

Abstract
Although great strides are being made in the care of individuals with cystic fibrosis (CF), this condition remains the most common fatal hereditary disease in North America. Numerous links exist between progression of CF lung disease and oxidative stress. The defect in CF is the loss of function of the transmembrane conductance regulator (CFTR) protein; recent evidence that CFTR expression and function are modulated by oxidative stress suggests that the loss may result in a poor adaptive response to oxidants. Pancreatic insufficiency in CF also increases susceptibility to deficiencies in lipophilic antioxidants. Finally the airway infection and inflammatory processes in the CF lung are potential sources of oxidants that can affect normal airway physiology and contribute to the mechanisms causing characteristic changes associated with bronchiectasis and loss of lung function. These multiple abnormalities in the oxidant/antioxidant balance raise several possibilities for therapeutic interventions that must be carefully assessed.
Introduction
Cystic fibrosis (CF) is the most common fatal disease caused by a single gene defect in Europe and North America [1,2]. The defective gene was identified in 1989 [3-5] and shown to encode the cystic fibrosis trans-membrane conductance regulator protein (CFTR). CFTR is a cyclic AMP-regulated anion channel with greatest selectivity for chloride, and bicarbonate [6-8]. Furthermore, CFTR regulates sodium transport across epithelial membranes through interactions with the epithelial sodium channel ENaC [9], and facilitates the trans-membrane export of the small organic anionic peptide glutathione [10]. The expression of CFTR is located in the apical membrane of epithelial cells that line mucous membranes and submucosal glands [11,12]. The tissues most affected by CFTR deficiency include the vas deferens, exocrine pancreas, liver, large intestine, sinuses, and the lower airways. The loss of water from secretions at the surface of these epithelial tissues is thought to be a key feature leading to mucus impaction and obstruction. Many CF infants develop signs of maldigestion manifested by failure to thrive. Obstruction of ducts in the exocrine pancreas traps proteins such as proteases and lipases in the pancreatic tissue, leading to failure of the exocrine pancreatic function and poor absorption of lipids [13]. Pancreatic insufficiency is present in 85% of patients with CF.
Oxidative stress in the lung
Oxidative stress may play a significant role in the pathophysiology of CF. The lung, the main organ responsible for morbidity and mortality in this disease, is particularly vulnerable to high levels of oxidative stress. It is exposed to 8000 L of oxygen-rich air per day as well as toxic particles, nitrogen dioxide, ozone, and other oxidants [14,15]. Further, there are large internal sources of oxidants including mitochondrial metabolic processes, peroxisomal fatty acid metabolism, cytochrome P450 reactions, phagocyte activation, and the nitric oxide synthase system (Fig. 1).
Fig. 1.
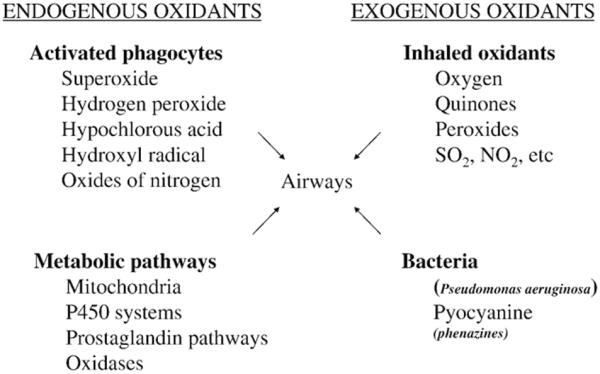
Potential sources of oxidative stress in the CF airways.
Oxidants are crucial for host defense. The respiratory tract is protected from infection by reactive oxygen species (ROS) generated by phagocytic and epithelial cells, and decreases in ROS can result in chronic bacterial infections [16]. In addition to killing bacteria, ROS stimulate wound repair, assist in protein folding, modify apoptosis, and promote signal transduction [17-19].
Although oxidants in the airway provide many benefits, an overabundance of oxidants can cause biomolecular damage that may play an important role in a number of lung diseases, including CF [20]. The reducing environment found in healthy lung cells prevents generation of an exaggerated inflammatory response to inhaled stresses. An oxidative environment, however, influences multiple intracellular signaling events, leading to apoptosis, increased synthesis and secretion of mucin [21], and alterations in ion transport including chloride secretion [22-26]. The result is a cascade of excessive inflammation, mediator release, and tissue injury-events that characterize CF lung disease. Beginning in infancy, individuals with CF are plagued by constant airway inflammation. Large numbers of activated neutrophils migrate into the airways to attack invading bacteria, and have the potential to release into their microenvironment vast amounts of oxidants, including superoxide anion, hydrogen peroxide (H2O2), and hypochlorous acid. Bacteria such as Pseudomonas aeruginosa that chronically infect the CF airways also generate oxygen radicals through the release of pyocyanin and other phenazine pigments [27]. Thus, the CF airway is exposed not only to the normal environmental oxidant burden, but also to oxidants derived from inflammatory and infectious processes, causing a net balance of excess oxidation, as demonstrated by protein and lipid oxidative markers [28-34].
Normal airways are able to cope with oxidative stress through a variety of mechanisms, including the absorption and/or biosynthesis of both nonenzymatic and enzymatic antioxidants. Higher levels of dietary or plasma antioxidants have been associated with better pulmonary function, as demonstrated by a higher level and lower rate of decline of the forced expiratory volume in 1 s (FEV1), and a decreased risk of chronic obstructive pulmonary disease (COPD) [35,36]. Major nonenzymatic antioxidants present at respiratory tract surfaces include ascorbate, urate, α-tocopherol, reduced glutathione (GSH), mucins, and albumin. Metal-binding proteins such as lactoferrin, transferrin, and ceruloplasmin also presumably function as intralumenal airway antioxidants. Although most major enzymatic antioxidants are primarily intracellular, superoxide dismutase and small amounts of catalase and glutathione peroxidase (GSHPx) have also been described in airway fluids [37,38].
Concentrations of the antioxidants that are available extra-cellularly vary widely by location (Table 1). In the normal lung, GSH and ascorbate, for example, are present in fairly high concentrations in bronchoalveolar lavage fluid (BALF), but in the upper airways, where CF disease would be prominent, concentrations are lower and mucin may represent the major scavenging antioxidant [39,40]. On the other hand, α-tocopherol is present in low concentrations throughout the airway surface fluid (ASF) compared to rather high levels in plasma, where its concentration is related to overall lipid levels [39].
Table 1.
Estimates of extracellular antioxidant concentrations in various respiratory tract lining fluids (RTLF) and plasma
| Nasal RTLF (NLF)* | Upper airways RTLF (ALF) | Lower lung RTLF (ELF) | Plasma | |
|---|---|---|---|---|
| Ascorbate (μM) | 28±19 | 2.4 | 40±18 | 67±25 |
| Urate (μM) | 225±105 | 52 | 207±167 | 387±132 |
| GSH (μM) | <0.5 | low | 109±64 | 1.0±0.7 |
| α-Tocopherol (μM) | low | low | 0.7±0.3 | 16±5 |
| Total protein (mg/ml) | low | low | 9.5±6.1 | 74±19 |
NLF, nasal lining fluid; ALF, airway lining fluid; ELF, epithelial lining fluid.
The antioxidants that are present in normal ASF are also found in CF ASF, although levels are sometimes altered. In order to understand how these numerous antioxidant systems can be overwhelmed in CF, it is important to examine the major antioxidant host defenses deployed in the CF airway.
Antioxidants in the airway lumen
Mucin
One of the most abundant proteins in sputum, mucin is both an antioxidant and an important determinant of mucus viscosity. The antioxidant properties of mucin are due to its large number of highly reactive cystine and cysteine residues and to the antioxidant properties of its carbohydrates [41]. Oxidative stress increases mucin gene expression [21] and secretion [42], resulting in increased antioxidant levels at respiratory tract surfaces.
Mucin viscosity is regulated by the movement of water, bicarbonate, and glutathione, all of which are influenced by CFTR [43]. A decrease in any of these factors, which follows from loss of CFTR function, increases the viscosity of the mucin. The thicker, more viscous mucus at the airway epithelial surface provides a stronger antioxidant barrier, although airway mucociliary clearance function is simultaneously compromised.
Mucin levels in CF sputum can be as concentrated as 40 mg/g sputum and may therefore represent much of the antioxidant potential in the CF airway. Too much mucin, however, can be counterproductive for the health of the airways; respiratory mucin actually protects pathogenic bacteria such as Pseudomonas aeruginosa from neutrophil-mediated killing [44]. Inhibition of mucin synthesis has been proposed as a potential therapeutic strategy in cystic fibrosis [45]. If this approach is developed, careful consideration will need to be given to the possibility that a decrease in mucin synthesis may also weaken the airway epithelial antioxidant barrier.
Glutathione
GSH, a tripeptide, is the major low molecular weight thiol compound in animals and has many different roles (Table 2). In the lung cell, GSH functions as an antioxidant in combination with GSHPx or peroxiredoxin. The resulting glutathione disulfide (GSSG) is either rapidly reduced by glutathione reductase and NADPH or utilized in the protein folding process in the endoplasmic reticulum. There, GSSG is recycled by protein disulfide isomerase to form GSH. Because of these recycling mechanisms, GSH is an extremely efficient intracellular buffer for oxidative stress. GSH can reach a concentration of 10 mM inside the cell and, coupled with GSSG, defines the redox state of the cell, although considerations must be given to the intracellular distribution of these species as the various subcellular compartments contain different ratios of this redox pair (e.g., cytosol compared to endoplasmic reticulum compartments).
Table 2.
Glutathione effects
| Effects of GSH sufficiency |
| Antioxidant |
| Anti-inflammatory |
| Mucolytic |
| Facilitates phagocytosis |
| Helps regulate apoptosis |
| Increases primary antibody response |
| Increases B-cell and T-cell proliferation |
| Increases IL-2 production |
| Increases antibody-dependent cellular cytotoxicity |
| Decreases IL-4 and IgE production |
| Detoxifies peroxides |
| Plays a role in posttranslational protein processing |
| Modifies protein function (e.g., protein glutathionylation) |
| Increased susceptibilities associated with GSH insufficiency [65] |
| Oxidant damage to organs and tissues |
| Lipid peroxidation |
| Inhibited ciliary beat function |
| Decreased lung surfactant |
| Bronchoconstriction |
| Increased viscoelasticity of mucus |
| Increased recruitment of neutrophils |
| Decreased S-nitrosoglutathione (GSNO) and nitric oxide (NO) |
| Dysregulation of inflammatory-immune system mediators |
GSH can also be found in the fluid surrounding the lung cell, either free or conjugated to xenobiotics, hydroxynonenal, and other nonforeign metabolites and oxidative products. Conjugation with GSH is a requirement for the externalization of many organic compounds. The extracellular GSH moiety of the secreted conjugates can be broken down by γ-glutamyl transpeptidase (GGT) and other enzymes on the cell surface to form components that can be reabsorbed. Most glutathione in the lung lining fluid, however, appears in the reduced state and acts as a mucolytic due to its ability to cleave disulfide bonds. The method by which GSH is maintained in the extracellular space is unknown, but rapid secretion and degradation are likely to be involved since neither reduction outside the cell nor uptake of GSSG are likely.
Increasing GSH synthesis rates is an important adaptive response to oxidative stress throughout the body [46-49]. Synthesis of GSH can be increased by raising levels of glutamate cysteine ligase (GCL) [48-50], the rate-limiting enzyme in GSH synthesis. GCL activity depends on the intracellular availability of cysteine, so increasing cysteine levels can partially overcome the normally tight regulatory feedback inhibition of GSH synthesis [42,47-49]. In addition, intracellular GSH levels can be raised by augmenting GGT activity, thereby increasing recruitment of cystine and cysteine from outside the cell through the scavenger pathway. This pathway is especially important in lung cells, which along with kidney cells are the only known net importers of GSH. This is useful because both cell types are exposed to high levels of oxidative stress.
Although GSH is not the most abundant antioxidant in human epithelial lining fluid (ELF)-both urate and mucin are present in larger amounts-it appears to play an important role there. In healthy subjects, ELF contains much higher levels of GSH (100-400 μM) than plasma (3 μM) [51]. In fact, GSH is the only antioxidant which is selectively elevated in ELF compared to plasma [40]. Chronic oxidative stress such as that caused by smoking can adaptively induce enzymes that result in a doubling of GSH levels in BALF over those in healthy nonsmokers [51]. The great majority of this is in the reduced state: 96% of the GSH in ELF of nonsmokers and 98% of that in smokers are reduced. In human epithelial cells, the GSH increase induced by oxidants and cigarette smoke is associated with a simultaneous decrease in CFTR function [26,52]. CFTR function is also decreased in healthy smokers as determined by nasal potential difference measurements [52], a response that may represent an antioxidant defense aimed at transiently increasing mucus viscosity and decreasing GSH loss from epithelial cells.
In the individual with CF, however, GSH levels are decreased to about 5-10% of normal in the ELF and 50% of normal in plasma [53]. A number of lung diseases are associated with low ELF GSH, such as idiopathic pulmonary fibrosis [54] and the acute respiratory distress syndrome [55], suggesting that chronic inflammation could be partly responsible [56]. Surprisingly, high levels of GSH have been observed in CF sputum, possibly reflecting the large number of airway inflammatory cells present [54,57]. These contrasting observations suggest that ELF and sputum represent different compartments. Infants and young children with CF-related pulmonary inflammation display less than half the normal amount of GSH in ELF even before there is extensive pulmonary disease [58]. However, GSH levels were shown to be increased in the peripheral blood lymphocytes of some individuals with decreasing lung function due to CF [59], demonstrating the complexity of the story.
Poor nutritional status, which affects the ability to maintain cellular and tissue GSH levels [60], may contribute to low GSH levels in CF [59]. Albumin, which can provide the cysteine needed for GSH production, can be low in individuals with CF due to poor nutrition and activation of inflammatory-immune processes [60]. However, it is likely that decreased GSH levels in the lower respiratory tract lining fluid of the person with CF are at least partially due to faulty CFTR. Healthy carriers of the ΔF508 CFTR mutation exhibit decreased ELF GSH compared to ELF GSH from healthy subjects without the CF mutation (Griese, personal communication). Likewise, the uninfected CFTR knock/out (k/o) mouse displays a 50% decrease in both reduced and disulfide forms of glutathione in lavage fluid [61]. When exposed to P. aeruginosa, the k/o mouse is unable to express a normal increase in ELF GSH [62] and suffers increased lung inflammation and mortality over wild type [63].
In addition to a reduction in extracellular GSH levels caused by loss of GSH transport through CFTR, CF lung cells apparently may reclaim spent extracellular glutathione faster, as evidenced by the increased GGT activity found in children with pulmonary inflammation due to CF [58]. GGT activity is typically increased by oxidative stress [64]: very young children with CF but without inflammation can have normal GSH levels, perhaps due to alternative, developmentally regulated mechanisms for GSH transport that are lost with age [58].
Regardless of the cause, depletion of extracellular lower respiratory tract GSH could contribute to the wide range of consequences of oxidative damage present in CF [65], or, alternatively, it could be a marker of this damage. A direct link between decreased extracellular GSH and these consequences, however, has not been established.
It is unclear whether CF causes changes in the intracellular GSH content of airway epithelial cells. The decreased ability of CF cells to transport GSH into the extracellular space could affect internal concentrations, and a resulting overabundance of intracellular GSH could account for the delay of apoptosis seen in CF lung epithelial cells [66]. On the other hand, decreased intracellular GSH would account for the augmentation of proinflammatory processes seen in CF, including those dependent on NFκB activation and arachidonic acid pathways. In fact, if GSH levels could be increased inside the cell, some NFκB activation by proinflammatory cytokines (such as TNFα) might be ameliorated [60]. Another potential benefit of augmenting intracellular GSH could be a reduction in Th2 response [67], resulting in a decrease in inflammation, as achieved in mice with P. aeruginosa lung infections [68,69]. Furthermore, increased GSH levels could augment levels of GSNO, a molecule that might improve maturation of CFTR and stimulate other chloride channels to increase chloride and GSH flux [70]. Unfortunately, there could also be dangers in altering the levels of such a pluripotent molecule. Increasing GSH could cause conformational changes in a variety of proteins, for example, resulting in alterations to vital cell responses such as immune modulation [71].
Human studies of GSH supplementation
Trials have been conducted in attempts to increase intracellular GSH by oral supplementation. Oral supplementation of healthy volunteers with up to 3 g GSH did not alter plasma levels of cysteine or GSH [72], perhaps because the intestinal mucosal epithelia and liver have very high levels of GGT and the supplemented GSH was rapidly metabolized. The half-life of GSH in human plasma is less than 3 min [73]. Attempts to increase GSH levels by increasing cysteine stores using lipoic acid [74] or whey protein [75,76] have met with some success. Lymphocyte GSH levels in healthy young adults rose 35% in one study using a cysteine donor [75] and in a report of one patient with COPD, increases in whole blood GSH levels correlated with clinical improvements in lung function (FVC and FEV1) [77]. In young adults with CF, 3 months of supplementation with a commercially available whey protein resulted in a 40% increase in lymphocyte GSH [78]. However, no improvements in muscle performance, sputum neutrophils, or lung function were seen, perhaps due to well-established disease, inadequate dose, or the short period of the study. Patients with COPD, for example, had decreased exhaled hydrogen peroxide levels only after 6 months of supplementation with a cysteine donor [79].
Attempts to normalize GSH in the ELF of individuals with CF have been made using inhaled GSH. This approach could carry more risk than an oral route. For example, inhaled GSH has been shown to induce bronchospasm in individuals with mild asthma [80]. Also, although BALF glutathione from healthy subjects is over 90% reduced, GSH that is aerosolized into the airway of CF patients and recovered by lavage is almost all oxidized, possibly by trace amounts of iron present in CF airway mucus [81]. GSH inhalation could therefore be associated with undesired effects: oxidized glutathione (GSSG) could affect cellular receptor and signaling activities by inducing changes in protein structure and function via effects on redox-active protein thiol groups [82]. Furthermore, the inflammatory cells recovered in the BALF in this study produced less superoxide anions than before therapy, suggesting the GSH treatment could potentially compromise airway antimicrobial host defenses [83]. Preliminary trials of inhaled GSH have shown no significant adverse clinical events in either healthy subjects or individuals with CF [84, 85], although the number of subjects in these studies was small.
Therapeutic use of inhaled GSH may be complicated by its rapid clearance from the lung; in one study of sheep, the alveolar lining fluid GSH returned to baseline levels within 2 h of a 600 mg aerosol dose [86]. Another important consideration is that inhaled GSH must reach the proper compartment which is the area between epithelial cells where neutrophils migrating into the lung are releasing myeloperoxidase to synthesize toxic hypochlorous acid [87].
To explore the potential usefulness of GSH aerosolization therapy, a trial was conducted using an over-the-counter preparation of GSH (pH 5.2; 66 mg/kg body weight divided into 4 inhalation sessions per day) for 8 weeks in 19 volunteers with CF, aged 6 to 19 [88]. The volunteers were assigned to either placebo or GSH, with 10 people in the GSH group. No significant differences occurred between treatment and placebo groups in FVC, FEV1, or FEF25-75. Peak flow and subjective feelings of wellness were significantly improved in the GSH treatment group, but because of the strong sulfur smell of GSH (quinine was added to the placebo), the effectiveness of the blinding can be questioned.
More rigorous testing was performed in a trial of inhaled GSH that examined the intrapulmonary deposition of the drug and assessed the tolerance and short-term effects of an optimized inhalation system (AKITA inhalation device, connected to a Pari LC Star nebulizer) [84]. Tests using inhaled 99mTc-labeled Fe3O4 aerosol particles in 6 subjects aged 35±2 years, with an average FEV1 of 69.6±7% (range 50-99%) demonstrated that the majority of material was deposited in the peripheral airways with a small fraction in the central airways. GSH tolerance and effects were assessed in 17 volunteers with CF, age 15-36 years with FEV1 of 61% predicted (range=43-104% predicted). Either 300 or 450 mg GSH (pH 7.0) was inhaled three times daily for 2 weeks. Bronchoalveolar lavage demonstrated increased levels of GSH and GSSG following inhalation. FEV1 and FVC both showed significant improvement following 2 weeks of GSH inhalation. No significant changes were seen in the numbers of neutrophils, lymphocytes, eosinophils, or macrophages in the lavage fluid, but CD4- and CD8-positive T cells with cytokine receptor 5 (CCR5) surface markers were significantly increased in the lavage fluid taken at the end of the 2-week treatment period. There was no apparent change in oxidative stress as measured by carbonyls, thiols, or lipid peroxides in the BAL fluid, but there was a very high level of oxidative stress in these subjects (at least 20-fold higher than controls). The levels of tested cytokines and chemokines (IL-1B, IL-2, IL-4, IL-5, IL-6, IL-10, TNFα, INFγ, G-CSF, MCP-1, and MIP-1β) did not change significantly following GSH inhalation.
Pulmonary function improvements following inhaled GSH treatments suggest that the therapy may be useful. However, any clinical trial investigating the effectiveness of inhaled GSH must consider half-life issues as well as appropriate concerns about the overall safety, optimal dose, duration of treatment, and efficacy.
Oral N-acetylcysteine
N-Acetylcysteine (2-acetamido-3-sulfanyl-propanoic acid, NAC) is absorbed by the intestine more readily than cysteine. Once taken up by cells NAC is deacylated to form cysteine, an essential precursor for GSH synthesis. Oral NAC added to a regimen of prednisone and azathioprine in patients with idiopathic pulmonary fibrosis, a chronic respiratory disease characterized by lung inflammation, high lung oxidant stress, and low ELF GSH, was associated with a slower deterioration in lung function over a 1-year treatment period [89]. Recently, a high-dose oral NAC trial in 18 CF subjects treated for 1 month demonstrated a statistically significant decrease in sputum elastase activity, IL-8, and neutrophil burden [90]. The number of subjects enrolled into the trial and the duration of the study were too small to allow the detection of any changes in clinical parameters such as lung function. The therapy was well tolerated and no adverse reactions were reported. While these results should encourage further studies, the authors caution against the uncontrolled use of oral NAC by CF patients since the nutraceutical status of commercial sources of NAC available over the counter prevents the proper quality control of most formulations.
Nacystelyn
Trials are also being conducted using cysteine-rich Nacystelyn (NAL), a mucolytic with antioxidant and anti-inflammatory activity [91-94]. NAL differs from N-acetylcysteine (Mucomyst), which has a long history of use in CF, by the addition of lysine, raising the pH from 2.2 to 6.0 and increasing the water solubility. The mucolytic effect of NAL is about the same as that of Pulmozyme and the effects are synergistic, as determined by an in vitro test of CF mucus transport [95]. In addition, NAL may have the potential to decrease oxidantinduced IL-8 production [93], decrease the oxidative burst from neutrophils, and increase intracellular GSH levels [91].
NAL may be administered in a dry powder inhaler, with 23.5% of the drug deposited in the lungs of adults and 16.5% in the lungs of children with CF [96]. Clinical trials have demonstrated the safety of NAL in healthy adults, smokers, and people with CF [96,97]. In a multicenter phase 2 trial, a placebo-controlled, double-blinded, randomized trial lasting 24 weeks demonstrated an improvement in mucus viscoelasticity in expectorated sputum and decreased self-reported respiratory infections with NAL aerosol administration (8 or 16 mg bid). Lung function did not improve, but the dose may have been suboptimal. A phase 1a double-blind and placebo-controlled trial in 12 patients receiving up to 80 mg NAL over 3 days showed that all doses were tolerated with no serious adverse events. Further clinical trials are now being planned.
Vitamin E (α-tocopherol)
Vitamin E, or α-tocopherol, is a powerful membrane-localized anti-lipid peroxidation antioxidant. As with other antioxidants, vitamin E has multiple roles in the body, including indirect stimulation of the transcription of certain genes encoding drug-metabolizing cytochrome P450 enzymes [98]. Consequently, high doses of vitamin E could potentially change the metabolism of many drugs.
Vitamin E deficiency has produced broad effects in animal models including modulation of innate and acquired immune functions, such as T- and B-cell mitogenesis, T-helper cell function, macrophage function, neutrophil chemotaxis, phagocytosis, and H2O2 production [99,100]. Prolonged deficiency of vitamin E in people can result in neuromuscular degeneration including cerebellar ataxia, neuropathy, or myopathy, particularly in children with cholestatic liver disease or intestinal resection [101-103]. Hemolytic anemia, retinal degeneration, and cognitive defects have also been observed [104,105].
Vitamin E, like other fat-soluble vitamins, is frequently deficient in CF. Because α-tocopherol is carried almost exclusively in circulating lipoproteins, serum levels depend on the level of circulating lipids, which are often low in individuals with CF. Consequently, vitamin E levels in the person with CF should be expressed as a ratio of vitamin E to total serum lipids. In infants, a ratio of at least 0.6 mg to tocopherol/g total lipids reflects adequate vitamin E; for older children and adults, the ratio should be at least 0.8 mg/g lipids. Using this method, 22.8% of infants identified with CF at 4-6 weeks of age through a newborn screening program demonstrated vitamin E deficiency [106]. In the only randomized study of newborn screening versus conventional diagnosis, there was no difference in the prevalence of vitamin E deficiency at diagnosis; vitamin E deficiency was seen in 49% of all patients. However, the duration of deficiency of this important antioxidant was 15 months longer in the patients diagnosed by the presence of symptoms than that in the newborn screened infants. Only 8 and 3% of patients had low α-tocopherol levels at 3 and 6 months after treatment, respectively [105]. In another report on vitamin E status in infants diagnosed by newborn screening, of the initial patients who were vitamin E sufficient, 12% subsequently became deficient despite supplementation, and 10% remained persistently deficient [107]. Numerous studies have shown that the great majority of children and adults with CF who do not receive vitamin E supplementation are deficient in vitamin E, even if they receive standard multiple vitamins and are on pancreatic replacement enzymes [107,108]. Unfortunately, no lung tissue or airway secretion determinations of vitamin E levels have been made in people with CF.
The recommended dietary intake of vitamin E is 6-10 IU/day in healthy children from birth to age 7 years and 15-25 IU/day in healthy individuals age 8 years to adult. High amounts of polyunsaturated fatty acids in the diet may increase the need for vitamin E to prevent lipid peroxidation. The current CF Foundation consensus conference on nutrition (Table 3) [109] recommends 40-400 IU/day (in addition to a multiple vitamin) to normalize plasma levels of vitamin E. Almost all individuals with CF are able to attain normal serum levels of vitamin E when given 5-10 IU/kg/day (approximately 400-800 IU/day for adults) of almost any form of vitamin E when given with food and pancreatic enzymes [110]. Low levels despite supplementation [106] may be due to lack of adherence, malabsorption despite pancreatic enzyme supplementation, intestinal resection, or liver disease. Individuals with cholestatic liver disease should be supplemented with D-α-tocopheryl- polyethylene glycol 1000 succinate (TPGS), the only truly water-soluble form available in the United States [111].
Table 3.
Current CF Foundation recommendations for antioxidant supplementation [109]
| Supplement | How often to measure levels | Recommended supplementation dose |
|---|---|---|
| β-Carotene | At physician's discretion | None |
| Vitamin A | Annually | 0 - 12 months 1500 IU |
| 1 - 3 years 5000 IU | ||
| 4 - 8 years 5 - 10,000 IU | ||
| >8 years 10,000 IU | ||
| Vitamin E | Annually | 0 - 12 months 40 - 50 IU |
| 1 - 3 years 80 - 150 IU | ||
| 4 - 8 years 100 - 200 IU | ||
| >8 years 200 - 400 IU | ||
| Zinc | Not recommended | Supplementation is reasonable in those with suboptimal vitamin A status or with night blindness that does not respond to vitamin A supplementation. A 6-month supplementation trial of Zn should be considered for children who are not growing well. |
| Other non-fat-soluble vitamins and minerals | A standard, age-appropriate dose of non-fat-soluble multivitamins should be given daily. |
Vitamin E supplementation has proven beneficial in certain populations: healthy elderly volunteers receiving 800 IU daily for 1 month significantly increased T-cell proliferation, IL-2 production, and response to DTH skin tests, and decreased PGE2 production [112-114]. Vitamin E supplementation also increased GSH levels in liver cells, possibly due to a “sparing” effect, with resultant benefits [115,116]. However, supplementation is not without potential risk. A recent metaanalysis of 19 clinical trials demonstrated an increased risk of mortality in trials using more than 150 IU/day in patients with a variety of diseases but with normal fat absorption [117], and the recent heart outcomes evaluation (HOPE) trial suggests that vitamin E supplementation may increase the risk for heart failure [118]. Adults who receive above 1500 IU/day and who are on coumadin or who are vitamin K deficient may have increased coagulopathy with a potential for decreased neutrophil function. This is especially significant in CF, as a number of patients are at least borderline deficient in vitamin K. Toxicity in children has not been sufficiently studied; some trials using large doses of iv vitamin E in premature infants resulted in increased risk of bacterial and fungal sepsis and necrotizing enterocolitis [119]. However, numerous trials of large oral doses of vitamin E in adults have not demonstrated any increased risk of infection.
The clinical results of vitamin E trials may be influenced by the nature and dose of the administered vitamin E supplement. The all-racemic form, or D,L-α-tocopherol is actually a mixture of eight stereoisomers and is the one commonly found in vitamin supplements because it is the least expensive form to manufacture. However, D-α (or RRR-α) tocopherol is more bioavailable. TPGS-vitamin E, which contains D-α tocopherol, is water soluble and highly bioavailable, but is found only in ADEKS drops and two liquid preparations (Liqui-E, Twinlabs, and Aqua-E, Yasoo Health) and is not present in the ADEKS or Vitamax tablets that are commonly used by the CF population.
No prospective trials have demonstrated an effect on pulmonary outcomes of vitamin E supplementation in individuals with CF. The level of lipid peroxidation as measured by 8-isoprostane-PGF2α decreases significantly with vitamin E supplementation in CF [120,121], but the clinical relevance of this is unknown. Nevertheless, preventing and correcting vitamin E deficiency in CF are essential in order to prevent neuromuscular degeneration that has been reported.
Carotenoids
Carotenoids found in the body are classified by their ability to be converted into vitamin A (retinol). Provitamin A carotenoids include β-carotene, the most prominent carotenoid in human nutrition, α-carotene, and β-cryptoxanthin. Nonprovitamin A carotenoids include lutein, zeaxanthin, and lycopene.
Carotenoids are present in carrots, tomatoes, watermelon, spinach, and many other foods. The preparation of the food significantly affects absorption and bioavailability of the carotenoids [122]. Raw fruits and vegetables such as carrots and tomatoes generally contain carotenoids with poor bioavailability because their crystalline structures are hard to break down. Mildly cooking the food or processing it with oil can increase the bioavailability significantly. The highest bioavailability is obtained from oil solution or water-dispersable beadlets of carotenoids [123].
Adverse clinical manifestations of carotenoid deficiency in man are unknown as long as vitamin A levels are normalized. Although higher levels of lutein and zeaxanthin in the diet were related to lower incidence of retinal pigmentary abnormalities [124], increasing the intake of these carotenoids has not been demonstrated to be protective against the development of age-related macular degeneration [125].
As with other lipophilic antioxidants, carotenoid and retinol levels are commonly decreased in CF. Children with CF display 9-24% of normal values [126-133]. The plasma β-carotene/cholesterol ratio is also very low, with a value of about 18% of normal [134]. Poor absorption, cholestatic liver disease, or rapid turnover of carotenoids due to active inflammation could all contribute to low carotenoid levels [135,136]. However, despite severe reduction of lutein and zeaxanthin in both plasma and retina of people with CF, no related abnormalities of visual function under daylight conditions have been found [137], though poor nocturnal vision in some individuals has been corrected by retinol supplementation [138].
The oxidant burden imposed by active CF respiratory tract disease could increase the importance of carotenoids. Several clinical features in CF may correlate with carotenoid concentrations including increased inflammation and decreased infection control [126]. Further, inflammatory markers in plasma correlate negatively with β-carotene, lycopene, and retinol [139].
Because of this close association, a number of trials have examined β-carotene supplementation in CF (Table 4) [128-130,132,140,141]. Plasma levels before supplementation were consistently very low. Plasma β-carotene levels achieved after supplementation ranged from 0.30 to 3.99 μmol/L, depending on dose and formula; water-miscible β-carotene is more effectively absorbed into the blood.
Table 4.
β-Carotene supplementation studies in cystic fibrosis
| References | Study period | Dose | n | CF patients |
|||
|---|---|---|---|---|---|---|---|
| Plasma β-carotene |
|||||||
| Months | mg kg-1 day-1 | mg day-1 | Baseline μmol/1 | Final μmol/1 | Healthy controls μmol/1 | ||
| Winklhofer-Roob et al. [128] | 3 | 0.5 | - | 32 | 0.09±0.06 | 1.07±0.86 | 0.99±0.41 |
| Winklhofer-Roob et al. [140] | 16 | 0.5 | - | 33 | 0.09±0.06 | 1.06±1.09 | 0.87±0.56 |
| Homnick et al. [141] | 12 | - | 144(60-240) | 5 | 0.09±0.02 | 0.62±0.19 | - |
| Homnick et al. [141] | 2 | - | 60 | 5 | 0.09±0.02 | 0.22±0.11a | - |
| Homnick et al. [141] | ? | - | 120 | 5 | 0.09±0.02 | 0.4±0.17a | - |
| Rust et al. [130] | 3 | 1 | Max 50 | 13 | 0.08±0.04 | 0.6±0.4 b | 0.30±0.1 |
| Rust et al. [130] | 3 | - | 10 | 13 | 0.08±0.04 | 0.3±0.2 b | 0.30±0.1 |
| Cobanoglu et al. [132] | 6 | 1.07±0.57 | - | 15 | 1.08c(0.3-2.9) | 2.4c(0.38-8.8) | 2.12c(0.14-2.9) |
| Lepage et al. [129] | 2 | - | 13.3 | 12 | 0.08±0.03 | 3.99±0.92 | 0.37±0.04 |
| Wood [133] | 2 | - | 25 | 24 | 0.00±0.02 | Δ0.1±0.04 | Nd |
Increase from baseline.
Subsequent study periods.
Serum concentrations (μg ml-1), median (minimum-maximum).
Carotenoid supplementation trials have generally demonstrated favorable results on biomarkers of oxidative stress. Malondialdehyde, a marker of lipid peroxidation which is elevated in children with CF [128], decreased in 5 out of the 6 β-carotene supplementation trials where it was studied; the one trial in which it did not decrease was a low-dose supplementation trial. β-Carotene supplementation improved resistance of LDL to oxidation [128,129] and decreased plasma TNFα [132,142] and neutrophil elastase/α-1-proteinase inhibitor complexes [142], markers of inflammation. Effects on other plasma micronutrient antioxidants are not entirely clear, but plasma retinol and α-tocopherol increased in at least one trial [132]. The increased plasma α-tocopherol could be due to increased adherence to vitamin supplementation due to the trial or a sparing effect of α-tocopherol by β-carotene.
Clinical outcomes have generally also been favorable. In an 8-year longitudinal study of 1194 French subjects from the general population, β-carotene was found to protect against the decline of FEV1 [143]. Furthermore, in a study of CF subjects, the number of days on antibiotics decreased significantly in a high-dose regimen study, although it did not change in the low-dose arm of that same study [144]. FEV1 increased significantly in some studies and FVC increased in those people with CF who experienced an increase in plasma carotenoid levels [132,144]. Unfortunately, the use of a combination of antioxidants in the last trial confounded interpretation of the results. Finally, weight and height z scores increased significantly in the two studies in which they were measured [142,145].
Side effects of β-carotene supplementation have been limited. In a 16-month trial of β-carotene supplementation in volunteers with CF there was a significant decrease in ALT and AST liver functions, but a significant increase in GGT (from 23.1±31 to 36.1±63.5) [146]. In addition, detrimental effects of β-carotene supplementation have been documented in heavy cigarette smokers in non-CF populations [147].
However, in individuals with CF, β-carotene supplementation has generally been associated with favorable results such as decreased levels of lipid peroxidation markers, circulating markers of inflammation, and number or severity of pulmonary exacerbations (Table 5). Increases in ex vivo resistance to LDL oxidation and improvements in lung function have also been demonstrated.
Table 5.
Reported effects of β-carotene supplementation in CF
| Final plasma β-carotene (μmol/l) | Study period | Oxidative stress |
Inflammation |
ATB, days | Lung function, FEV1 | Weight* height† z score | Ref. No. | |||
|---|---|---|---|---|---|---|---|---|---|---|
| (months) | MDA | LDL | 8-iso-PGF2α | NE/α1-PI | TNFα | |||||
| 1.07±0.86 | 3 | ↓ | ↓ | - | - | - | - | - | - | [104] |
| 1.07±0.19 | 18 | ↓ | - | - | ↓ | ↓ | - | - | ↑* | [118] |
| 3.99±0.92 | 2 | ↓ | - | - | - | - | - | - | - | [105] |
| 0.60±0.40 | 3 | ↓ | - | - | - | - | ↓ | - | - | [119] |
| 0.30±0.20 | 6 | NS | - | - | - | - | NS | NS | ↑† | [119] |
| 0.07 to 1.63 | 6 | ↓ | - | - | - | ↓ | - | ↑ | - | [108] |
| Δ0.1±0.04 | 2 | - | - | NS | - | - | NS | NS | - | [109] |
MDA, malondialdehyde; LDL, low-density lipoprotein oxidation; 8-iso-PGF2α, 8-iso-prostaglandin F(2)α; NE/α1-PI, neutrophil elastase/α1-PI; TNFα, tumor necrosis factor α; ATB, antibiotic treatment; FEV1, forced expiratory volume in 1 s.
Coenzyme Q-10
Coenzyme Q-10 (CoQ) or ubiquinone is a lipid-soluble component of the mitochondrial electron transport chain. CoQ also helps reduce vitamin E, maintaining it as a viable antioxidant [148], and is an antioxidant in its own right. Diet provides more than 25% of the circulating CoQ pool; the remainder is synthesized through the cholesterol pathway. Because CoQ is primarily found in tissues containing high percentages of mitochondria, meats, poultry, fish, and liver are good sources of this antioxidant. The typical daily intake of CoQ is 3-5 mg.
Malabsorption or oxidation of CoQ may result in decreased circulating levels in people with CF, possibly resulting in decreased ATP production or protection against oxidative stress or both. In a study of 274 patients with CF, over 50% of subjects had below normal levels of total (oxidized and reduced) CoQ (0.4 μg/ml in children under 8 years; 0.3 μg/ml in individuals over 8 years of age) (R. Sokol, personal communication). As expected, pancreatic insufficient patients demonstrated much lower levels of CoQ (mean=0.31 μg/ml) than pancreatic sufficient patients (mean=0.49 μg/ml, P<0.0001). Because decreased CoQ levels may reflect decreased lipid levels in the serum, CoQ levels should be interpreted in the context of cholesterol levels and, if possible, triglyceride and phospholipid levels as well [149-151].
Despite a deficiency in CoQ levels in individuals with CF, in a retrospective study there did not appear to be a relationship between CoQ and pulmonary function (as expressed by days hospitalized, number of admissions, FVC % predicted and FEV1% predicted, R. Sokol, pers. commun.) [152]. Also, CoQ levels did not appear to correlate with Pseudomonas infection and weight and height z scores.
Enzymatic antioxidants
The nonenzymatic antioxidants described thus far have low rate constants (105) and are only moderately efficient. In contrast, the enzymatic antioxidant extracellular superoxide dismutase (EC-SOD), which is found at high concentrations in the normal lung [153], has a much higher rate constant (109) and is very efficient. EC-SOD is essential for protecting NO from oxidation and therefore is essential for protecting the lung from inflammation. The concentration of EC-SOD in the CF lung is unknown.
Recombinant human Cu2+/Zn2+ superoxide dismutase has been delivered to the sheep airway epithelial surface by aerosol and shown to retain its activity against superoxide for several hours [154]. However, clinical studies of this approach have not been reported.
A mimetic of EC-SOD with a long half-life and broad antioxidant properties has been developed. This new molecule was successful in reducing neutrophil migration into the lungs of rats exposed to 8 weeks of oxidative stress in the form of tobacco smoke [155]. The mimetic also reduced eosinophilia and inflammatory mediators in a mouse model of asthma [156]. Its usefulness in CF has not been explored.
Zinc
Zinc (Zn) is an essential trace element that functions as a cofactor for more than 200 intracellular and extracellular enzymes. As such, it is involved in many cell processes including growth, differentiation, and signaling. Zn is important in antioxidant defenses due to its association with EC-SOD and a cytosolic copper-zinc-SOD. Further, there is evidence that Zn stabilizes lipid membranes and prevents peroxidation. Some alternative chloride channels that are Zn sensitive might even be targets for CF therapy [157].
Severe Zn deficiency causes loss of appetite and impaired growth. In mouse models of asthma, Zn deficiency correlated very strongly with high levels of eosinophilia and increased Th2 responses [158,159]. That may be especially relevant to the CF population, where there is often a large Th2 response. There is no robust marker, however, for Zn deficiency. Plasma levels of Zn can be apparently normal and the individual may still be clinically deficient such that Zn supplementation improves growth and resistance to infection. Dozens of trials of oral Zn supplementation have demonstrated decreases in infectious diarrhea or lower respiratory tract infections without large changes in circulating Zn levels. Zn levels in red blood cells provide a more accurate picture, but this test is not routinely performed, and Zn content in hair or nails is too variable to be of use.
Individuals with CF can experience a wide range of Zn deficiencies. Twenty to 30% of infants with CF identified by newborn screening have insufficient plasma levels of Zn by 2 months of age [160], and some infants with CF can have severe Zn deficiency by that time [161,162], resulting in rash, diarrhea, and a potentially fatal decrease in resistance to infection. Because Zn pools in the body depend on fat absorption [160], pancreatic enzyme administration greatly increases Zn levels [163], but supplementation is still needed for normalizing plasma levels in many individuals with CF.
Zn supplementation could produce multiple benefits in individuals with CF. In individuals without CF in developing countries, Zn supplementation can decrease mortality due to infection and increase linear growth in children who are more than 2 standard deviations (SD) below the mean [164]. Such a response in linear growth would be helpful in the CF population, since stature correlates with survival and the average adult with CF is 5 inches below predicted height.
Because clinical deficiencies can exist in the presence of normal plasma levels, Zn supplementation should be tried especially in individuals who are 2 SD below the norm in height or weight. Most vitamins used in the CF population, except for Vitamax, already contain Zn, although at doses lower than those used in the Zn trials in developing countries. As with other nutrients, higher levels may be needed in the individual with CF due to absorption difficulties. It is not known whether supersupplementation of Zn, beyond that required to normalize plasma levels, would improve health.
Clinical trials should be conducted before Zn supplementation is mandated for all individuals with CF. Because one study has shown an association between Zn supplementation and a very mild developmental delay [165], these trials must be conducted extremely carefully. Furthermore, a biomarker (for Zn levels or oxidative stress) may not be readily available for a proof of concept trial.
Selenium
Selenium (Se) is an essential component of a number of enzymes including GSHPxs, thioredoxin reductases, thyroid hormones, and circulating selenoprotein-P. In addition to its antioxidant functions, Se is needed for a variety of other functions including neutrophil responses to pathogens [166], cell cycling [167], and metabolism of carcinogens [168].
Se levels in the body reflect the varying content of Se in foods, depending in part on where the food is grown. The average intake of Se from food in the United States is 108 μg/day, resulting in an average Se serum level of 1.57 nmol/L in adults and 1.52-1.54 nmol/L in children 9-18 years of age [169]. In the United Kingdom, the average plasma Se level is considerably lower (0.86-0.88 nmol/L in 4-18 year olds) [170]. Factors other than dietary intake may affect Se plasma levels; Se levels in the UK study increased with age and were lower in lower social classes and in children of parents who smoked.
Se deficiency results in reduced GSHPx activity, which can be restored by Se supplementation. In fact, the Recommended Daily Intake (RDI) for Se (70 μg/day for men and 55 μg/day for women) is based on the amount needed to maintain GSHPx activity, plus a 30% safety factor inflation. Plasma levels of selenium respond quickly to supplementation. GSHPx activity can be used to measure Se status, but it saturates at low levels and is only an approximate characterization of suboptimal intake.
Increasing plasma levels of Se may have important health implications. Higher plasma levels of Se are associated with decreased risk of asthma [171] and with higher FEV1, particularly in the face of chronic oxidative stress caused by active or passive exposure to cigarette smoke [172]. Selenium supplementation has been associated with a reduction in cancer risk [173]. In individuals with CF, plasma levels of Se are generally 1 SD below normal (0.65 μm/L Se vs 0.85 μm/L Se [174]). The reasons for low levels are unclear, but may include differences in dietary intake, absorption, distribution, or excretion. Although these levels do not indicate Se deficiency, they are low enough that supplementation would be expected to increase GSHPx activity. However, the consequence of that response is not clear.
Se supplementation studies in individuals with CF suggest benefit. Use of a Se-containing pancreatic enzyme supplement increased plasma Se levels and GSHPx activity [174]. Also, a randomized controlled trial in 46 children with CF, 10-12 years of age, compared a supplement containing 10 mg vitamin E and 500 μg vitamin A with an antioxidant cocktail containing 90 μg Se (delivered as Se-methionine), 200 mg vitamin E, 300 mg vitamin C, 25 mg β-carotene, and 500 μg vitamin A over 8 weeks [133]. Although the baseline level of plasma Se in this study (1.0 μm/L) was already higher than in many of the earlier studies, plasma Se increased 0.60 μm/L (P<0.001). No change occurred in oxidative stress as a result of treatment, as measured by F2-isoprostanes, but the increase in plasma Se levels correlated with an increase in percentage predicted FEV1. Thus, this short-term study showed both statistical and biological significance of supplementation.
The heterogeneity of Se status in CF in relation to disease progression is not known, and should be examined to determine the kinds of patients who are most likely to benefit from supplementation. In a cancer prevention trial in a non-CF population, individuals with lower initial Se plasma levels received greater benefit from supplementation [173], suggesting many individuals with CF may benefit, since plasma selenium levels in these individuals are often low. However, caution is needed in designing a supplementation trial due to possible toxic effects. The upper limit of Se intake is set at 400 μg/day, and Se content in the pancreatic enzyme preparations in use must be considered.
Iron
Iron is a metal ion essential for the synthesis of numerous heme-centered enzymes and other proteins involved in oxygen transport, oxidative metabolism, and host defenses. However, iron that is not bound to proteins can undergo reduction to the ferrous (Fe2+) state, a process catalyzed by superoxide anions. The Fe2+ ion greatly accelerates autoxidation of catechols, conversion of hydrogen peroxide to hydroxyl radicals, and lipid peroxidation. Most iron in biological compartments is bound in the ferric (Fe(III)) state to proteins such as ferritin, transferrin, and lactoferrin. In this form it is not readily available to catalyze ROS formation [175]. Iron-catalyzed ROS synthesis is also decreased by the presence of ceruloplasmin, a copper-rich protein synthesized in the liver and secreted in plasma. The ferroxidase activity of ceruloplasmin oxidizes Fe2+ to the Fe (III), thus preventing hydroxyl radical formation [176]. Transferrin and to a lesser extent ceruloplasmin in the ELF of healthy individuals can prevent iron-mediated lipid peroxidation [177]. Unfortunately individuals with CF whose lungs are colonized by Pseudomonas aeruginosa are at an increased risk of iron-mediated ROS formation because elastase from the bacteria cleaves transferrin and lactoferin [81]. Indeed, lactoferrin and transferin cleavage products appear in the ELF of most individuals with CF and Pseudomonas aeruginosa [178]. The iron released from these cleaved proteins can catalyze hydroxyl radical formation. Although no proven effective therapy is currently available to prevent iron-catalyzed ROS formation in the lung, these data point to the importance of preventing and treating airway Pseudomonas aeruginosa infection in the CF lung as an antioxidant strategy.
History of clinical trials of antioxidants in other diseases
Before designing clinical trials of antioxidant supplements in CF, it is useful to examine data from animal models and human trials in other diseases. Studies of high efficiency antioxidants in more than 20 animal models of inflammatory diseases demonstrate that severe inflammatory stress is diminished by high antioxidant levels, and accelerated by low antioxidant levels. Powerful antioxidants have not been available for human trial, but supplementation with weaker antioxidants has been tested for prevention of a variety of diseases. Results have been mixed. In the National Health and Nutrition Examination Survey iv trial, levels of high-sensitivity C-reactive protein, an inflammatory marker, were inversely related to the levels of serum antioxidant micronutrients including Se [179]. However, results of antioxidant supplementation trials have largely been disappointing, demonstrating little effect of Se and vitamins E and C on gastric cancer [180] and no effect of vitamin E and β-carotene on the prevention of heart disease [181]. Some randomized, controlled trials have actually displayed adverse effects of supplementation with antioxidants such as β-carotene and vitamins E or A on lung cancer and heart disease [118,182,183], demonstrating a need for caution. In other trials, the results have been mixed: Se supplementation correlated with an increased risk of some cancers and a reduced risk of others [173].
Thus, despite the clear relationship between antioxidant-rich fruit and vegetable consumption and health, results from a number of large, well-designed trials of antioxidant supplements do not overtly support their use. A host of factors may cause the conflicting results, including wrong formulations or doses of the supplements and perhaps suboptimal delivery of antioxidants to the sites of inflammation-related oxidative stress. Alternatively, in these populations, which are not deficient in antioxidants, the body may simply excrete or metabolize the surplus nutrients.
Future clinical trials of antioxidants in CF
Arguments for antioxidant trials in CF appear compelling despite disappointing results in reducing risk of some other diseases associated with lower degrees of oxidative stress [183]. Levels of various antioxidants in extracellular fluids are significantly decreased in the individual with established CF lung disease. It seems possible that restoring these levels to restore redox balance could, in theory, modulate inflammatory and immune processes, reduce inflammation-related oxidative stresses, and decrease oxidative biomolecular damage with minimal detrimental effects on intracellular signaling pathways.
Recommendations
Supplementation for normalization of serum levels of vitamins A and E should be a part of routine CF care. These lipid-soluble antioxidants, which have profound cytoprotective effects on bronchoepithelial cells, are often dramatically decreased in plasma of individuals with CF. The known extrapulmonary clinical deficiency states for these two nutrients require prevention and correction of low serum levels. The reported relationships between plasma levels and improved clinical outcomes for other lipid soluble antiox-idants are preliminary but because the evidence suggests that supplementation may be beneficial, carefully controlled trials should be a priority.
Individuals with CF should ingest a minimum of the RDI for water-soluble antioxidants such as ascorbate, zinc, and selenium. Studies to determine optimal intakes for these and for antioxidants for which there are no RDIs, such as coenzyme Q-10, should be encouraged.
Interventional studies using antioxidant “cocktails” or of individual antioxidants should include measurements of biomarkers of inflammation and oxidative stress, as well as scoring for clinically relevant outcomes.
A working group should be established to study optimal levels for nutritional antioxidants in CF and to standardize the way levels are expressed, including the use of lipid correction and documentation of pancreatic function. This group should also recommend that biologic markers of antioxidant stress and inflammation to be used in future trials of antioxidants in people with CF.
A carefully designed, proof of concept human trial of inhaled GSH prepared in a good manufacturing procedures (GMP) facility, which includes measures of safety and biologic markers of antioxidant effect, should be conducted to determine if a larger trial is warranted. Similar investigation should be conducted into the potential benefits of oral supplementation with lipid-soluble antioxidants, perhaps in combination.
Individuals with CF should avoid exposures that can increase oxidant stress, such as environmental tobacco smoke or excessive levels of air pollutants.
Adherence to routine CF care, including preventive care and surveillance for and treatment of pulmonary exacerbations, is important to control lung inflammation and the resultant oxidative stress.
Conclusions
Many questions must be answered to understand the potential of systemic or aerosolized antioxidant treatment in CF:
Do oxidants at the respiratory tract surface cause injury, or are they simply markers of airway inflammatory-immune system activations?
Can antioxidant substances at respiratory tract surfaces actually be increased and can we target antioxidants to that surface?
If antioxidants can be augmented at the airway surface, what effects will there be on the intracellular redox state and on levels of enzymatic antioxidants in the inflammatoryimmune system cells and epithelial cells at the respiratory surfaces?
What effects might the presence of augmented antioxidants have on antimicrobial and repair processes at airway surfaces?
Can antioxidant therapy prevent the oxidative inactivation of key proteins involved in host defenses such as alpha-1 antiproteinase?
Will augmentation of antioxidants at these surfaces amelio-rate the relentless and progressive respiratory tract injury in CF?
Any clinical trial must consider the pleiotropic effects of antioxidants and ensure that important oxidative host defenses are not compromised. The answers to these questions could potentially have significant effects on treatment strategies for the individual with CF.
Acknowledgments
We thank Dr. Preston Campbell of the Cystic Fibrosis Foundation for critical reading of the manuscript and insightful suggestions. List of Participants: Frank Accurso, M.D., Department of Pediatrics, University of Colorado School of Medicine and The Children's Hospital, Denver, CO; Melissa A. Ashlock, M.D., Cystic Fibrosis Foundation, Bethesda, MD; Robert J. Beall, Ph.D., Cystic Fibrosis Foundation, Bethesda, MD; Clark Bishop, M.D., Intermountain Health Care, Provo, UT; Drucy Borowitz, M.D., Pediatric Pulmonology, State University of New York at Buffalo, Buffalo, NY; Tim Byers, M.D., M.P.H., Department of Preventative Medicine and Biometrics, University of Colorado, Denver, CO; Preston W. Campbell, III, M.D., Cystic Fibrosis Foundation, Bethesda, MD; André M. Cantin, M.D., Pulmonary Division, University of Sherbrooke, Sherbrooke, Quebec; Patricia A. Cassano, Ph. D., Division of Nutritional Sciences, Cornell University, Ithaca, NY; James D. Crapo, M.D., Department of Medicine, National Jewish Medical and Research Center, Denver, CO; Carroll E. Cross, M.D., Pulmonary-Critical Care Medicine, UC Davis Medical Center, Sacramento, CA; Brian J. Day, Ph.D., Department of Medicine and Pharmaceutical Sciences, National Jewish Medical and Research Center, Denver, CO; Henry Jay Forman, Ph.D., Division of Natural Sciences, University of California, Merced, CA; Matthias Griese, M.D., Kinderklinik und Kinderpoliklinik IM, University of Munich, Munich, Germany; Carolyn Habbersett, M.A., Cystic Fibrosis Foundation, Bethesda, MD; Leslie Hazle, M.S., R.N., Cystic Fibrosis Foundation, Bethesda, MD; Michael W. Konstan, M.D., Department of Pediatrics, Rainbow Babies and Children's Hospital, Cleveland, OH; Norman Krinsky, Ph.D., Department of Biochemistry, Tufts University School of Medicine, Boston, MA; Larry C. Lands, M.D., Ph.D., McGill University Health Centre, Montreal Children's Hospital, Montreal, Quebec; Bruce Marshall, M.D., Cystic Fibrosis Foundation, Bethesda, MD; Philip O'Reilly, M.D., Division of Pulmonary/Allergy/Critical Care Medicine, University of Alabama School of Medicine, Birmingham, AL; Christopher M. Penland, Ph.D., Cystic Fibrosis Foundation, Bethesda, MD; Ronald J. Sokol, M.D., Department of Pediatrics, University of Colorado School of Medicine and The Children's Hospital, Denver, CO; Allison M. Tobin, Cystic Fibrosis Foundation, Bethesda, MD; David A. Waltz, M.D., Department of Medicine, Children's Hospital, Boston, MA; Diana R. Wetmore, Ph.D., Cystic Fibrosis Foundation, Bethesda, MD; Terry B. White, Ph.D. Cystic Fibrosis Foundation, Bethesda, MD; Brigitte M. Winklhofer-Roob, M.D., Institute of Molecular Biology, Biochemistry, and Microbiology, Karl-Franzens University of Graz, Graz, Austria; Dayong Wu, M.D., Ph.D., Nutritional Immunology Laboratory, Tufts University, Boston, MA.
Abbreviations
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- ASF
airway surface fluid
- BALF
bronchoalveolar lavage fluid
- cAMP
adenosine 3,5-cyclic phosphoric acid
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane conductance regulator
- COPD
chronic obstructive pulmonary disease
- CoQ
coenzyme Q-10
- ELF
epithelial lining fluid
- ENaC
epithelial sodium channel
- EC-SOD
extracellular superoxide dismutase
- FEV1
forced expiratory volume in 1 s
- FVC
forced vital capacity
- FEF25-75
forced expiratory flow at 25-75% of forced vital capacity
- GGT
gamma glutamyl transpeptidase
- GSH
glutathione
- GSNO
S-nitrosoglutathione
- GSSG
glutathione disulfide
- GSHPx
glutathione peroxidase
- GMP
good manufacturing procedures
- IL-8
interleukin-8
- IL-2
interleukin-2
- iv
intravenous
- LDL
low-density lipoproteins
- NAL
Nacystelyn
- NFκB
nuclear factor kappa B
- PGE2
prostaglandin E2
- RDI
recommended daily intake
- ROS
reactive oxygen species
- SD
standard deviation
- Se
selenium
- SOD
superoxide dismutase
- TPGS
D-α-tocopheryl-polyethylene glycol 1000 succinate
Footnotes
A list of participants may be found in the Acknowledgments.
References
- [1].Davis PB, Drumm M, Konstan MW. Cystic fibrosis. Am. J. Respir. Crit. Care Med. 1996;154:1229–1256. doi: 10.1164/ajrccm.154.5.8912731. [DOI] [PubMed] [Google Scholar]
- [2].Rosenstein BJ, Cutting GR. The diagnosis of cystic fibrosis: a consensus statement. Cystic fibrosis foundation consensus panel. J. Pediatr. 1998;132:589–595. doi: 10.1016/s0022-3476(98)70344-0. [DOI] [PubMed] [Google Scholar]
- [3].Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, Rozmahel R, Cole JL, Kennedy D, Hidaka N, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245:1059–1065. doi: 10.1126/science.2772657. [DOI] [PubMed] [Google Scholar]
- [4].Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA [published erratum appears in Science 245 (4925):1437; 1989] Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- [5].Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989;245:1073–1080. doi: 10.1126/science.2570460. [DOI] [PubMed] [Google Scholar]
- [6].Pilewski JM, Frizzell RA. Role of CFTR in airway disease. Physiol. Rev. 1999;79:S215–S255. doi: 10.1152/physrev.1999.79.1.S215. [DOI] [PubMed] [Google Scholar]
- [7].Hug MJ, Tamada T, Bridges RJ. CFTR and bicarbonate secretion to epithelial cells. News Physiol. Sci. 2003;18:38–42. doi: 10.1152/nips.01412.2002. [DOI] [PubMed] [Google Scholar]
- [8].Ballard ST, Trout L, Bebok Z, Sorscher EJ, Crews A. CFTR involvement in chloride, bicarbonate, and liquid secretion by airway submucosal glands. Am. J. Physiol. 1999;277:L694–L699. doi: 10.1152/ajplung.1999.277.4.L694. [DOI] [PubMed] [Google Scholar]
- [9].Mall M, Bleich M, Kuehr J, Brandis M, Greger R, Kunzelmann K. CFTR-mediated inhibition of epithelial Na+ conductance in human colon is defective in cystic fibrosis. Am. J. Physiol. 1999;277:G709–G716. doi: 10.1152/ajpgi.1999.277.3.G709. [DOI] [PubMed] [Google Scholar]
- [10].Linsdell P, Hanrahan JW. Glutathione permeability of CFTR. Am. J. Physiol. 1998;275:C323–C326. doi: 10.1152/ajpcell.1998.275.1.C323. [DOI] [PubMed] [Google Scholar]
- [11].Engelhardt JF, Yankaskas JR, Ernst SA, Yang Y, Marino CR, Boucher RC, Cohn JA, Wilson JM. Submucosal glands are the predominant site of CFTR expression in the human bronchus. Nat. Genet. 1992;2:240–248. doi: 10.1038/ng1192-240. [DOI] [PubMed] [Google Scholar]
- [12].Trezise AE, Buchwald M. In vivo cell-specific expression of the cystic fibrosis transmembrane conductance regulator. Nature. 1991;353:434–437. doi: 10.1038/353434a0. [DOI] [PubMed] [Google Scholar]
- [13].Durie PR, Forstner GG. The exocrine pancreas. In: Yankaskas JR, Knowles MR, editors. Cystic Fibrosis in Adults. Lippincott-Raven. 1999. pp. 261–287. [Google Scholar]
- [14].Health effects of outdoor air pollution committee of the environmental and occupational health assembly of the american thoracic society. Am. J. Respir. Crit. Care Med. 1996;153:3–50. doi: 10.1164/ajrccm.153.1.8542133. [DOI] [PubMed] [Google Scholar]
- [15].Health effects of outdoor air pollution: part 2 Committee of the environmental and occupational health assembly of the american thoracic society. Am. J. Respir. Crit. Care Med. 1996;153:477–498. doi: 10.1164/ajrccm.153.2.8564086. [DOI] [PubMed] [Google Scholar]
- [16].Johnston RB., Jr. Clinical aspects of chronic granulomatous disease. Curr. Opin. Hematol. 2001;8:17–22. doi: 10.1097/00062752-200101000-00004. [DOI] [PubMed] [Google Scholar]
- [17].Sen CK. The general case for redox control of wound repair. Wound Repair Regen. 2003;11:431–438. doi: 10.1046/j.1524-475x.2003.11607.x. [DOI] [PubMed] [Google Scholar]
- [18].Powis G, Oblong J, Briehl M. Redox signalling and the control of cell growth and death. Pharmacol. Ther. 1995;68:149–173. doi: 10.1016/0163-7258(95)02004-7. [DOI] [PubMed] [Google Scholar]
- [19].Tu BP, Weissman JS. Oxidative protein folding in eukaryotes: mechanisms and consequences. J. Cell Biol. 2004;164:341–346. doi: 10.1083/jcb.200311055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Halliwell B, Cross CE, Gutteridge JMC. Free radicals, antioxidants, and human disease: Where are we now? J. Lab. Clin. Med. 1992;119:598–620. [PubMed] [Google Scholar]
- [21].Takeyama K, Dabbagh K, Jeong Shim J, Dao-Pick T, Ueki IF, Nadel JA. Oxidative stress causes mucin synthesis via transactivation of epidermal growth factor receptor: role of neutrophils. J. Immunol. 2000;164:1546–1552. doi: 10.4049/jimmunol.164.3.1546. [DOI] [PubMed] [Google Scholar]
- [22].Nguyen TD, Canada AT. Modulation of human colonic T84 cell secretion by hydrogen peroxide. Biochem. Pharmacol. 1994;47:403–410. doi: 10.1016/0006-2952(94)90032-9. [DOI] [PubMed] [Google Scholar]
- [23].Tamai H, Kachur JF, Baron DA, Grisham MB, Gaginella TS. Monochloramine, a neutrophil-derived oxidant, stimulates rat colonic secretion. J. Pharmacol. Exp. Ther. 1991;257:887–894. [PubMed] [Google Scholar]
- [24].Jung JS, Lee JY, Oh SO, Jang PG, Bae HR, Kim YK, Lee SH. Effect of t-butylhydroperoxide on chloride secretion in rat tracheal epithelia. Pharmacol. Toxicol. 1998;82:236–242. doi: 10.1111/j.1600-0773.1998.tb01431.x. [DOI] [PubMed] [Google Scholar]
- [25].Cowley EA, Linsdell P. Oxidant stress stimulates anion secretion from the human airway epithelial cell line Calu-3: implications for cystic fibrosis lung disease. J. Physiol. 2002;543:201–209. doi: 10.1113/jphysiol.2002.022400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Cantin AM, Bilodeau G, Ouellet C, Liao J, Hanrahan JW. Oxidant stress suppresses CFTR expression. Am. J. Physiol. Cell Physiol. 2006;290:C262–C270. doi: 10.1152/ajpcell.00070.2005. [DOI] [PubMed] [Google Scholar]
- [27].O'Malley YQ, Reszka KJ, Britigan BE. Direct oxidation of 2,7-dichlorodihydrofluorescein by pyocyanin and other redox-active compounds independent of reactive oxygen species production. Free Radic. Biol. Med. 2004;36:90–100. doi: 10.1016/j.freeradbiomed.2003.09.021. [DOI] [PubMed] [Google Scholar]
- [28].Kharitonov SA, Barnes PJ. Biomarkers of some pulmonary diseases in exhaled breath. Biomarkers. 2002;7:1–32. doi: 10.1080/13547500110104233. [DOI] [PubMed] [Google Scholar]
- [29].Balint B, Kharitonov SA, Hanazawa T, Donnelly LE, Shah PL, Hodson ME, Barnes PJ. Increased nitrotyrosine in exhaled breath condensate in cystic fibrosis. Eur. Respir. J. 2001;17:1201–1207. doi: 10.1183/09031936.01.00072501. [DOI] [PubMed] [Google Scholar]
- [30].Jones KL, Hegab AH, Hillman BC, Simpson KL, Jinkins PA, Grisham MB, Owens MW, Sato E, Robbins RA. Elevation of nitrotyrosine and nitrate concentrations in cystic fibrosis sputum. Pediatr. Pulmonol. 2000;30:79–85. doi: 10.1002/1099-0496(200008)30:2<79::aid-ppul1>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- [31].Kettle AJ, Chan T, Osberg I, Senthilmohan R, Chapman AL, Mocatta TJ, Wagener JS. Myeloperoxidase and protein oxidation in the airways of young children with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2004;170:1317–1323. doi: 10.1164/rccm.200311-1516OC. [DOI] [PubMed] [Google Scholar]
- [32].Montuschi P, Kharitonov SA, Ciabattoni G, Corradi M, van Rensen L, Geddes DM, Hodson ME, Barnes PJ. Exhaled 8-isoprostane as a new non-invasive biomarker of oxidative stress in cystic fibrosis. Thorax. 2000;55:205–209. doi: 10.1136/thorax.55.3.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Paredi P, Kharitonov SA, Leak D, Shah PL, Cramer D, Hodson ME, Barnes PJ. Exhaled ethane is elevated in cystic fibrosis and correlates with carbon monoxide levels and airway obstruction. Am. J. Respir. Crit. Care Med. 2000;161:1247–1251. doi: 10.1164/ajrccm.161.4.9906122. [DOI] [PubMed] [Google Scholar]
- [34].Van Der Vliet A, Nguyen MN, Shigenaga MK, Eiserich JP, Marelich GP, Cross CE. Myeloperoxidase and protein oxidation in cystic fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2000;279:L537–L546. doi: 10.1152/ajplung.2000.279.3.L537. [DOI] [PubMed] [Google Scholar]
- [35].Tabak C, Smit HA, Heederik D, Ocke MC, Kromhout D. Diet and chronic obstructive pulmonary disease: independent beneficial effects of fruits, whole grains, and alcohol (the MORGEN study) Clin. Exp. Allergy. 2001;31:747–755. doi: 10.1046/j.1365-2222.2001.01064.x. [DOI] [PubMed] [Google Scholar]
- [36].Watson L, Margetts B, Howarth P, Dorward M, Thompson R, Little P. The association between diet and chronic obstructive pulmonary disease in subjects selected from general practice. Eur. Respir. J. 2002;20:313–318. doi: 10.1183/09031936.02.00256402. [DOI] [PubMed] [Google Scholar]
- [37].Cantin AM, Fells GA, Hubbard RC, Crystal RG. Antioxidant macromolecules in the epithelial lining fluid of the normal human lower respiratory tract. J. Clin. Invest. 1990;86:962–971. doi: 10.1172/JCI114798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].DiSilvestro RA, Pacht E, Davis WB, Jarjour N, Joung H, Trela- Fulop K. BAL fluid contains detectable superoxide dismutase 1 activity. Chest. 1998;113:401–404. doi: 10.1378/chest.113.2.401. [DOI] [PubMed] [Google Scholar]
- [39].van der Vliet A, O'Neill CA, Cross CE, Koostra JM, Volz WG, Halliwell B, Louie S. Determination of low-molecular-mass antioxidant concentrations in human respiratory tract lining fluids. Am. J. Physiol. 1999;276:L289–L296. doi: 10.1152/ajplung.1999.276.2.L289. [DOI] [PubMed] [Google Scholar]
- [40].Schock BC, Koostra J, Kwack S, Hackman RM, Van Der Vliet A, Cross CE. Ascorbic acid in nasal and tracheobronchial airway lining fluids. Free Radic. Biol. Med. 2004;37:1393–1401. doi: 10.1016/j.freeradbiomed.2004.07.023. [DOI] [PubMed] [Google Scholar]
- [41].Cross CE, Halliwell B, Allen A. Antioxidant protection: a function of tracheobronchial and gastrointestinal mucus. Lancet. 1984;1:1328–1330. doi: 10.1016/s0140-6736(84)91822-1. [DOI] [PubMed] [Google Scholar]
- [42].Wright DT, Fischer BM, Li C, Rochelle LG, Akley NJ, Adler KB. Oxidant stress stimulates mucin secretion and PLC in airway epithelium via a nitric oxide-dependent mechanism. Am. J. Physiol. 1996;271:L854–L861. doi: 10.1152/ajplung.1996.271.5.L854. [DOI] [PubMed] [Google Scholar]
- [43].Perez-Vilar J, Boucher RC. Reevaluating gel-forming mucins' roles in cystic fibrosis lung disease. Free Radic. Biol. Med. 2004;37:1564–1577. doi: 10.1016/j.freeradbiomed.2004.07.027. [DOI] [PubMed] [Google Scholar]
- [44].Vishwanath S, Ramphal R, Guay CM, DesJardins D, Pier GB. Respiratory-mucin inhibition of the opsonophagocytic killing of Pseudomonas aeruginosa. Infect. Immun. 1988;56:2218–2222. doi: 10.1128/iai.56.9.2218-2222.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Walker NM, Simpson JE, Levitt RC, Boyle KT, Clarke LL. Talniflumate increases survival in a cystic fibrosis mouse model of distal intestinal obstructive syndrome. J. Pharmacol. Exp. Ther. 2006;317:275–283. doi: 10.1124/jpet.105.094847. [DOI] [PubMed] [Google Scholar]
- [46].Rahman I, MacNee W. Oxidative stress and regulation of glutathione in lung inflammation. Eur. Respir. J. 2000;16:534–554. doi: 10.1034/j.1399-3003.2000.016003534.x. [DOI] [PubMed] [Google Scholar]
- [47].Ray S, Watkins DN, Misso NL, Thompson PJ. Oxidant stress induces gamma-glutamylcysteine synthetase and glutathione synthesis in human bronchial epithelial NCI-H292 cells. Clin. Exp. Allergy. 2002;32:571–577. doi: 10.1046/j.0954-7894.2002.01294.x. [DOI] [PubMed] [Google Scholar]
- [48].Shi MM, Iwamoto T, Forman HJ. Gamma-glutamylcysteine synthetase and GSH increase in quinone-induced oxidative stress in BPAEC. Am. J. Physiol. 1994;267:L414–L421. doi: 10.1152/ajplung.1994.267.4.L414. [DOI] [PubMed] [Google Scholar]
- [49].Shi MM, Kugelman A, Iwamoto T, Tian L, Forman HJ. Quinone-induced oxidative stress elevates glutathione and induces gamma-glutamylcysteine synthetase activity in rat lung epithelial L2 cells. J. Biol. Chem. 1994;269:26512–26517. [PubMed] [Google Scholar]
- [50].Dickinson DA, Iles KE, Watanabe N, Iwamoto T, Zhang H, Krzywanski DM, Forman HJ. 4-hydroxynonenal induces glutamate cysteine ligase through JNK in HBE1 cells. Free Radic. Biol. Med. 2002;33:974. doi: 10.1016/s0891-5849(02)00991-7. [DOI] [PubMed] [Google Scholar]
- [51].Cantin AM, North SL, Hubbard RC, Crystal RG. Normal alveolar epithelial lining fluid contains high levels of glutathione. J. Appl. Physiol. 1987;63:152–157. doi: 10.1152/jappl.1987.63.1.152. [DOI] [PubMed] [Google Scholar]
- [52].Cantin AM, Hanrahan JW, Bilodeau G, Ellis L, Dupuis A, Liao J, Zielenski J, Durie P. Cystic fibrosis transmembrane conductance regulator function is suppressed in cigarette smokers. Am. J. Respir. Crit. Care Med. 2006;173:1139–1144. doi: 10.1164/rccm.200508-1330OC. [DOI] [PubMed] [Google Scholar]
- [53].Roum JH, Buhl R, McElvaney NG, Borok Z, Crystal RG. Systemic deficiency of glutathione in cystic fibrosis. J. Appl. Physiol. 1993;75:2419–2424. doi: 10.1152/jappl.1993.75.6.2419. [DOI] [PubMed] [Google Scholar]
- [54].Cantin AM, Hubbard RC, Crystal RG. Glutathione deficiency in the epithelial lining fluid of the lower respiratory tract in idiopathic pulmonary fibrosis. Am. Rev. Respir. Dis. 1989;139:370–372. doi: 10.1164/ajrccm/139.2.370. [DOI] [PubMed] [Google Scholar]
- [55].Bunnell E, Pacht ER. Oxidized glutathione is increased in the alveolar fluid of patients with the adult respiratory distress syndrome. Am. Rev. Respir. Dis. 1993;148:1174–1178. doi: 10.1164/ajrccm/148.5.1174. [DOI] [PubMed] [Google Scholar]
- [56].Arsalane K, Dubois CM, Muanza T, Begin R, Boudreau F, Asselin C, Cantin AM. Transforming growth factor-beta1 is a potent inhibitor of glutathione synthesis in the lung epithelial cell line A549: transcriptional effect on the GSH rate-limiting enzyme gamma-glutamyl-cysteine synthetase. Am. J. Respir. Cell Mol. Biol. 1997;17:599–607. doi: 10.1165/ajrcmb.17.5.2833. [DOI] [PubMed] [Google Scholar]
- [57].Kogan I, Ramjeesingh M, Li C, Kidd JF, Wang Y, Leslie EM, Cole SP, Bear CE. CFTR directly mediates nucleotide-regulated glutathione flux. EMBO J. 2003;22:1981–1989. doi: 10.1093/emboj/cdg194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Hull J, Vervaart P, Grimwood K, Phelan P. Pulmonary oxidative stress response in young children with cystic fibrosis. Thorax. 1997;52:557–560. doi: 10.1136/thx.52.6.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Lands LC, Grey V, Smountas AA, Kramer VG, McKenna D. Lymphocyte glutathione levels in children with cystic fibrosis. Chest. 1999;116:201–205. doi: 10.1378/chest.116.1.201. [DOI] [PubMed] [Google Scholar]
- [60].Cantin AM, Paquette B, Richter M, Larivee P. Albumin-mediated regulation of cellular glutathione and nuclear factor kappa B activation. Am. J. Respir. Crit. Care Med. 2000;162:1539–1546. doi: 10.1164/ajrccm.162.4.9910106. [DOI] [PubMed] [Google Scholar]
- [61].Velsor LW, van Heeckeren A, Day BJ. Antioxidant imbalance in the lungs of cystic fibrosis transmembrane conductance regulator protein mutant mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2001;281:L31–L38. doi: 10.1152/ajplung.2001.281.1.L31. [DOI] [PubMed] [Google Scholar]
- [62].Day BJ, van Heeckeren A, Velsor LW. Elevation of Lung CFTR, MRP-2 and epithelial lining fluid glutathione in mice with Pseudomonas aeruginosa lung infection. Am. J. Respir. Crit. Care Med. 2003;167:916A. [Google Scholar]
- [63].Heeckeren A, Walenga R, Konstan MW, Bonfield T, Davis PB, Ferkol T. Excessive inflammatory response of cystic fibrosis mice to bronchopulmonary infection with Pseudomonas aeruginosa. J. Clin. Invest. 1997;100:2810–2815. doi: 10.1172/JCI119828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Kugelman A, Choy HA, Liu R, Shi MM, Gozal E, Forman HJ. g-Glutamyl transpeptidase is increased by oxidative stress in rat alveolar L2 epithelial cells. Am. J. Respir. Cell Mol. Biol. 1994;11:586–592. doi: 10.1165/ajrcmb.11.5.7946387. [DOI] [PubMed] [Google Scholar]
- [65].Hudson VM. Rethinking cystic fibrosis pathology: the critical role of abnormal reduced glutathione (GSH) transport caused by CFTR mutation. Free Radic. Biol. Med. 2001;30:1440–1461. doi: 10.1016/s0891-5849(01)00530-5. [DOI] [PubMed] [Google Scholar]
- [66].Jungas T, Motta I, Duffieux F, Fanen P, Stoven V, Ojcius DM. Glutathione levels and BAX activation during apoptosis due to oxidative stress in cells expressing wild-type and mutant cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 2002;277:27912–27918. doi: 10.1074/jbc.M110288200. [DOI] [PubMed] [Google Scholar]
- [67].Peterson JD, Herzenberg LA, Vasquez K, Waltenbaugh C. Glutathione levels in antigen-presenting cells modulate Th1 versus Th2 response patterns. Proc. Natl. Acad. Sci. U.S.A. 1998;95:3071–3076. doi: 10.1073/pnas.95.6.3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Moser C, Jensen PO, Kobayashi O, Hougen HP, Song Z, Rygaard J, Kharazmi A. N, H. b. Improved outcome of chronic Pseudomonas aeruginosa lung infection is associated with induction of a Th1-dominated cytokine response. Clin. Exp. Immunol. 2002;127:206–213. doi: 10.1046/j.1365-2249.2002.01731.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Moser C, Johansen HK, Song Z, Hougen HP, Rygaard J, Hoiby N. Chronic Pseudomonas aeruginosa lung infection is more severe in Th2 responding BALB/c mice compared to Th1 responding C3H/HeN mice. Apmis. 1997;105:838–842. [PubMed] [Google Scholar]
- [70].Howard M, Fischer H, Roux J, Santos BC, Gullans SR, Yancey PH, Welch WJ. Mammalian osmolytes and S-nitrosoglutathione promote Delta F508 cystic fibrosis transmembrane conductance regulator (CFTR) protein maturation and function. J. Biol. Chem. 2003;278:35159–35167. doi: 10.1074/jbc.M301924200. [DOI] [PubMed] [Google Scholar]
- [71].Gringhuis SI, Papendrecht-van der Voort EA, Leow A, Nivine Levarht EW, Breedveld FC, Verweij CL. Effect of redox balance alterations on cellular localization of LAT and downstream T-cell receptor signaling pathways. Mol. Cell. Biol. 2002;22:400–411. doi: 10.1128/MCB.22.2.400-411.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Witschi A, Reddy S, Stofer B, Lauterburg BH. The systemic availability of oral glutathione. Eur. J. Clin. Pharmacol. 1992;43:667–669. doi: 10.1007/BF02284971. [DOI] [PubMed] [Google Scholar]
- [73].Wendel A, Cikryt P. The level and half-life of glutathione in human plasma. FEBS Lett. 1980;120:209–211. doi: 10.1016/0014-5793(80)80299-7. [DOI] [PubMed] [Google Scholar]
- [74].Busse E, Zimmer G, Schopohl B, Kornhuber B. Influence of alpha-lipoic acid on intracellular glutathione in vitro and in vivo. Arzneimit-telforschung. 1992;42:829–831. [PubMed] [Google Scholar]
- [75].Lands LC, Grey VL, Smountas AA. Effect of supplementation with a cysteine donor on muscular performance. J. Appl. Physiol. 1999;87:1381–1385. doi: 10.1152/jappl.1999.87.4.1381. [DOI] [PubMed] [Google Scholar]
- [76].Micke P, Beeh KM, Schlaak JF, Buhl R. Oral supplementation with whey proteins increases plasma glutathione levels of HIV-infected patients. Eur. J. Clin. Invest. 2001;31:171–178. doi: 10.1046/j.1365-2362.2001.00781.x. [DOI] [PubMed] [Google Scholar]
- [77].Lothian B, Grey V, Kimoff RJ, Lands LC. Treatment of obstructive airway disease with a cysteine donor protein supplement: a case report. Chest. 2000;117:914–916. doi: 10.1378/chest.117.3.914. [DOI] [PubMed] [Google Scholar]
- [78].Grey V, Mohammed SR, Smountas AA, Bahlool R, Lands LC. Improved glutathione status in young adult patients with cystic fibrosis supplemented with whey protein. J. Cyst. Fibros. 2003;2:195–198. doi: 10.1016/S1569-1993(03)00097-3. [DOI] [PubMed] [Google Scholar]
- [79].Kasielski M, Nowak D. Long-term administration of N-acetylcysteine decreases hydrogen peroxide exhalation in subjects with chronic obstructive pulmonary disease. Respir. Med. 2001;95:448–456. doi: 10.1053/rmed.2001.1066. [DOI] [PubMed] [Google Scholar]
- [80].Marrades RM, Roca J, Barbera JA, de Jover L, MacNee W, Rodriguez-Roisin R. Nebulized glutathione induces bronchoconstriction in patients with mild asthma. Am. J. Respir. Crit. Care Med. 1997;156:425–430. doi: 10.1164/ajrccm.156.2.9611001. [DOI] [PubMed] [Google Scholar]
- [81].Britigan BE, Edeker BL. Pseudomonas and neutrophil products modify transferrin and lactoferrin to create conditions that favor hydroxyl radical formation. J. Clin. Invest. 1991;88:1092–1102. doi: 10.1172/JCI115408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Jonas CR, Gu LH, Nkabyo YS, Mannery YO, Avissar NE, Sax HC, Jones DP, Ziegler TR. Glutamine and KGF each regulate extracellular thiol/disulfide redox and enhance proliferation in Caco-2 cells. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003;285:R1421–R1429. doi: 10.1152/ajpregu.00702.2002. [DOI] [PubMed] [Google Scholar]
- [83].Roum JH, Borok Z, McElvaney NG, Grimes GJ, Bokser AD, Buhl R, Crystal RG. Glutathione aerosol suppresses lung epithelial surface inflammatory cell-derived oxidants in cystic fibrosis. J. Appl. Physiol. 1999;87:438–443. doi: 10.1152/jappl.1999.87.1.438. [DOI] [PubMed] [Google Scholar]
- [84].Griese M, Ramakers J, Krasselt A, Starosta V, Van Koningsbruggen S, Fischer R, Ratjen F, Mullinger B, Huber RM, Maier K, Rietschel E, Scheuch G. Improvement of alveolar glutathione and lung function but not oxidative state in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2004;169:822–828. doi: 10.1164/rccm.200308-1104OC. [DOI] [PubMed] [Google Scholar]
- [85].Hartl D, Starosta V, Maier K, Beck-Speier I, Rebhan C, Becker BF, Latzin P, Fischer R, Ratjen F, Huber RM, Rietschel E, Krauss- Etschmann S, Griese M. Inhaled glutathione decreases PGE2 and increases lymphocytes in cystic fibrosis lungs. Free Radic. Biol. Med. 2005;39:463–472. doi: 10.1016/j.freeradbiomed.2005.03.032. [DOI] [PubMed] [Google Scholar]
- [86].Buhl R, Vogelmeier C, Critenden M, Hubbard RC, Hoyt RF, Jr., Wilson EM, Cantin AM, Crystal RG. Augmentation of glutathione in the fluid lining the epithelium of the lower respiratory tract by directly administering glutathione aerosol. Proc. Natl. Acad. Sci. U.S.A. 1990;87:4063–4067. doi: 10.1073/pnas.87.11.4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Venglarik CJ, Giron-Calle J, Wigley AF, Malle E, Watanabe N, Forman HJ. Hypochlorous acid alters bronchial epithelial cell membrane properties and prevention by extracellular glutathione. J. Appl. Physiol. 2003;95:2444–2452. doi: 10.1152/japplphysiol.00002.2003. [DOI] [PubMed] [Google Scholar]
- [88].Bishop C, Hudson VM, Hilton SC, Wilde C. A pilot study of the effect of inhaled buffered reduced glutathione on the clinical status of patients with cystic fibrosis. Chest. 2005;127:308–317. doi: 10.1378/chest.127.1.308. [DOI] [PubMed] [Google Scholar]
- [89].Demedts M, Behr J, Buhl R, Costabel U, Dekhuijzen R, Jansen HM, MacNee W, Thomeer M, Wallaert B, Laurent F, Nicholson AG, Verbeken EK, Verschakelen J, Flower CD, Capron F, Petruzzelli S, Vuyst DeP., van den Bosch JM, Rodriguez-Becerra E, Corvasce G, Lankhorst I, Sardina M, Montanari M. High-dose acetylcysteine in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2005;353:2229–2242. doi: 10.1056/NEJMoa042976. [DOI] [PubMed] [Google Scholar]
- [90].Tirouvanziam R, Conrad CK, Bottiglieri T, Herzenberg LA, Moss RB, Herzenberg LA. High-dose oral N-acetylcysteine, a glutathione prodrug, modulates inflammation in cystic fibrosis. Proc. Natl. Acad. Sci. U.S.A. 2006;103:4628–4633. doi: 10.1073/pnas.0511304103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Gillissen A, Jaworska M, Orth M, Coffiner M, Maes P, App EM, Cantin AM, Schultze-Werninghaus G. Nacystelyn, a novel lysine salt of N-acetylcysteine, to augment cellular antioxidant defence in vitro. Respir. Med. 1997;91:159–168. doi: 10.1016/s0954-6111(97)90052-4. [DOI] [PubMed] [Google Scholar]
- [92].Dasgupta B, King M. Reduction in viscoelasticity in cystic fibrosis sputum in vitro using combined treatment with Nacystelyn and rhDNase. Pediatr. Pulmonol. 1996;22:161–166. doi: 10.1002/(SICI)1099-0496(199609)22:3<161::AID-PPUL4>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- [93].Antonicelli F, Parmentier M, Drost EM, Hirani N, Rahman I, Donaldson K, MacNee W. Nacystelyn inhibits oxidant-mediated interleukin-8 expression and NF-kappaB nuclear binding in alveolar epithelial cells. Free Radic. Biol. Med. 2002;32:492–502. doi: 10.1016/s0891-5849(01)00820-6. [DOI] [PubMed] [Google Scholar]
- [94].Antonicelli F, Brown D, Parmentier M, Drost EM, Hirani N, Rahman I, Donaldson K, MacNee W. Regulation of LPS-mediated inflammation in vivo and in vitro by the thiol antioxidant Nacystelyn. Am. J. Physiol. Lung Cell Mol. Physiol. 2004;286:L1319–L1327. doi: 10.1152/ajplung.00329.2003. [DOI] [PubMed] [Google Scholar]
- [95].Sun F, Tai S, Lim T, Baumann U, King M. Additive effect of dornase alfa and Nacystelyn on transportability and viscoelasticity of cystic fibrosis sputum. Can. Respir. J. 2002;9:401–406. doi: 10.1155/2002/508942. [DOI] [PubMed] [Google Scholar]
- [96].Vanderbist F, Wery B, Baran D, Van Gansbeke B, Schoutens A, Moes AJ. Deposition of Nacystelyn from a dry powder inhaler in healthy volunteers and cystic fibrosis patients. Drug Dev. Ind. Pharm. 2001;27:205–212. doi: 10.1081/ddc-100000238. [DOI] [PubMed] [Google Scholar]
- [97].App EM, Baran D, Dab I, Malfroot A, Coffiner M, Vanderbist F, King M. Dose-finding and 24-h monitoring for efficacy and safety of aerosolized Nacystelyn in cystic fibrosis. Eur. Respir. J. 2002;19:294–302. doi: 10.1183/09031936.02.00025802. [DOI] [PubMed] [Google Scholar]
- [98].Pascussi JM, Gerbal-Chaloin S, Drocourt L, Maurel P, Vilarem MJ. The expression of CYP2B6, CYP2C9 and CYP3A4 genes: a tangle of networks of nuclear and steroid receptors. Biochim. Biophys. Acta. 2003;1619:243–253. doi: 10.1016/s0304-4165(02)00483-x. [DOI] [PubMed] [Google Scholar]
- [99].Baehner RL, Boxer LA. Role of membrane vitamin E and cytoplasmic glutathione in the regulation of phagocytic functions of neutrophils and monocytes. Am. J. Pediatr. Hematol. Oncol. 1979;1:71–76. [PubMed] [Google Scholar]
- [100].Beharka A, Redican S, Leka L, Meydani SN. Vitamin E status and immune function. Methods Enzymol. 1997;282:247–263. doi: 10.1016/s0076-6879(97)82112-x. [DOI] [PubMed] [Google Scholar]
- [101].Ouahchi K, Arita M, Kayden H, Hentati F, Ben Hamida M, Sokol R, Arai H, Inoue K, Mandel JL, Koenig M. Ataxia with isolated vitamin E deficiency is caused by mutations in the alpha-tocopherol transfer protein. Nat. Genet. 1995;9:141–145. doi: 10.1038/ng0295-141. [DOI] [PubMed] [Google Scholar]
- [102].Sokol RJ. Fat-soluble vitamins and their importance in patients with cholestatic liver diseases. Gastroenterol. Clin. North Am. 1994;23:673–705. [PubMed] [Google Scholar]
- [103].Sokol RJ. Vitamin E and neurologic function in man. Free Radic. Biol. Med. 1989;6:189–207. doi: 10.1016/0891-5849(89)90117-2. [DOI] [PubMed] [Google Scholar]
- [104].Wilfond BS, Farrell PM, Laxova A, Mischler E. Severe hemolytic anemia associated with vitamin E deficiency in infants with cystic fibrosis. Implications for neonatal screening. Clin. Pediatr. (Phila.) 1994;33:2–7. doi: 10.1177/000992289403300101. [DOI] [PubMed] [Google Scholar]
- [105].Koscik RL, Farrell PM, Kosorok MR, Zaremba KM, Laxova A, Lai HC, Douglas JA, Rock MJ, Splaingard ML. Cognitive function of children with cystic fibrosis: deleterious effect of early malnutrition. Pediatrics. 2004;113:1549–1558. doi: 10.1542/peds.113.6.1549. [DOI] [PubMed] [Google Scholar]
- [106].Feranchak AP, Sontag MK, Wagener JS, Hammond KB, Accurso FJ, Sokol RJ. Prospective, long-term study of fat-soluble vitamin status in children with cystic fibrosis identified by newborn screen. J. Pediatr. 1999;135:601–610. doi: 10.1016/s0022-3476(99)70059-4. [DOI] [PubMed] [Google Scholar]
- [107].Mischler EH, Marcus MS, Sondel SA, Laxova A, Carey P, Langhough R, Farrell PM. Nutritional assessment of infants with cystic fibrosis diagnosed through screening. Pediatr. Pulmonol. Suppl. 1991;7:56–63. doi: 10.1002/ppul.1950110712. [DOI] [PubMed] [Google Scholar]
- [108].Madarasi A, Lugassi A, Greiner E, Holics K, Biro L, Mozsary E. Antioxidant status in patients with cystic fibrosis. Ann. Nutr. Metab. 2000;44:207–211. doi: 10.1159/000046685. [DOI] [PubMed] [Google Scholar]
- [109].Borowitz D, Baker RD, Stallings VA. Consensus report on nutrition for pediatric patients with cystic fibrosis. J. Pediatr. Gastroenterol. Nutr. 2002;35:246–259. doi: 10.1097/00005176-200209000-00004. [DOI] [PubMed] [Google Scholar]
- [110].Winklhofer-Roob BM, Tuchschmid PE, Molinari L, Shmerling DH. Response to a single oral dose of all-rac-alpha-tocopheryl acetate in patients with cystic fibrosis and in healthy individuals. Am. J. Clin. Nutr. 1996;63:717–721. doi: 10.1093/ajcn/63.5.717. [DOI] [PubMed] [Google Scholar]
- [111].Sokol RJ, Butler-Simon N, Conner C, Heubi JE, Sinatra FR, Suchy FJ, Heyman MB, Perrault J, Rothbaum RJ, Levy J, et al. Multicenter trial of d-alpha-tocopheryl polyethylene glycol 1000 succinate for treatment of vitamin E deficiency in children with chronic cholestasis. Gastroenterology. 1993;104:1727–1735. doi: 10.1016/0016-5085(93)90652-s. [DOI] [PubMed] [Google Scholar]
- [112].Pallast EG, Schouten EG, de Waart FG, Fonk HC, Doekes G, von Blomberg BM, Kok FJ. Effect of 50- and 100-mg vitamin E supplements on cellular immune function in noninstitutionalized elderly persons. Am. J. Clin. Nutr. 1999;69:1273–1281. doi: 10.1093/ajcn/69.6.1273. [DOI] [PubMed] [Google Scholar]
- [113].Meydani SN, Barklund MP, Liu S, Meydani M, Miller RA, Cannon JG, Morrow FD, Rocklin R, Blumberg JB. Vitamin E supplementation enhances cell-mediated immunity in healthy elderly subjects. Am. J. Clin. Nutr. 1990;52:557–563. doi: 10.1093/ajcn/52.3.557. [DOI] [PubMed] [Google Scholar]
- [114].Meydani SN, Meydani M, Blumberg JB, Leka LS, Siber G, Loszewski R, Thompson C, Pedrosa MC, Diamond RD, Stollar BD. Vitamin E supplementation and in vivo immune response in healthy elderly subjects. a randomized controlled trial. JAMA. 1997;277:1380–1386. doi: 10.1001/jama.1997.03540410058031. [DOI] [PubMed] [Google Scholar]
- [115].Scott DL, Kelleher J, Losowsky MS. The influence of dietary selenium and vitamin E on glutathione peroxidase and glutathione in the rat. Biochim. Biophys. Acta. 1977;497:218–224. doi: 10.1016/0304-4165(77)90154-4. [DOI] [PubMed] [Google Scholar]
- [116].Pascoe GA, Fariss MW, Olafsdottir K, Reed DJ. A role of vitamin E in protection against cell injury. Maintenance of intracellular glutathione precursors and biosynthesis. Eur. J. Biochem. 1987;166:241–247. doi: 10.1111/j.1432-1033.1987.tb13508.x. [DOI] [PubMed] [Google Scholar]
- [117].Miller III ER, Pastor-Barriuso R, Dalal D, Riemersma RA, Appel LJ, Guallar E. Meta-analysis: high-dosage vitamin E supp-lementation may increase all-cause mortality. Ann. Intern. Med. 2005;142:37–46. doi: 10.7326/0003-4819-142-1-200501040-00110. [DOI] [PubMed] [Google Scholar]
- [118].Lonn E, Bosch J, Yusuf S, Sheridan P, Pogue J, Arnold JM, Ross C, Arnold A, Sleight P, Probstfield J, Dagenais GR. Effects of long-term vitamin E supplementation on cardiovascular events and cancer: a randomized controlled trial. JAMA. 2005;293:1338–1347. doi: 10.1001/jama.293.11.1338. [DOI] [PubMed] [Google Scholar]
- [119].Johnson L, Bowen FW, Jr., Abbasi S, Herrmann N, Weston M, Sacks L, Porat R, Stahl G, Peckham G, Delivoria-Papadopoulos M, et al. Relationship of prolonged pharmacologic serum levels of vitamin E to incidence of sepsis and necrotizing enterocolitis in infants with birth weight 1,500 grams or less. Pediatrics. 1985;75:619–638. [PubMed] [Google Scholar]
- [120].Ciabattoni G, Davi G, Collura M, Iapichino L, Pardo F, Ganci A, Romagnoli R, Maclouf J, Patrono C, Montuschi P, Collins JV, Lazzeri N, Corradi M, Kharitonov SA, Barnes PJ, Cipollone F, Patrignani P, Pasquale M, Gregorio DiD., Bucciarelli T, Cuccurullo F, Panara MR, Tacconelli S, Seta F, Alessandrini P, Mezzetti A, Santini G, Sciulli MG, Gallina P, Bon GB, van Rensen L, Geddes DM, Hodson ME. In vivo lipid peroxidation and platelet activation in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2000;162:1195–1201. doi: 10.1164/ajrccm.162.4.9911071. [DOI] [PubMed] [Google Scholar]
- [121].Mezzetti A, Cipollone F, Cuccurullo F. Oxidative stress and cardiovascular complications in diabetes: isoprostanes as new markers on an old paradigm. Cardiovasc. Res. 2000;47:475–488. doi: 10.1016/s0008-6363(00)00118-8. [DOI] [PubMed] [Google Scholar]
- [122].Yeum KJ, Russell RM. Carotenoid bioavailability and bioconversion. Annu. Rev. Nutr. 2002;22:483–504. doi: 10.1146/annurev.nutr.22.010402.102834. [DOI] [PubMed] [Google Scholar]
- [123].West CE, Eilander A, van Lieshout M. Consequences of revised estimates of carotenoid bioefficacy for dietary control of vitamin A deficiency in developing countries. J. Nutr. 2002;132:2920S–2926S. doi: 10.1093/jn/132.9.2920S. [DOI] [PubMed] [Google Scholar]
- [124].Mares-Perlman JA, Fisher AI, Klein R, Palta M, Block G, Millen AE, Wright JD. Lutein and zeaxanthin in the diet and serum and their relation to age-related maculopathy in the third national health and nutrition examination survey. Am. J. Epidemiol. 2001;153:424–432. doi: 10.1093/aje/153.5.424. [DOI] [PubMed] [Google Scholar]
- [125].Krinsky NI, Landrum JT, Bone RA. Biologic mechanisms of the protective role of lutein and zeaxanthin in the eye. Annu. Rev. Nutr. 2003;23:171–201. doi: 10.1146/annurev.nutr.23.011702.073307. [DOI] [PubMed] [Google Scholar]
- [126].Homnick DN, Cox JH, DeLoof MJ, Ringer TV. Carotenoid levels in normal children and in children with cystic fibrosis. J. Pediatr. 1993;122:703–707. doi: 10.1016/s0022-3476(06)80008-9. [DOI] [PubMed] [Google Scholar]
- [127].Kawchak DA, Sowell AL, Hofley PM, Zemel BS, Scanlin TF, Stallings VA. Longitudinal analysis shows serum carotenoid concen-trations are low in children with cystic fibrosis. J. Am. Diet. Assoc. 1999;99:1569–1572. doi: 10.1016/S0002-8223(99)00386-7. [DOI] [PubMed] [Google Scholar]
- [128].Winklhofer-Roob BM, Puhl H, Khoschsorur G, van't Hof MA, Esterbauer H, Shmerling DH. Enhanced resistance to oxidation of low density lipoproteins and decreased lipid peroxide formation during beta-carotene supplementation in cystic fibrosis. Free Radic. Biol. Med. 1995;18:849–859. doi: 10.1016/0891-5849(94)00203-v. [DOI] [PubMed] [Google Scholar]
- [129].Lepage G, Champagne J, Ronco N, Lamarre A, Osberg I, Sokol RJ, Roy CC. Supplementation with carotenoids corrects increased lipid peroxidation in children with cystic fibrosis. Am. J. Clin. Nutr. 1996;64:87–93. doi: 10.1093/ajcn/64.1.87. [DOI] [PubMed] [Google Scholar]
- [130].Rust P, Eichler I, Renner S, Elmadfa I. Effects of long-term oral beta-carotene supplementation on lipid peroxidation in patients with cystic fibrosis. Int. J. Vitam. Nutr. Res. 1998;68:83–87. [PubMed] [Google Scholar]
- [131].Benabdeslam H, Abidi H, Garcia I, Bellon G, Gilly R, Revol A. Lipid peroxidation and antioxidant defenses in cystic fibrosis patients. Clin. Chem. Lab. Med. 1999;37:511–516. doi: 10.1515/CCLM.1999.082. [DOI] [PubMed] [Google Scholar]
- [132].Cobanoglu N, Ozcelik U, Gocmen A, Kiper N, Dogru D. Antioxidant effect of beta-carotene in cystic fibrosis and bronchiectasis: clinical and laboratory parameters of a pilot study. Acta Paediatr. 2002;91:793–798. doi: 10.1080/08035250213212. [DOI] [PubMed] [Google Scholar]
- [133].Wood LG, Fitzgerald DA, Lee AK, Garg ML. Improved antioxidant and fatty acid status of patients with cystic fibrosis after antioxidant supplementation is linked to improved lung function. Am. J. Clin. Nutr. 2003;77:150–159. doi: 10.1093/ajcn/77.1.150. [DOI] [PubMed] [Google Scholar]
- [134].Portal BC, Richard MJ, Faure HS, Hadjian AJ, Favier AE. Altered antioxidant status and increased lipid peroxidation in children with cystic fibrosis. Am. J. Clin. Nutr. 1995;61:843–847. doi: 10.1093/ajcn/61.4.843. [DOI] [PubMed] [Google Scholar]
- [135].Schweigert FJ. Inflammation-induced changes in the nutritional biomarkers serum retinol and carotenoids. Curr. Opin. Clin. Nutr. Metab. Care. 2001;4:477–481. doi: 10.1097/00075197-200111000-00002. [DOI] [PubMed] [Google Scholar]
- [136].Kritchevsky SB, Bush AJ, Pahor M, Gross MD. Serum carotenoids and markers of inflammation in nonsmokers. Am. J. Epidemiol. 2000;152:1065–1071. doi: 10.1093/aje/152.11.1065. [DOI] [PubMed] [Google Scholar]
- [137].Schupp C, Olano-Martin E, Gerth C, Morrissey BM, Cross CE, Werner JS. Lutein, zeaxanthin, macular pigment, and visual function in adult cystic fibrosis patients. Am. J. Clin. Nutr. 2004;79:1045–1052. doi: 10.1093/ajcn/79.6.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Huet F, Semama D, Maingueneau C, Charavel A, Nivelon JL. Vitamin A deficiency and nocturnal vision in teenagers with cystic fibrosis. Eur. J. Pediatr. 1997;156:949–951. doi: 10.1007/s004310050749. [DOI] [PubMed] [Google Scholar]
- [139].McGrath LT, Mallon P, Dowey L, Silke B, McClean E, McDonnell M, Devine A, Copeland S, Elborn S. Oxidative stress during acute respiratory exacerbations in cystic fibrosis. Thorax. 1999;54:518–523. doi: 10.1136/thx.54.6.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Winklhofer-Roob BM, van't Hof MA, Shmerling DH. Response to oral beta-carotene supplementation in patients with cystic fibrosis: a 16-month follow-up study. Acta Paediatr. 1995;84:1132–1136. doi: 10.1111/j.1651-2227.1995.tb13512.x. [DOI] [PubMed] [Google Scholar]
- [141].Homnick DN, Spillers CR, Cox SR, Cox JH, Yelton LA, DeLoof MJ, Oliver LK, Ringer TV. Single- and multiple-dose-response relationships of beta-carotene in cystic fibrosis. J. Pediatr. 1995;127:491–494. doi: 10.1016/s0022-3476(95)70089-7. [DOI] [PubMed] [Google Scholar]
- [142].Winklhofer-Roob BM, Schlegel-Haueter SE, Khoschsorur G, van't Hof MA, Suter S, Shmerling DH. Neutrophil elastase/alpha 1-proteinase inhibitor complex levels decrease in plasma of cystic fibrosis patients during long-term oral beta-carotene supplementation. Pediatr. Res. 1996;40:130–134. doi: 10.1203/00006450-199607000-00022. [DOI] [PubMed] [Google Scholar]
- [143].Guenegou A, Leynaert B, Pin I, Le Moel G, Zureik M, Neukirch F. Serum carotenoids, vitamins A and E, and 8 year lung function decline in a general population. Thorax. 2006;61:320–326. doi: 10.1136/thx.2005.047373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Rust P, Eichler I, Renner S, Elmadfa I. Long-term oral beta-carotene supplementation in patients with cystic fibrosis-effects on antioxidative status and pulmonary function. Ann. Nutr. Metab. 2000;44:30–37. doi: 10.1159/000012818. [DOI] [PubMed] [Google Scholar]
- [145].Renner S, Rath R, Rust P, Lehr S, Frischer T, Elmadfa I, Eichler I. Effects of beta-carotene supplementation for six months on clinical and laboratory parameters in patients with cystic fibrosis. Thorax. 2001;56:48–52. doi: 10.1136/thorax.56.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Winklhofer-Roob BM, Khoschsorur G, Shmerling DH. Effect of long-term oral beta-carotene supplementation on indices of liver function in cystic fibrosis. Gastroenterology. 1995;108:A1199. [Google Scholar]
- [147].Pryor WA, Stahl W, Rock CL. Beta carotene: from biochemistry to clinical trials. Nutr. Rev. 2000;58:39–53. doi: 10.1111/j.1753-4887.2000.tb07810.x. [DOI] [PubMed] [Google Scholar]
- [148].Kagan VE, Tyurina YY. Recycling and redox cycling of phenolic antioxidants. Ann. N.Y. Acad. Sci. 1998;854:425–434. doi: 10.1111/j.1749-6632.1998.tb09921.x. [DOI] [PubMed] [Google Scholar]
- [149].Hargreaves IP. Ubiquinone: cholesterol's reclusive cousin. Ann. Clin. Biochem. 2003;40:207–218. doi: 10.1258/000456303321610493. [DOI] [PubMed] [Google Scholar]
- [150].Colquhoun DM, Jackson R, Walters M, Hicks BJ, Goldsmith J, Young P, Strakosch C, Kostner KM. Effects of simvastatin on blood lipids, vitamin E, coenzyme Q10 levels and left ventricular function in humans. Eur. J. Clin. Invest. 2005;35:251–258. doi: 10.1111/j.1365-2362.2005.01486.x. [DOI] [PubMed] [Google Scholar]
- [151].Mancini A, Corbo GM, Gaballo A, Valente S, Gigliotti P, Cimino V, De Marinis L, Principi F, Littarru GP. Relationships between plasma CoQ10 levels and thyroid hormones in chronic obstructive pulmonary disease. Biofactors. 2005;25:201–204. doi: 10.1002/biof.5520250124. [DOI] [PubMed] [Google Scholar]
- [152].Sokol RJ, Osberg I, Sontag M, Wagener JS, Accurso FJ. Reduced serum coenzyme Q-10 concentrations in children with cystic fibrosis (abstract) Pediatr. Pulmonol. 2003;36:338. [Google Scholar]
- [153].Marklund SL. Human copper-containing superoxide dismutase of high molecular weight. Proc. Natl. Acad. Sci. U.S.A. 1982;79:7634–7638. doi: 10.1073/pnas.79.24.7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Gillissen A, Roum JH, Hoyt RF, Crystal RG. Aerosolization of superoxide dismutase. Augmentation of respiratory epithelial lining fluid antioxidant screen by aerosolization of recombinant human Cu++/Zn++ superoxide dismutase. Chest. 1993;104:811–815. doi: 10.1378/chest.104.3.811. [DOI] [PubMed] [Google Scholar]
- [155].Smith KR, Uyeminami DL, Kodavanti UP, Crapo JD, Chang LY, Pinkerton KE. Inhibition of tobacco smoke-induced lung inflammation by a catalytic antioxidant. Free Radic. Biol. Med. 2002;33:1106–1114. doi: 10.1016/s0891-5849(02)01003-1. [DOI] [PubMed] [Google Scholar]
- [156].Chang LY, Crapo JD. Inhibition of airway inflammation and hyperreactivity by an antioxidant mimetic. Free Radic. Biol. Med. 2002;33:379–386. doi: 10.1016/s0891-5849(02)00919-x. [DOI] [PubMed] [Google Scholar]
- [157].Zsembery A, Fortenberry JA, Liang L, Bebok Z, Tucker TA, Boyce AT, Braunstein GM, Welty E, Bell PD, Sorscher EJ, Clancy JP, Schwiebert EM. Extracellular zinc and ATP restore chloride secretion across cystic fibrosis airway epithelia by triggering calcium entry. J. Biol. Chem. 2004;279:10720–10729. doi: 10.1074/jbc.M313391200. [DOI] [PubMed] [Google Scholar]
- [158].Truong-Tran AQ, Ruffin RE, Foster PS, Koskinen AM, Coyle P, Philcox JC, Rofe AM, Zalewski PD. Altered zinc homeostasis and caspase-3 activity in murine allergic airway inflammation. Am. J. Respir. Cell. Mol. Biol. 2002;27:286–296. doi: 10.1165/rcmb.2001-0014OC. [DOI] [PubMed] [Google Scholar]
- [159].Richter M, Bonneau R, Girard MA, Beaulieu C, Larivee P. Zinc status modulates bronchopulmonary eosinophil infiltration in a murine model of allergic inflammation. Chest. 2003;123:446S. doi: 10.1378/chest.123.3_suppl.446s. [DOI] [PubMed] [Google Scholar]
- [160].Krebs NF, Westcott JE, Arnold TD, Kluger BM, Accurso FJ, Miller LV, Hambidge KM. Abnormalities in zinc homeostasis in young infants with cystic fibrosis. Pediatr. Res. 2000;48:256–261. doi: 10.1203/00006450-200008000-00022. [DOI] [PubMed] [Google Scholar]
- [161].Hansen RC, Lemen R, Revsin B. Cystic fibrosis manifesting with acrodermatitis enteropathica-like eruption. Association with essential fatty acid and zinc deficiencies. Arch. Dermatol. 1983;119:51–55. [PubMed] [Google Scholar]
- [162].Crone J, Huber WD, Eichler I, Granditsch G. Acrodermatitis enteropathica-like eruption as the presenting sign of cystic fibrosis-case report and review of the literature. Eur. J. Pediatr. 2002;161:475–478. doi: 10.1007/s00431-002-0982-0. [DOI] [PubMed] [Google Scholar]
- [163].Easley D, Krebs N, Jefferson M, Miller L, Erskine J, Accurso F, Hambidge KM. Effect of pancreatic enzymes on zinc absorption in cystic fibrosis. J. Pediatr. Gastroenterol. Nutr. 1998;26:136–139. doi: 10.1097/00005176-199802000-00003. [DOI] [PubMed] [Google Scholar]
- [164].Brown KH, Peerson JM, Rivera J, Allen LH. Effect of supplemental zinc on the growth and serum zinc concentrations of prepubertal children: a meta-analysis of randomized controlled trials. Am. J. Clin. Nutr. 2002;75:1062–1071. doi: 10.1093/ajcn/75.6.1062. [DOI] [PubMed] [Google Scholar]
- [165].Hamadani JD, Fuchs GJ, Osendarp SJ, Khatun F, Huda SN, Grantham-McGregor SM. Randomized controlled trial of the effect of zinc supplementation on the mental development of Bangladeshi infants. Am. J. Clin. Nutr. 2001;74:381–386. doi: 10.1093/ajcn/74.3.381. [DOI] [PubMed] [Google Scholar]
- [166].Arthur JR, McKenzie RC, Beckett GJ. Selenium in the immune system. J. Nutr. 2003;133:1457S–1459S. doi: 10.1093/jn/133.5.1457S. [DOI] [PubMed] [Google Scholar]
- [167].Swede H, Dong Y, Reid M, Marshall J, Ip C. Cell cycle arrest biomarkers in human lung cancer cells after treatment with selenium in culture. Cancer Epidemiol. Biomarkers Prev. 2003;12:1248–1252. [PubMed] [Google Scholar]
- [168].El-Bayoumy K, Sinha R. Mechanisms of mammary cancer chemo-prevention by organoselenium compounds. Mutat. Res. 2004;551:181–197. doi: 10.1016/j.mrfmmm.2004.02.023. [DOI] [PubMed] [Google Scholar]
- [169].Niskar AS, Paschal DC, Kieszak SM, Flegal KM, Bowman B, Gunter EW, Pirkle JL, Rubin C, Sampson EJ, McGeehin M. Serum selenium levels in the US population: Third National Health and Nutrition Examination Survey, 1988-1994. Biol. Trace Elem. Res. 2003;91:1–10. doi: 10.1385/BTER:91:1:1. [DOI] [PubMed] [Google Scholar]
- [170].Bates CJ, Thane CW, Prentice A, Delves HT, Gregory J. Selenium status and associated factors in a British national diet and nutrition survey: young people aged 4-18 y. Eur. J. Clin. Nutr. 2002;56:873–881. doi: 10.1038/sj.ejcn.1601405. [DOI] [PubMed] [Google Scholar]
- [171].Rubin RN, Navon L, Cassano PA. Relationship of serum antioxidants to asthma prevalence in youth. Am. J. Respir. Crit. Care Med. 2004;169:393–398. doi: 10.1164/rccm.200301-055OC. [DOI] [PubMed] [Google Scholar]
- [172].Hu G, Cassano PA. Antioxidant nutrients and pulmonary function: the Third National Health and Nutrition Examination Survey (NHANES III) Am. J. Epidemiol. 2000;151:975–981. doi: 10.1093/oxfordjournals.aje.a010141. [DOI] [PubMed] [Google Scholar]
- [173].Duffield-Lillico AJ, Reid ME, Turnbull BW, Combs GF, Jr., Slate EH, Fischbach LA, Marshall JR, Clark LC. Baseline characteristics and the effect of selenium supplementation on cancer incidence in a randomized clinical trial: a summary report of the Nutritional Prevention of Cancer Trial. Cancer Epidemiol. Biomarkers Prev. 2002;11:630–639. [PubMed] [Google Scholar]
- [174].Winklhofer-Roob BM, Tiran B, Tuchschmid PE, van't Hof MA, Shmerling DH. Effects of pancreatic enzyme preparations on erythrocyte glutathione peroxidase activities and plasma selenium concentrations in cystic fibrosis. Free Radic. Biol. Med. 1998;25:242–249. doi: 10.1016/s0891-5849(98)00061-6. [DOI] [PubMed] [Google Scholar]
- [175].Halliwell B, Gutteridge JMC. Free radicals in biology and medicine. Oxford University Press; Oxford: 1999. [Google Scholar]
- [176].Hellman NE, Gitlin JD. Ceruloplasmin metabolism and function. Annu. Rev. Nutr. 2002;22:439–458. doi: 10.1146/annurev.nutr.22.012502.114457. [DOI] [PubMed] [Google Scholar]
- [177].Pacht ER, Davis WB. Role of transferrin and ceruloplasmin in antioxidant activity of lung epithelial lining fluid. J. Appl. Physiol. 1988;64:2092–2099. doi: 10.1152/jappl.1988.64.5.2092. [DOI] [PubMed] [Google Scholar]
- [178].Britigan BE, Hayek MB, Doebbeling BN, Fick RB., Jr. Transferrin and lactoferrin undergo proteolytic cleavage in the Pseudo-monas aeruginosa-infected lungs of patients with cystic fibrosis. Infect. Immun. 1993;61:5049–5055. doi: 10.1128/iai.61.12.5049-5055.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].King DE, Egan BM, Geesey ME. Relation of dietary fat and fiber to elevation of C-reactive protein. Am. J. Cardiol. 2003;92:1335–1339. doi: 10.1016/j.amjcard.2003.08.020. [DOI] [PubMed] [Google Scholar]
- [180].Blot WJ, Li JY, Taylor PR, Guo W, Dawsey SM, Li B. The Linxian trials: mortality rates by vitamin-mineral intervention group. Am. J. Clin. Nutr. 1995;62:1424S–1426S. doi: 10.1093/ajcn/62.6.1424S. [DOI] [PubMed] [Google Scholar]
- [181].Shekelle PG, Morton SC, Jungvig LK, Udani J, Spar M, Tu W, Suttorp MJ, Coulter I, Newberry SJ, Hardy M. Effect of supplemental vitamin E for the prevention and treatment of cardiovas-cular disease. J. Gen. Intern. Med. 2004;19:380–389. doi: 10.1111/j.1525-1497.2004.30090.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Omenn GS, Goodman GE, Thornquist MD, Balmes J, Cullen MR, Glass A, Keogh JP, Meyskens FL, Jr., Valanis B, Williams JH, Jr., Barnhart S, Cherniack MG, Brodkin CA, Hammar S. Risk factors for lung cancer and for intervention effects in CARET, the Beta-Carotene and Retinol Efficacy Trial. J. Natl. Cancer Inst. 1996;88:1550–1559. doi: 10.1093/jnci/88.21.1550. [DOI] [PubMed] [Google Scholar]
- [183].Miller ER, III, Pastor-Barriuso R, Dalal D, Riemersma RA, Appel LJ, Guallar E. Meta-analysis: high-dosage vitamin E supple-mentation may increase all-cause mortality. Ann. Intern. Med. 2005;142:37–46. doi: 10.7326/0003-4819-142-1-200501040-00110. [DOI] [PubMed] [Google Scholar]