

Abstract
H2O2 produced by stimulation of the macrophage NADPH oxidase is involved both in bacterial killing and as a second messenger in these cells. Protein tyrosine phosphatases (PTPs) are targets for H2O2 signaling through oxidation of their catalytic cysteine, resulting in inhibition of their activity. Here, we show that, in the rat alveolar macrophage NR8383 cell line, H2O2 produced through the ADP-stimulated respiratory burst induces the formation of a disulfide bond between PTP1B and GSH that was detectable with an antibody to glutathione-protein complexes and was reversed by DTT addition. PTP1B glutathionylation was dependent on H2O2 as the presence of catalase at the time of ADP stimulation inhibited the formation of the conjugate. Interestingly, other PTPs, i.e., SHP-1 and SHP-2, did not undergo glutathionylation in response to ADP stimulation of the respiratory burst, although glutathionylation of these proteins could be shown by reaction with 25 mM glutathione disulfide in vitro. While previous studies have suggested the reversible oxidation of PTP1B during signaling or showed PTP1B glutathionylation in vitro, the present study directly demonstrates that physiological stimulation of H2O2 production results in PTP1B glutathionylation in intact cells, which may affect downstream signaling.
Keywords: Protein tyrosine phosphatase 1B, Hydrogen peroxide, Glutathione, Glutathione disulfide
Introduction
It is now well accepted that reactive oxygen and nitrogen species can act as second messengers in signal transduction [1-5]. In particular, H2O2, which is mostly derived from superoxide (O2•-) produced by stimulation of the NADPH oxidase (NOX2) in macrophages and other phagocytes (respiratory burst) [6] or by homologous NOXs in many other cell types, has been shown to be required for various cellular responses to several stimuli such as insulin, angiotensin, and growth factors or stimuli of the respiratory burst. While the mechanisms through which H2O2 regulates pathways and responses are still uncertain in many cases [5,7-11], “glutathionylation” of signaling proteins, which is one of the posttranslational modifications that occurs in cells in response to environmental or endogenous stimulation, has been implicated in cell signaling [12-14]. During this process, a critical cysteine in a particular signaling protein is sensitive to oxidation by H2O2 or other hydroperoxides and the oxidized cysteine can then form a disulfide bond with glutathione (GSH), resulting in altered activity. Several signaling proteins, have been shown to undergo glutathionylation, including enzymes such as protein tyrosine phosphatases (PTPs) and transcription factors [12-17].
PTPs constitute a large family of physiologically important regulatory enzymes that control several biological functions, including the state of tyrosine phosphorylation of proteins, which, at any moment, is the result of differences established by the activities of protein tyrosine kinase (PTK) versus PTPs. Protein tyrosine phosphorylation is one of the mechanisms that enable the cell to respond to several extracellular stimuli, having a role in the regulation of most cellular functions such as growth, proliferation, differentiation, and metabolism [18,19]. While PTKs have been extensively studied, the past few years have seen an increase in interest in PTPs because of their recognized role as regulators of signal transduction under normal and pathophysiological conditions [20,21].
PTP1B is “the prototypical enzyme” of the PTP family, according to Tonks and co-workers [22] and numerous studies have focused attention on PTP1B because of its role as a critical physiological regulator of metabolism. In fact, PTP1B knockout mice do not develop diabetes and obesity when placed on a calorie-rich diet, contrary to wild-type mice [23,24]. Besides its role as a negative regulator in insulin and leptin signaling, PTP1B is implicated in several other signaling pathways such as growth factor [25-27] and integrin signaling [27,28]. As PTP1B seems to regulate multiple cellular processes, it is important to understand how the function of this protein can be modulated in the cell. Previous work demonstrated that PTP1B can undergo glutathionylation in vitro. Treatment of PTP1B with diamide and reduced glutathione or with glutathione disulfide (GSSG) alone resulted in glutathionylation [12].
In the present study, we report that in the NR8383 cell line, which is derived from rat alveolar macrophages, ADP stimulation of H2O2 production through the respiratory burst induces the reversible H2O2-dependent formation of glutathionylated PTP1B. In contrast, two other PTPs, SHP-1 and SHP-2 that could be glutathionylated in vitro by addition of a nonphysiological concentration of glutathione disulfide (GSSG), were not glutathionylated by ADP stimulation of the respiratory burst, indicating specificity.
Materials and methods
Materials and reagents
Unless otherwise noted, all chemicals were from Sigma (St. Louis, MO). Monoclonal anti-PTP1B was from Calbiochem (EMD Biosciences, Inc., San Diego, CA), monoclonal anti-SHP-1 and anti-SHP-2 were from Transduction Laboratories (Lexington, KY). Monoclonal antibody to glutathione-protein complexes was from Virogen (Watertown, MA), protein A-Sepharose was from Amersham Biosciences (Piscataway, NJ).
Cell culture
The NR8383 rat alveolar macrophage cell line [29] was kindly provided to us by Dr. G.H. Zhang, University of Texas Health Science Center at San Antonio. Cells were cultured in F-12K medium (Life Technologies, Grand Island, NY) supplemented with 15% heat-inactivated fetal bovine serum (Omega Scientific inc., Tarzana, CA), penicillin (100 U/ml), and streptomycin (100 μg/ml) and maintained at a concentration of 1 × 106/ml in a humidified atmosphere containing 5% CO2 and 95% air at 37°C. Cells were collected and resuspended in fresh medium twice a week.
Immunoprecipitation
Cells were seeded at a density of 1 × 106/ml in fresh medium the day before each experiment when cells (5 × 106) were washed with ice-cold phosphate-buffered saline (PBS) and treated under various conditions before lysis in 1 ml buffer containing 50 mM Hepes (pH 7.4), 150 mM NaCl, 1% Triton X-100, 0.5% NP40, 50 mM β-glycerophosphate, 1 mM sodium orthovanadate, 10 mM sodium pyrophosphate, 1 mM EGTA, 1 mM PMSF, 10% glycerol, 100 mM sodium fluoride, 10 μM DETAPAC, 5 mM GSH, 5 mM NEM, 10 ng/ml leupeptin, and 10 ng/ml aprotinin. NEM (5 mM) was added to the dishes for 2 min before lysis. The protein concentration of whole cell lysates was measured using the Bio-Rad protein assay (Bio-Rad, Hercules, CA) and adjusted to 500 μg before immunoprecipitation with protein A-Sepharose-conjugated antibodies to PTP1B, SHP-1, or SHP-2 at 4°C overnight. The antibodies had been previously crosslinked to protein A-Sepharose beads using dimethyl pimelimidate (DMP) as described by Schneider et al. [30] and stored at 4°C until used. The immuno-complexes were washed 4 times with the cell lysis buffer, resuspended in 2× sample buffer containing sodium dodecyl sulfate (SDS), Tris base, pH 6.5, glycerol, and pyronin Y, and heated for 10 min at 95°C. DTT was added to the sample buffer when indicated.
Western blotting
Proteins were resolved on a 4-20% Tris-glycine acrylamide gel (Invitrogen, Carlsbad, CA) under denaturing conditions before being transferred electrophoretically onto a polyvinylidene difluoride (PVDF) membrane (Immobilon P; Millipore, Bedford, MA). Membranes were blocked with 5% nonfat dry milk (NFDM) at room temperature for 1 h and then incubated overnight at 4°C with primary antibody diluted in 5% NFDM in Tris buffer saline (TBS) as indicated (1:1000 anti-glutathione-protein complexes; 1:1000 anti-PTP1B; 1:500 anti-SHP-1; 1:1000 anti-SHP-2). After being washed with TBS containing 0.05% Tween 20, the membrane was incubated with goat antimouse IgG conjugated to horseradish peroxidase (1:10,000) at room temperature for 2 h. The blots were developed by the enhanced chemiluminescence technique (ECL Plus, Amersham, Arlington Heights, IL) according to the manufacturer's instructions. The bands of interest were imaged with a Kodak Instant Imager 2.0.
Results
Exogenous H2O2 stimulates PTP1B glutathionylation in rat alveolar macrophages
Although previous work suggested that PTP1B could be glutathionylated in vitro under nonphysiological conditions and some reversibly oxidized but unidentified form of PTP1B could be produced in cells [12], the actual formation of glutathionylated PTP1B had not previously been directly demonstrated in intact cells under physiological conditions. The availability of an antibody that specifically recognizes proteins complexed with GSH and can be used for Western blotting provided a possible methodology to detect glutathionylation in intact cells. As a proof of principle, we treated NR8383 cells with a bolus of a nontoxic concentration of H2O2 that produced oxidizing conditions for PTP1B glutathionylation without damaging the cells. PTP1B was then immunoprecipitated and the immunoprecipitates were separated by SDS-PAGE followed by Western blot analysis under nonreducing conditions to detect the formation of PTP1B mixed disulfide. Exposure to 100 μM H2O2 resulted in the time-dependent increase in glutathionylated PTP1B and addition to the lysis buffer of DTT, a reducing agent, eliminated the detection of the glutathionylated protein band (Fig. 1, upper blots), supporting its identification as PTP1B-SSG. The same membrane was stripped and reprobed with an antibody against the PTP1B protein to check the efficiency of the immunoprecipitation (Fig. 1, lower blots). Two bands were observed: a lower band, which had the same mobility as PTP1B-SSG, and an upper band, most likely corresponding to native PTP1B. In samples to which DTT was added, the lower band disappeared while the density of the upper band increased, consistent with reduction of PTP1B-SSG to PTP1B-S-, the thiolate form.
Fig. 1.
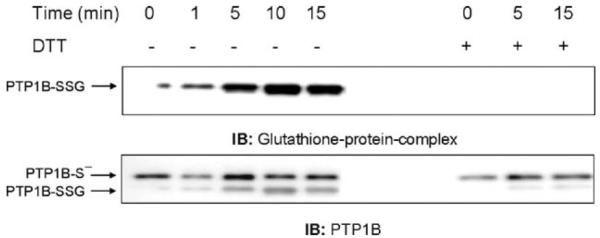
Glutathionylation of PTP1B is induced by the addition of exogenous H2O2. NR8383 cells were exposed to 100 μM H2O2 and then lysed with a buffer containing N-ethylmaleimide (NEM). GSH was added to remove excess NEM. Whole cell extracts were immunoprecipitated with an antibody to PTP1B. Membranes were immunoblotted with antibodies to either glutathione-protein complexes or PTP1B. Based on the densitometry of IP from two experiments, the maximum percentage PTP1B-SSG in respect to total PTP1B was 31.3 ± 19.1%, which was at 10 min. The results followed the same pattern of increase to a maximum for both experiments. Despite the variability, which is largely due to the semiquantitative nature of densitometry, the pattern of change over time in both experiments was the same.
ADP-induced respiratory burst causes PTP1B glutathionylation
We next examined whether H2O2 produced through ADP stimulation of the respiratory burst could similarly induce PTP1B glutathionylation in NR8383 cells. At various times after treating NR8383 cells with 400 μM ADP, we immunoprecipitated PTP1B and determined glutathionylation by immunoblotting with the antibody to glutathione-protein complexes. As shown in Fig. 2 (upper blot), PTP1B glutathionylation increased over several minutes in stimulated cells, reached a maximum at 10 min, and then declined toward the control value that was reached by approximately 30 min (data not shown), indicating that the process of PTP1B glutathionylation induced by the respiratory burst was reversible. As with bolus addition of 100 μM H2O2, PTP1B was not fully glutathionylated after ADP stimulation. Reprobing the membrane with the PTP1B antibody showed the relative extent of PTP1B glutathionylation by comparison of the two bands, one of which (PTP1B-SSG) was completely converted to the other (PTP1B-S-) by DTT treatment (Fig. 2, lower blot).
Fig. 2.
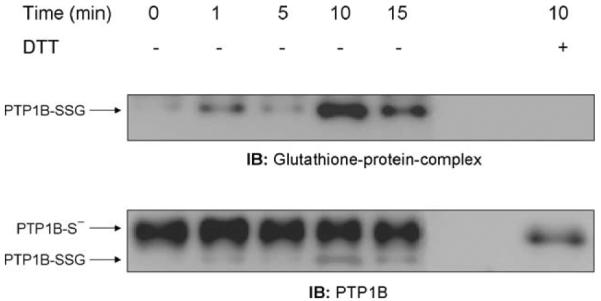
ADP stimulation of the respiratory burst induces glutathionylation of PTP1B. NR8383 cells were incubated at 37°C for different lengths of time in the presence or absence of 400 μM ADP. Aliquots of whole cellular extract were immunoprecipitated with an antibody to PTP 1B, and the immunoprecipitates were probed by immunoblotting with antibodies to glutathione-protein complexes or PTP1B. DTT was added to the loading buffer of some samples to reverse glutathionylation. Quantification of the glutathionylated PTP1B, for this experiment, was plotted as percentage of PTP1B-SSG of total PTP1B. Based on the densitometry of IP of different experiments (n = 5) the mean of the percentage of PTP1B-SSG in respect to the total PTP1B as a function of time after stimulation with ADP was 32.1 ± 15.5% for control, 36.4 ± 21.02% 5 min, 57.9 ± 27.5% 10 min, 41.7 ± 29.1% 15 min, and 44.5 ± 23.6% 20 min. Despite the variability, which is largely due to the semiquantitative nature of densitometry, the pattern of change over time in all four experiments was the same.
Exogenous catalase inhibits ADP-stimulated PTP1B glutathionylation
To further demonstrate that the H2O2 produced during the respiratory burst induced by ADP treatment was critical for PTP1B glutathionylation, we treated the NR83838 cells in the absence or presence of extracellular catalase during stimulation. ADP stimulation of the respiratory burst results from activation of the NADPH oxidase (NOX2) at the plasma membrane and production of O2•- on the outer surface of the cell where it nonenzymatically dismutates to form H2O2 and O2. In contrast to O2•-, the uncharged H2O2 can readily enter the cell and participate in cell signaling. Formation of H2O2 on the outside of the cell allows the use of exogenously added catalase as a tool for examining the involvement of H2O2 in signaling. Fig. 3 shows that 100 U/ml catalase added before ADP stimulation inhibited PTP1B glutathionylation, indicating its dependence on H2O2 produced by the phagocyte NADPH oxidase.
Fig. 3.
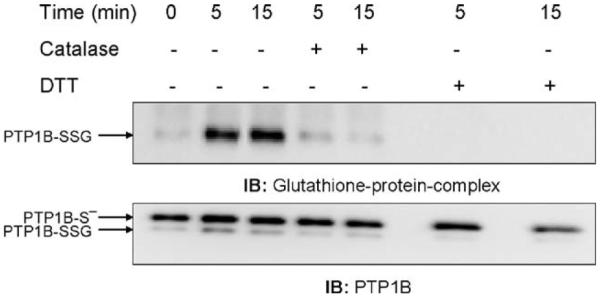
Catalase inhibits the glutathionylation of PTP1B induced by the ADP-stimulated respiratory burst. After 5 min preincubation with 100 U/ml catalase, NR8383 cells were exposed to ADP as in Fig. 2 and then lysed. Proteins (500 μg) were subjected to immunoprecipitation with the antibody to PTP1B and the immunoprecipitates were separated by electrophoresis and immunoblotted. The membranes were probed with the antibody to glutathione-protein complexes and then reprobed with the antibody to PTP1B.
BCNU treatment alone induces glutathionylation in the presence and absence of ADP
One proposed mechanism by which the glutathionylation of proteins may occur is thiol/disulfide exchange. It is, in fact, possible to observe a reaction of glutathionylation in vitro between PTP1B and GSSG [12]. This nonenzymatic reaction occurs slowly and requires a GSSG concentration that would not be possible in vivo under normal physiological conditions due to the rapid NADPH-dependent reduction of GSSG by glutathione reductase that maintains a low GSSG concentration. Thus, using an inhibitor of glutathione reductase such as BCNU would increase the levels of GSSG in cells. NR8383 cells were incubated for 2 h with BCNU before stimulation with ADP and samples were treated as above. Fig. 4 shows that the glutathionylation of PTP1B occurred under these conditions, even in the absence of ADP stimulation. Thus, it is possible that during pathophysiologic conditions induced by oxidative stress, when the GSH/GSSG ratio is markedly diminished, PTP1B could be glutathionylated by thiol-disulfide exchange.
Fig. 4.
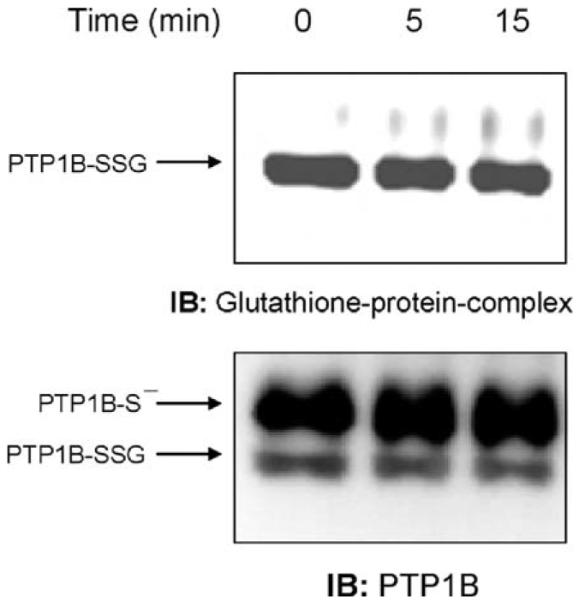
Inhibition of glutathione reductase by BCNU induces PTP1B glutathionylation. After preincubation for 2 h with 100 μM BCNU, cells were immunoprecipitated with the antibody to PTP1B and the immunoprecipitates were separated by electrophoresis and immunoblotted with the antibody to glutathione-protein complexes. The membranes were probed with the antibody to glutathione-protein complexes and then reprobed with the antibody to PTP1B.
ADP-induced respiratory burst does not cause SHP-1 and SHP-2 glutathionylation
We next investigated whether other protein tyrosine phosphatases could be glutathionylated under the same physiological conditions used for PTP1B. SHP-1 and SHP-2, which are expressed in NR8383 cells [31], have previously been shown to be sensitive to oxidation by H2O2 [32,33], and thus, were chosen for these studies. After stimulation with ADP, the total cytosolic extract from NR8383 cells was immunoprecipitated with antibodies to either SHP-1 or SHP-2 for various lengths of incubation. The immunoprecipitates were separated as previously and protein glutathionylation was visualized by immunoblotting with the glutathione-protein complexes antibody. No glutathionylation of SHP-1 or SHP-2 was observed after ADP stimulation of the respiratory burst (Fig. 5A), although the two proteins were efficiently immunoprecipitated, as shown by reprobing the membrane with antibodies against either the SHP-1 or SHP-2 proteins (Fig. 5A). To verify that this negative result was not due to a failure to detect the formation of glutathionylated proteins, we forced the disulfide bond formation between the proteins and the glutathione by adding 25 mM GSSG to the lysis buffer. Two bands at ~68 and ~72 kDa were observed that comigrated with the SHP-1 and SHP-2 proteins, respectively, and disappeared when exogenous DTT was added (Fig. 5B). The data demonstrate that glutathionylation of SHP-1 and SHP-2 can be detected by our method but does not occur after ADP stimulation of the respiratory burst in NR8383 cells.
Fig. 5.

H2O2 produced by the ADP-stimulated respiratory burst does not cause glutathionylation of SHP-1 and SHP-2 in NR8383 cells. (A) NR8383 cells were exposed to 400 μM ADP for various times. SHP-1 and SHP-2 were immunoprecipitated with their respective antibodies, and the immunoprecipitates were separated by electrophoresis and immunoblotted with the antibody to glutathione-protein complexes. The membranes were stripped and reprobed with the antibodies to SHP-1 or SHP-2. (B) Disulfide bond formation between SHP-1 and SHP-2 and glutathione was induced by adding 25 mM GSSG in the lysis buffer. Aliquots of lysate were immunoprecipitated with antibodies to SHP-1 or SHP-2. In some samples, DTT was added to the loading buffer to reverse glutathionylation. The immunoprecipitates were separated by electrophoresis and immunoblotted with the antibody to glutathione-protein complexes. The membranes were stripped and reprobed with the antibodies to SHP-1 or SHP-2.
Discussion
Reactive species of oxygen (ROS) are produced during the respiratory burst by the NADPH oxidase of macrophages and neutrophils (NOX2) and in other cells by the homologues NOX proteins. It is now accepted that H2O2 plays a role as a second messenger in cells by its ability to modulate the activity of signaling proteins and we have hypothesized that redox signaling may be at least as important a physiological role for the respiratory burst as the microbicidal activity [6]. One of the mechanisms by which H2O2 acts is by causing the formation of disulfide bonds between a target protein cysteine residue and the glutathione. This posttranslational modification called glutathionylation is able to modify the activity of proteins such as PTP that contains an essential cysteine residue in its active site [22].
The present work supports the key role of H2O2 in the reversible regulation under physiological conditions of PTP1B, the prototypical enzyme of the PTP family. The observation that PTP1B activity is modulated by glutathionylation was first made by Barrett et al. [12] in experiments on purified human PTP1B incubated in vitro with a strong oxidant, diamide, in the presence of GSH or with a high concentration of GSSG. Although reversible inhibition of PTP1B by H2O2 has been demonstrated in cells and glutathionylation has been demonstrated in vitro, the mechanism through which H2O2 induces glutathionylation of PTP1B is not entirely clear [12,34-36]. Denu and Tanner first demonstrated reversible oxidation of the catalytic cysteine of PTP1B, hypothesizing a chemical mechanism involving a cysteine sulfenic acid intermediate [34]. Barrett et al. then proposed that the cysteine oxidized to a sulfenic acid could either further oxidize to form the sulfinic and sulfonic acid forms or react with GSH to form a disulfide [12]. More recent studies have shown by crystallization of isolated PTP1B treated with H2O2 the presence of a sulfenyl amide that, in the absence of GSH, is most likely formed by reaction of the sulfenic acid form of PTP1B with an amide from the protein backbone [37]. They suggested that the sulfenyl amide is the intermediate that reacts with GSH to form the disulfide [35,36]. The formation of a PTP1B sulfenic acid intermediate, however, has been not demonstrated in cells and glutathionylation could, in theory, occur in cells without this intermediate derivative through reaction of the enzyme with GSSG as shown in vitro [12]. Regardless of the mechanism, the present study demonstrates the formation of the disulfide with GSH under physiological stimulation. Although the glutathionylation of other proteins has been shown in cells before, the experimental models usually reflect a condition of oxidative stress rather than redox signaling [14,38]. In fact, glutathionylation may serve as an adaptive response against severe stress conditions such as oxidative stress in which the GSH/GSSG ratio is perturbed. Evidence for this mechanism has been obtained in a Parkinson disease experimental model by Kil and Park who showed increases in glutathionylated isocitric dehydrogenase in mouse brain [14]. Similar evidence was suggested by the data here where addition of an inhibitor of glutathione reductase that shifts the balance of GSH/GSSG toward GSSG, mimicking a pathophysiological condition, resulted in glutathionylation of PTP1B, even without ADP stimulation.
Our results demonstrate that glutathionylation of PTP1B occurs in cells under physiological stimulation of the production of H2O2 through the ADP-stimulated respiratory burst and dismutation and could be an important mechanism by which macrophages fine-tune downstream signaling pathways. Glutathionylation of PTP1B is a reversible event as its extent started to decrease after 10 min. Most significantly, we show that the glutathionylation caused by ADP stimulation is dependent on H2O2. Furthermore, the ADP-stimulated glutathionylation of PTP1B in this system depends on the generation of extracellular H2O2 as catalase added in the media abolishes the glutathionylation. Catalase added on the outside does not affect the intracellular H2O2 concentration when H2O2 is generated intracellularly. This is because the external media represents an extremely large sink into which H2O2 can diffuse and thus, removal of H2O2 that diffuses to the outside would make no difference in the internal concentration. On the other hand, if H2O2 is generated on the outside, then catalase can destroy it before it can enter the cell as it did here. In accordance with our results, Mahadev et al. provided evidence that in adipocyte cells stimulated by insulin, the catalytic activity of PTP1B is inhibited by NOX4-induced ROS production [10]. It is likely that the activation of the different NOX isoforms in several cell types is a general mechanism by which the H2O2-induced glutathionylation inhibits PTP1B activity and since this inactivation is transient, it can act as a switch that is able to turn on and off the enzymatic function in response to stimuli.
It is important to note that ADP stimulation did not result in glutathionylation of SHP-1 and SHP-2, even though the glutathionylation of these PTPs was detected in the presence of a high concentration of GSSG. This result provides evidence that glutathionylation can be a specific mechanism of modification of PTPs. Singh et al. [33] and Meng et al. [32] have demonstrated the regulation of SHP-1 and SHP-2, respectively, through oxidation by H2O2. Interestingly, oxidation and inactivation of SHP-2 occurred only for that fraction of SHP-2 that was recruited into a complex with the PDGF receptor [32]. Similarly, SHP-1 oxidation and inhibition of activity was found to be restricted to the BCR-associated pool of SHP-1 [33]. Thus, according to these and the results here, it appears that the action of H2O2 is dependent upon the localization of its production and its target, as is consistent with other signaling mechanisms. It is likely that in our experimental model, SHP-1 and SHP-2 are not glutathionylated because they are not in proximity to the NOX2 that is activated through ADP stimulation. Localization of H2O2 action provides the specificity that characterizes signal transduction as opposed to the random action of H2O2 that typifies oxidative stress.
Acknowledgment
This work was supported by NIH Grant HL37556.
Abbreviations
- PTK
protein tyrosine kinase
- PMSF
phenylmethylsulfonyl fluoride
- DETAPAC
diethylenetriaminepentaacetic acid
- NEM
N-ethylmaleimide
- DMP
dimethyl pimelimidate
- BCNU
1,3-bis-(2-chloroethyl)-1-nitrosourea
- DTT
dithiothreitol
- PTP
protein tyrosine phosphatase
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- NFDM
nonfat dry milk
- ROS
reactive oxygen species
References
- [1].Droge W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- [2].Wink DA, Mitchell JB. Chemical biology of nitric oxide: insights into regulatory, cytotoxic, and cytoprotective mechanisms of nitric oxide. Free Radic. Biol. Med. 1998;25:434–456. doi: 10.1016/s0891-5849(98)00092-6. [DOI] [PubMed] [Google Scholar]
- [3].Rhee SG. Redox signaling: hydrogen peroxide as intracellular messenger. Exp. Mol. Med. 1999;31:53–59. doi: 10.1038/emm.1999.9. [DOI] [PubMed] [Google Scholar]
- [4].Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat. Rev., Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- [5].Bokoch GM, Knaus UG. NADPH oxidases: not just for leukocytes anymore! Trends Biochem. Sci. 2003;28:502. doi: 10.1016/S0968-0004(03)00194-4. [DOI] [PubMed] [Google Scholar]
- [6].Forman HJ, Torres M. Redox signaling in macrophages. Mol. Aspects Med. 2001;22:189–216. doi: 10.1016/s0098-2997(01)00010-3. [DOI] [PubMed] [Google Scholar]
- [7].Finkel T. Signal transduction by reactive oxygen species in nonphagocytic cells. J. Leukocyte Biol. 1999;65:337–340. doi: 10.1002/jlb.65.3.337. [DOI] [PubMed] [Google Scholar]
- [8].Gorin Y, Kim NH, Feliers D, Bhandari B, Choudhury GG, Abboud HE. Angiotensin II activates Akt/protein kinase B by an arachidonic acid/redox-dependent pathway and independent of phosphoinositide 3-kinase. FASEB J. 2001;15:1909–1920. doi: 10.1096/fj..01-0165com. [DOI] [PubMed] [Google Scholar]
- [9].Forman HJ, Torres M. Reactive oxygen species and cell signaling: respiratory burst in macrophage signaling. Am. J. Respir. Crit. Care Med. 2002;166:S4–S8. doi: 10.1164/rccm.2206007. [DOI] [PubMed] [Google Scholar]
- [10].Mahadev K, Motoshima H, Wu X, Ruddy JM, Arnold RS, Cheng G, Lambeth JD, Goldstein BJ. The NAD(P)H oxidase homolog Nox4 modulates insulin-stimulated generation of H2O2 and plays an integral role in insulin signal transduction. Mol. Cell. Biol. 2004;24:1844–1854. doi: 10.1128/MCB.24.5.1844-1854.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sundaresan M, Yu Z-X, Ferrans VJ. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- [12].Barrett WC, DeGnore JP, Konig S, Fales HM, Keng YF, Zhang ZY, Yim MB, Chock PB. Regulation of PTP1B via glutathionylation of the active site cysteine 215. Biochemistry. 1999;38:6699–6705. doi: 10.1021/bi990240v. [DOI] [PubMed] [Google Scholar]
- [13].Lee SR, Kwon KS, Kim SR, Rhee SG. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J. Biol. Chem. 1998;273:15366–15372. doi: 10.1074/jbc.273.25.15366. [DOI] [PubMed] [Google Scholar]
- [14].Kil IS, Park JW. Regulation of mitochondrial NADP+-dependent isocitrate dehydrogenase activity by glutathionylation. J. Biol. Chem. 2005;280:10846–10854. doi: 10.1074/jbc.M411306200. [DOI] [PubMed] [Google Scholar]
- [15].Klatt P, Lamas S. c-Jun regulation by S-glutathionylation. Methods Enzymol. 2002;348:157–174. doi: 10.1016/s0076-6879(02)48635-1. [DOI] [PubMed] [Google Scholar]
- [16].Brar SS, Grigg C, Wilson KS, Holder WD, Jr., Dreau D, Austin C, Foster M, Ghio AJ, Whorton AR, Stowell GW, Whittall LB, Whittle RR, White DP, Kennedy TP. Disulfiram inhibits activating transcription factor/cyclic AMP-responsive element binding protein and human melanoma growth in a metal-dependent manner in vitro, in mice and in a patient with metastatic disease. Mol. Cancer Ther. 2004;3:1049–1060. [PubMed] [Google Scholar]
- [17].Fratelli M, Goodwin LO, Orom UA, Lombardi S, Tonelli R, Mengozzi M, Ghezzi P. Gene expression profiling reveals a signaling role of glutathione in redox regulation. Proc. Natl. Acad. Sci. U. S. A. 2005;102:13998–14003. doi: 10.1073/pnas.0504398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hunter T. The Croonian Lecture 1997. The phosphorylation of proteins on tyrosine: its role in cell growth and disease. Philos. Trans. R. Soc. Lond., B Biol. Sci. 1998;353:583–605. doi: 10.1098/rstb.1998.0228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Tonks NK, Neel BG. Combinatorial control of the specificity of protein tyrosine phosphatases. Curr. Opin. Cell Biol. 2001;13:182–195. doi: 10.1016/s0955-0674(00)00196-4. [DOI] [PubMed] [Google Scholar]
- [20].Tonks NK. PTP1B: from the sidelines to the front lines! FEBS Lett. 2003;546:140–148. doi: 10.1016/s0014-5793(03)00603-3. [DOI] [PubMed] [Google Scholar]
- [21].Dube N, Tremblay ML. Beyond the metabolic function of PTP1B. Cell Cycle. 2004;3:550–553. [PubMed] [Google Scholar]
- [22].Barford D, Jia Z, Tonks NK. Protein tyrosine phosphatases take off. Nat. Struct. Biol. 1995;2:1043–1053. doi: 10.1038/nsb1295-1043. [DOI] [PubMed] [Google Scholar]
- [23].Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy AL, Normandin D, Cheng A, Himms-Hagen J, Chan CC, Ramachandran C, Gresser MJ, Tremblay ML, Kennedy BP. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science. 1999;283:1544–1548. doi: 10.1126/science.283.5407.1544. [DOI] [PubMed] [Google Scholar]
- [24].Klaman LD, Boss O, Peroni OD, Kim JK, Martino JL, Zabolotny JM, Moghal N, Lubkin M, Kim YB, Sharpe AH, Stricker-Krongrad A, Shulman GI, Neel BG, Kahn BB. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol. Cell. Biol. 2000;20:5479–5489. doi: 10.1128/mcb.20.15.5479-5489.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liu F, Chernoff J. Protein tyrosine phosphatase 1B interacts with and is tyrosine phosphorylated by the epidermal growth factor receptor. Biochem. J. 1997;327:139–145. doi: 10.1042/bj3270139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Haj FG, Markova B, Klaman LD, Bohmer FD, Neel BG. Regulation of receptor tyrosine kinase signaling by protein tyrosine phosphatase-1B. J. Biol. Chem. 2003;278:739–744. doi: 10.1074/jbc.M210194200. [DOI] [PubMed] [Google Scholar]
- [27].Liang F, Lee SY, Liang J, Lawrence DS, Zhang ZY. The role of protein-tyrosine phosphatase 1B in integrin signaling. J. Biol. Chem. 2005;280:24857–24863. doi: 10.1074/jbc.M502780200. [DOI] [PubMed] [Google Scholar]
- [28].Liu F, Sells MA, Chernoff J. Protein tyrosine phosphatase 1B negatively regulates integrin signaling. Curr. Biol. 1998;8:173–176. doi: 10.1016/s0960-9822(98)70066-1. [DOI] [PubMed] [Google Scholar]
- [29].Helmke RJ, Boyd RL, German VF, Mangos JA. From growth factor dependence to growth factor responsiveness: the genesis of an alveolar macrophage cell line. In Vitro Cell. Dev. Biol. 1987;23:567–574. doi: 10.1007/BF02620974. [DOI] [PubMed] [Google Scholar]
- [30].Schneider C, Newman RA, Sutherland DR, Asser U, Greaves MF. A one-step purification of membrane proteins using a high efficiency immunomatrix. J. Biol. Chem. 1982;257:10766–10769. [PubMed] [Google Scholar]
- [31].Torres M, Forman HJ. Vanadate inhibition of protein tyrosine phosphatases mimics hydrogen peroxide in the activation of the ERK pathway in alveolar macrophages. Ann. N. Y. Acad. Sci. 2002;973:345–348. doi: 10.1111/j.1749-6632.2002.tb04663.x. [DOI] [PubMed] [Google Scholar]
- [32].Meng TC, Fukada T, Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol. Cell. 2002;9:387–399. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- [33].Singh DK, Kumar D, Siddiqui Z, Basu SK, Kumar V, Rao KV. The strength of receptor signaling is centrally controlled through a cooperative loop between Ca2+ and an oxidant signal. Cell. 2005;121:281–293. doi: 10.1016/j.cell.2005.02.036. [DOI] [PubMed] [Google Scholar]
- [34].Denu JM, Tanner KG. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry. 1998;37:5633–5642. doi: 10.1021/bi973035t. [DOI] [PubMed] [Google Scholar]
- [35].Salmeen A, Andersen JN, Myers MP, Meng TC, Hinks JA, Tonks NK, Barford D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature. 2003;423:769–773. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- [36].van Montfort RL, Congreve M, Tisi D, Carr R, Jhoti H. Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature. 2003;423:773–777. doi: 10.1038/nature01681. [DOI] [PubMed] [Google Scholar]
- [37].Sivaramakrishnan S, Keerthi K, Gates KS. A chemical model for redox regulation of protein tyrosine phosphatase 1B (PTP1B) activity. J. Am. Chem. Soc. 2005;127:10830–10831. doi: 10.1021/ja052599e. [DOI] [PubMed] [Google Scholar]
- [38].Cross JV, Templeton DJ. Thiol oxidation of cell signaling proteins: controlling an apoptotic equilibrium. J. Cell. Biochem. 2004;93:104–111. doi: 10.1002/jcb.20202. [DOI] [PubMed] [Google Scholar]