

Abstract
γ-Glutamyl transpeptidase (GGT) plays critical roles in glutathione homeostasis and metabolism. Rat GGT is a single-copy gene from which seven types of GGT mRNA with a common protein encoding sequence, but different 5′-untranslated regions, may be transcribed. We previously showed that type V-2 was the predominant form of GGT mRNA in rat L2 epithelial cells, and that it could be induced by 4-hydroxynonenal (HNE) through the electrophile response element (EpRE) located in GGT promoter 5 (GP5). Here, we report transcription factors binding to GP5 EpRE and the involved signaling pathways. Immunodepletion gel shift assays demonstrated that GP5 EpRE bound JunB, c-Jun, FosB, and Fra2 from unstimulated cells, and that after exposure to HNE, EpRE binding complexes contained nuclear factor erythroid 2-related factor (Nrf) 1, Nrf2, JunB, c-Jun, FosB, c-Fos, Fra1, and Fra2. HNE-induced binding of Nrf2 and c-Jun in GP5 EpRE was confirmed by chromatin immunoprecipitation assays. Using reporter assays and specific inhibitors, we found that HNE induction of rat GGT mRNA V-2 was dependent on activation of extracellular signal-regulated kinase (ERK) and p38 mitogen-activated protein kinase (MAPK), but not protein kinase C or phosphatidylinositol 3-kinase. Pretreatment with ERK and p38MAPK inhibitors also blocked HNE-increased EpRE binding. HNE-increased nuclear content of Nrf1, Nrf2, and c-Jun in L2 cells was partially blocked by inhibition of either ERK1/2 or p38MAPK and completely blocked by simultaneous inhibition of both MAPKs. In conclusion, HNE induces GGT mRNA V-2 through altered EpRE transcription factor binding mediated by both ERK and p38MAPK.
Keywords: electrophile response element, γ-glutamyl transpeptidase, glutathione, 4-hydroxynonenal, nuclear factor erythroid 2-related factor 2
γ-Glutamyl transpeptidase (γ-glutamyl transferase; GGT) plays key roles in the maintenance of glutathione homeostasis (1-8), detoxification of xenobiotics, and metabolism of endogenous biomolecules (9). Studies with GGT-deficient cells and animal models have revealed that GGT is an integral part of the antioxidant and detoxification system in the lung (3-6). Like several other stress response enzymes, GGT is also responsive to oxidative stress with increase of its activity and mRNA levels. Substances that generate reactive oxygen species/reactive nitrogen species (ROS/RNS) and/or perturb the redox balance can increase GGT expression in various cells and tissues, including lung (10-13). We have previously reported that GGT was induced by redox-cycling quinones and the lipid peroxidation product 4-hydroxynonenal (HNE) in rat lung type II (L2) cells (14-18), and have been interested in defining the underlying mechanism(s). The rat GGT gene exists as a single copy, the expression of which is regulated by five tandemly arranged promoters that generate seven transcripts having different 5′-untranslated regions but the same protein coding sequence (19). Recently, we found that mRNA V-2 was the major form of GGT mRNAs that were induced by HNE in rat L2 cells (18), and its induction was mediated through the electrophile response element (EpRE) located in the proximal region of GGT promoter 5 (GP5) (Zhang and colleagues, unpublished data).
EpRE (also called antioxidant response element) was first identified in the promoter of rat glutathione S-transferase Ya (alpha form) (20, 21). It is now known that EpRE plays a critical role in the constitutive and oxidative/electrophilic stress-induced expression of many antioxidant and phase II enzymes, including reduced nicotinamide adenine dinucleotide phosphate: quinone oxidoreductase-1 (22), heme oxygenase-1 (23), and glutamate cysteine ligase (GCL) (24, 25). Many transcription factors have been reported to bind to EpRE, including nuclear factor erythroid 2-related factor (Nrf) 1, Nrf2, c-Jun, JunB, JunD, c-Fos, Fra1, Fra2, and small avian musculoaponeurotic fibrosarcoma (Maf) proteins (26-30). Among them, Nrf2 is a basic leucine zipper protein belonging to the cap “n” collar family of transcription factors and is a well established player in EpRE signaling. Under unstimulated conditions, Nrf2 is sequestered in the cytosol through association with Kelch-like ECH-associated protein1 (Keap1), which anchors it to the cytoskeleton. Recently, it was found that Keap1 could function as ubiquitin-E3 ligase and mediate the constant degradation of Nrf2 by the proteosome system (31, 32). Upon exposure of cells to electrophiles, Nrf2 is dissociated from Keap1 and escapes degradation. Released Nrf2 is then translocated to the nucleus, where it forms heterodimers with other leucine zipper proteins, such as c-Jun and small Mafs, and binds to EpRE (33-35). Another Nrf family member, Nrf1, has also been found to be involved in phase II and antioxidant gene induction (36, 37). Nrf1 contains an Nrf2-ECH homology 2 (Neh2) domain that is the same domain through which Nrf2 associates with Keap1, suggesting the possibility that it may also associate with Keap1 and follow regulatory pathways similar to those followed by Nrf2.
Several signaling pathways, including extracellular signal-regulated kinase (ERK), p38 mitogen-activated protein kinase (MAPK), c-Jun N-terminal kinase (JNK), protein kinase C (PKC) and phosphatidylinositol 3-kinase (PI3K), have been shown to be involved in EpRE-mediated gene induction (34, 38-43). These signaling kinases may regulate EpRE activity through Nrf2. Some reports suggest that the phosphorylation of Nrf2 by PKC is critical for Nrf2-Keap1 dissociation and Nrf2 activation (40, 44). As for the involvement of MAPK pathways in Nrf2 activation, Zipper and Mulcahy (45) found that ERK and p38MAPK were not involved in Nrf2 dissociation, but only mediated Nrf2 nuclear translocation, and this was unrelated to Nrf2 phosphorylation. They propose that, instead of acting on Nrf2 itself, MAPK may indirectly increase Nrf2 nuclear translocation by phosphorylating unknown factors related to Nrf2 nuclear translocation.
Driven by our previous finding that GGT mRNA V-2 was induced through EpRE, we continue here to investigate the transcription factors that bind to the electrophile response element in rat GP5 EpRE in response to HNE treatment, and to define the upstream signaling pathways leading to GP5 EpRE activation. Our results suggest that Nrf1, Nrf2, c-Jun, JunB, FosB, c-Fos, Fra1, and Fra2 are components of the GP5 EpRE/protein complex after HNE exposure in L2 cells, and that the increased nuclear content of Nrf1, Nrf2, and c-Jun is mediated through both ERK and p38MAPK pathways.
MATERIALS AND METHODS
Chemicals and Reagents
Unless otherwise noted, all chemicals were from Sigma (St. Louis, MO). HNE was purchased from Cayman Chemical (Ann Arbor, MI). Antibodies were from Santa Cruz (Santa Cruz, CA). Basic pGL3 luciferase vector, competent cells, gel shift assay kit, luciferase activity assay kit, and restriction enzymes were from Promega (Madison, WI). Chromatin immunoprecipitation (ChIP) assay kit was from Upstate (Chicago, IL). FuGENE 6 transfection reagent was from Roche (Indianapolis, IN). M-PER mammalian protein extraction reagent and NE-PER nuclear extraction reagent were from Pierce (Rockford, IL). Kinase inhibitors were purchased from Calbiochem (La Jolla, CA). All chemicals used were at least analytical grade.
Cell Culture and Treatments
L2 cells (from the American Type Culture Collection) were cultured in F-12K medium (Life Technologies, Grand Island, NY) supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin in a humidified incubator containing 5% CO2 at 37°C.
HNE was dissolved in ethanol, and the final concentration of ethanol in the medium was 0.05%. L2 cells were treated at ~90% confluence with HNE. Cells were rinsed with cold PBS before being harvested using rubber policemen.
Western Blotting Assay
Western blotting was performed as described previously (46). Briefly, protein was extracted, and 25 μg protein was heated for 15 min at 95°C in a 2× loading buffer containing SDS (Tris base, pH 6.5, glycerol, DTT, and pyronin Y), electrophoresed under denaturing conditions on a 10% Tris-glycine acrylamide gel (Invitrogen, Carlsbad, CA), and then electroblotted onto a polyvinylidene difluoride (PVDF) membrane (Immobilon P; Millipore, Bedford, MA). Membranes were blocked with 5% fat-free milk at room temperature for 1 h, and then incubated overnight at 4°C with appropriate primary antibody in 5% milk in Trisbuffered saline (TBS). After being washed with TBS containing 0.05% Tween 20 (TTBS), the membrane was incubated with appropriate secondary antibody at room temperature for 2 h. After TTBS washing, the membrane was treated with an enhanced chemiluminescence (ECL Plus; Amersham, Arlington Heights, IL) reagent mixture for 5 min. The target bands were imaged on a Kodak Image Station 2000R (Eastman Kodak Co., New Haven CT).
Plasmids
A DNA fragment of GP5 was amplified using primer pairs with specific nuclease digestion site, with DNA from L2 cells as the PCR template. The reverse primer was 5′-GCTAGATCTTGTCTT TGTGCTACTG-3′ (Bgl II), and the forward primer was 5′-CTACGC GTGGATGGATA GATAGATA-3′ (Mlu I). GP5 (-645/+18)-Luc was made by cloning the digested PCR product into basic pGL-3 luciferase vector.
Transfection Procedure and Assay of Luciferase and β-Galactosidase Activity
Cells (70-80% confluence) were transfected with plasmids by using FuGENE 6 transfection reagent (Roche), and β-galactosidase plasmid (1/10 of total amount of plasmids) was cotransfected as an internal control. Twelve hours after transfection, the medium was replaced; 24 h later, the cells were treated with and without HNE. The cell pellet was lysated with M-PER mammalian protein extraction reagent (Pierce) and centrifuged at 12,500 × g for 5 min. The supernatant was then used for determination of the activity of luciferase and β-galactosidase.
To determine the β-galactosidase activity, 25 μl of supernatant was added to a reaction mixture containing 300 μM 4-methyllumbelliferyl β-D-galactoside. After incubation at room temperature for 20 min with shaking, β-galactosidase activity was determined in a fluorescence microplate reader (Molecular Device Corp., Sunnyvale, CA) at an excitation wavelength of 360 nm and an emission wavelength of 450 nm.
For the luciferase assay, a luciferase assay kit (Promega) was used. Briefly, 20 μl of cell lysate was added to the reaction mixture provided in the kit, and luciferase activity was determined in a luminometor (Berthold Detection Systems, Pforzheim, Germany). The final luciferase activity was normalized with the activity of cotransfected β-galactosidase.
Electrophoretic Mobility Shift Assay
Electrophoretic mobility shift assay (EMSA) was performed as described previously (47). Briefly, nuclear extracts were prepared from L2 cells treated with or without HNE using NE-PER nuclear extraction reagent (Pierce). A total of 8 μg nuclear extract was preincubated in a gel shift binding reaction containing 4% glycerol, 1 mM MgCl2, 0.5 mM EDTA, 4 mM DTT, 50 mM NaCl, 10 mM Tris-HCl (pH 7.5), and 0.2 μg poly (dI-dC) at room temperature for 10 min before 32P-γ-ATP end-labeled double-stranded oligonucleotides were added. Samples were then incubated for an additional 20 min at room temperature. The samples were electrophoresed in 6% DNA retardation gel at 150 V for 2-3 h. Gels were dried and scanned with the Cyclone Storage Phosphor System, and the total counts were quantified with OptiQuant Image analysis software (Packark Instrument Co., Meriden, CT). The sequence of the sense oligonucleotide used was 5′-GTAC CCACAAT GACACAGCAAGAAAGCCT-3′.
Immunodepletion EMSA Assay
To determine the EpRE binding proteins, 4 μg of antibodies against specific proteins were added to the EMSA reaction mixture and incubated for 1 h at room temperature before radiolabeled oligonucleotides were added. Because the addition of antibodies produced bands that have principally decreased intensity of DNA binding (immunodepletion) rather than producing a clear shift, the decrease in the intensity of the bands was used for quantitation (47, 48). An antibody to the p65 component of NF-κB was used to demonstrate that the decrease in binding was not a result of nonspecific interaction.
ChIP Assay
ChIP assays were performed by following a protocol provided with the kit from Upstate. Briefly, cells were incubated with formaldehyde by directly adding it into the medium (1% final concentration) at room temperature (49) for 10 min. The cell pellet was then lysed on ice for 10 min and sonicated under conditions that cause DNA to be broken into 200- to 800-bp fragments. Sonicated cell lysate was precleared with 75 μl of salmon sperm DNA/agarose, and the supernatant was used for immunoprecipitation with antibodies to specific transcription factors overnight at 4°C. The protein/DNA complex was eluted from agarose in elution buffer; the DNA/protein complex was reversed by adding 5 M NaCl and incubating the mixture at 65°C for 4 h. The DNA was extracted with phenol:chloroform:isoamyl alcohol (25:24:1). Primers used for PCR in the ChIP assay were forward, 5′-CAGTACGTGGAAATCC TTATCA-3′,and reverse, 5′-GTGGA ATAGAGTGGGAGCAT-3′.
Statistical Analysis
SigmaStat software (SPSS Science, Chicago, IL) was used for statistical analysis, and statistical significance was accepted when P < 0.05. Comparison of variants between experimental groups was performed with ANOVA and Tukey's test.
RESULTS
Identification of Transcription Factors Binding to GP5 EpRE by Immunodepletion EMSA Assay
Previously, we demonstrated that mRNA V-2, one of the major GGT transcripts expressed in rat lung, was induced by HNE through the EpRE motif in the proximal region of GP5 EpRE in rat lung L2 cells (Zhang and colleagues, unpublished data).
By using antibodies against transcription factors known to bind EpRE in other genes, we performed an immunodepletion gel shift assay to determine the nuclear proteins binding GP5 EpRE. In this assay, an interaction between the antibody and the protein interferes with the ability of the protein to bind DNA, and this causes a decrease in the EpRE complex when the target protein is present (immunodepletion). Under resting conditions, the intensity of the DNA/protein complex was reduced by the presence of antibodies against JunB, c-Jun, FosB, and Fra2 (Figure 1A), suggesting that these transcription factors may constitutively bind GP5 EpRE. The intensity of the HNE-induced EpRE complex was significantly decreased by antibodies against Nrf1, Nrf2, FosB, c-Fos, JunB, c-Jun, Fra1, and Fra2 (Figure 1A), indicating their presence in the complex after HNE stimulation. If the immunodepletion efficiency is assumed to be 100% (because the addition of antibody could completely abrogate DNA binding capability of the target protein), the percentage of immunodepletion should represent the relative amount of the protein in the EpRE complex. Thus, more than 90% of the HNE-stimulated complex contained Nrf1 and Nrf2 together; ~ 75% of the HNE-stimulated complex contained JunB and c-Jun, and 60% of the HNE-stimulated complex contained Fra2, a Fos family member (Figure 1B). Bands were not depleted completely by the antibodies because the protein complexes bound to the EpRE are composed of a mixture of dimers. Thus, dimers containing combinations of transcription factors other than the one with which the antibody reacts are not depleted. So, our assumption was that any antibody to a particular transcription factor, X, was 100% effective in decreasing complexes containing X, whereas any remaining band contained complexes that did not contain X.
Figure 1.

Analysis of GP5 EpRE binding proteins. (A) Immunodepletion EMSA. L2 cells were treated with vehicle or 15 μM HNE for 1 h. Nuclear extracts were incubated with the appropriate antibody for 1 h at room temperature, and the complex was analyzed by EMSA. DNA binding activity was decreased by preincubation with an antibody that interferes with the binding of the target protein. (B) Semiquantitative summary based on graphic values generated by Cyclone Storage Phosphor System from four independent experiments; the immunodepletion effect of the antibody is plotted as the percentage of immunodepletion (of the DNA binding complex). Values plotted are relative means ± SE (control, white bars;15 μM, gray bars). *P < 0.05 compared with no antibody control. #P < 0.05 compared with no antibody HNE. Ab, antibody.
Confirmation of EpRE Binding Proteins In Vivo by ChIP Assay
The gel shift assay (EMSA) determines the potential protein association with a particular DNA cis-element sequence ex vivo, but it may not reflect what actually occurs at the cis-element in a specific promoter in vivo. To confirm the binding of Nrf2 and c-Jun to GP5 EpRE in vivo, we performed ChIP assays. As shown in Figure 2A, after HNE exposure, the recruitment of Nrf2 and the binding of c-Jun to the promoter (GP5 EpRE) were increased significantly. These data confirm that Nrf2 and c-Jun are two of the proteins bound to GP5 EpRE, and their binding was increased after HNE treatment. Figure 2B shows that, in a negative control experiment, activating transcription factor (ATF) 2 was not part of the HNE-induced EpRE complex, but that Nrf2 was.
Figure 2.
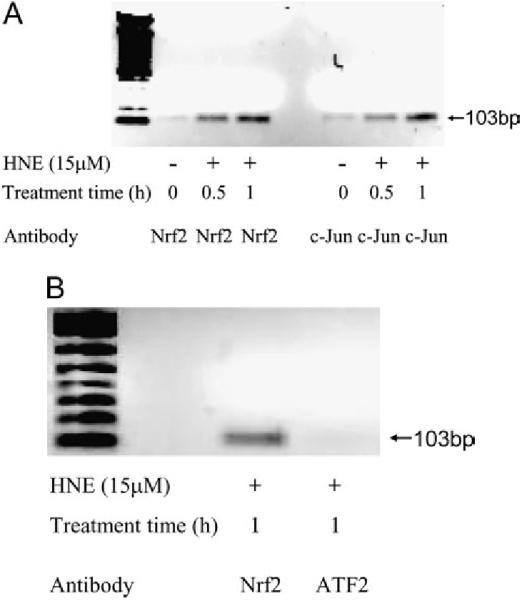
Recruitment of Nrf 2 and c-Jun to EpRE of GP5 in HNE-stimulated L2 cells. After stimulation with 15 μM HNE, cells were fixed with 1% formaldehyde, lysed; and sonicated to shear chromatin in 0.2- to 0.8-kb fragments, which were then immunoprecipitated with anti-Nrf2 antibody. Nrf2-coprecipitating DNA was analyzed by PCR with specific primers amplifying GP5. (A) Binding of Nrf2 and c-Jun to GP5 EpRE in vivo;(B) negative control.
PD98059 and SB203580 Inhibit HNE-Induced GP5/reporter Transgene Expression
We reported previously that both ERK and p38MAPK pathways were involved in HNE-mediated GGT induction in L2 cells, and that V-2 was the major type of GGT mRNA induced by HNE (18); however, the direct connection between ERK, p38MAPK, and HNE induction of GGT mRNA V-2 has not been established. To investigate this, GP5 (-645/+18)-Luc, a reporter plasmid that contains the proximal region of rat GP5 and has been shown to respond to HNE treatment (Zhang and colleagues, unpublished data) was used. L2 cells transiently transfected with GP5 (-645/+18)-Luc were pretreated with and without MAPK inhibitors for 1 h before HNE exposure. PD98059 and SB203580, an ERK inhibitor and a p38MAPK inhibitor, respectively, had no inhibitory effect on basal luciferase expression, but significantly reduced HNE-induced luciferase activity from 1.7-fold to 1.3-fold (Figure 3A). In cells pretreated with PD98059 and SB203580 simultaneously, HNE-induced luciferase activity was completely eliminated. At a concentration that completely inhibits JNK activity (18), a c-Jun N-terminal kinase inhibitor, JNKi, had no effect on HNE-induced transgene expression. Although PKC and PI3K are involved in EpRE-mediated gene induction in other systems, various PKC and PI3K inhibitors had no inhibitory effect on HNE-mediated transgene induction in L2 cells (Figure 3B). These data reveal that both ERK and p38MAPK, but not PKC or PI3K, are involved in HNE-mediated GP5 activation.
Figure 3.

Signaling pathways involved in HNE-induced activation of rat GP5. PD: PD98059, 50 μM; SB: SB203580, 10 μM; JNK inhibitor (JNKi), 10 μM. (A) PD98059 and SB203580 abrogated HNE-induced transgene expression of rat GP5. *P < 0.01 compared with control; #P < 0.01 compared with HNE alone, n = 3. (B) PKC and PI3K inhibitors have no effect on HNE-mediated transgene induction. L2 cells were transiently transfected with GP5 (-645/+18)-Luc for 24 h and pretreated with inhibitors for 1 h before being treated with and without HNE. Luciferase activity was determined 24 h after HNE treatment, and transfection efficiency was controlled by normalization with cotransfected β-galactosidase activity. (C PD98059 and SB203580 eliminated the HNE-mediated increase of the EpRE complex. After pretreatment with inhibitors, L2 cells were treated with and without 15 μM HNE for 1 h and nuclear extracts were prepared. Synthetic oligonucleotides containing GP5 EpRE were end-labeled and incubated with nuclear extract, and the DNA/protein complex was analyzed by EMSA. MAPKi, MAPK inhibitor.
Because EpRE is essential for HNE-mediated GP5 activition (Zhang and colleagues, unpublished data), we next determined whether ERK and p38MAPK participate by regulating the alteration of the GP5 EpRE complex. Without inhibitor pretreatment, HNE induced a significant increase in the EpRE DNA/protein complex. Pretreatment with either PD98059 or SB203580 markedly reduced HNE-mediated increase of the EpRE DNA/protein complex (Figure 3C). This suggests that MAPKs may regulate EpRE signaling through affecting the binding of transcription factors to EpRE.
Effects of MAPK Inhibitors on the Nuclear Content of EpRE Binding Proteins
To study how ERK and p38MAPK are involved in GP5 EpRE signaling and which EpRE binding proteins are affected by activation of these MAPKs, we further investigated the nuclear content of some EpRE binding proteins with and without MAPK inhibitor pretreatment. As shown in Figure 4, exposure to 15 μM HNE significantly increased the content of Nrf1, Nrf2, and c-Jun in the nucleus, but had no effect on that of JunB, Fra1, and Fra2. Pretreatment with either 50 μM PD98059 or 10 μM SB203580 partially reduced the HNE-mediated increase in nuclear Nrf1, Nrf2, and c-Jun. When L2 cells were simultaneously pretreated with PD98059 and SB203580, the HNE-mediated nuclear increase of Nrf1, Nrf2, and c-Jun was abrogated. Neither inhibitor affected the nuclear content of JunB, Fra1, and Fra2, which were involved in the EpRE complex in response to HNE. These data suggest that ERK and p38MAPK may regulate GP5 EpRE activation by affecting the nuclear content of Nrf1, Nrf2, and c-Jun in L2 cells.
Figure 4.
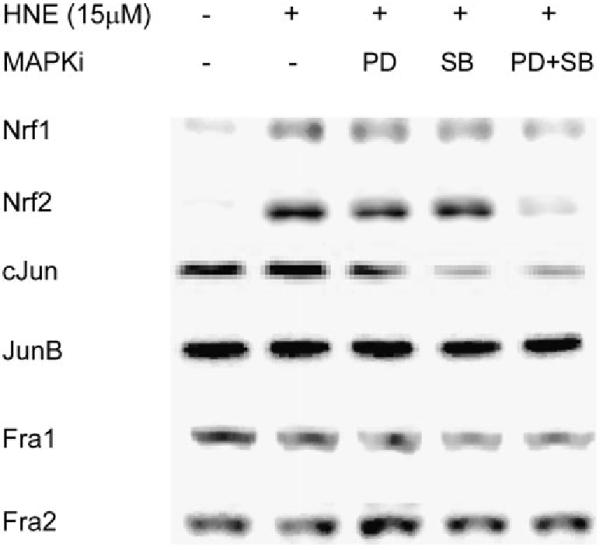
Effect of ERK and p38MAPK inhibitors on the nuclear content of EpRE binding proteins. Cells were pretreated with inhibitors for 1 h before being exposed to 15 μM HNE. Nuclear extracts were prepared 1 h after HNE treatment, and proteins were determined with Western blotting. PD, PD98059, 50 μM; SB, SB203580, 10 μM.
DISCUSSION
In the present study, we used immunodepletion gel shift assays to show that the transcription factors binding to EpRE of rat GP5 in unstimulated conditions include JunB, c-Jun, FosB, and Fra2, and that HNE exposure caused a significant increase in EpRE binding activity and changes in the composition of the binding complex, which was composed of Nrf1, Nrf2, JunB, c-Jun, c-Fos, FosB, Fra1, and Fra2 (Figure 1). We also confirmed the increased binding of Nrf2 and c-Jun to GP5 EpRE after HNE treatment in vivo (Figure 2). All of these transcription factors have previously been reported to bind EpRE in other genes. Among them, Nrf1, Nrf2, JunB, and c-Jun have been reported to positively regulate gene expression; there is evidence that c-Fos and Fra1 are negative regulators of gene expression (28, 50). Nrf1 and Nrf2 are basic leucine zipper proteins that do not form a homodimer or heterodimer with each other, and need other leucine zipper proteins to be active (51, 52). For example, Nrf1 and Nrf2 have been reported to associate with Jun proteins and positively regulate EpRE activity (50). Fra2 accounts for 60% of the GP5 EpRE complex, suggesting that Fra2 may associate with Nrf, Jun, or other proteins in the complex. Previously, Fra2 has been found to heterodimerize with small Maf proteins (Maf F/K/G) and bind to EpRE (53, 54), although its role in EpRE-mediated gene induction remains to be determined. Compared with other Fos proteins in the EpRE complex (Fra1 and Fra2), FosB and c-Fos together contribute ~40% of the complex after HNE stimulation. Whether FosB and c-Fos positively regulate GP5 EpRE, however, needs further study.
Using reporter assays, we demonstrate that both ERK1/2 and p38MAPK were positively involved in EpRE-mediated induction of promoter 5 activity by HNE (Figure 3A). We also found that PD98059 and SB203580, which are ERK and p38MAPK pathway inhibitors, respectively, decreased HNE-induced EpRE/protein complexes (Figure 3C). Both results suggest the involvement of ERK1/2 and p38MAPK in the regulation of HNE-induced GP5 EpRE activity in L2 cells. This finding is consistent with a report by Zipper and colleagues (34), who found that inhibition of ERK1/2 or p38MAPK additively blocked pyrrolidine dithiocarbamate-induced EpRE/protein binding complex and GCL transgene expression in human hepatocellular liver carcinoma cell line (HepG2) cells. In another study, Yu and coworkers also showed that the EpRE signaling could be attenuated by PD98059 and dominant negative ERK2 in HepG2 cells (55). Although p38MAPK was found to negatively regulate EpRE-mediated expression of murine glutathione S-transferase A1 in HepG2 and Hepa1c1c7 cells (56), its negative role in the regulation of EpRE was not consistently observed in other reports. Another MAPK, JNK, which has been shown to be activated by HNE (18, 57-60) and to be involved in EpRE-mediated gene induction (46), is not responsible for HNE induction of GGT in L2 cells (18). Although PKC and PI3K have been reported to be involved in EpRE-mediated gene induction (39, 43, 61-63), they are not involved in HNE induction of GP5 EpRE in L2 cells (Figure 3B).
Upon exposure to HNE, Nrf2 is activated (as measured by increased nuclear content) through both the ERK and p38MAPK pathways (Figure 4), but not the PKC or PI3K pathways (data not shown). The process of Nrf2 activation includes dissociation from Keap1, nuclear translocation, dimerization, and binding to EpRE. All these steps are thought to be regulated in a concerted way for Nrf2 activation. The two mechanisms that have been proposed for Nrf2-Keap1 dissociation are Nrf2 phosphorylation and Keap1 modification. Huang and colleagues found that inhibition of PKC blocked Nrf2 phosphorylation and nuclear accumulation induced by tert-butylhydro-quinone in HepG2 cells (44) without an effect on Nrf2 nuclear translocation (64). A mechanism for this was defined in a report from Bloom and Jaiswal (40), who showed that phosphorylation of Nrf2 at Ser40 by PKC was necessary for release of Nrf2 from Keap1, but that PKC was not required for Nrf2 stabilization/accumulation. The alternative hypothesis of Nrf2 liberation, via modification of Keap1, proposes that the active thiol groups on Keap1 could act as oxidative stress sensors and that modification of the thiol groups by ROS or electrophiles disrupts the Nrf2-Keap1 complex and leads to Nrf2 activation. This hypothesis is supported by evidence of constitutive Nrf2 activation when the active cysteine residues on Keap1 are mutated (35, 65, 66), and by the conjugate formation between Keap1 and electrophiles through the proposed sulfhydryl groups (67). The HNE-induced Nrf2 activation in L2 cells could be explained by the Keap1 modification theory. Although Numazawa and coworkers showed that atypical PKC iota was involved in Nrf2 phosphorylation and nuclear translocation by HNE (63), pretreatment with PKC inhibitors decreased neither GP5 EpRE-mediated gene induction (Figure 3B) nor HNE-stimulated Nrf2 nuclear translocation (data not shown) in L2 cells. Although PKC has been reported to be involved in Nrf2-Keap1 dissociation, Keap1-HNE conjugation (67) may not require participation of PKC.
Our finding that ERK1/2 and p38MAPK were involved in HNE-induced Nrf2 nuclear accumulation in L2 cells (Figure 4) is similar to that of a previous report by Zipper and Mulcahy (45), who found that pyrrolidine dithiocarbamate-induced Nrf2 nuclear localization could be diminished by inhibiting either ERK or p38MAPK alone, and could be abrogated by inhibiting both kinases. Because ERK and p38MAPK are not involved in the release of Nrf2 from Keap1 (68), and phosphorylation of Nrf2 was not required for Nrf2 transactivation (45, 69), a new target of signaling kinases, cAMP response element binding protein (CREB)-binding protein (CBP, or CREB-binding protein) was proposed. Cotransfection of CBP and MAPK kinase synergistically increased Nrf2 activity (69). Indeed, CBP has been reported to bind to Nrf2 transactivation domain (70), and it could be phosphorylated by ERK (71-76). CBP also functions as the coactivitor of c-Jun (77), another EpRE binding protein identified in this report. Interestingly, phosphorylation of CREB, the coactivator of CBP, could be mediated by ERK/ribosome S6 kinase or p38MAPK/MAPK-activated protein kinase 2 pathways; inhibiting either pathway partially, and inhibiting both pathways simultaneously, completely abrogated CREB phosphorylation and activation (78). However, currently, roles for CBP in Nrf2 nuclear translocation and for CREB in EpRE signaling are hypothetical. Collectively, ERK1/2 and p38MAPK are both involved in HNE-mediated Nrf2 nuclear translocation in L2 cells, but the underlying mechanism needs further investigation.
In this study, we also found that the nuclear content of another Nrf family member, Nrf1, was also increased by HNE stimulation (Figure 4). Nrf1 has been found to play a critical role in embryo development (79), and is involved in the regulation of antioxidant genes, such as GCL (36, 37). Nrf1 contains a Neh2 domain (Nrf2 associates with Keap1 through an Neh2 domain), suggesting that Nrf1 may also associate with Keap1 and be regulated in a manner similar to that in which Nrf2 is regulated. Indeed, as with Nrf2, ERK and p38MAPK are also involved in Nrf1 nuclear accumulation after HNE exposure. Examining the association between Nrf1 and Keap1 in vivo, using techniques such as ChIP assays, may provide further information on the mechanism of Nrf1 activation by ROS or electrophiles.
Aside from directly targeting Nrf2, stress signals can also regulate EpRE function by targeting Nrf2 partners or other EpRE binding proteins; for example, it was reported that Jun proteins (JunB, c-Jun, and JunD) were critical for EpRE-mediated human quinone oxidoreductase-1 gene induction in HepG2 cells (50). In the present research, c-Jun was significantly increased in the nucleus after HNE exposure, and this could be partially decreased by inhibiting either ERK1/2 or p38MAPK, and completely blocked by inhibiting both ERK1/2 and p38MAPK (Figure 4). The transcription response of c-Jun is mediated by several cis elements, including ATF1, myocyte enhancer factor 2, and ATF c-Jun binding sites (80-82). Although the intracellular pathways linking HNE to c-Jun transcription are not completely known, several reports have demonstrated that ERK and/or p38MAPK is involved in c-Jun expression (83-87) through phosphorylating and activating ATF1 (79, 85, 88), myocyte enhancer factor 2 (87), and/or ATF2 (89, 90).
In summary, we have identified the proteins that bind to EpRE of rat GP5, and the signaling pathways involved. In resting conditions, the EpRE complex included JunB, c-Jun, FosB, and Fra2; after HNE stimulation, the transcription factors in the complex were Nrf1/2, JunB, c-Jun, FosB, c-Fos, Fra1, and Fra2. The HNE induction of GGT mRNA V-2 is mediated through ERK and p38MAPK, which are responsible for the increased nuclear content of Nrf1/2 and c-Jun after HNE stimulation.
Footnotes
Conflict of Interest Statement: None of the authors has a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Hanigan MH, Ricketts WA. Extracellular glutathione is a source of cysteine for cells that express gamma-glutamyl transpeptidase. Biochemistry. 1993;32:6302–6306. doi: 10.1021/bi00075a026. [DOI] [PubMed] [Google Scholar]
- 2.Griffith OW, Meister A. Excretion of cysteine and gamma-glutamylcysteine moieties in human and experimental animal gamma-glutamyl transpeptidase deficiency. Proc Natl Acad Sci USA. 1980;77:3384–3387. doi: 10.1073/pnas.77.6.3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lieberman MW, Wiseman AL, Shi ZZ, Carter BZ, Barrios R, Ou CN, Chevez-Barrios P, Wang Y, Habib GM, Goodman JC, et al. Growth retardation and cysteine deficiency in gamma-glutamyl transpeptidase-deficient mice. Proc Natl Acad Sci USA. 1996;93:7923–7926. doi: 10.1073/pnas.93.15.7923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rojas E, Valverde M, Kala SV, Kala G, Lieberman MW. Accumulation of DNA damage in the organs of mice deficient in gamma-glutamyltranspeptidase. Mutat Res. 2000;447:305–316. doi: 10.1016/s0027-5107(99)00191-8. [DOI] [PubMed] [Google Scholar]
- 5.Jean JC, Liu Y, Brown LA, Marc RE, Klings E, Joyce-Brady M. Gammaglutamyl transferase deficiency results in lung oxidant stress in normoxia. Am J Physiol Lung Cell Mol Physiol. 2002;283:L766–L776. doi: 10.1152/ajplung.00250.2000. [DOI] [PubMed] [Google Scholar]
- 6.Barrios R, Shi ZZ, Kala SV, Wiseman AL, Welty SE, Kala G, Bahler AA, Ou CN, Lieberman MW. Oxygen-induced pulmonary injury in gamma-glutamyl transpeptidase-deficient mice. Lung. 2001;179:319–330. doi: 10.1007/s004080000071. [DOI] [PubMed] [Google Scholar]
- 7.Meister A. The gamma-glutamyl cycle: diseases associated with specific enzyme deficiencies. Ann Intern Med. 1974;81:247–253. doi: 10.7326/0003-4819-81-2-247. [DOI] [PubMed] [Google Scholar]
- 8.Stark AA, Porat N, Volohonsky G, Komlosh A, Bluvshtein E, Tubi C, Steinberg P. The role of gamma-glutamyl transpeptidase in the biosynthesis of glutathione. Biofactors. 2003;17:139–149. doi: 10.1002/biof.5520170114. [DOI] [PubMed] [Google Scholar]
- 9.Paolicchi A, Sotiropuolou M, Perego P, Daubeuf S, Visvikis A, Lorenzini E, Franzini M, Romiti N, Chieli E, Leone R, et al. Gamma-glutamyl transpeptidase catalyses the extracellular detoxification of cisplatin in a human cell line derived from the proximal convoluted tubule of the kidney. Eur J Cancer. 2003;39:996–1003. doi: 10.1016/s0959-8049(03)00067-4. [DOI] [PubMed] [Google Scholar]
- 10.Griffiths SA, Good VM, Gordon LA, Hudson EA, Barrett MC, Munks RJ, Manson MM. Characterization of a promoter for gamma-glutamyl transpeptidase activated in rat liver in response to aflatoxin B1 and ethoxyquin. Mol Carcinog. 1995;14:251–262. doi: 10.1002/mc.2940140405. [DOI] [PubMed] [Google Scholar]
- 11.Takahashi Y, Oakes SM, Williams MC, Takahashi S, Miura T, Joyce-Brady M. Nitrogen dioxide exposure activates gamma-glutamyl transferase gene expression in rat lung. Toxicol Appl Pharmacol. 1997;143:388–396. doi: 10.1006/taap.1996.8087. [DOI] [PubMed] [Google Scholar]
- 12.van Klaveren RJ, Hoet PH, Pype JL, Demedts M, Nemery B. Increase in gamma-glutamyltransferase by glutathione depletion in rat type II pneumocytes. Free Radic Biol Med. 1997;22:525–534. doi: 10.1016/s0891-5849(96)00375-9. [DOI] [PubMed] [Google Scholar]
- 13.Knickelbein RG, Ingbar DH, Seres T, Snow K, Johnston RB, Jr, Fayemi O, Gumkowski F, Jamieson JD, Warshaw JB. Hyperoxia enhances expression of gamma-glutamyl transpeptidase and increases protein S-glutathiolation in rat lung. Am J Physiol. 1996;270:L115–L122. doi: 10.1152/ajplung.1996.270.1.L115. [DOI] [PubMed] [Google Scholar]
- 14.Kugelman A, Choy HA, Liu R, Shi MM, Gozal E, Forman HJ. Gamma-glutamyl transpeptidase is increased by oxidative stress in rat alveolar L2 epithelial cells. Am J Respir Cell Mol Biol. 1994;11:586–592. doi: 10.1165/ajrcmb.11.5.7946387. [DOI] [PubMed] [Google Scholar]
- 15.Liu RM, Hu H, Robison TW, Forman HJ. Increased gamma-glutamylcysteine synthetase and gamma-glutamyl transpeptidase activities enhance resistance of rat lung epithelial L2 cells to quinone toxicity. Am J Respir Cell Mol Biol. 1996;14:192–197. doi: 10.1165/ajrcmb.14.2.8630270. [DOI] [PubMed] [Google Scholar]
- 16.Liu RM, Hu H, Robison TW, Forman HJ. Differential enhancement of gamma-glutamyl transpeptidase and gamma-glutamylcysteine synthetase by tert-butylhydroquinone in rat lung epithelial L2 cells. Am J Respir Cell Mol Biol. 1996;14:186–191. doi: 10.1165/ajrcmb.14.2.8630269. [DOI] [PubMed] [Google Scholar]
- 17.Liu RM, Shi MM, Giulivi C, Forman HJ. Quinones increase gamma-glutamyl transpeptidase expression by multiple mechanisms in rat lung epithelial cells. Am J Physiol. 1998;274:L330–L336. doi: 10.1152/ajplung.1998.274.3.L330. [DOI] [PubMed] [Google Scholar]
- 18.Zhang H, Dickinson DA, Liu RM, Forman HJ. 4-Hydroxynonenal increases gamma-glutamyl transpeptidase gene expression through mitogen-activated protein kinase pathways. Free Radic Biol Med. 2005;38:463–471. doi: 10.1016/j.freeradbiomed.2004.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chikhi N, Holic N, Guellaen G, Laperche Y. Gamma-glutamyl transpeptidase gene organization and expression: a comparative analysis in rat, mouse, pig and human species. Comp Biochem Physiol B Biochem Mol Biol. 1999;122:367–380. doi: 10.1016/s0305-0491(99)00013-9. [DOI] [PubMed] [Google Scholar]
- 20.Rushmore TH, King RG, Paulson KE, Pickett CB. Regulation of glutathione S-transferase Ya subunit gene expression: identification of a unique xenobiotic-responsive element controlling inducible expression by planar aromatic compounds. Proc Natl Acad Sci, USA. 1990;87:3826–3830. doi: 10.1073/pnas.87.10.3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rushmore TH, Pickett CB. Transcriptional regulation of the rat glutathione S-transferase Ya subunit gene. J Biol Chem. 1990;265:14648–14653. [PubMed] [Google Scholar]
- 22.Favreau LV, Pickett CB. The rat quinone reductase antioxidant response element: identification of the nucleotide sequence required for basal and inducible activity and detection of antioxidant response element-binding proteins in hepatoma and non-hepatoma cell lines. J Biol Chem. 1995;270:24468–24474. doi: 10.1074/jbc.270.41.24468. [DOI] [PubMed] [Google Scholar]
- 23.Alam J, Cook JL. Transcriptional regulation of the heme oxygenase-1 gene via the stress response element pathway. Curr Pharm Des. 2003;9:2499–2511. doi: 10.2174/1381612033453730. [DOI] [PubMed] [Google Scholar]
- 24.Moinova HR, Mulcahy RT. An electrophile responsive element (EpRE) regulates beta-naphthoflavone induction of the human gamma-glutamylcysteine synthetase regulatory subunit gene: constitutive expression is mediated by an adjacent AP-1 site. J Biol Chem. 1998;273:14683–14689. doi: 10.1074/jbc.273.24.14683. [DOI] [PubMed] [Google Scholar]
- 25.Mulcahy RT, Gipp JJ. Identification of a putative antioxidant response element in the 5′-flanking region of the human gamma-glutamylcysteine synthetase heavy subunit gene. Biochem Biophys Res Commun. 1995;209:227–233. doi: 10.1006/bbrc.1995.1493. [DOI] [PubMed] [Google Scholar]
- 26.Li Y, Jaiswal AK. Identification of jun-B as third member in human antioxidant response element-nuclear proteins complex. Biochem Biophys Res Commun. 1992;188:992–996. doi: 10.1016/0006-291x(92)91329-o. [DOI] [PubMed] [Google Scholar]
- 27.Li Y, Jaiswal AK. Regulation of human NAD(P)H:quinone oxidoreductase gene: role of AP1 binding site contained within human antioxidant response element. J Biol Chem. 1992;267:15097–15104. [PubMed] [Google Scholar]
- 28.Venugopal R, Jaiswal AK. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc Natl Acad Sci USA. 1996;93:14960–14965. doi: 10.1073/pnas.93.25.14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jeyapaul J, Jaiswal AK. Nrf2 and c-Jun regulation of antioxidant response element (ARE) -mediated expression and induction of gamma-glutamylcysteine synthetase heavy subunit gene. Biochem Pharmacol. 2000;59:1433–1439. doi: 10.1016/s0006-2952(00)00256-2. [DOI] [PubMed] [Google Scholar]
- 30.Dhakshinamoorthy S, Jaiswal AK. Small Maf (MafG and MafK) proteins negatively regulate antioxidant response element-mediated expression and antioxidant induction of the NAD(P)H:quinone oxidoreductase1 gene. J Biol Chem. 2000;275:40134–40141. doi: 10.1074/jbc.M003531200. [DOI] [PubMed] [Google Scholar]
- 31.Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol. 2003;23:8137–8151. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang DD, Lo SC, Cross JV, Templeton DJ, Hannink M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol. 2004;24:10941–10953. doi: 10.1128/MCB.24.24.10941-10953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zipper LM, Mulcahy RT. Inhibition of ERK and p38 MAP kinases inhibits binding of Nrf2 and induction of GCS genes. Biochem Biophys Res Commun. 2000;278:484–492. doi: 10.1006/bbrc.2000.3830. [DOI] [PubMed] [Google Scholar]
- 35.Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, Yamamoto M, Talalay P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci USA. 2002;99:11908–11913. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kwong M, Kan YW, Chan JY. The CNC basic leucine zipper factor, Nrf1, is essential for cell survival in response to oxidative stressinducing agents: role for Nrf1 in gamma-gcs(l) and gss expression in mouse fibroblasts. J Biol Chem. 1999;274:37491–37498. doi: 10.1074/jbc.274.52.37491. [DOI] [PubMed] [Google Scholar]
- 37.Myhrstad MC, Husberg C, Murphy P, Nordstrom O, Blomhoff R, Moskaug JO, Kolsto AB. TCF11/Nrf1 overexpression increases the intracellular glutathione level and can transactivate the gamma-glutamylcysteine synthetase (GCS) heavy subunit promoter. Biochim Biophys Acta. 2001;1517:212–219. doi: 10.1016/s0167-4781(00)00276-1. [DOI] [PubMed] [Google Scholar]
- 38.Alam J, Wicks C, Stewart D, Gong P, Touchard C, Otterbein S, Choi AM, Burow ME, Tou J. Mechanism of heme oxygenase-1 gene activation by cadmium in MCF-7 mammary epithelial cells: role of p38 kinase and Nrf2 transcription factor. J Biol Chem. 2000;275:27694–27702. doi: 10.1074/jbc.M004729200. [DOI] [PubMed] [Google Scholar]
- 39.Lee JM, Hanson JM, Chu WA, Johnson JA. Phosphatidylinositol 3-kinase, not extracellular signal-regulated kinase, regulates activation of the antioxidant-responsive element in IMR-32 human neuroblastoma cells. J Biol Chem. 2001;276:20011–20016. doi: 10.1074/jbc.M100734200. [DOI] [PubMed] [Google Scholar]
- 40.Bloom DA, Jaiswal AK. Phosphorylation of Nrf2 at Ser40 by protein kinase C in response to antioxidants leads to the release of Nrf2 from INrf2, but is not required for Nrf2 stabilization/accumulation in the nucleus and transcriptional activation of antioxidant response element-mediated NAD(P)H:quinone oxidoreductase-1 gene expression. J Biol Chem. 2003;278:44675–44682. doi: 10.1074/jbc.M307633200. [DOI] [PubMed] [Google Scholar]
- 41.Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol. 2003;23:7198–7209. doi: 10.1128/MCB.23.20.7198-7209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gong P, Hu B, Cederbaum AI. Diallyl sulfide induces heme oxygenase-1 through MAPK pathway. Arch Biochem Biophys. 2004;432:252–260. doi: 10.1016/j.abb.2004.09.024. [DOI] [PubMed] [Google Scholar]
- 43.Martin D, Rojo AI, Salinas M, Diaz R, Gallardo G, Alam J, De Galarreta CM, Cuadrado A. Regulation of heme oxygenase-1 expression through the phosphatidylinositol 3-kinase/Akt pathway and the Nrf2 transcription factor in response to the antioxidant phytochemical carnosol. J Biol Chem. 2004;279:8919–8929. doi: 10.1074/jbc.M309660200. [DOI] [PubMed] [Google Scholar]
- 44.Huang HC, Nguyen T, Pickett CB. Regulation of the antioxidant response element by protein kinase C-mediated phosphorylation of NF-E2-related factor 2. Proc Natl Acad Sci USA. 2000;97:12475–12480. doi: 10.1073/pnas.220418997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zipper LM, Mulcahy RT. Erk activation is required for Nrf2 nuclear localization during pyrrolidine dithiocarbamate induction of glutamate cysteine ligase modulatory gene expression in HepG2 cells. Toxicol Sci. 2003;73:124–134. doi: 10.1093/toxsci/kfg083. [DOI] [PubMed] [Google Scholar]
- 46.Dickinson DA, Iles KE, Watanabe N, Iwamoto T, Zhang H, Krzywanski DM, Forman HJ. 4-Hydroxynonenal induces glutamate cysteine ligase through JNK in HBE1 cells. Free Radic Biol Med. 2002;33:974. doi: 10.1016/s0891-5849(02)00991-7. [DOI] [PubMed] [Google Scholar]
- 47.Dickinson DA, Iles KE, Zhang H, Blank V, Forman HJ. Curcumin alters EpRE and AP-1 binding complexes and elevates glutamate-cysteine ligase gene expression. FASEB J. 2003;17:473–475. doi: 10.1096/fj.02-0566fje. [DOI] [PubMed] [Google Scholar]
- 48.Tyulmenkov VV, Klinge CM. Selectivity of antibodies to estrogen receptors alpha and beta (ERalpha and ERbeta) for detecting DNA-bound ERalpha and ERbeta in vitro. Steroids. 2000;65:505–512. doi: 10.1016/s0039-128x(00)00109-4. [DOI] [PubMed] [Google Scholar]
- 49.Orlando V. Mapping chromosomal proteins in vivo by formaldehydecrosslinked-chromatin immunoprecipitation. Trends Biochem Sci. 2000;25:99–104. doi: 10.1016/s0968-0004(99)01535-2. [DOI] [PubMed] [Google Scholar]
- 50.Venugopal R, Jaiswal AK. Nrf2 and Nrf1 in association with Jun proteins regulate antioxidant response element-mediated expression and coordinated induction of genes encoding detoxifying enzymes. Oncogene. 1998;17:3145–3156. doi: 10.1038/sj.onc.1202237. [DOI] [PubMed] [Google Scholar]
- 51.Chan JY, Han XL, Kan YW. Isolation of cDNA encoding the human NF-E2 protein. Proc Natl Acad Sci USA. 1993;90:11366–11370. doi: 10.1073/pnas.90.23.11366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moi P, Chan K, Asunis I, Cao A, Kan YW. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc Natl Acad Sci USA. 1994;91:9926–9930. doi: 10.1073/pnas.91.21.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kataoka K, Igarashi K, Itoh K, Fujiwara KT, Noda M, Yamamoto M, Nishizawa M. Small Maf proteins heterodimerize with Fos and may act as competitive repressors of the NF-E2 transcription factor. Mol Cell Biol. 1995;15:2180–2190. doi: 10.1128/mcb.15.4.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kepa JK, Ross D. DT-diaphorase activity in NSCLC and SCLC cell lines: a role for fos/jun regulation. Br J Cancer. 1999;79:1679–1684. doi: 10.1038/sj.bjc.6690268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu R, Lei W, Mandlekar S, Weber MJ, Der CJ, Wu J, Kong AT. Role of a mitogen-activated protein kinase pathway in the induction of phase II detoxifying enzymes by chemicals. J Biol Chem. 1999;274:27545–27552. doi: 10.1074/jbc.274.39.27545. [DOI] [PubMed] [Google Scholar]
- 56.Yu R, Mandlekar S, Lei W, Fahl WE, Tan TH, Kong AT. p38 Mitogenactivated protein kinase negatively regulates the induction of phase II drug-metabolizing enzymes that detoxify carcinogens. J Biol Chem. 2000;275:2322–2327. doi: 10.1074/jbc.275.4.2322. [DOI] [PubMed] [Google Scholar]
- 57.Soh Y, Jeong KS, Lee IJ, Bae MA, Kim YC, Song BJ. Selective activation of the c-Jun N-terminal protein kinase pathway during 4-hydroxynonenal-induced apoptosis of PC12 cells. Mol Pharmacol. 2000;58:535–541. doi: 10.1124/mol.58.3.535. [DOI] [PubMed] [Google Scholar]
- 58.Camandola S, Poli G, Mattson MP. The lipid peroxidation product 4-hydroxy-2,3-nonenal increases AP-1-binding activity through caspase activation in neurons. J Neurochem. 2000;74:159–168. doi: 10.1046/j.1471-4159.2000.0740159.x. [DOI] [PubMed] [Google Scholar]
- 59.Tamagno E, Robino G, Obbili A, Bardini P, Aragno M, Parola M, Danni O. H2O2 and 4-hydroxynonenal mediate amyloid beta-induced neuronal apoptosis by activating JNKs and p38MAPK. Exp Neurol. 2003;180:144–155. doi: 10.1016/s0014-4886(02)00059-6. [DOI] [PubMed] [Google Scholar]
- 60.Bruckner SR, Estus S. JNK3 contributes to c-jun induction and apoptosis in 4-hydroxynonenal-treated sympathetic neurons. J Neurosci Res. 2002;70:665–670. doi: 10.1002/jnr.10437. [DOI] [PubMed] [Google Scholar]
- 61.Kang KW, Choi SH, Kim SG. Peroxynitrite activates NF-E2-related factor 2/antioxidant response element through the pathway of phosphatidylinositol 3-kinase: the role of nitric oxide synthase in rat glutathione S-transferase A2 induction. Nitric Oxide. 2002;7:244–253. doi: 10.1016/s1089-8603(02)00117-9. [DOI] [PubMed] [Google Scholar]
- 62.Kim SG, Kim SO. PKC downstream of PI3-kinase regulates peroxynitrite formation for Nrf2-mediated GSTA2 induction. Arch Pharm Res. 2004;27:757–762. doi: 10.1007/BF02980145. [DOI] [PubMed] [Google Scholar]
- 63.Numazawa S, Ishikawa M, Yoshida A, Tanaka S, Yoshida T. Atypical protein kinase C mediates activation of NF-E2-related factor 2 in response to oxidative stress. Am J Physiol Cell Physiol. 2003;285:C334–C342. doi: 10.1152/ajpcell.00043.2003. [DOI] [PubMed] [Google Scholar]
- 64.Huang HC, Nguyen T, Pickett CB. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J Biol Chem. 2002;277:42769–42774. doi: 10.1074/jbc.M206911200. [DOI] [PubMed] [Google Scholar]
- 65.Kang MI, Kobayashi A, Wakabayashi N, Kim SG, Yamamoto M. Scaffolding of Keap1 to the actin cytoskeleton controls the function of Nrf2 as key regulator of cytoprotective phase 2 genes. Proc Natl Acad Sci USA. 2004;101:2046–2051. doi: 10.1073/pnas.0308347100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wakabayashi N, Dinkova-Kostova AT, Holtzclaw WD, Kang MI, Kobayashi A, Yamamoto M, Kensler TW, Talalay P. Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc Natl Acad Sci USA. 2004;101:2040–2045. doi: 10.1073/pnas.0307301101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Levonen AL, Landar A, Ramachandran A, Ceaser EK, Dickinson DA, Zanoni G, Morrow JD, Darley-Usmar VM. Cellular mechanisms of redox cell signalling: role of cysteine modification in controlling antioxidant defences in response to electrophilic lipid oxidation products. Biochem J. 2004;378:373–382. doi: 10.1042/BJ20031049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zipper LM, Mulcahy RT. The Keap1 BTB/POZ dimerization function is required to sequester Nrf2 in cytoplasm. J Biol Chem. 2002;277:36544–36552. doi: 10.1074/jbc.M206530200. [DOI] [PubMed] [Google Scholar]
- 69.Shen G, Hebbar V, Nair S, Xu C, Li W, Lin W, Keum YS, Han J, Gallo MA, Kong AN. Regulation of Nrf2 transactivation domain activity: the differential effects of mitogen-activated protein kinase cascades and synergistic stimulatory effect of Raf and CREB-binding protein. J Biol Chem. 2004;279:23052–23060. doi: 10.1074/jbc.M401368200. [DOI] [PubMed] [Google Scholar]
- 70.Katoh Y, Itoh K, Yoshida E, Miyagishi M, Fukamizu A, Yamamoto M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells. 2001;6:857–868. doi: 10.1046/j.1365-2443.2001.00469.x. [DOI] [PubMed] [Google Scholar]
- 71.Liu YZ, Chrivia JC, Latchman DS. Nerve growth factor up-regulates the transcriptional activity of CBP through activation of the p42/p44(MAPK) cascade. J Biol Chem. 1998;273:32400–32407. doi: 10.1074/jbc.273.49.32400. [DOI] [PubMed] [Google Scholar]
- 72.Ait-Si-Ali S, Carlisi D, Ramirez S, Upegui-Gonzalez LC, Duquet A, Robin P, Rudkin B, Harel-Bellan A, Trouche D. Phosphorylation by p44 MAP Kinase/ERK1 stimulates CBP histone acetyl transferase activity in vitro. Biochem Biophys Res Commun. 1999;262:157–162. doi: 10.1006/bbrc.1999.1132. [DOI] [PubMed] [Google Scholar]
- 73.Liu YZ, Thomas NS, Latchman DS. CBP associates with the p42/p44 MAPK enzymes and is phosphorylated following NGF treatment. Neuroreport. 1999;10:1239–1243. doi: 10.1097/00001756-199904260-00016. [DOI] [PubMed] [Google Scholar]
- 74.See RH, Calvo D, Shi Y, Kawa H, Luke MP, Yuan Z. Stimulation of p300-mediated transcription by the kinase MEKK1. J Biol Chem. 2001;276:16310–16317. doi: 10.1074/jbc.M008113200. [DOI] [PubMed] [Google Scholar]
- 75.Gusterson R, Brar B, Faulkes D, Giordano A, Chrivia J, Latchman D. The transcriptional co-activators CBP and p300 are activated via phenylephrine through the p42/p44 MAPK cascade. J Biol Chem. 2002;277:2517–2524. doi: 10.1074/jbc.M104626200. [DOI] [PubMed] [Google Scholar]
- 76.Sang N, Stiehl DP, Bohensky J, Leshchinsky I, Srinivas V, Caro J. MAPK signaling up-regulates the activity of hypoxia-inducible factors by its effects on p300. J Biol Chem. 2003;278:14013–14019. doi: 10.1074/jbc.M209702200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bannister AJ, Oehler T, Wilhelm D, Angel P, Kouzarides T. Stimulation of c-Jun activity by CBP: c-Jun residues Ser63/73 are required for CBP induced stimulation in vivo and CBP binding in vitro. Oncogene. 1995;11:2509–2514. [PubMed] [Google Scholar]
- 78.Xing J, Kornhauser JM, Xia Z, Thiele EA, Greenberg ME. Nerve growth factor activates extracellular signal-regulated kinase and p38 mitogen-activated protein kinase pathways to stimulate CREB serine 133 phosphorylation. Mol Cell Biol. 1998;18:1946–1955. doi: 10.1128/mcb.18.4.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Farmer SC, Sun CW, Winnier GE, Hogan BL, Townes TM. The bZIP transcription factor LCR-F1 is essential for mesoderm formation in mouse development. Genes Dev. 1997;11:786–798. doi: 10.1101/gad.11.6.786. [DOI] [PubMed] [Google Scholar]
- 80.Han TH, Lamph WW, Prywes R. Mapping of epidermal growth factor-, serum-, and phorbol ester-responsive sequence elements in the c-jun promoter. Mol Cell Biol. 1992;12:4472–4477. doi: 10.1128/mcb.12.10.4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.van Dam H, Duyndam M, Rottier R, Bosch A, de Vries-Smits L, Herrlich P, Zantema A, Angel P, van der Eb AJ. Heterodimer formation of cJun and ATF-2 is responsible for induction of c-jun by the 243 amino acid adenovirus E1A protein. EMBO J. 1993;12:479–487. doi: 10.1002/j.1460-2075.1993.tb05680.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Clarke N, Arenzana N, Hai T, Minden A, Prywes R. Epidermal growth factor induction of the c-jun promoter by a Rac pathway. Mol Cell Biol. 1998;18:1065–1073. doi: 10.1128/mcb.18.2.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Marinissen MJ, Chiariello M, Pallante M, Gutkind JS. A network of mitogen-activated protein kinases links G protein-coupled receptors to the c-jun promoter: a role for c-Jun NH2-terminal kinase, p38s, and extracellular signal-regulated kinase 5. Mol Cell Biol. 1999;19:4289–4301. doi: 10.1128/mcb.19.6.4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Marinissen MJ, Chiariello M, Gutkind JS. Regulation of gene expression by the small GTPase Rho through the ERK6 (p38 gamma) MAP kinase pathway. Genes Dev. 2001;15:535–553. doi: 10.1101/gad.855801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gupta P, Prywes R. ATF1 phosphorylation by the ERK MAPK pathway is required for epidermal growth factor-induced c-jun expression. J Biol Chem. 2002;277:50550–50556. doi: 10.1074/jbc.M209799200. [DOI] [PubMed] [Google Scholar]
- 86.Leppa S, Saffrich R, Ansorge W, Bohmann D. Differential regulation of c-Jun by ERK and JNK during PC12 cell differentiation. EMBO J. 1998;17:4404–4413. doi: 10.1093/emboj/17.15.4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Han J, Jiang Y, Li Z, Kravchenko VV, Ulevitch RJ. Activation of the transcription factor MEF2C by the MAP kinase p38 in inflammation. Nature. 1997;386:296–299. doi: 10.1038/386296a0. [DOI] [PubMed] [Google Scholar]
- 88.Deak M, Clifton AD, Lucocq LM, Alessi DR. Mitogen- and stressactivated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J. 1998;17:4426–4441. doi: 10.1093/emboj/17.15.4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Raingeaud J, Gupta S, Rogers JS, Dickens M, Han J, Ulevitch RJ, Davis RJ. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J Biol Chem. 1995;270:7420–7426. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- 90.Raingeaud J, Whitmarsh AJ, Barrett T, Derijard B, Davis RJ. MKK3 and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol Cell Biol. 1996;16:1247–1255. doi: 10.1128/mcb.16.3.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]