

Abstract
Mitochondrial Ca2+ accumulation is a tightly controlled process, in turn regulating functions as diverse as aerobic metabolism and induction of cell death. The link between Ca2+ (dys)regulation, mitochondria and cellular derangement is particularly evident in neurodegenerative disorders, in which genetic models and environmental factors allowed to identify common traits in the pathogenic routes. We will here summarize: i) the current view of mechanisms and functions of mitochondrial Ca2+ homeostasis, ii) the basic principles of organelle Ca2+ transport, iii) the role of Ca2+ in neuronal cell death, and iv) the new information on the pathogenesis of Alzheimer's, Huntington's and Parkinson's diseases, highlighting the role of Ca2+ and mitochondria.
Keywords: Mitochondria, Calcium, Neurodegenerative disease, Alzheimer's disease, Parkinson's disease, Huntington's disease
1. Mitochondria in calcium signalling
The notion that mitochondria are active players in cellular calcium homeostasis dates back to the demonstration of the chemiosmotic theory, based on the concept of a major proton electrochemical gradient that could drive the rapid accumulation of cations across the ion-impermeant mitochondrial inner membrane. Such a notion was corroborated by the direct measurement of Ca2+ uptake by isolated mitochondria, and the functional, albeit not molecular, elucidation of the transporters [1].
However, the experiments that in the 80s drove massive interest into calcium as a ubiquitous second messenger also led to the gradual decline in the attention on mitochondrial Ca2+ homeostasis. On the one hand, it appeared clear that the endoplasmic reticulum (ER), through resident Ca2+ channels (those sensitive to inositol 1,4,5 trisphosphate, IP3R, and to the plant alkaloid ryanodine, RyR) acts as the intracellular pool of Ca2+ mobilized upon cell stimulation. On the other, the development of accurate fluorescent indicators for the measurement of Ca2+ concentration in living cells showed that cytosolic Ca2+ concentration fluctuates between approx. 0.1 μM at rest to 2–3 μM at the peak of the rise elicited by the opening of plasma membrane or ER Ca2+ channels. Under those conditions, the low affinity of the mitochondrial Ca2+ transporters should allow little Ca2+ uptake into the organelle. Thus, the prevalent notion was that mitochondrial Ca2+ accumulation is negligible in physiological conditions, and could become relevant only upon massive Ca2+ overload, that could take place in severe cellular dysfunction.
This situation was completely reversed when tools were developed that allowed the selective measurement of mitochondrial Ca2+ concentration ([Ca2+]m) in living cells. Targeting to mitochondria the Ca2+-sensitive photoprotein aequorin [2] demonstrated that a rapid [Ca2+]m peak, reaching values well above those of the bulk cytosol, parallels the [Ca2+] rise evoked in the cytoplasm by cell stimulation [3]. Similar conclusions could be reached also with fluorescent indicators, such as the positively charged Ca2+ indicator rhod 2 (that accumulates within the organelle) [4] and the more recently developed GFP-based fluorescent indicators [5]. With the latter probes, endowed with a much stronger signal than the photoprotein, single-cell imaging of organelle Ca2+ could be carried out [6]. With these tools in hands, not only the notion was confirmed that mitochondria promptly respond to cytosolic [Ca2+] rises, but also that the [Ca2+]c oscillations, the typical response to agonists of many cell types, are paralleled by rapid spiking of [Ca2+]m, thus providing a frequency-mediated signal specifically decoded within the mitochondria, as clearly shown in hepatocytes [7], cardiomyocytes [8], and HeLa cells [9]. The apparent discrepancy between the promptness and amplitude of the mitochondrial Ca2+ response and the low affinity of the organelle transporters was reconciled by the demonstration that mitochondria are in close contact with the source of the cytosolic Ca2+ rise (i.e. the ER via IP3Rs and RyRs and the plasma membrane via a wide variety of voltage- and agonist-sensitive Ca2+ channels). Thus, upon cell stimulation they are exposed to microdomains of high [Ca2+] that greatly exceed the values measured in the cytosol and well match the affinity of mitochondrial Ca2+ transporters [10].
As soon as the concept was established that a Ca2+ rise in the cytosol is paralleled by a cycle of mitochondrial Ca2+ uptake, and subsequent release (through the pathways that will be briefly described later), the identification of the functional significance of this process became a primary goal. Also in this task, the fine biochemical work carried out in the 70s provided a hypothesis to test: three key metabolic enzymes (the pyruvate, ketoglutarate and isocitrate dehydrogenases) were shown to be activated by Ca2+ by different mechanisms: in the case of pyruvate dehydrogenase through a Ca2+-dependent dephosphorylation step, in the latter two cases through the direct binding of Ca2+ to the enzyme complex [11]. Recently, also some metabolite transporters were shown to be regulated by Ca2+ and participate in the enhancement of aerobic metabolism upon cell stimulation [12]. Thus, an obvious role for mitochondrial Ca2+ accumulation could be inferred, i.e. that of rapidly upregulating mitochondrial ATP production in stimulated cells. The possibility of directly monitoring, in parallel, Ca2+ and ATP levels within mitochondria and in the cytosol proved that this is indeed the case [13]. Interestingly, this route for controlling mitochondrial metabolic output proved to be affected in mitochondrial genetic disorders. In cybrids harboring the tRNAlys mutation of MERRF (myoclonic epilepsy with ragged-red fibers), mitochondrial Ca2+ responses were reduced, and, accordingly, ATP production upon cell stimulation, and the pharmacological correction of the Ca2+ alteration also restored the metabolic dysfunction [14,15].
In addition to the function of metabolic coupling, mitochondrial Ca2+ accumulation was shown to underlie a role for these organelles in shaping the spatio/temporal patterning of cytosolic Ca2+ rises. Mitochondria, distinctly from cytosolic proteins, are highly sophisticated, “tunable” buffers that vary their activity in different phases and functional states of the cell; indeed, their number, shape, distribution [16] and most likely their responsiveness to Ca2+ [17] are controlled by converging signalling pathways. This Ca2+ buffering activity influences cytosolic Ca2+ signals in two conceptually different ways, i.e., 1) by acting as high-capacity sinks placed on the way of a propagating Ca2+ wave and 2) by clearing Ca2+ in restricted microdomains (such as the microenvironment of a Ca2+channel). In the first case, spatial clusters of mitochondria have been demonstrated to isolate functionally distinct domains of polarized cells, namely, a mitochondrial “firewall” was shown to prevent the spread of Ca2+ signals from the apical (secretory) region of pancreatic acinar cell from the basolateral region, containing the nucleus [18]. Similarly, neuronal mitochondria have been shown to buffer [Ca2+] increases in defined cellular regions, e.g., the presynaptic motoneuron ending [19]. As to the second case, a thoroughly investigated example is the regulation of Ca2+ release through IP3Rs. In Xenopus oocytes, the mitochondria energization state (and thus the capacity to accumulate Ca2+) was shown to modify the propagating Ca2+ waves induced by IP3 [20]. In permeabilized blowfly salivary glands, it was observed that perfusion of IP3 induced ER [Ca2+] oscillations, the frequency of which increased with the dose of IP3. Such an effect was observed only upon energization of mitochondria, implying a primary role of these organelles in regulating Ca2+ microdomains in the proximity of IP3Rs and thus the oscillatory pace of stimulated cells [21]. In mammals, this effect has been seen in many cell systems, including hepatocytes, HeLa cells, astrocytes, and BHK cells. As to the cellular consequence, very different effects were observed given the bell-shaped sensitivity of IP3Rs to Ca2+ concentration on the cytosolic side. In astrocytes and hepatocytes, cytosolic excitability appeared enhanced when mitochondrial Ca2+ uptake was inhibited, indicating that mitochondrial clearance of the Ca2+ microdomain reduced the positive Ca2+ feedback on the IP3R and/or buffered substantial Ca2+ loads [22]. Conversely, in BHK cells, inhibition of mitochondrial Ca2+ uptake resulted in reduction of ER Ca2+ release [23], thus indicating that mitochondria prevent the Ca2+-dependent inactivation of the channel.
The interest in the process of mitochondrial Ca2+ homeostasis dramatically increased when it became apparent that also cell death is causally linked to organelle Ca2+ loading. On the one hand, it was clear that cellular Ca2+ overload, such as that caused by hyperstimulation of ionotropic glutamate receptors, leads to Ca2+ cycling across the mitochondrial membranes, collapse of the proton gradient and bioenergetic catastrophe, thus leading to cell death by necrosis, as discussed in more detail later in this review. On the other hand, Ca2+ proved to sensitize cells to apoptotic challenges, acting on the mitochondrial checkpoint. This notion, subsequently confirmed by the study of other anti- and pro-apoptotic proteins, emerged from the analysis of the effect of Bcl-2 on Ca2+ signalling. We refer to other specifically focused reviews for a detailed coverage of this topic [24]. Briefly, we here summarize that Bcl-2, by partially emptying the ER Ca2+ store, reduces the release from this organelle and the loading of mitochondria. As a consequence, the efficacy of apoptotic challenges in opening the permeability transition pore (PTP), causing mitochondrial morphological alterations and releasing caspase cofactors, such as cytochrome c, is greatly reduced [25].
2. The basics of mitochondrial Ca2+ transport
For entering the mitochondrial matrix, Ca2+ needs to cross two lipid bilayers, the outer and inner mitochondrial membranes (Fig. 1). The outer mitochondrial membrane (OMM) is permeable to ions and small proteins (MW<10 kDa) due to the abundance of a large conductance channel, known as mitochondrial porin or voltage-dependent anion channel (VDAC). It should be noted, however, that the channel appears to be gated in vivo, and permeability is controlled by ATP and other regulatory factors [26]. Ca2+ diffusion through the OMM was thus traditionally considered not to be a limiting factor in mitochondrial Ca2+ uptake. Recent data showed that the availability and selective placement of VDAC channels at ER/mitochondria contact sites facilitate mitochondrial Ca2+ accumulation, in keeping with the idea that the latter process requires the fast and efficient transfer of Ca2+ microdomains from the mouth of the Ca2+ channels located in neighbouring ER or plasma membranes to the transporters of the ion-impermeant inner membrane (IMM) [27]. The IMM is an ion-impermeable membrane, with a much larger extension of the OMM (and consequent formation of foldings into the internal space, known as cristae). The activity of respiratory chain complexes allows the translocation of H+ in the space between the two membranes, which consequently generates an electrochemical gradient (ΔμH) composed of a chemical (ΔpH) and electrical (ΔψH) component. In mitochondria, most of the ΔμH established by the respiratory chain is supposed to be in the form of ΔψH (around −180 mV), which provides a huge driving force for Ca2+ entry into the organelle. Indeed, collapse of the ΔψH by protonophores, such as p-[trifluoromethoxyl]-phenyl-hydrazone (FCCP), abolishes mitochondrial Ca2+ uptake.
Fig. 1.
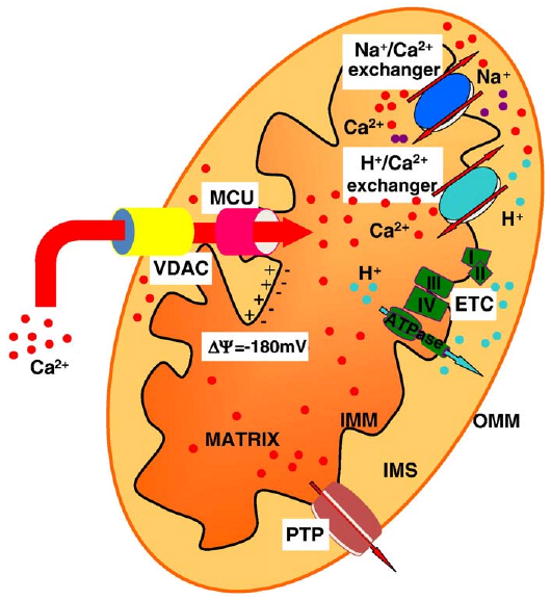
Schematic map of mitochondrial Ca2+ transporters. Mitochondria accumulate Ca2+ in the matrix via an electrogenic Ca2+ uniporter (MCU) that acts to equilibrate Ca2+ according the electrochemical gradient generated by the respiratory chain (ETC). Voltage Dependent Anion Channel (VDAC) controls the Ca2+ diffusion through the outer mitochondrial membrane (OMM), thus facilitating mitochondrial Ca2+ accumulation. As to the efflux pathways, a Na+/Ca2+ and a H+/Ca2+ exchangers have been shown to operate. The permeability transition pore (PTP) opening plays different roles: its brief opening could allow rapid Ca2+ release, but its long-lasting openings (potentiated by apoptotic stimuli and Ca2+ itself) could trigger cell death process. IMS, intermembrane space; IMM, inner mitochondrial membrane.
Mitochondrial Ca2+ traffic takes place essentially through two pathways: i) an electrogenic pathway, the mitochondrial Ca2+ uniporter (MCU), corresponding to the channel activity demonstrated by Clapham et al. [28] that acts to equilibrate Ca2+ with its electrochemical gradient, and thus accumulates the cation into the matrix; ii) two exchangers (with H+ and Na+, mostly expressed in nonexcitable and excitable cells, respectively), that utilize the electrochemical gradient of the monovalent cations to prevent the attainment of electrical equilibrium (that would imply, for a mitochondrial membrane potential, Δψm, of −180 mV and a cytosolic Ca2+ concentration of 0.1 μM, accumulation of Ca2+ into the matrix up to 0.1 M).
Numerous works has given the biochemical properties of these two pathways, and it is possible to summarize as it follows:
Given the huge driving force for accumulation, studies in isolated organelles with clamped ΔΨ (by establishing K+ diffusion potentials with valinomycin in non-respiring mitochondria) allowed to estimate a Vmax>1400 nmol Ca2+ (mg protein)−1 min−1 and an apparent Km<10 μM in sucrose-based media [29]. Also in isolated mitochondria, a number of inhibitors were identified. Ruthenium compounds (typically Ruthenium Red, RR), represent a class of non-competitive inhibitors, but unfortunately their poor permeability across cellular membranes has made it of little use in studies in intact cells. A second class of inhibitors is divalent cations that are themselves transported by the uniporter (e.g. Sr2+, Mn2+, Ba2+ and lanthanides).
As to the efflux pathways, studies in isolated organelles allowed to estimate their properties. The mitochondrial Na+/Ca2+ exchanger (mNCX) has a Vmax ranging between 2.6 (liver) and 18 nmol Ca2+ (mg protein)−1 min−1 (heart). The dependence on Na+ is sigmoidal, with typical Km values of about 8–10 mM Na+. Ca2+ efflux is inhibited by Sr2+, Ba2+, Mg2+ or Mn2+, and by a variety of compounds of pharmacological interest such as diltiazem, verapamil and other blockers of the voltage-dependent calcium channels, and more specifically by CGP37157 [30]. As to the H+/Ca2+ exchanger (mHCX), (1) it saturates at Ca2+ loads of 25 nmol (mg protein)−1; (2) its Vmax is not influenced by the concentration of inorganic phosphate and does not exceed a rate of 1.2 nmol Ca2+ (mg protein)−1 min−1; and (3) it extrudes Ca2+ against a gradient that is much higher than thermodynamically permissible for an electroneutral H+/Ca2+ exchanger [31]. Thus, either Ca2+ efflux occurs via a nH+/Ca2+ exchanger with n>2, or it has an active component. Accordingly, Ca2+ efflux is inhibited rather than stimulated by small depolarizations [32].
2.1. The permeability transition pore
This high-conductance channel (PTP) mediating mitochondrial swelling, postulated on the basis of experimental evidence dating back more than 40 years [33], has attracted enormous interest in the last decade, when its role in mitochondrial dysfunction and mitochondria-dependent cell routes has become clear. It is a high-conductance, non-selective channel that exhibits a prominent dependence on matrix Ca2+ and is inhibited by Cyclosporin A (CsA). Reversible PTP openings have been resolved both in individual isolated mitochondria [34] and in intact cells [35]. PTP could play a role in various conditions. Long-lasting openings, triggered by apoptotic challenges and potentiated by Ca2+-mediated cellular signals [36] cause morphological transitions of mitochondria underlying the release of caspase cofactors into the cytosol, and the initiation of the cell death process. Conversely, given the large size and lack of selectivity of the PTP (providing charge compensation within the channel and allowing maximal Ca2+ flux at zero potential) brief PTP openings could allow at least in principle rapid Ca2+ release from the organelle [37].
3. Calcium routes to neurodegeneration
It was hypothesized already in the 70s that prolonged stimulation of N-methyl-d-aspartate (NMDA) ionotropic glutamate receptors can induce massive cell death in the brain (excitotoxicity), by causing Ca2+ and Na+ overload in post-synaptic neurons [38]. Excitotoxicity plays a central role in promoting cell death during neuronal ischemia and substantial work has been placed in clarifying the different phases of the process, and their reversibility, and the potential pharmacological targets. It is now accepted that a primary Ca2+ increase occurs, as a consequence of direct entry through NMDA receptors, but also following depolarization, and hence opening of voltage-gated Ca2+ channels, release from internal stores and reversal of the plasma membrane Na+/Ca2+ exchanger (NCX). Cell death, however, does not depend on this initial Ca2+ rise, but rather invariably follows a delayed massive accumulation of Ca2+, occurring a few hours after the toxic challenge and representing a no-return transition into the death process. Recent work has highlighted events occurring in the interphase between the two Ca2+ rises, and suggested a progression route into the secondary Ca2+ overload and delayed cell death. Specifically, Bano et al. [39] showed that calpain-mediated cleavage on NCX is a critical step for allowing the delayed Ca2+ deregulation: inhibition of NCX cleavage protects from excitotoxic challenges, and conversely downregulation of the exchanger sensitizes to sub-lethal stimuli. As to mitochondria, direct measurement of matrix Ca2+ showed that the impairment of their Ca2+ uptake capacity is downstream of the delayed Ca2+ dysregulation, and not the cause of it. Thus, the delayed Ca2+ increase is not due to the sudden discharge from the mitochondrial buffer that still retains the capacity to accumulate Ca2+. Rather, the delayed, massive influx of Ca2+ into the cell leads to organelle Ca2+ overload, collapse of the electrochemical proton gradient and bioenergetic catastrophe, leading to necrotic cell death [40].
Interestingly, such a commitment mechanism appears to closely match an emerging paradigm clarified in a number of apoptotic routes. Indeed, we and other showed that mitochondrial involvement in apoptosis (morphological transitions and release of caspase cofactors) often utilizes Ca2+ as a key sensitizing factor. In agreement with this notion, Bcl-2 reduces the state of filling of intracellular Ca2+ stores, thereby reducing mitochondrial loading upon physiological and pathological challenges and protecting cells from apoptotic death [25]. The pro-apoptotic protein Bax antagonizes this signalling effect, by augmenting the state of filling of intracellular Ca2+ stores [41]. In addition, closely mimicking the data described above in excitotoxicity, early cleavage of plasma membrane Ca2+ pumps (PMCA) by caspases was shown to greatly enhance the efficacy of apoptotic challenges in neurons [39] and hepatocytes [42]. In this context, one can envision a common route to neurodegeneration in neurons, in which Ca2+ plays an important regulatory role and eventually, by affecting the mitochondrial checkpoint, renders cell death obligatory. Thus, while the fate into a rapid necrotic outcome or a more controlled apoptotic elimination will depend on the intensity of the insult and on the bioenergetic balance of the cell, the observation of common signalling themes and molecular targets highlights the extensive crosstalk between the different death pathways and the potential strategies for pharmacological intervention.
4. Looking into complex models: the pathogenesis of neurodegenerative disorders
4.1. Alzheimer's disease (AD)
Alzheimer's disease (AD) is a devastating neurological disorder clinically characterized by impairment of cognitive function and changes in behavior and personality. Morphological hallmarks of the pathology are cortical and hippocampal atrophy, accumulation of abnormal fibers in neuronal cell bodies (neurofibrillary tangles composed of a hyperphosphorylated form of the microtubular protein tau), and the presence, in the extracellular space, of senile plaques, the major component of which is the peptide β-amyloid (Aβ) in two most frequent forms, Aβ40 and Aβ42. These derive from the transmembrane protein APP (Amyloid Precursor Protein), that can be alternatively processed by three different enzymes, named α, β, and γ secretases. The combined action of β and γ secretases leads to the formation of a soluble fragment (sAPPβ) and of Aβ, together with its cytosolic counterpart AICD (APP Intra-Cellular Domain) [43].
Although the majority of AD cases are sporadic, a significant fraction of AD is inherited in a dominant pattern. Mutations in the genes encoding for APP and for Presenilin-1 and -2 (PS1 and PS2), two proteins belonging to the γ-secretase enzymatic complex, have been linked to the familial form of AD (FAD; see [44] for a recent review). Since the majority of FAD mutations have been found to increase the Aβ42/Aβ40 ratio, the initial hypothesis was that the disease was dependent on the enhanced fibrillization of the more amyloidogenic Aβ42 [45].
Although this concept has never been questioned, about a decade ago various experimental observations suggested that an alteration in intracellular Ca2+ homeostasis could also contribute to the development of FAD, and more in general to the pathogenesis of AD. Indeed, PSs mutations were shown to alter the expression, or the sensitivity, of ER Ca2+ release channels (RyR and IP3R) in different cell models [46–49] and in neurons from Tg AD mice [50,51], leading to the “Ca2+ overload” hypothesis [45,52], i.e. the idea that exaggerated ER Ca2+ release affects cellular targets such as mitochondria, favouring cellular demise (Fig. 2). The Ca2+ overload mechanism has however remained largely mysterious until it was found that wt PSs, but not the FAD mutants, can form Ca2+ permeable leak channels in the ER [53,54], thus providing a clear case for enhanced Ca2+ release in the pathological model. Although the data are straightforward, and support a very appealing model, the following work in other laboratories was not entirely consistent with the first formulation of the hypothesis (see for example [55–59]). Specifically, Zatti et al. [60–62] showed that some FAD-linked PS2 mutations caused a reduction, not an increase, in ER/Golgi Ca2+ levels. This experimental discrepancy, while not disproving the Ca2+ hypothesis, may thus allow two possible conclusions. The first is that the system is likely to be very complex, with additional unidentified regulatory elements, and the use of different cell systems and experimental approaches (e.g. silencing or overexpression, knock-out or knock-in models) may trigger equally different compensatory mechanisms and calcium effects in the cells. Secondly, given that the discrepancies mostly refer to PS2, a speculative, but appealing, possibility is that PS2 and PS1 play distinct roles in ER/Golgi Ca2+ handling. In particular, FAD-PS1 mutations, that cause an increase in the ER Ca2+, unavoidably exacerbate cell death. Conversely, PS2 mutants, by favouring low ER Ca2+ levels, might confer relative protection to other routes of cell intoxication, such as Aβ peptides and oxidative damage. This hypothesis would be consistent with the above described role of the ER/mitochondrial Ca2+ relationship, and with the clinical observation that FAD-linked PS2 mutations have been associated to milder phenotypes [60–62].
Fig. 2.
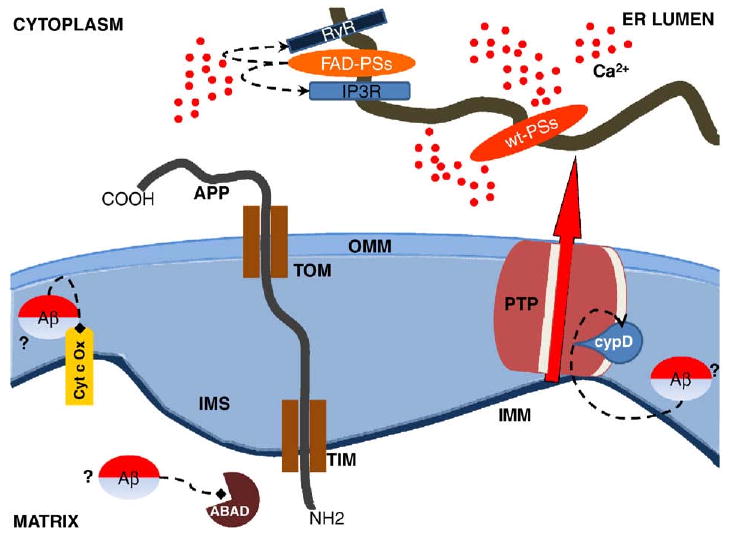
Presenilins (PSs), Amyloid Precursor Protein (APP) and peptide β-amyloid (Aβ) can affect mitochondrial functionality by different means. FAD-linked PSs mutations may alter the expression/sensitivity of ER Ca2+ release channels (RyR and IP3R) leading to an exaggerated ER Ca2+ release that in turn may cause abnormal mitochondrial Ca2+ uptake. wt PSs, but not the FAD mutants, were reported to form Ca2+ permeable leak channels in the ER providing a clear explanation for the enhanced Ca2+ release found in different AD models. APP can interact with the mitochondrial TIM/TOM protein import complex. The presence of an acidic domain within APP sequence may be responsible for an incomplete mitochondrial translocation that, in turn, inhibited the entry of nuclear-encoded mitochondrial proteins. Mitochondrial Aβ accumulation has been correlated with impaired enzymatic activities of cytochrome c oxidase (Cyt c-OX) and inhibition of mitochondrial Aβ-binding alcohol dehydrogenase (ABAD), leading to mitochondrial oxidative damage. Intra-mitochondrial Aβ was demonstrated to directly interact with cyclophilin D (CypD), the PTP component that binds CsA and renders the channel more sensitive to Ca2+, making AD mitochondria more sensitive to PTP opening. The origin of intra-mitochondrial Aβ peptides is however unclear. ER, endoplasmic reticulum; OMM outer mitochondrial membrane. IMS, intermembrane space; IMM, inner mitochondrial membrane.
No matter how the synergistic “Ca2+ hit” occurs, mitochondrial dysfunction appears an obligatory downstream step in the pathogenesis of AD. Decreased cytochrome c oxidase activity, increased free-radical generation leading to oxidative stress and reduced energy metabolism was described in the brain of AD patients before Aβ plaques formation [63–67]. Moreover, electron microscopy analysis of mitochondria in various regions of AD brain revealed significant morphological organelle alterations, such as reduced size of mitochondrial cristae [68].
As to the mechanism, converging evidence points to a role for Aβ peptides and the PTP and, possibly, to a Ca2+-sensitization step. Endogenous as well as ectopically expressed wt or FAD-linked Swedish APP have been found to localize to mitochondria in different cell types [69–71]. In isolated mitochondria, Aβ peptides were shown to inhibit mitochondrial respiration [72,73] and, in the presence of Ca2+, cause the opening of PTP. The involvement of PTP was further reinforced by the analysis of mouse models in which cyclophilin D, (CypD), the PTP component that binds CsA and renders the channel more sensitive to Ca2+, was knocked out [74,75]. Interestingly, this genetic alteration substantially improves the cognitive abilities of a mouse model of AD and alleviates Aβ-mediated reduction of long-term potentiation [76]. Moreover, intra-mitochondrial Aβ was demonstrated to directly interact with CypD, thus providing a molecular basis for this pathogenic mechanism [76]. Other putative damaging effects of Aβ were reported. In PC12 cells, Aβ blocked the entry of nuclear-encoded proteins into mitochondria causing decreased mitochondrial membrane potential, increased ROS production, oxygen glucose deprivation and altered mitochondrial morphology [77]. In another study, Aβ increased neuronal ROS production, activated mitochondrial fission proteins Drp1 and Fis1 and caused mitochondrial fragmentation [78]. Finally, in the mitochondrial matrix Aβ peptides were shown to interact with and inhibit the activity of mitochondrial Aβ-binding alcohol dehydrogenase (ABAD), leading to mitochondrial oxidative damage, increased carbonylation of mitochondrial proteins and impaired activity of respiratory complexes. Some of these alterations were found to occur in AD mouse models before the development of senile plaques, suggesting that mitochondrial dysfunction is an early event in the pathogenesis of the disease [79].
Thus calcium and mitochondria may represent the common theme linking the various reported aspects of the pathogenesis of AD. Several aspects remain however to be solved, and we wish to point out two crucial ones. The first is a clear understanding of the dysregulation of calcium signalling, as discussed above. The second one refers to the origin of intra-mitochondrial Aβ, if a primary role is attributed to this molecule. Experiments of limited trypsin proteolysis and chemical cross-linking showed that APP interacts with the mitochondrial TIM/TOM protein import complex and its orientation is such that the N-terminal resides inside the mitochondria while the large C-terminal part of the protein faces the cytosol [69,71]. Thus, although the presence within mitochondria of γ-secretase constituents and functional γ-secretase activity has been reported [80], it is unlikely that Aβ is being produced on the matrix side. Moreover, the mitochondrial presence of functional β-secretase, essential for the generation of the C99 peptide, substrate for γ-secretase, has not been reported yet. As to direct permeation from the ER, the Golgi complex or secretory vesicles to the cytosol, and then across the outer and the inner mitochondrial membranes (the latter notoriously highly impermeable to most solutes, including ions), this audacious possibility awaits experimental confirmation.
4.2. Huntington's disease (HD)
Huntington's disease (HD) is a progressive neurodegenerative disorder characterized by motor, cognitive and psychiatric symptoms including depression, personality changes, weight loss, dementia and motor disturbances. The latter are characterized by uncontrolled movements (chorea), developing in the terminal stage into severe akinesia. The disease is inherited in an autosomal dominant fashion and the mutation responsible for the disease was identified as a CAG-triplet expansion in exon 1 of huntingtin gene. The CAG sequence codes for glutamine and in HD, an expansion of the polyglutamine (polyQ) stretch beyond 35 glutamines results in pathogenicity and causes the selective death of the GABAergic spiny neurons of the striatum. The length of the polyQ stretch inversely correlates with the age of onset symptoms. Mutant Huntingtin (polyQ-Htt) forms intracellular aggregated, particularly in cell nuclei, and, to a lesser extent, in the cytoplasm, neurites, and terminals [81]. The aggregates have both been suggested to be toxic as well as neuroprotective to the cells. The exact function of Htt is still unknown: it is considered a scaffolding protein mediating protein–protein interactions and to play a role in processes as diverse as axonal transport, regulation of transcription, exocytosis, Ca2+ homeostasis, bioenergetic metabolism and prevention of apoptosis [82]. Dysfunctions in any one of these processes could be involved in the etiology of HD. Moreover, a series of seminal, independent observations have highlighted a possible role for Ca2+ and mitochondria also in this neurodegenerative disorder (Fig. 3). The first experimental evidence linking neurodegeneration in HD to mitochondrial impairment was the observation that treatment of mice with 3-nitropropionic acid (3-NPA), an inhibitor of complex II of the respiratory chain, induces a degeneration of striatal neurons in vivo, recapitulating HD pathogenesis [83]. In addition, the activities of the respiratory complexes II, III and IV were shown to be reduced in HD [84]. Then, Panov et al. showed that mutant but not wild type Htt directly impairs mitochondrial function. They also proposed that mutant Htt localize on mitochondrial membranes in the neurons of mutant mice, suggesting that a direct relationship may occur between the observed defects and the disease [85]. Finally, Bezprozvanny et al. demonstrated that Htt forms a ternary complex with Htt-associated protein-1A (HAP-1A) and type 1 IP3Rs. In this complex, polyQ-Htt, but not the wild type Htt, facilitates Ca2+ release from the ER and renders neurons more sensitive to Ca2+-mediated cellular dysfunction [86]. As to the mechanistic link, mitochondrial Ca2+ overload appeared the decisive commitment step [87]. In keeping with this view, Choo et al. showed that mutated, but not wild type, Htt induces PTP opening in isolated mitochondria [88], and we recently demonstrated a facilitated opening of PTP in permeabilized polyQ-Htt expressing cells [89]. Thus, also in this case PTP appears to be the final commitment step in a number of cellular stress conditions, with Ca2+ acting as a potent sensitizing factor [89]. As to the stress signals converging on mitochondria, logical candidates are ROS, putatively involved in age-related disorders and in conditions of mitochondrial impairment. Interestingly, the coordinated genomic program mediated by Peroxisome proliferator-activated receptor-coactivator (PGC-1α), a transcription factor that upregulates ROS-scavenging systems upon oxidative stress [90], appears dysfunctional in HD striata [91], and also in cellular models of the disease [92]. Indeed, in both cases the level of the PGC-1α transcript is downregulated, and, at least in part, the scavenging enzymes are accordingly reduced. This observation appears very appealing, as recent work has highlighted PGC-1α transcriptional regulation as a promising drug target [93].
Fig. 3.
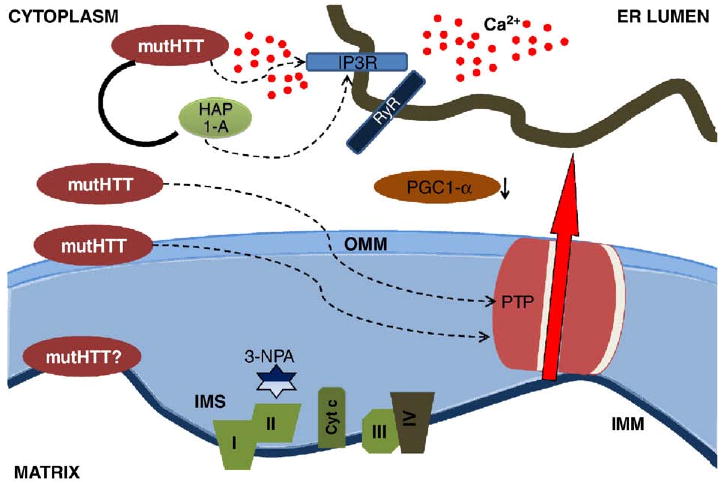
Mutant Huntingtin (mutHtt) impairs mitochondrial function by transcriprional and non-trascriptional mechanisms. The transcriptional effects are mediated by nuclear translocation of mutHtt. One important consequence of the regulation of gene transcription is the downregulation of PGC1α, and thus the reduced expression of nuclear-encoded mitochondrial proteins involved in the respiratory chains and in the oxidative-stress defense. MutHtt also associates with the outer mitochondrial membrane (OMM) directly affecting the PTP opening susceptibility and making striatal neurons more vulnerable to excitotoxic stimuli. MutHtt reduces complex II activity and the treatment with the complex II inhibitor 3-nitropropionic acid (3-NPA) induces striatal neurodegeneration in vitro and in vivo. The association of mutHtt with HAP-1A and with the IP3R type I facilitates ER Ca2+ release, thus making mitochondria more susceptible to Ca2+ overload. ER, endoplasmic reticulum; IMS, intermembrane space; IMM, inner mitochondrial membrane.
Another important way in which mitochondrial function could be impaired in HD is through abnormal axonal trafficking of mitochondria to and from the synapse. PolyQ-Htt has been proposed to inhibit axonal transport through several mechanisms and, recently, it was shown that fragments of Htt associate with mitochondria thus interfering with their microtubule-associated transport [94]. Overall, the data so far obtained appear very coherent, and highlight a straightforward pathogenic route, based on the triggering of a calcium-dependent mitochondrial dysfunction. What remains to be assessed is how the putative damaging effects of polyQ-Htt are coordinately regulated. Are mitochondrial respiratory deficiencies and calcium-mediated organelle dysfunction two independent effects of mutated Htt, or are they causally linked? Or does the latter gradually induce the former, in a vicious cycle of ROS production and genetic damage that can be further exacerbated by toxic challenges affecting the brain? And in this picture, why isn't PGC-1α and its anti-oxidant program upregulated? Relative to the respiratory deficiencies, recent work on cultured striatal neurons transfected with a polyQ-Htt showed downregulation of complex II components at the protein, but not at the mRNA, level [95]. Complex II deficiency appears thus due to a post-transcriptional (proteolytic) effect. Whether a calcium-sensitive protease (e.g. calpains) is involved, as was reported in other non genetic models of neurodegeneration [39], has yet to be demonstrated.
4.3. Parkinson's disease (PD)
Parkinson's disease (PD) is a chronic progressive neurodegenerative disorder clinically characterized by motor impairments involving resting tremor, progressive rigidity, bradykinesia and postural instability. PD pathology is characterized most prominently by loss of dopaminergic neurons in the substantia nigra and formation of intraneuronal protein aggregates called Lewy body.
Mitochondrial involvement in PD has been an established notion for many years, since the recognition of the mechanism of action of MPTP (1-methyl 4-phenyl-1,2,3,6-tetrahydropyridine). This compound is formed during production of synthetic heroine, and its metabolite MPP+ is a mitochondrial toxin, that blocks complex I of the respiratory chain. In drug abusers, it causes a form of PD that is clinically indistinguishable from the sporadic variety. Further studies corroborated this observation. Biochemical analysis of PD samples revealed the presence of a mild, systemic defect of complex I, and chronic exposure to the most classical complex I inhibitor (rotenone) accurately recapitulated the pathological, biochemical, and behavioral features of PD. The mechanism of complex I inhibition toxicity probably involves oxidative stress, caused by the block of the respiratory chain, but the selective vulnerability of dopaminergic neurons still remain elusive to explain [96].
The recent identification of a cohort of genes involved in the familial forms of PD further corroborates the notion of mitochondrial involvement, as apparently unrelated proteins seem to share this organelle as a common theme, and possibly point to a signalling role for Ca2+ (Fig. 4). Specifically, mutations were reported in genes encoding for α-synuclein, DJ-1, Leucin Rich-Repeated Kinase (LRRK2), ubiquitin C-terminal hydrolase L1 (UCHL1), phosphatase-tensin homologue (PTEN)-induced kinase 1 (PINK1), and parkin, as well as within the mtDNA (for a recent review, see [97]). Mutations in the gene encoding the mitochondrial serine protease HtrA2 have also been linked to PD in several families.
Fig. 4.
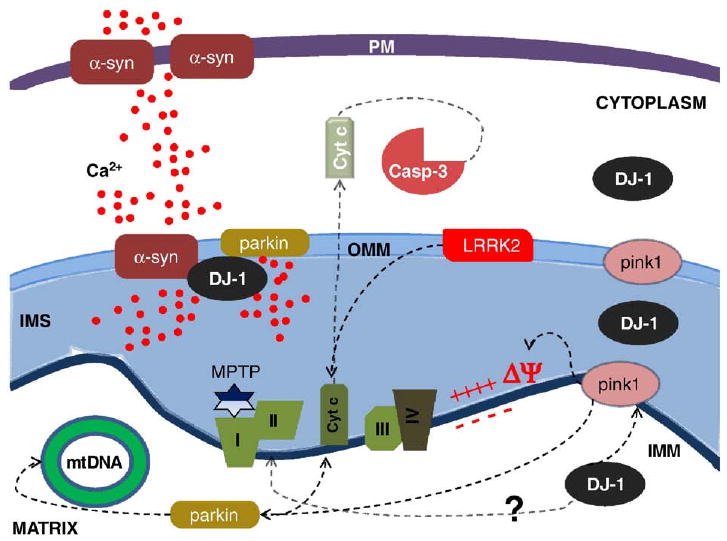
Mutations in familial PD-linked genes encoding α-synuclein (α-syn), parkin, pink1, DJ-1 and LRRK2 cause mitochondrial dysfunctions through common intersecting pathways. Mutant α-syn promotes Ca2+ influx and mitochondrial Ca2+ overload and mutant parkin exacerbates this effect. DJ-1, PINK1 and Parkin may act in series on the same protein targets: genetic data suggest that pink1 is upstream of parkin, and that all of them exert a protective role preserving mitochondrial functions, morphology and preventing mitochondrial-induced apoptosis. LRRK2 mutations induce apoptotic death through cytochrome c release and activation of caspase-3. Complex I activity is reduced in PD and its inhibition by MPTP or rotenone causes dopaminergic degeneration. Mutations in mtDNA-encoded complex I subunits also cause PD. PM, plasma membrane; OMM outer mitochondrial membrane; IMS, intermembrane space; IMM, inner mitochondrial membrane.
α-synuclein mutations are linked to autosomal dominant familiar PD. Inclusions immunopositive for α-synuclein are found in Levy's body, raising to the possibility that the toxicity of the protein is due to an abnormal form of aggregation or fibril formation, in analogy on what happens in AD. In a recent study [98] the different levels of α-synuclein oligomerization have been linked to cell death. In particular, an heterogeneous mixture of small oligomers of α-synuclein can lead to Ca2+ dysregulation, probably via a pore-forming mechanism. Calpain, a major Ca2+-activated protease, can cleave α-synuclein, producing a truncated form more prone to aggregate, thus leading to formation of protofibrils [99]. Overall, these observations suggest that a vicious cycle of Ca2+ dysregulation (coordinately activated by the oligomers and, possibly, excitotoxic stimulation) and fibril deposition eventually leads to severe Ca2+ overload, to the point of mitochondrial permeability transition and commitment to neuronal cell death.
Parkin has been identified as a ubiquitin-protein ligase (E3) that acts along with the ubiquitin-conjugating enzymes (E2s) in selectivity of ubiquitination and recognition of substrates [100]. Inactivation of parkin leads to reduction in UPS-mediated degradation of its substrates, among which a glycosylated form of α-synuclein. Interestingly, Drosophila parkin null mutants exhibited defects in mitochondrial function with signs of increased oxidative stress, muscle degeneration and male sterility. Reduced levels of mitochondrial proteins involved in mitochondrial oxidative phosphorylation were also reported in parkin-knock-out mice, which exhibited normal brain morphology, but increased striatal extracellular dopamine levels [101]. Overexpression of parkin in cultured cells prevents mitochondrial swelling and stress-induced apoptosis. The protein appears localized in the mitochondrial matrix, where it enhances mitochondrial gene transcription and biogenesis in proliferating cells, but the exact mechanism for the protective function of parkin in mitochondria is unknown [102]. Recently, parkin has been shown to promote the selective clearance of damaged mitochondria through the mitophagic process [103] further reinforcing the concept that it has a neuroprotective role.
PINK1 is highly conserved protein, containing a catalytic serine-threonine kinase domain, ubiquitously expressed in the human brain. It is unambiguously localized to mitochondrial membranes, and its overexpression protects cells from mitochondrial depolarization and apoptosis induced by the proteosomal inhibitor MG132, and from mitochondrially-induced apoptosis triggered by staurosporine [104]. Loss of function mutations are responsible for male sterility, and muscle and dopaminergic neuronal degeneration in Drosophila [105]. Overexpression of parkin was shown to rescue the mitochondria dysfunction caused by PINK1 deficiency (but did not rescue the increased sensitivity of PINK1 mutant flies to apoptosis), suggesting that the two proteins could cooperate (probably regulating the balance between mitochondrial fission and fusion, [106]) in preserving mitochondrial integrity in various stress conditions [107]. Interestingly, mutant PINK1 has been shown to exacerbate mitochondrial alterations (disturbing the mitochondrial Ca2+ fluxes) promoted by mutant α-synuclein, thus suggesting a cooperative role of these two proteins [108].
LRRK2 is a recently found gene, mutated in familiar late-onset PD. Its product is a kinase [109] and a significant fraction of the protein is associated with mitochondria [110]. Pathogenic PD mutations are linked to activation of the intrinsic apoptotic pathway, with cytochrome c released into the cytosol and activation of caspase-3 [111].
Finally, little is known of the function of the DJ-1 gene product. DJ-1 expression is particularly abundant in brain region where it is largely cytoplasmic except for a pool localized in mitochondria. In particular, DJ-1 was reported to be located in the mitochondrial intermembrane space and in the matrix, while very little, if any, is associated with outer or inner mitochondrial membranes. This finding suggests that DJ-1 may act within mitochondria but its precise neuroprotective mechanism remains obscure (for a review, see [112]). Recent findings suggest that DJ-1 is an atypical peroxiredoxin-like peroxidase [113] indicating that it may play a role in reducing mitochondrial oxidative stress.
These data highlight mitochondria involvement in PD. Obviously, an intriguing possibility is that mitochondrial Ca2+ homeostasis, and thus the sensitivity to Ca2+-mediated challenges, are directly or indirectly controlled by these proteins. Work on this topic is under way in several laboratories, including ours, and it is easy to predict that also in this model of neurodegeneration the concerted action of Ca2+ and toxic challenges on mitochondria will prove to represent the mechanism leading through time to neuronal dysfunction. Indeed, preliminary evidence from our laboratory, demonstrating an alteration in mitochondrial Ca2+ homeostasis in a PD model, supports this possibility. Specifically, by using recombinant aequorin to evaluate Ca2+ fluxes [3] we observed an alteration of mitochondrial Ca2+ signals in SH-SY5Y cells overexpressing wild type or mutated (G2019S and R1441C) LRRK2, associated with PD (Celsi in preparation).
Overall, the picture emerging from the study of the pathogenesis of the neurodegenerative disorders appears terribly complex, with many uncertainties and gaps to fill. Nevertheless, the common role of mitochondria dysfunction (metabolic, morphologic or dynamic) appears very clear, and provides not only a leading theme for further studies, but also a promising pharmacological target in these devastating diseases.
Acknowledgments
Experimental work in the authors' laboratories is supported by grants from the Italian Ministry of Education (Prin 2005), Italian Ministry of University (FIRB grant no. RBIN042Z2Y), The European Union (FP7 “MyoAGE”), the Emilia-Romagna region, the Italian Association for Cancer Research (AIRC), local funds from Ferrara University, Telethon, The Italian Multiple Sclerosis Foundation (FISM), the Italian Space Agency (ASI), NIH (Grant #1P01AG025532-01A1) and the United Mitochondrial Disease Foundation (UMDF).
References
- 1.Carafoli E. Historical review: mitochondria and calcium: ups and downs of an unusual relationship. Trends Biochem Sci. 2003;28:175. doi: 10.1016/S0968-0004(03)00053-7. [DOI] [PubMed] [Google Scholar]
- 2.De Giorgi F, Brini M, Bastianutto C, Marsault R, Montero M, Pizzo P, Rossi R, Rizzuto R. Targeting aequorin and green fluorescent protein to intracellular organelles. Gene. 1996;173:113. doi: 10.1016/0378-1119(95)00687-7. [DOI] [PubMed] [Google Scholar]
- 3.Pinton P, Rimessi A, Romagnoli A, Prandini A, Rizzuto R. Biosensors for the detection of calcium and pH. In: Liza APaE., editor. Methods in Cell BiologyMitochondria. 2nd. Academic Press; 2007. pp. 297–325. [DOI] [PubMed] [Google Scholar]
- 4.Babcock DF, Herrington J, Goodwin PC, Park YB, Hille B. Mitochondrial participation in the intracellular Ca2+ network. J Cell Biol. 1997;136:833. doi: 10.1083/jcb.136.4.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Knöpfel T, Dìez-Garcìa J, Akemann W. Optical probing of neuronal circuit dynamics: genetically encoded versus classical fluorescent sensors. Trends Neurosci. 2006;29:160. doi: 10.1016/j.tins.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 6.Rudolf R, Mongillo M, Rizzuto R, Pozzan T. Looking forward to seeing calcium. Nat Rev Mol Cell Biol. 2003;4:579. doi: 10.1038/nrm1153. [DOI] [PubMed] [Google Scholar]
- 7.Robb-Gaspers LD, Burnett P, Rutter GA, Denton RM, Rizzuto R, Thomas AP. Integrating cytosolic calcium signals into mitochondrial metabolic responses. EMBO J. 1998;17:4987. doi: 10.1093/emboj/17.17.4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dedkova EN, Blatter LA. Mitochondrial Ca2+ and the heart. Cell Calcium. 2008;44:77. doi: 10.1016/j.ceca.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 9.Pinton P, Brini M, Bastianutto C, Tuft RA, Pradier L, Rizzuto R. New light on mitochondrial calcium. BioFactors. 1998;8:243. doi: 10.1002/biof.5520080312. [DOI] [PubMed] [Google Scholar]
- 10.Rizzuto R, Pozzan T. Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev. 2006;86:369. doi: 10.1152/physrev.00004.2005. [DOI] [PubMed] [Google Scholar]
- 11.Denton RM, McCormack JG. Ca2+ as a second messenger within mitochondria of the heart and other tissues. Annu Rev Physiol. 1990;52:451. doi: 10.1146/annurev.ph.52.030190.002315. [DOI] [PubMed] [Google Scholar]
- 12.Lasorsa FM, Pinton P, Palmieri L, Fiermonte G, Rizzuto R, Palmieri F. Recombinant expression of the Ca2+-sensitive aspartate/glutamate carrier increases mitochondrial ATP production in agonist-stimulated chinese hamster ovary cells. J Biol Chem. 2003;278:38686. doi: 10.1074/jbc.M304988200. [DOI] [PubMed] [Google Scholar]
- 13.Jouaville LS, Pinton P, Bastianutto C, Rutter GA, Rizzuto R. Regulation of mitochondrial ATP synthesis by calcium: evidence for a long-term metabolic priming. Proc Natl Acad Sci U S A. 1999;96:13807. doi: 10.1073/pnas.96.24.13807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brini M, Pinton P, King MP, Davidson M, Schon EA, Rizzuto R. A calcium signaling defect in the pathogenesis of a mitochondrial DNA inherited oxidative phosphorylation deficiency. Nat Med. 1999;5:951. doi: 10.1038/11396. [DOI] [PubMed] [Google Scholar]
- 15.Visch HJ, Rutter GA, Koopman WJH, Koenderink JB, Verkaart S, de Groot T, Varadi A, Mitchell KJ, van den Heuvel LP, Smeitink JAM, Willems PHGM. Inhibition of mitochondrial Na+–Ca2+ exchange restores agonist-induced ATP production and Ca2+ handling in human complex I deficiency. J Biol Chem. 2004;279:40328. doi: 10.1074/jbc.M408068200. [DOI] [PubMed] [Google Scholar]
- 16.Herzig S, Martinou JC. Mitochondrial dynamics: to be in good shape to survive. Curr Mol Med. 2008;8:131. doi: 10.2174/156652408783769625. [DOI] [PubMed] [Google Scholar]
- 17.Hoek JB, Farber JL, Thomas AP, Wang X. Calcium ion-dependent signalling and mitochondrial dysfunction: mitochondrial calcium uptake during hormonal stimulation in intact liver cells and its implication for the mitochondrial permeability transition. Biochim Biophys Acta. 1995;1271:93. doi: 10.1016/0925-4439(95)00015-v. [DOI] [PubMed] [Google Scholar]
- 18.Tinel H, Cancela JM, Mogami H, Gerasimenko JV, Gerasimenko OV, Tepikin AV, Petersen OH. Active mitochondria surrounding the pancreatic acinar granule region prevent spreading of inositol trisphosphate-evoked local cytosolic Ca(2+) signals. EMBO J. 1999;18:4999. doi: 10.1093/emboj/18.18.4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.David G, Barrett JN, Barrett EF. Evidence that mitochondria buffer physiological Ca2+ loads in lizard motor nerve terminals. J Physiol. 1998;509:59. doi: 10.1111/j.1469-7793.1998.059bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Falcke M, Hudson JL, Camacho P, Lechleiter JD. Impact of mitochondrial Ca2+ cycling on pattern formation and stability. Biophys J. 1999;77:37. doi: 10.1016/S0006-3495(99)76870-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zimmermann B. Control of InsP3-induced Ca2+ oscillations in permeabilized blowfly salivary gland cells: contribution of mitochondria. J Physiol. 2000;525(Pt 3):707–19. doi: 10.1111/j.1469-7793.2000.t01-1-00707.x. 707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boitier E, Rea R, Duchen MR. Mitochondria exert a negative feedback on the propagation of intracellular Ca2+ waves in rat cortical astrocytes. J Cell Biol. 1999;145:795. doi: 10.1083/jcb.145.4.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Landolfi B, Curci S, Debellis L, Pozzan T, Hofer AM. Ca2+ homeostasis in the agonist-sensitive internal store: functional interactions between mitochondria and the ER measured in situ in intact cells. J Cell Biol. 1998;142:1235. doi: 10.1083/jcb.142.5.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hetz C, Glimcher L. The daily job of night killers: alternative roles of the BCL-2 family in organelle physiology. Trends Cell Biol. 2008;18:38. doi: 10.1016/j.tcb.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 25.Pinton P, Ferrari D, Rapizzi E, Di VF, Pozzan T, Rizzuto R. The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: significance for the molecular mechanism of Bcl-2 action. EMBO J. 2001;20:2690. doi: 10.1093/emboj/20.11.2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rostovtseva T, Bezrukov S. VDAC regulation: role of cytosolic proteins and mitochondrial lipids. J Bioenerg Biomembranes. 2008;40:163. doi: 10.1007/s10863-008-9145-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Szabadkai G, Bianchi K, Varnai P, De Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T, Rizzuto R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol. 2006;175:901. doi: 10.1083/jcb.200608073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427:360. doi: 10.1038/nature02246. [DOI] [PubMed] [Google Scholar]
- 29.Bragadin M, Pozzan T, Azzone GF. Kinetics of Ca2+ carrier in rat liver mitochondria. Biochemistry. 1979;18:5972. doi: 10.1021/bi00593a033. [DOI] [PubMed] [Google Scholar]
- 30.Cox DA, Matlib MA. Modulation of intramitochondrial free Ca2+ concentration by antagonists of Na(+)–Ca2+ exchange. Trends Pharmacol Sci. 1993;14:408. doi: 10.1016/0165-6147(93)90063-P. [DOI] [PubMed] [Google Scholar]
- 31.Wingrove DE, Gunter TE. Kinetics of mitochondrial calcium transport. II. A kinetic description of the sodium-dependent calcium efflux mechanism of liver mitochondria and inhibition by ruthenium red and by tetraphenylphosphonium. J Biol Chem. 1986;261:15166. [PubMed] [Google Scholar]
- 32.Bernardi P, Azzone GF. Regulation of Ca2+ efflux in rat liver mitochondria. Role of membrane potential. Eur J Biochem. 1983;134:377. doi: 10.1111/j.1432-1033.1983.tb07578.x. [DOI] [PubMed] [Google Scholar]
- 33.Azzone GF, Azzi A. Volume changes induced by inorganic phosphate in liver mitochondria. Biochem J. 1965;94:10C. doi: 10.1042/bj0940010c. [DOI] [PubMed] [Google Scholar]
- 34.Huser J, Rechenmacher CE, Blatter LA. Imaging the permeability pore transition in single mitochondria. Biophys J. 1998;74:2129. doi: 10.1016/S0006-3495(98)77920-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Petronilli V, Miotto G, Canton M, Brini M, Colonna R, Bernardi P, Di LF. Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophys J. 1999;76:725. doi: 10.1016/S0006-3495(99)77239-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Szalai G, Krishnamurthy R, Hajnoczky G. Apoptosis driven by IP(3)-linked mitochondrial calcium signals. EMBO J. 1999;18:6349. doi: 10.1093/emboj/18.22.6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bernardi P, Petronilli V. The permeability transition pore as a mitochondrial calcium release channel: a critical appraisal. J Bioenerg Biomembr. 1996;28:131. doi: 10.1007/BF02110643. [DOI] [PubMed] [Google Scholar]
- 38.Olney JW. Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science. 1969;164:719. doi: 10.1126/science.164.3880.719. [DOI] [PubMed] [Google Scholar]
- 39.Bano D, Young KW, Guerin CJ, Lefeuvre R, Rothwell NJ, Naldini L, Rizzuto R, Carafoli E, Nicotera P. Cleavage of the plasma membrane Na+/Ca2+ exchanger in excitotoxicity. Cell. 2005;120:275. doi: 10.1016/j.cell.2004.11.049. [DOI] [PubMed] [Google Scholar]
- 40.Mattson MP. Calcium and neurodegeneration. Aging Cell. 2007;6:337. doi: 10.1111/j.1474-9726.2007.00275.x. [DOI] [PubMed] [Google Scholar]
- 41.Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science. 2003;300:135. doi: 10.1126/science.1081208. [DOI] [PubMed] [Google Scholar]
- 42.Chami M, Prandini A, Campanella M, Pinton P, Szabadkai G, Reed JC, Rizzuto R. Bcl-2 and Bax exert opposing effects on Ca2+ signaling, which do not depend on their putative pore-forming region. J Biol Chem. 2004;279:54581. doi: 10.1074/jbc.M409663200. [DOI] [PubMed] [Google Scholar]
- 43.Thinakaran G, Koo EH. Amyloid precursor protein trafficking, processing, and function. J Biol Chem. 2008;283:29615. doi: 10.1074/jbc.R800019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bertram L, Tanzi RE. Thirty years of Alzheimer's disease genetics: the implications of systematic meta-analyses. Nat Rev Neurosci. 2008;9:768. doi: 10.1038/nrn2494. [DOI] [PubMed] [Google Scholar]
- 45.Thinakaran G, Sisodia SS. Presenilins and Alzheimer disease: the calcium conspiracy. Nat Neurosci. 2006;9:1354. doi: 10.1038/nn1106-1354. [DOI] [PubMed] [Google Scholar]
- 46.Guo Q, Furukawa K, Sopher BL, Pham DG, Xie J, Robinson N, Martin GM, Mattson MP. Alzheimer's PS-1 mutation perturbs calcium homeostasis and sensitizes PC12 cells to death induced by amyloid beta-peptide. NeuroReport. 1996;8:379. doi: 10.1097/00001756-199612200-00074. [DOI] [PubMed] [Google Scholar]
- 47.Leissring MA, Parker I, LaFerla FM. Presenilin-2 mutations modulate amplitude and kinetics of inositol 1, 4,5-trisphosphate-mediated calcium signals. J Biol Chem. 1999;274:32535. doi: 10.1074/jbc.274.46.32535. [DOI] [PubMed] [Google Scholar]
- 48.Chan SL, Mayne M, Holden CP, Geiger JD, Mattson MP. Presenilin-1 mutations increase levels of ryanodine receptors and calcium release in PC12 cells and cortical neurons. J Biol Chem. 2000;275:18195. doi: 10.1074/jbc.M000040200. [DOI] [PubMed] [Google Scholar]
- 49.Lee SY, Hwang DY, Kim YK, Lee JW, Shin IC, Oh KW, Lee MK, Lim JS, Yoon DY, Hwang SJ, Hong JT. PS2 mutation increases neuronal cell vulnerability to neurotoxicants through activation of caspase-3 by enhancing of ryanodine receptor-mediated calcium release. FASEB J. 2006;20:151. doi: 10.1096/fj.05-4017fje;1. [DOI] [PubMed] [Google Scholar]
- 50.Leissring MA, Parker I, LaFerla FM. Presenilin-2 mutations modulate amplitude and kinetics of inositol 1, 4,5-trisphosphate-mediated calcium signals. J Biol Chem. 1999;274:32535. doi: 10.1074/jbc.274.46.32535. [DOI] [PubMed] [Google Scholar]
- 51.Smith IF, Green KN, LaFerla FM. Calcium dysregulation in Alzheimer's disease: recent advances gained from genetically modified animals. Cell Calcium. 2005;38:427. doi: 10.1016/j.ceca.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 52.LaFerla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer's disease. Nat Rev Neurosci. 2002;3:862. doi: 10.1038/nrn960. [DOI] [PubMed] [Google Scholar]
- 53.Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH, Serneels L, De SB, Yu G, Bezprozvanny I. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer's disease-linked mutations. Cell. 2006;126:981. doi: 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nelson O, Tu H, Lei T, Bentahir M, De SB, Bezprozvanny I. Familial Alzheimer disease-linked mutations specifically disrupt Ca2+ leak function of presenilin 1. J Clin Invest. 2007;117:1230. doi: 10.1172/JCI30447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kasri NN, Kocks SL, Verbert L, Hebert SS, Callewaert G, Parys JB, Missiaen L, De SH. Up-regulation of inositol 1,4,5-trisphosphate receptor type 1 is responsible for a decreased endoplasmic-reticulum Ca2+ content in presenilin double knock-out cells. Cell Calcium. 2006;40:41. doi: 10.1016/j.ceca.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 56.Green KN, Demuro A, Akbari Y, Hitt BD, Smith IF, Parker I, LaFerla FM. SERCA pump activity is physiologically regulated by presenilin and regulates amyloid beta production. J Cell Biol. 2008;181:1107. doi: 10.1083/jcb.200706171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, Staufenbiel M, Konnerth A, Garaschuk O. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer's disease. Science. 2008;321:1686. doi: 10.1126/science.1162844. [DOI] [PubMed] [Google Scholar]
- 58.Cheung KH, Shineman D, Muller M, Cardenas C, Mei L, Yang J, Tomita T, Iwatsubo T, Lee VM, Foskett JK. Mechanism of Ca2+ disruption in Alzheimer's disease by presenilin regulation of InsP3 receptor channel gating. Neuron. 2008;58:871. doi: 10.1016/j.neuron.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fedrizzi L, Lim D, Carafoli E, Brini M. Interplay of the Ca2+-binding protein DREAM with presenilin in neuronal Ca2+ signaling. J Biol Chem. 2008;283:27494. doi: 10.1074/jbc.M804152200. [DOI] [PubMed] [Google Scholar]
- 60.Giacomello M, Barbiero L, Zatti G, Squitti R, Binetti G, Pozzan T, Fasolato C, Ghidoni R, Pizzo P. Reduction of Ca2+ stores and capacitative Ca2+ entry is associated with the familial Alzheimer's disease presenilin-2 T122R mutation and anticipates the onset of dementia. Neurobiol Dis. 2005;18:638. doi: 10.1016/j.nbd.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 61.Zatti G, Burgo A, Giacomello M, Barbiero L, Ghidoni R, Sinigaglia G, Florean C, Bagnoli S, Binetti G, Sorbi S, Pizzo P, Fasolato C. Presenilin mutations linked to familial Alzheimer's disease reduce endoplasmic reticulum and Golgi apparatus calcium levels. Cell Calcium. 2006;39:539. doi: 10.1016/j.ceca.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 62.Zatti G, Ghidoni R, Barbiero L, Binetti G, Pozzan T, Fasolato C, Pizzo P. The presenilin 2 M239I mutation associated with familial Alzheimer's disease reduces Ca2+ release from intracellular stores. Neurobiol Dis. 2004;15:269. doi: 10.1016/j.nbd.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 63.Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:759. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 64.Parker WD., Jr Cytochrome oxidase deficiency in Alzheimer's disease. Ann N Y Acad Sci. 1991;640:59. doi: 10.1111/j.1749-6632.1991.tb00191.x. [DOI] [PubMed] [Google Scholar]
- 65.Maurer I, Zierz S, Moller HJ. A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol Aging. 2000;21:455. doi: 10.1016/s0197-4580(00)00112-3. [DOI] [PubMed] [Google Scholar]
- 66.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 67.Reddy PH, Beal MF. Are mitochondria critical in the pathogenesis of Alzheimer's disease? Brain Res Brain Res Rev. 2005;49:618. doi: 10.1016/j.brainresrev.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 68.Baloyannis SJ. Mitochondrial alterations in Alzheimer's disease. J Alzheimers Dis. 2006;9:119. doi: 10.3233/jad-2006-9204. [DOI] [PubMed] [Google Scholar]
- 69.Anandatheerthavarada HK, Biswas G, Robin MA, Avadhani NG. Mitochondrial targeting and a novel transmembrane arrest of Alzheimer's amyloid precursor protein impairs mitochondrial function in neuronal cells. J Cell Biol. 2003;161:41. doi: 10.1083/jcb.200207030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Keil U, Bonert A, Marques CA, Scherping I, Weyermann J, Strosznajder JB, Muller-Spahn F, Haass C, Czech C, Pradier L, Muller WE, Eckert A. Amyloid beta-induced changes in nitric oxide production and mitochondrial activity lead to apoptosis. J Biol Chem. 2004;279:50310. doi: 10.1074/jbc.M405600200. [DOI] [PubMed] [Google Scholar]
- 71.Park HJ, Kim SS, Seong YM, Kim KH, Goo HG, Yoon EJ, Min dS, Kang S, Rhim H. Beta-amyloid precursor protein is a direct cleavage target of HtrA2 serine protease. Implications for the physiological function of HtrA2 in the mitochondria. J Biol Chem. 2006;281:34277. doi: 10.1074/jbc.M603443200. [DOI] [PubMed] [Google Scholar]
- 72.Casley CS, Land JM, Sharpe MA, Clark JB, Duchen MR, Canevari L. Beta-amyloid fragment 25–35 causes mitochondrial dysfunction in primary cortical neurons. Neurobiol Dis. 2002;10:258. doi: 10.1006/nbdi.2002.0516. [DOI] [PubMed] [Google Scholar]
- 73.Crouch PJ, Blake R, Duce JA, Ciccotosto GD, Li QX, Barnham KJ, Curtain CC, Cherny RA, Cappai R, Dyrks T, Masters CL, Trounce IA. Copper-dependent inhibition of human cytochrome c oxidase by a dimeric conformer of amyloid-beta1–42. J Neurosci. 2005;25:672. doi: 10.1523/JNEUROSCI.4276-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 75.Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci U S A. 2005;102:12005. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Du H, Guo L, Fang F, Chen D, Sosunov A, McKhann M, Yan Y, Wang C, Zhang H, Molkentin JD, Gunn-Moore FJ, Vonsattel JP, Arancio O, Chen JX, Yan SD. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat Med. 2008;14:1097. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sirk D, Zhu Z, Wadia JS, Shulyakova N, Phan N, Fong J, Mills LR. Chronic exposure to sub-lethal beta-amyloid (Abeta) inhibits the import of nuclear-encoded proteins to mitochondria in differentiated PC12 cells. J Neurochem. 2007;103:1989. doi: 10.1111/j.1471-4159.2007.04907.x. [DOI] [PubMed] [Google Scholar]
- 78.Barsoum MJ, Yuan H, Gerencser AA, Liot G, Kushnareva Y, Graber S, Kovacs I, Lee WD, Waggoner J, Cui J, White AD, Bossy B, Martinou JC, Youle RJ, Lipton SA, Ellisman MH, Perkins GA, Bossy-Wetzel E. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 2006;25:3900. doi: 10.1038/sj.emboj.7601253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH. Mitochondria are a direct site of A beta accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. 2006;15:1437. doi: 10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- 80.Hansson CA, Frykman S, Farmery MR, Tjernberg LO, Nilsberth C, Pursglove SE, Ito A, Winblad B, Cowburn RF, Thyberg J, Ankarcrona M. Nicastrin, presenilin, APH-1, and PEN-2 form active gamma-secretase complexes in mitochondria. J Biol Chem. 2004;279:51654. doi: 10.1074/jbc.M404500200. [DOI] [PubMed] [Google Scholar]
- 81.Sapp E, Schwarz C, Chase K, Bhide PG, Young AB, Penney J, Vonsattel JP, Aronin N, DiFiglia M. Huntingtin localization in brains of normal and Huntington's disease patients. Ann Neurol. 1997;42:604. doi: 10.1002/ana.410420411. [DOI] [PubMed] [Google Scholar]
- 82.Cattaneo E, Zuccato C, Tartari M. Normal huntingtin function: an alternative approach to Huntington's disease. Nat Rev Neurosci. 2005;6:919. doi: 10.1038/nrn1806. [DOI] [PubMed] [Google Scholar]
- 83.Brouillet E, Jacquard C, Bizat N, Blum D. 3-Nitropropionic acid: a mitochondrial toxin to uncover physiopathological mechanisms underlying striatal degeneration in Huntington's disease. J Neurochem. 2005;95:1521. doi: 10.1111/j.1471-4159.2005.03515.x. [DOI] [PubMed] [Google Scholar]
- 84.Milakovic T, Johnson GV. Mitochondrial respiration and ATP production are significantly impaired in striatal cells expressing mutant huntingtin. J Biol Chem. 2005;280:30773. doi: 10.1074/jbc.M504749200. [DOI] [PubMed] [Google Scholar]
- 85.Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ, Greenamyre JT. Early mitochondrial calcium defects in Huntington's disease are a direct effect of polyglutamines. Nat Neurosci. 2002;5:731. doi: 10.1038/nn884. [DOI] [PubMed] [Google Scholar]
- 86.Tang TS, Tu H, Chan EY, Maximov A, Wang Z, Wellington CL, Hayden MR, Bezprozvanny I. Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1. Neuron. 2003;39:227. doi: 10.1016/s0896-6273(03)00366-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bezprozvanny I, Hayden MR. Deranged neuronal calcium signaling and Huntington disease. Biochem Biophys Res Commun. 2004;322:1310. doi: 10.1016/j.bbrc.2004.08.035. [DOI] [PubMed] [Google Scholar]
- 88.Choo YS, Johnson GV, MacDonald M, Detloff PJ, Lesort M. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum Mol Genet. 2004;13:1407. doi: 10.1093/hmg/ddh162. [DOI] [PubMed] [Google Scholar]
- 89.Lim D, Fedrizzi L, Tartari M, Zuccato C, Cattaneo E, Brini M, Carafoli E. Calcium homeostasis and mitochondrial dysfunction in striatal neurons of Huntington disease. J Biol Chem. 2008;283:5780. doi: 10.1074/jbc.M704704200. [DOI] [PubMed] [Google Scholar]
- 90.St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S, Handschin C, Zheng K, Lin J, Yang W, Simon DK, Bachoo R, Spiegelman BM. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 91.Weydt P, Pineda VV, Torrence AE, Libby RT, Satterfield TF, Lazarowski ER, Gilbert ML, Morton GJ, Bammler TK, Strand AD, Cui L, Beyer RP, Easley CN, Smith AC, Krainc D, Luquet S, Sweet IR, Schwartz MW, La Spada AR. Thermoregulatory and metabolic defects in Huntington's disease transgenic mice implicate PGC-1alpha in Huntington's disease neurodegeneration. Cell Metab. 2006;4:349. doi: 10.1016/j.cmet.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 92.Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 93.Arany Z, Wagner BK, Ma Y, Chinsomboon J, Laznik D, Spiegelman BM. Gene expression-based screening identifies microtubule inhibitors as inducers of PGC-1alpha and oxidative phosphorylation. Proc Natl Acad Sci U S A. 2008;105:4721. doi: 10.1073/pnas.0800979105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Orr AL, Li S, Wang CE, Li H, Wang J, Rong J, Xu X, Mastroberardino PG, Greenamyre JT, Li XJ. N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J Neurosci. 2008;28:2783. doi: 10.1523/JNEUROSCI.0106-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Majumder P, Raychaudhuri S, Chattopadhyay B, Bhattacharyya NP. Increased caspase-2, calpain activations and decreased mitochondrial complex II activity in cells expressing exogenous huntingtin exon 1 containing CAG repeat in the pathogenic range. Cell Mol Neurobiol. 2007;27:1127. doi: 10.1007/s10571-007-9220-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Greenamyre JT, Sherer TB, Betarbet R, Panov AV. Complex I and Parkinson's disease. IUBMB Life. 2001;52:135. doi: 10.1080/15216540152845939. [DOI] [PubMed] [Google Scholar]
- 97.Schapira AH. Mitochondria in the aetiology and pathogenesis of Parkinson's disease. Lancet Neurol. 2008;7:97. doi: 10.1016/S1474-4422(07)70327-7. [DOI] [PubMed] [Google Scholar]
- 98.Danzer KM, Haasen D, Karow AR, Moussaud S, Habeck M, Giese A, Kretzschmar H, Hengerer B, Kostka M. Different species of alpha-synuclein oligomers induce calcium influx and seeding. J Neurosci. 2007;27:9220. doi: 10.1523/JNEUROSCI.2617-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dufty BM, Warner LR, Hou ST, Jiang SX, Gomez-Isla T, Leenhouts KM, Oxford JT, Feany MB, Masliah E, Rohn TT. Calpain-cleavage of alpha-synuclein: connecting proteolytic processing to disease-linked aggregation. Am J Pathol. 2007;170:1725. doi: 10.2353/ajpath.2007.061232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dawson TM. Parkin and defective ubiquitination in Parkinson's disease. J Neural Transm Suppl. 2006:209. doi: 10.1007/978-3-211-45295-0_32. [DOI] [PubMed] [Google Scholar]
- 101.Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, Klose J, Shen J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem. 2004;279:18614. doi: 10.1074/jbc.M401135200. [DOI] [PubMed] [Google Scholar]
- 102.Jiang H, Ren Y, Zhao J, Feng J. Parkin protects human dopaminergic neuroblastoma cells against dopamine-induced apoptosis. Hum Mol Genet. 2004;13:1745. doi: 10.1093/hmg/ddh180. [DOI] [PubMed] [Google Scholar]
- 103.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wood-Kaczmar A, Gandhi S, Yao Z, Abramov AS, Miljan EA, Keen G, Stanyer L, Hargreaves I, Klupsch K, Deas E, Downward J, Mansfield L, Jat P, Taylor J, Heales S, Duchen MR, Latchman D, Tabrizi SJ, Wood NW. PINK1 is necessary for long term survival and mitochondrial function in human dopaminergic neurons. PLoS ONE. 2008;3:e2455. doi: 10.1371/journal.pone.0002455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, Chung J. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- 106.Deng H, Dodson MW, Huang H, Guo M. The Parkinson's disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci U S A. 2008;105:14503. doi: 10.1073/pnas.0803998105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yang Y, Gehrke S, Imai Y, Huang Z, Ouyang Y, Wang JW, Yang L, Beal MF, Vogel H, Lu B. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci U S A. 2006;103:10793. doi: 10.1073/pnas.0602493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Marongiu R, Spencer B, Crews L, Adame A, Patrick C, Trejo M, Dallapiccola B, Valente EM, Masliah E. Mutant Pink1 induces mitochondrial dysfunction in a neuronal cell model of Parkinson's disease by disturbing calcium flux. J Neurochem. 2009 doi: 10.1111/j.1471-4159.2009.05932.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A. 2005;102:16842. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Biskup S, Moore DJ, Celsi F, Higashi S, West AB, Andrabi SA, Kurkinen K, Yu SW, Savitt JM, Waldvogel HJ, Faull RL, Emson PC, Torp R, Ottersen OP, Dawson TM, Dawson VL. Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann Neurol. 2006;60:557. doi: 10.1002/ana.21019. [DOI] [PubMed] [Google Scholar]
- 111.Iaccarino C, Crosio C, Vitale C, Sanna G, Carri MT, Barone P. Apoptotic mechanisms in mutant LRRK2-mediated cell death. Hum Mol Genet. 2007;16:1319. doi: 10.1093/hmg/ddm080. [DOI] [PubMed] [Google Scholar]
- 112.Dodson MW, Guo M. Pink1, Parkin, DJ-1 and mitochondrial dysfunction in Parkinson's disease. Curr Opin Neurobiol. 2007;17:331. doi: 10.1016/j.conb.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 113.Andres-Mateos E, Perier C, Zhang L, Blanchard-Fillion B, Greco TM, Thomas B, Ko HS, Sasaki M, Ischiropoulos H, Przedborski S, Dawson TM, Dawson VL. DJ-1 gene deletion reveals that DJ-1 is an atypical peroxiredoxin-like peroxidase. Proc Natl Acad Sci U S A. 2007;104:14807. doi: 10.1073/pnas.0703219104. [DOI] [PMC free article] [PubMed] [Google Scholar]