2006) could provide an additional driving force to expand pores and permit the release of the HIV-1 core. It is thus possible that cellular factors involved in membrane trafficking are responsible for the strong virus' preference for endocytic entry.
Experimental Procedures
BlaM assay for virus-cell fusion
TZM-bl cells (4·104 cells/well) were grown overnight in 96-well plates in a phenol red-free growth medium. Viruses were added to cells at MOI 0.7-1.0 and centrifuged at 2095g, 4°C for 30 min. The cells were washed with a cold medium to remove free viruses, and fusion was initiated by shifting to 37°C. After an indicated time at 37°C, fusion was stopped by adding C52L (1 µM) or other fusion inhibitors. All samples were maintained at 37°C for a total of 90 min (unless indicated otherwise), chilled by briefly placing on ice, loaded with the CCF2-AM substrate (GeneBLAzer in vivo Detection Kit, Invitrogen), and incubated overnight at 13.5°C (or as indicated). In the temperature block protocol, cells were placed on ice until the end of the experiment and then loaded with the BlaM substrate. Fusion of HIV-1 pseudotyped with VSV G was carried out as described above, but stopped at indicated times by either treating cells with pronase, placing cells on ice (TB) or by adding 50 mM NH4Cl. The temperature chase experiments were carried out by introducing C52L peptide after 20 min of incubation, as in the standard fusion protocol. The BlaM signal was then chased by placing cells on ice either immediately or at the indicated times of incubation at 37°C in the presence of C52L. The BlaM activity was quantified using Synergy HT fluorescence plate reader (Bio-Tek Instr., Germany). The extent of virus-cell fusion was determined from the ratio of blue (440-480 nm) and green (518-538 nm) emission upon exciting the cells at 405-415 nm. HIV-1 fusion with CEMss cells was carried out by re-suspending the cells in media containing HXB2 pseudoviruses (MOI 1-1.5) and centrifuging at 2095g, 4°C for 30 min. The free viruses were washed off, and fusion was initiated by shifting to 37°C and stopped after indicated times either by adding C52L peptide or by placing cells on ice. The cells were then loaded with the CCF2-AM substrate, transferred into a 96-well plate (1·105 cells/well), and the BlaM activity was determined as described above.
Single particle imaging and analysis
Viruses were centrifuged onto TZM-bl cells cultured on a cover glass (2095g for 1 hr at 12°C). The cells were washed to remove unbound viruses and transferred into an imaging chamber. Virus-cell fusion was triggered by quickly and locally raising the temperature to 37°C using a home-built temperature-jump setup (Melikyan et al., 2000) and visualized using a Zeiss LSM 510 Meta confocal microscope. Unless noted otherwise, samples were simultaneously excited at 488 and 633 nm, and the emitted light was collected by a C-Apo 40x/1.2 water immersion or a Neofluar 40x/1.3 oil immersion objective, split and passed through 505-550 nm band-pass and 650 nm long-pass filters. To minimize photobleaching, only 3-4 Z-stacks spaced by 2.5 μm were acquired every 6-7 sec for 35-40 min. Single particle tracking was performed using Volocity image analysis software (Improvision, Perkin Elmer). Briefly, the total number of cell-associated viruses containing detectable amounts of DiD and GFP-based content marker was determined by identifying contiguous pixels with fluorescence intensity at least 3-fold greater than background. The waiting times for fusion were estimated as the time interval from raising the temperature to 37°C to the point when the signal from either the membrane or content marker dropped to the background level. Three-dimensional tracking of particles over time was performed by adjusting the intensity threshold and the maximal particle displacement between consecutive frames.
Acknowledgments
The authors are grateful to Drs. C. Aiken, M. Alizon, J. Binley, J. Cunningham, M. Kielian, T. Kirchhausen, G. Lewis, M. Lu, W. Mothes, J. Strizki, L. Wang and J. Young, as well as the NIH AIDS Research and Reference Reagent Program for reagents, expression vectors, and cell lines. We thank Dr. M. Reitz for his help in designing the MA-GFP-CA construct, and Drs. L. Chernomordik and R. Dutch for stimulating discussions. This work was supported by NIH R01 GM054787 and AI053668 grants to GM. YK was partially supported by the SDG from the American Heart Association.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors have no conflict of interest to declare.
References
- Aiken C. Pseudotyping human immunodeficiency virus type 1 (HIV-1) by the glycoprotein of vesicular stomatitis virus targets HIV-1 entry to an endocytic pathway and suppresses both the requirement for Nef and the sensitivity to cyclosporin A. J Virol. 1997;71:5871–5877. doi: 10.1128/jvi.71.8.5871-5877.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer N, Schober D, Prchla E, Murphy RF, Blaas D, Fuchs R. Effect of bafilomycin A1 and nocodazole on endocytic transport in HeLa cells: implications for viral uncoating and infection. J Virol. 1998;72:9645–9655. doi: 10.1128/jvi.72.12.9645-9655.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beer C, Andersen DS, Rojek A, Pedersen L. Caveola-dependent endocytic entry of amphotropic murine leukemia virus. J Virol. 2005;79:10776–10787. doi: 10.1128/JVI.79.16.10776-10787.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt SM, Mariani R, Holland AU, Hope TJ, Landau NR. Association of chemokine-mediated block to HIV entry with coreceptor internalization. J Biol Chem. 2002;277:17291–17299. doi: 10.1074/jbc.M108232200. [DOI] [PubMed] [Google Scholar]
- Campbell EM, Nunez R, Hope TJ. Disruption of the actin cytoskeleton can complement the ability of Nef to enhance human immunodeficiency virus type 1 infectivity. J Virol. 2004;78:5745–5755. doi: 10.1128/JVI.78.11.5745-5755.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavrois M, De Noronha C, Greene WC. A sensitive and specific enzyme-based assay detecting HIV-1 virion fusion in primary T lymphocytes. Nat Biotechnol. 2002;20:1151–1154. doi: 10.1038/nbt745. [DOI] [PubMed] [Google Scholar]
- Cavrois M, Neidleman J, Yonemoto W, Fenard D, Greene WC. HIV-1 virion fusion assay: uncoating not required and no effect of Nef on fusion. Virology. 2004;328:36–44. doi: 10.1016/j.virol.2004.07.015. [DOI] [PubMed] [Google Scholar]
- Chernomordik LV, Kozlov MM. Membrane hemifusion: crossing a chasm in two leaps. Cell. 2005;123:375–382. doi: 10.1016/j.cell.2005.10.015. [DOI] [PubMed] [Google Scholar]
- Daecke J, Fackler OT, Dittmar MT, Krausslich HG. Involvement of clathrin-mediated endocytosis in human immunodeficiency virus type 1 entry. J Virol. 2005;79:1581–1594. doi: 10.1128/JVI.79.3.1581-1594.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damke H, Baba T, Warnock DE, Schmid SL. Induction of mutant dynamin specifically blocks endocytic coated vesicle formation. J Cell Biol. 1994;127:915–934. doi: 10.1083/jcb.127.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y, Zheng Q, Ketas TJ, Moore JP, Lu M. Protein design of a bacterially expressed HIV-1 gp41 fusion inhibitor. Biochemistry. 2007;46:4360–4369. doi: 10.1021/bi7001289. [DOI] [PubMed] [Google Scholar]
- Doms RW, Trono D. The plasma membrane as a combat zone in the HIV battlefield. Genes Dev. 2000;14:2677–2688. doi: 10.1101/gad.833300. [DOI] [PubMed] [Google Scholar]
- Eckert DM, Kim PS. Mechanisms of Viral Membrane Fusion and Its Inhibition. Annu Rev Biochem. 2001;70:777–810. doi: 10.1146/annurev.biochem.70.1.777. [DOI] [PubMed] [Google Scholar]
- Fredericksen BL, Wei BL, Yao J, Luo T, Garcia JV. Inhibition of endosomal/lysosomal degradation increases the infectivity of human immunodeficiency virus. J Virol. 2002;76:11440–11446. doi: 10.1128/JVI.76.22.11440-11446.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey S, Marsh M, Gunther S, Pelchen-Matthews A, Stephens P, Ortlepp S, Stegmann T. Temperature dependence of cell-cell fusion induced by the envelope glycoprotein of human immunodeficiency virus type 1. J Virol. 1995;69:1462–1472. doi: 10.1128/jvi.69.3.1462-1472.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallay P, Hope T, Chin D, Trono D. HIV-1 infection of nondividing cells through the recognition of integrase by the importin/karyopherin pathway. Proc Natl Acad Sci U S A. 1997;94:9825–9830. doi: 10.1073/pnas.94.18.9825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson HI, Hope TJ. The temperature arrested intermediate of virus-cell fusion is a functional step in HIV infection. Virol J. 2006;3:36. doi: 10.1186/1743-422X-3-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katen LJ, Januszeski MM, Anderson WF, Hasenkrug KJ, Evans LH. Infectious entry by amphotropic as well as ecotropic murine leukemia viruses occurs through an endocytic pathway. J Virol. 2001;75:5018–5026. doi: 10.1128/JVI.75.11.5018-5026.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Chang S. Ever-expanding network of dynamin-interacting proteins. Mol Neurobiol. 2006;34:129–136. doi: 10.1385/MN:34:2:129. [DOI] [PubMed] [Google Scholar]
- Kolokoltsov AA, Deniger D, Fleming EH, Roberts NJ, Jr, Karpilow JM, Davey RA. Small interfering RNA profiling reveals key role of clathrin-mediated endocytosis and early endosome formation for infection by respiratory syncytial virus. J Virol. 2007;81:7786–7800. doi: 10.1128/JVI.02780-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruchten AE, McNiven MA. Dynamin as a mover and pincher during cell migration and invasion. J Cell Sci. 2006;119:1683–1690. doi: 10.1242/jcs.02963. [DOI] [PubMed] [Google Scholar]
- Lakadamyali M, Rust MJ, Zhuang X. Endocytosis of influenza viruses. Microbes Infect. 2004;6:929–936. doi: 10.1016/j.micinf.2004.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell. 2006;10:839–850. doi: 10.1016/j.devcel.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Maddon PJ, McDougal JS, Clapham PR, Dalgleish AG, Jamal S, Weiss RA, Axel R. HIV infection does not require endocytosis of its receptor, CD4. Cell. 1988;54:865–874. doi: 10.1016/s0092-8674(88)91241-x. [DOI] [PubMed] [Google Scholar]
- Marechal V, Prevost MC, Petit C, Perret E, Heard JM, Schwartz O. Human immunodeficiency virus type 1 entry into macrophages mediated by macropinocytosis. J Virol. 2001;75:11166–11177. doi: 10.1128/JVI.75.22.11166-11177.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markosyan RM, Cohen FS, Melikyan GB. Time-resolved imaging of HIV-1 Env-mediated lipid and content mixing between a single virion and cell membrane. Mol Biol Cell. 2005;16:5502–5513. doi: 10.1091/mbc.E05-06-0496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh M, Bron R. SFV infection in CHO cells: cell-type specific restrictions to productive virus entry at the cell surface. J Cell Sci. 1997;110(Pt 1):95–103. doi: 10.1242/jcs.110.1.95. [DOI] [PubMed] [Google Scholar]
- Marsh M, Helenius A. Virus entry: open sesame. Cell. 2006;124:729–740. doi: 10.1016/j.cell.2006.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matlin KS, Reggio H, Helenius A, Simons K. Pathway of vesicular stomatitis virus entry leading to infection. J Mol Biol. 1982;156:609–631. doi: 10.1016/0022-2836(82)90269-8. [DOI] [PubMed] [Google Scholar]
- McClure MO, Marsh M, Weiss RA. Human immunodeficiency virus infection of CD4-bearing cells occurs by a pH-independent mechanism. Embo J. 1988;7:513–518. doi: 10.1002/j.1460-2075.1988.tb02839.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikyan GB. Common principles and intermediates of viral protein-mediated fusion: the HIV-1 paradigm. Retrovirology. 2008;5:111. doi: 10.1186/1742-4690-5-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikyan GB, Markosyan RM, Hemmati H, Delmedico MK, Lambert DM, Cohen FS. Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J Cell Biol. 2000;151:413–424. doi: 10.1083/jcb.151.2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mkrtchyan SR, Markosyan RM, Eadon MT, Moore JP, Melikyan GB, Cohen FS. Ternary complex formation of human immunodeficiency virus type 1 Env, CD4, and chemokine receptor captured as an intermediate of membrane fusion. J Virol. 2005;79:11161–11169. doi: 10.1128/JVI.79.17.11161-11169.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mothes W, Boerger AL, Narayan S, Cunningham JM, Young JAT. Retroviral entry mediated by receptor priming and low pH triggering of an envelope glycoprotein. Cell. 2000;103:679–689. doi: 10.1016/s0092-8674(00)00170-7. [DOI] [PubMed] [Google Scholar]
- Pauza CD, Price TM. Human immunodeficiency virus infection of T cells and monocytes proceeds via receptor-mediated endocytosis. J Cell Biol. 1988;107:959–968. doi: 10.1083/jcb.107.3.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelchen-Matthews A, Clapham P, Marsh M. Role of CD4 endocytosis in human immunodeficiency virus infection. J Virol. 1995;69:8164–8168. doi: 10.1128/jvi.69.12.8164-8168.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelish HE, Peterson JR, Salvarezza SB, Rodriguez-Boulan E, Chen JL, Stamnes M, Macia E, Feng Y, Shair MD, Kirchhausen T. Secramine inhibits Cdc42-dependent functions in cells and Cdc42 activation in vitro. Nat Chem Biol. 2006;2:39–46. doi: 10.1038/nchembio751. [DOI] [PubMed] [Google Scholar]
- Peters C, Baars TL, Buhler S, Mayer A. Mutual control of membrane fission and fusion proteins. Cell. 2004;119:667–678. doi: 10.1016/j.cell.2004.11.023. [DOI] [PubMed] [Google Scholar]
- Pizzato M, Helander A, Popova E, Calistri A, Zamborlini A, Palu G, Gottlinger HG. Dynamin 2 is required for the enhancement of HIV-1 infectivity by Nef. Proc Natl Acad Sci U S A. 2007;104:6812–6817. doi: 10.1073/pnas.0607622104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan A, McGeachie AB, Keating DJ, van Dam EM, Rusak J, Chau N, Malladi CS, Chen C, McCluskey A, Cousin MA, Robinson PJ. Myristyl trimethyl ammonium bromide and octadecyl trimethyl ammonium bromide are surface-active small molecule dynamin inhibitors that block endocytosis mediated by dynamin I or dynamin II. Mol Pharmacol. 2007;72:1425–1439. doi: 10.1124/mol.107.034207. [DOI] [PubMed] [Google Scholar]
- Schaeffer E, Soros VB, Greene WC. Compensatory link between fusion and endocytosis of human immunodeficiency virus type 1 in human CD4 T lymphocytes. J Virol. 2004;78:1375–1383. doi: 10.1128/JVI.78.3.1375-1383.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein BS, Gowda SD, Lifson JD, Penhallow RC, Bensch KG, Engleman EG. pH-independent HIV entry into CD4-positive T cells via virus envelope fusion to the plasma membrane. Cell. 1987;49:659–668. doi: 10.1016/0092-8674(87)90542-3. [DOI] [PubMed] [Google Scholar]
- Sun X, Yau VK, Briggs BJ, Whittaker GR. Role of clathrin-mediated endocytosis during vesicular stomatitis virus entry into host cells. Virology. 2005;338:53–60. doi: 10.1016/j.virol.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Wei BL, Denton PW, O'Neill E, Luo T, Foster JL, Garcia JV. Inhibition of lysosome and proteasome function enhances human immunodeficiency virus type 1 infection. J Virol. 2005;79:5705–5712. doi: 10.1128/JVI.79.9.5705-5712.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, Saag MS, Wu X, Shaw GM, Kappes JC. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob Agents Chemother. 2002;46:1896–1905. doi: 10.1128/AAC.46.6.1896-1905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder A, Yu D, Dong L, Iyer SR, Xu X, Kelly J, Liu J, Wang W, Vorster PJ, Agulto L, et al. HIV envelope-CXCR4 signaling activates cofilin to overcome cortical actin restriction in resting CD4 T cells. Cell. 2008;134:782–792. doi: 10.1016/j.cell.2008.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu P, Liu J, Bess J, Jr, Chertova E, Lifson JD, Grise H, Ofek GA, Taylor KA, Roux KH. Distribution and three-dimensional structure of AIDS virus envelope spikes. Nature. 2006;441:847–852. doi: 10.1038/nature04817. [DOI] [PubMed] [Google Scholar]