

Abstract
There is no clinical treatment that reduces acinar injury during pancreatitis. Human immunodeficiency virus (HIV) protease inhibitors (PI), including nelfinavir (NFV) and ritonavir (RTV), may reduce the rate of pancreatitis in HIV-infected patients. Since permeability transition pore (PTPC)-mediated mitochondrial dysfunction occurs during pancreatitis, and we have shown that PI prevents PTPC opening, we studied its effects in a model of pancreatitis. The effect of NFV plus RTV (NFV/RTV) or vehicle on caerulein-induced pancreatitis in mice was compared by measuring changes in mitochondrial membrane potential in vitro and cytochrome c leakage in vivo. Histological and inflammatory makers were also compared. NFV/RTV improved DiOC6 retention in acini exposed to caerulein in vitro. In vivo NFV prevented cytosolic leakage of cytochrome c and reduced pancreatic acinar injury, active caspase-3 staining, TUNEL-positive acinar cells, and serum amylase (P < 0.05). Conversely, trypsin activity, serum cytokine levels, and pancreatic and lung inflammation were unaffected. NFV/RTV reduces pancreatic injury and acinar cell death in experimental mouse caerulein-induced pancreatitis but does not impact inflammation.
Keywords: permeability transition pore inhibition, necrosis, apoptosis, protease inhibitors
the mechanisms leading to cell death in acute pancreatitis are controversial (15, 17), and theories include abnormal vesicular trafficking leading to activation of zymogens (39), toxic effects of intracellular calcium (17, 27), and fatty acid ethyl esters (14), as well as activation of transcription factors (4), inflammatory mediators (6, 36, 50), cytoskeletal rearrangement (45), production of reactive oxygen species (5, 42), and mitochondrial damage (9, 11, 12). During experimental mouse pancreatitis, both necrosis and apoptosis occur (3, 10, 12, 13), although their roles during pancreatitis are controversial. Some studies suggest that apoptosis may be protective during pancreatitis (3); agents such as 1-cyano-2-hydroxy-3-butene (CHB), which induce apoptosis, also cause pancreatitis (47), suggesting a more causal role for apoptosis during pancreatitis.
A common organelle involved in both necrosis and apoptosis is the mitochondrion (7, 37, 43). During apoptosis, mitochondria lose transmembrane potential (9), resulting in cytochrome c release into the cytoplasm and formation of the apoptosome (12, 18, 26). The mitochondrial permeability transition pore complex (PTPC) controls transmembrane potential during apoptosis (13). PTPC opening is also implicated in necrotic death since cyclophilin D (a part of the PTPC) knockout mice are resistant to necrotic cell death induced by ischemia reperfusion injury (1, 28), reactive oxygen species, and calcium (1, 28). Since the human immunodeficiency virus (HIV) protease inhibitors (PI) nelfinavir (NFV) and retonavir (RTV) prevent loss of mitochondrial transmembrane potential (29) by binding to and preventing opening of the mitochondrial PTPC (49), they may also prevent cell death. Indeed, NFV plus RTV (NFV/RTV) treatments reduce cell death and improve functional outcomes during mouse models of Fas-induced hepatitis, cerebral ischemia (49), sepsis (cecal ligation and perforation) (48), and retinal degeneration following retinal detachment (22).
Additional clinical clues that PI might benefit pancreatitis come from observations that the incidence of drug-induced pancreatitis decreased coincident with introduction of PI therapy, including NFV (8, 32). For these reasons we opted to assess whether PI might alter the outcome of experimental mouse pancreatitis.
MATERIALS AND METHODS
All experiments were approved by the Institutional Animal Care and Use Committee at Mayo Clinic, Rochester, MN. Male C57/Bl6 mice (Harlan Laboratories, Indianapolis, IN), 18–20 g, were housed and fed under standard conditions. The CCK analog caerulein and Boc-Glu-Ala-Arg-methyl-coumaryl-7-amide were purchased from Research Plus (Bayonne, NJ). All other reagents were purchased from Sigma (St. Louis, MO). Terminal deoxynucleotide dUTP transferase nick-end labeling (TUNEL) and active caspase 3 staining were performed at Molecular Histology (Little Rock, AR).
PI treatment and Z-VAD-fmk treatment.
NFV has a short half-life in mice. RTV, another PI and CYP 3A inhibitor, increases plasma levels of NFV in mice with its own levels being undetectable; therefore we used NFV/RTV (48, 49). At 17 h before the first caerulein injection, animals were given 125 mg/kg of pediatric NFV suspension from Agouron Pharmaceuticals (La Jolla, CA) and 13 mg/kg of liquid RTV from Abbott Pharmaceuticals (Abbott Park, IL). NFV/RTV in distilled water or vehicle control (water) was given every 8 h by oral gavage, as previously described (48). Water given by gavage was used as the vehicle. Z-VAD-fmk was dissolved in DMSO at 10 mg/ml and administered intraperitoneally 30 min at 5 mg/kg (0.1 ml) as previously published (24).
Blood and tissue preparation.
Animals were given hourly intraperitoneal injections of saline (control) or caerulein (50 μg/kg) in saline for 12 h, as previously described (41). They were euthanized 1 h after the last injection, and blood and tissue samples were harvested and frozen or fixed in formalin. For lung histology, lungs were inflated with neutral buffered formalin via tracheal puncture at 20 cm water and clamped for 10 min in situ before being harvested. For MPO determination, the right ventricle was perfused with saline and the left ventricle was drained to remove loose blood in the pulmonary circulation that may interfere with the assay.
Morphological examination.
The 5-μm sections of paraffin-embedded pancreas or lung tissue were stained with hematoxylin and eosin (H&E), active caspase 3, or TUNEL and examined by an experienced morphologist blinded to the sample identity. Acinar cell injury and/or necrosis and TUNEL positivity were quantitated by morphometry as described (3). Briefly, these and active caspase-3 were measured by imaging the whole section sequentially with a ×10 lens and storing JPEG images by a person blinded to its identity and the morphologist examining these. Nonacinar areas were excluded, and extent of injured acinar area was expressed as the percent of the total acinar tissue. Injury criteria, as previously described (2, 16), were the presence of acinar-cell ghosts with loss of cellular outline, spillage of intracellular contents, appearance of a diffuse pinkish appearance, and/or and the destruction of the histoarchitecture of whole or parts of the acini.
Assays.
Serum amylase activity was measured colorimetrically using the Phadebas assay (Pharmacia Diagnostics, Portage, MI). Neutrophil sequestration in pancreas and lung was quantitated by measuring tissue MPO activity as described previously (20). Trypsin activity was measured fluorometrically, as described previously (23). Cytokines were measured by the Cytokine Core Laboratory (University of Maryland). For cytochrome c, pancreatic subcellular fractions were prepared, as previously described (26), and analyzed by Western blot using cytochrome c antibody from Oncogene (Cambridge, MA). Heat shock protein (HSP)70 was used as a mitochondrial marker, and PCNA as a cytoplasmic marker, both from Oncogene (Cambridge, MA) as previously described (29, 30).
Preparation of acini and measurement of DiOC6 retention.
Acini were harvested in 20 mM HEPES (pH 7.4), 120 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 10 mM glucose, 10 mM sodium pyruvate, 0.1% bovine serum albumin, and 0.01% soybean trypsin inhibitor, as previously described (19), and filtered through a 70-μm mesh, then a 40-μm mesh, to get clusters 40–70 μm in size. Viability prior to use was >98% by Trypan blue exclusion. NFV or DMSO was added 30 min before 10 nM caerulein or saline (control) and incubated for 4 h. Retention of 3,3′-dihexyloxacarbocyanine (DiOC6) from Molecular Probes (Carlsbad, CA) was used for measuring mitochondrial membrane potential as previously described (12), despite theoretical concerns that it might not be as sensitive as other assays of mitochondrial potential.
Measurement of TNF-α and KC mRNA.
RNA was extracted from mouse acini by use of the ToTally RNA kit from Ambion (Austin, Texas). Semiquantitative PCR was done on cDNA extracted from 5 μg RNA by use of the Platinum PCR supermix purchased from Invitrogen (Carlsbad, CA). Universal 18S internal standards [yielding a 315-bp band, used in case of keratinocyte chemoattractant (KC)], and Classic 18S internal standards (yielding a 489-bp band, used in case of TNF-α) obtained from Ambion (Austin, TX) were used at a 1.7 ratio along with KC and TNF-α primers. The KC and TNF-α primers (see sequences below) were synthesized at Sigma-Genosys (St. Louis, MO). The primers used for KC were forward 5′-GACGAGACCAGGAGAAACAGGGTT, reverse 5′-AACGGAGAAAGAAGACAGACTGCT. For TNF-α, the forward and reverse primers were 5′-TCCCTCTCATCAGTTCTATGGCCC and 5′-AACCTGGGAGTAGACAAGGTACAAC, respectively. PCR was done in a 24-well thermocycler purchased from Perkin Elmer (Waltham, MA), as described previously (38), using 28 cycles (based on optimization protocol recommended in the 18S RNA Kit) with an annealing temperature of 60°C. Cyber Gold purchased from BioWhittaker (Walkersville, MD) was used to stain the bands and the intensity was measured by using The Gel Doc system from Bio-Rad (Hercules, CA).
Analysis of data.
The results reported represent means ± SE of values obtained from three or more in vitro experiments and compared by Student's t-test when the data consisted of only two groups or ANOVA when comparing three or more groups. Results from in vivo experiments were generated by use of six mice per group. If ANOVA indicated a significant difference, the data were analyzed by using Tukey's method as a post hoc test for the difference between groups. A P value of 0.05 was considered significant.
RESULTS
NFV prevents caerulein-induced loss of mitochondrial membrane potential in mice acinar cells.
We treated isolated acini with NFV alone or vehicle, followed by caerulein (10 nM), and measured mitochondrial transmembrane potential using DiOC6, as previously described (34). Although NFV did not alter mitochondrial transmembrane potential in resting acini (92.2 ± 8.0% of controls, P = 0.48), it prevented caerulein-induced (60 ± 2.6% control) loss of mitochondrial transmembrane potential (Fig. 1, P < 0.0001).
Fig. 1.

Nelfinavir (NFV) stabilizes mitochondrial membrane potential in acinar cells. Acini were preincubated with saline (CON) or 30 micromolar NFV for 30 min (solid bars) followed by addition of 10 nM caerulein (Cer) for 3 h as indicated. 3, 3′-Dihexyloxacarbocyanine (DiOC6) retention was measured as described in materials and methods and expressed as means ± SE from 3 different experiments. *P value <0.05 compared with vehicle-treated group. There was no difference induced by vehicle treatment compared with caerulein alone.
NFV prevents cytochrome c leakage and protects from caerulein injury. We next assessed whether NFV/RTV treatment of mice altered the postmitochondrial signaling events of caerulein-induced pancreatitis in vivo. Six mice per group were analyzed.
First, we measured active caspase 3 generation 12 h following caerulein since at this time point caspase 3 activation has been previously shown to be increased (10). Under control conditions there was negligible active caspase 3 staining (Fig. 2A), which increased to 13.5 ± 1.8% cells in caerulein treated mice (Fig. 2B) vs. 0.1 ± 0.02% in controls (P < 0.0001). Vehicle did not significantly affect this increase (14.4 ± 1.2%, P = 0.88, Fig. 2C); however, both Z-VAD-fmk (Fig. 2D) and NFV (Fig. 2E) reduced this significantly to 5.4 ± 1.9% (P < 0.02) and 7.6 ± 1.9% (P < 0.005), respectively (Fig. 2F).
Fig. 2.
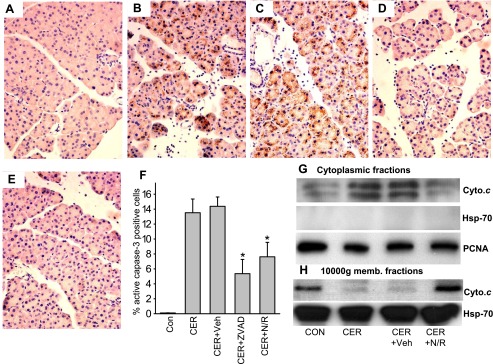
NFV reduces active caspase 3 staining and cytochrome c (Cyto.c) leakage into the cytosol during pancreatitis. Pancreas was harvested 1 h after the last of a series of 12 hourly intraperitoneal injections of saline (A), 50 μg/kg caerulein (B), animals pretreated with water before administration of caerulein (C), and animals pretreated with NFV/ritonovir (RTV) (N/R; D) or Z-VAD-fmk (ZVAD; E) before administration of caerulein. These were fixed and stained for active caspase-3 and accessed as described in materials and methods. Representative ×400 images are shown. F: % active caspase-3 positive cells expressed as means ± SE. Immunoblots of cytoplasmic fractions (G) and mitochondrial fractions (H) blotted for cytochrome c (top) and then stripped and blotted for HSP70 (G, middle and H, bottom) which was used a control for lane loading of the mitochondrial fractions. Since there was no leakage of heat shock protein (HSP)70 into the cytosolic fractions, PCNA was used as a control for lane loading of the cytosolic fractions as described in materials and methods (G, bottom). Each column represents pancreatic protein amounts in the respective fractions of pancreatic tissue after the different treatments. The 4 different treatments were control animals, 3-h caerulein pancreatitis, 3-h pancreatitis prophylaxed with vehicle, and 3-h pancreatitis prophylaxed with NFV/RTV.
Next, we measured cytochrome c release into the cytosol (Fig. 2, G and H) by Western blots of mitochondrial and cytoplasmic fractions of pancreatic tissues. PCNA and HSP70 were used to confirm the purity of the cytoplasmic and mitochondrial fractions. Cytochrome c was minimally present in the cytosolic fractions of control cells yet significantly increased in the tissues from mice with caerulein-induced pancreatitis. Prior administration of NFV/RTV reduced cytochrome c release to the level of untreated control cells (Fig. 2, G and H).
NFV/RTV also decreased the percentage of TUNEL-positive cells (2.78%) compared with the vehicle-treated animals (7.04%, P < 0.0005) or caerulein-only groups (Fig. 3B) (6.71%, P < 0.04). As a positive control for inhibiting apoptosis, ZVAD-fmk also reduced the number of TUNEL-positive cells to 0.5% (P < 0.02) (Figs. 3, A–F).
Fig. 3.

Terminal deoxynucleotide dUTP transferase nick-end labeling (TUNEL) staining and histology [hematoxylin and eosin (H&E)] showing pancreatic acinar apoptosis and injury are reduced by NFV pretreatment. Pancreas was harvested 1 h after the last of a series of 12 hourly intraperitoneal injections of saline (A), 50 μg/kg caerulein (B), animals pretreated with water before administration of caerulein (C), and animals pretreated with Z-VAD-fmk (D) or NFV/RTV (E) before administration of caerulein. These were fixed and stained for TUNEL as described in materials and methods. F: quantitation of these apoptotic nuclei depicted as means ± SE. G–L: acinor injury as determined by H & E staining as indicated. *Significant (P < 0.05) reduction compared with untreated caerulein- or vehicle-treated groups.
In addition to reducing TUNEL positivity, acinar injury as determined by H & E staining was reduced (P < 0.03) by NFV/RTV treatment (7.6%), compared with the caerulein (24.9%), or caerulein + vehicle (26.8%)-treated groups (Figs. 3, G–L).
NFV's protection from acinar cell injury is independent of proinflammatory cascades and trypsin generation. The pathogenesis of pancreatitis is felt to involve local pancreatic injury followed by local and systemic inflammation. Having shown that NFV/RTV protect against acinar injury, we next studied their effects on systemic amylase levels and on inflammation. Reductions in cell death were accompanied by a reduction in serum amylase from 17.1 ± 2.4-fold control in caerulein and 19.0 ± 2.6-fold in caerulein+vehicle-treated animals to 12.4 ± 1.5-fold in NFV-treated animals (P < 0.05) (Fig. 4A).
Fig. 4.

NFV/RTV reduce serum amylase without affecting the increase in trypsin, pancreatic water content, or neutrophil infiltration. Serum amylase (A), pancreatic water content (B), or pancreatic MPO levels (C) were measured in control animals or those which received 50 μg/kg caerulein intraperitoneal every hour for 12 h, caerulein after pretreatment with vehicle (Veh) before the induction of pancreatitis, or pretreatment with Z-VAD-fmk NFV/RTV. *P value <0.05 compared with vehicle-treated group. D: intrapancreatic trypsin activity measured in control mice or 30 min after receiving 50 μg/kg ip caerulein, or those pretreated with vehicle, Z-VAD-fmk, or NFV/RTV before the induction of pancreatitis. *P value <0.05 compared with vehicle-treated group.
NFV/RTV and Z-VAD-fmk did not alter the increase in pancreatic MPO levels (Fig. 4C) or pancreatic edema (Fig. 4B) during pancreatitis. In addition, since trypsin contributes to the pathogenesis of pancreatitis (35), we measured its activity in pancreatic homogenates and found that trypsin activity was unaffected (Fig. 4D) either by NFV/RTV or, predictably, by Z-VAD-fmk. In vitro, trypsin activity was not impacted by NFV (data not shown). Also, NFV/RTV did not affect the caerulein-induced degradation of IK-Bα (results not shown).
Systemic inflammation was measured by analyzing serum levels of IL-6, TNF-α, MCP-1, IL-10, IL-1β, or KC (data not shown). In all cases, serum levels of these cytokines were not altered by NFV/RTV. Inflammation in a distal site was assessed in the lung by histology and MPO activation. Lung histologies were similar in caerulein-treated animals with or without Z-VAD or NFV/RTV (Fig. 5, A–E). Likewise, lung MPO activity remained similar in caerulein-treated animals irrespective of whether they received ZVAD or NFV/RTV (Fig. 5F).
Fig. 5.
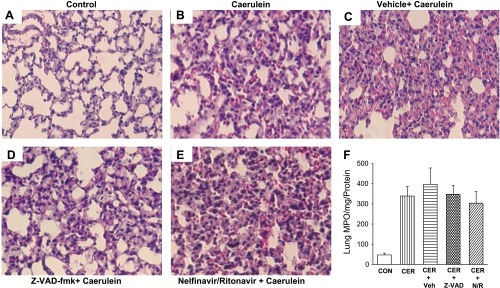
Lung inflammation as seen on histology and neutrophil infiltration as measured by MPO are unaffected by NFV/RTV. Representative ×250 images of lungs harvested, fixed, and stained with H&E 1 h after the last of a series of 12 hourly intraperitoneal injections of saline (A), 50 μg/kg caerulein (B), animals pretreated with vehicle before administration of caerulein (C), and animals pretreated with NFV/RTV (D) or Z-VAD-fmk (E) before administration of caerulein. F: bar graph of MPO activity per mg protein. The different groups are saline (CON), 50 μg/kg caerulein, animals pretreated with vehicle before administration of caerulein, and animals pretreated with Z-VAD-fmk or NFV/RTV before administration of caerulein.
DISCUSSION
Although it remains controversial what role apoptosis vs. necrosis play in the pathogenesis of pancreatitis, it is apparent that both processes do occur, although to different degrees in different experimental models. In this study we have used ZVAD-fmk treatments as a means of inhibiting the consequences of caspase activation that occur during apoptosis. Consistent with ZVAD-fmk selectively inhibiting caspases, mice receiving ZVAD-fmk in addition to caerulein had less caspase 3 activation, TUNEL positivity, and overall pancreatic injury compared with caerulein alone. These data suggest that, in this model, apoptosis contributes to pancreatic injury. Moreover, and consistent with our underlying hypothesis, treatment with PI of isolated acini in vitro, or mice in vivo, results in reduced acinar injury, caspase 3 activation, and TUNEL positivity. These observations are also consistent with observations in patients receiving PI: 1) The use of HIV PI such as NFV has reduced the incidence of pancreatitis in HIV-infected patients from as high as 14% in the pre-highly active antiretroviral therapy era to rates as low as 0.13, 0.35, and 0.85% since 1996 (8, 31, 33, 40). 2) Nucleoside reverse transcriptase inhibitors such as didanosine (ddI), stauvidine (4dT), and agents such as azathioprine that are mitochondriotoxic cause pancreatitis (25, 44). 3) NFV reduces incidence of pancreatitis caused by these agents (e.g., from 4.93% with ddI/d4T/efavirenz to 0% with NFV included, and from 6.25% in all regimens with ddI/d4T to 2.05% with NFV included in these) (32).
In addition to our observations concerning the effect of NFV/RTV on acinar injury, we observed, somewhat unexpectedly, no effect on pancreatic inflammation, edema, serum cytokines, and pulmonary inflammation. This suggests that the processes that promote acinar cell injury and inflammation are separable, as has been previously speculated (46). This is further supported by observations that stimuli that cause inflammation do not necessarily cause pancreatitis (see Supplementary Fig. S1).1 It is therefore tempting to speculate that stimuli that induce pancreatic cell death might also independently induce inflammation in an unrelated way. This might be tested, for example, by determining whether caerulein causes systemic inflammation in mice who had previously received pancreatectomies.
In summary, NFV protects pancreatitis by preventing loss of mitochondrial membrane potential, thus reducing acinar cell injury, without interfering with caerulein-induced inflammatory pathways.
GRANTS
A. Badley is supported by grants from National Institute of Allergy and Infectious Diseases (R01 AI62261-01-1 and R01 AI40384) and by the Burroughs Wellcome Award for Translational Research (ID no. 1005160). S. Rizza is supported by a grant from the National Institute of Diabetes and Digestive and Kidney Diseases (R21DK 77575-1).
Supplementary Material
Acknowledgments
We acknowledge Jonathan Henriksen at the Department of Pathology, University of Minnesota, MN for help in scanning images.
Footnotes
Supplemental data for this article are available online at the American Journal of Physiology Gastrointestinal and Liver Physiology website.
REFERENCES
- 1.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434: 658–662, 2005. [DOI] [PubMed] [Google Scholar]
- 2.Bhatia M, Saluja AK, Hofbauer B, Frossard JL, Lee HS, Castagliuolo I, Wang CC, Gerard N, Pothoulakis C, Steer ML. Role of substance P and the neurokinin 1 receptor in acute pancreatitis and pancreatitis-associated lung injury. Proc Natl Acad Sci USA 95: 4760–4765, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bhatia M, Wallig MA, Hofbauer B, Lee HS, Frossard JL, Steer ML, Saluja AK. Induction of apoptosis in pancreatic acinar cells reduces the severity of acute pancreatitis. Biochem Biophys Res Commun 246: 476–483, 1998. [DOI] [PubMed] [Google Scholar]
- 4.Chen X, Ji B, Han B, Ernst SA, Simeone D, Logsdon CD. NF-kappaB activation in pancreas induces pancreatic and systemic inflammatory response. Gastroenterology 122: 448–457, 2002. [DOI] [PubMed] [Google Scholar]
- 5.Dabrowski A, Boguslowicz C, Dabrowska M, Tribillo I, Gabryelewicz A. Reactive oxygen species activate mitogen-activated protein kinases in pancreatic acinar cells. Pancreas 21: 376–384, 2000. [DOI] [PubMed] [Google Scholar]
- 6.Denham W, Yang J, Fink G, Denham D, Carter G, Ward K, Norman J. Gene targeting demonstrates additive detrimental effects of interleukin 1 and tumor necrosis factor during pancreatitis. Gastroenterology 113: 1741–1746, 1997. [DOI] [PubMed] [Google Scholar]
- 7.Ding SP, Li JC, Jin C. A mouse model of severe acute pancreatitis induced with caerulein and lipopolysaccharide. World J Gastroenterol 9: 584–589, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dutta SK, Ting CD, Lai LL. Study of prevalence, severity, and etiological factors associated with acute pancreatitis in patients infected with human immunodeficiency virus. Am J Gastroenterol 92: 2044–2048, 1997. [PubMed] [Google Scholar]
- 9.Ehlers RA, Hernandez A, Bloemendal LS, Ethridge RT, Farrow B, Evers BM. Mitochondrial DNA damage and altered membrane potential (delta psi) in pancreatic acinar cells induced by reactive oxygen species. Surgery 126: 148–155, 1999. [PubMed] [Google Scholar]
- 10.Frossard JL, Rubbia-Brandt L, Wallig MA, Benathan M, Ott T, Morel P, Hadengue A, Suter S, Willecke K, Chanson M. Severe acute pancreatitis and reduced acinar cell apoptosis in the exocrine pancreas of mice deficient for the Cx32 gene. Gastroenterology 124: 481–493, 2003. [DOI] [PubMed] [Google Scholar]
- 11.Gerasimenko JV, Gerasimenko OV, Palejwala A, Tepikin AV, Petersen OH, Watson AJ. Menadione-induced apoptosis: roles of cytosolic Ca2+ elevations and the mitochondrial permeability transition pore. J Cell Sci 115: 485–497, 2002. [DOI] [PubMed] [Google Scholar]
- 12.Gukovskaya AS, Gukovsky I, Jung Y, Mouria M, Pandol SJ. Cholecystokinin induces caspase activation and mitochondrial dysfunction in pancreatic acinar cells. Roles in cell injury processes of pancreatitis. J Biol Chem 277: 22595–22604, 2002. [DOI] [PubMed] [Google Scholar]
- 13.Gukovskaya AS, Gukovsky I, Zaninovic V, Song M, Sandoval D, Gukovsky S, Pandol SJ. Pancreatic acinar cells produce, release, and respond to tumor necrosis factor-alpha. Role in regulating cell death and pancreatitis. J Clin Invest 100: 1853–1862, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gukovskaya AS, Mouria M, Gukovsky I, Reyes CN, Kasho VN, Faller LD, Pandol SJ. Ethanol metabolism and transcription factor activation in pancreatic acinar cells in rats. Gastroenterology 122: 106–118, 2002. [DOI] [PubMed] [Google Scholar]
- 15.Gukovskaya AS, Pandol SJ. Cell death pathways in pancreatitis and pancreatic cancer. Pancreatology 4: 567–586, 2004. [DOI] [PubMed] [Google Scholar]
- 16.Gukovskaya AS, Perkins P, Zaninovic V, Sandoval D, Rutherford R, Fitzsimmons T, Pandol SJ, Poucell-Hatton S. Mechanisms of cell death after pancreatic duct obstruction in the opossum and the rat. Gastroenterology 110: 875–884, 1996. [DOI] [PubMed] [Google Scholar]
- 17.Halangk W, Lerch MM. Early events in acute pancreatitis. Gastroenterol Clin North Am 33: 717–731, 2004. [DOI] [PubMed] [Google Scholar]
- 18.Halestrap AP The mitochondrial permeability transition pore in reperfusion injury and cardioprotection. Cardiovasc J S Afr 15: S5, 2004. [DOI] [PubMed] [Google Scholar]
- 19.Han B, Ji B, Logsdon CD. CCK independently activates intracellular trypsinogen and NF-κB in rat pancreatic acinar cells. Am J Physiol Cell Physiol 280: C465–C472, 2001. [DOI] [PubMed] [Google Scholar]
- 20.Haqqani AS, Sandhu JK, Birnboim HC. A myeloperoxidase-specific assay based upon bromide-dependent chemiluminescence of luminol. Anal Biochem 273: 126–132, 1999. [DOI] [PubMed] [Google Scholar]
- 21.Hietaranta AJ, Saluja AK, Bhagat L, Singh VP, Song AM, Steer ML. Relationship between NF-kappaB and trypsinogen activation in rat pancreas after supramaximal caerulein stimulation. Biochem Biophys Res Commun 280: 388–395, 2001. [DOI] [PubMed] [Google Scholar]
- 22.Hisatomi T, Nakazawa T, Noda K, Almulki L, Miyahara S, Nakao S, Ito Y, She H, Kohno R, Michaud N, Ishibashi T, Hafezi-Moghadam A, Badley AD, Kroemer G, Miller JW. HIV protease inhibitors provide neuroprotection through inhibition of mitochondrial apoptosis in mice. J Clin Invest 118: 2025–2038, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hofbauer B, Saluja AK, Lerch MM, Bhagat L, Bhatia M, Lee HS, Frossard JL, Adler G, Steer ML. Intra-acinar cell activation of trypsinogen during caerulein-induced pancreatitis in rats. Am J Physiol Gastrointest Liver Physiol 275: G352–G362, 1998. [DOI] [PubMed] [Google Scholar]
- 24.Iwata A, Nishio K, Winn RK, Chi EY, Henderson WR Jr, Harlan JM. A broad-spectrum caspase inhibitor attenuates allergic airway inflammation in murine asthma model. J Immunol 170: 3386–3391, 2003. [DOI] [PubMed] [Google Scholar]
- 25.Kakuda TN Pharmacology of nucleoside and nucleotide reverse transcriptase inhibitor-induced mitochondrial toxicity. Clin Ther 22: 685–708, 2000. [DOI] [PubMed] [Google Scholar]
- 26.Mareninova OA, Sung KF, Hong P, Lugea A, Pandol SJ, Gukovsky I, Gukovskaya AS. Cell death in pancreatitis: caspases protect from necrotizing pancreatitis. J Biol Chem 281: 3370–3381, 2006. [DOI] [PubMed] [Google Scholar]
- 27.Mooren F, Hlouschek V, Finkes T, Turi S, Weber IA, Singh J, Domschke W, Schnekenburger J, Kruger B, Lerch MM. Early changes in pancreatic acinar cell calcium signaling after pancreatic duct obstruction. J Biol Chem 278: 9361–9369, 2003. [DOI] [PubMed] [Google Scholar]
- 28.Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 434: 652–658, 2005. [DOI] [PubMed] [Google Scholar]
- 29.Phenix BN, Lum JJ, Nie Z, Sanchez-Dardon J, Badley AD. Antiapoptotic mechanism of HIV protease inhibitors: preventing mitochondrial transmembrane potential loss. Blood 98: 1078–1085, 2001. [DOI] [PubMed] [Google Scholar]
- 30.Prasanth SG, Prasanth KV, Siddiqui K, Spector DL, Stillman B. Human Orc2 localizes to centrosomes, centromeres and heterochromatin during chromosome inheritance. EMBO J 23: 2651–2663, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reisler RB, Han C, Burman WJ, Tedaldi EM, Neaton JD. Grade 4 events are as important as AIDS events in the era of HAART. J Acquir Immune Defic Syndr Hum Retrovirol 34: 379–386, 2003. [DOI] [PubMed] [Google Scholar]
- 32.Reisler RB, Murphy RL, Redfield RR, Parker RA. Incidence of pancreatitis in HIV-1-infected individuals enrolled in 20 adult AIDS clinical trials group studies: lessons learned. J Acquir Immune Defic Syndr Hum Retrovirol 39: 159–166, 2005. [PMC free article] [PubMed] [Google Scholar]
- 33.Riedel DJ, Gebo KA, Moore RD, Lucas GM. A ten-year analysis of the incidence and risk factors for acute pancreatitis requiring hospitalization in an urban HIV clinical cohort. AIDS Patient Care STDS 22: 113–121, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rottenberg H, Wu S. Quantitative assay by flow cytometry of the mitochondrial membrane potential in intact cells. Biochim Biophys Acta 1404: 393–404, 1998. [DOI] [PubMed] [Google Scholar]
- 35.Saluja AK, Bhagat L, Lee HS, Bhatia M, Frossard JL, Steer ML. Secretagogue-induced digestive enzyme activation and cell injury in rat pancreatic acini. Am J Physiol Gastrointest Liver Physiol 276: G835–G842, 1999. [DOI] [PubMed] [Google Scholar]
- 36.Sandoval D, Gukovskaya A, Reavey P, Gukovsky S, Sisk A, Braquet P, Pandol SJ, Poucell-Hatton S. The role of neutrophils and platelet-activating factor in mediating experimental pancreatitis. Gastroenterology 111: 1081–1091, 1996. [DOI] [PubMed] [Google Scholar]
- 37.Schild L, Matthias R, Stanarius A, Wolf G, Augustin W, Halangk W. Induction of permeability transition in pancreatic mitochondria by cerulein in rats. Mol Cell Biochem 195: 191–197, 1999. [DOI] [PubMed] [Google Scholar]
- 38.Singh VP, Bhagat L, Navina S, Sharif R, Dawra RK, Saluja AK. Protease-activated receptor-2 protects against pancreatitis by stimulating exocrine secretion. Gut 56: 958–964, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Singh VP, Saluja AK, Bhagat L, van Acker GJ, Song AM, Soltoff SP, Cantley LC, Steer ML. Phosphatidylinositol 3-kinase-dependent activation of trypsinogen modulates the severity of acute pancreatitis. J Clin Invest 108: 1387–1395, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith CJ, Olsen CH, Mocroft A, Viard JP, Staszewski S, Panos G, Staub T, Blaxhult A, Vetter N, Lundgren JD. The role of antiretroviral therapy in the incidence of pancreatitis in HIV-positive individuals in the EuroSIDA study. AIDS 22: 47–56, 2008. [DOI] [PubMed] [Google Scholar]
- 41.Song AM, Bhagat L, Singh VP, Van Acker GG, Steer ML, Saluja AK. Inhibition of cyclooxygenase-2 ameliorates the severity of pancreatitis and associated lung injury. Am J Physiol Gastrointest Liver Physiol 283: G1166–G1174, 2002. [DOI] [PubMed] [Google Scholar]
- 42.Soybir G, Koksoy F, Ekiz F, Yalcin O, Fincan K, Haklar G, Yuksel M. The effects of free oxygen radical scavenger and platelet-activating factor antagonist agents in experimental acute pancreatitis. Pancreas 19: 143–149, 1999. [DOI] [PubMed] [Google Scholar]
- 43.Tani S, Itoh H, Okabayashi Y, Nakamura T, Fujii M, Fujisawa T, Koide M, Otsuki M. New model of acute necrotizing pancreatitis induced by excessive doses of arginine in rats. Dig Dis Sci 35: 367–374, 1990. [DOI] [PubMed] [Google Scholar]
- 44.Tapner MJ, Jones BE, Wu WM, Farrell GC. Toxicity of low dose azathioprine and 6-mercaptopurine in rat hepatocytes. Roles of xanthine oxidase and mitochondrial injury. J Hepatol 40: 454–463, 2004. [DOI] [PubMed] [Google Scholar]
- 45.Torgerson RR, McNiven MA. The actin-myosin cytoskeleton mediates reversible agonist-induced membrane blebbing. J Cell Sci 111: 2911–2922, 1998. [DOI] [PubMed] [Google Scholar]
- 46.Van Acker GJ, Perides G, Weiss ER, Das S, Tsichlis PN, Steer ML. Tumor progression locus-2 is a critical regulator of pancreatic and lung inflammation during acute pancreatitis. J Biol Chem 282: 22140–22149, 2007. [DOI] [PubMed] [Google Scholar]
- 47.Walgren JL, Mitchell MD, Whiteley LO, Thompson DC. Identification of novel peptide safety markers for exocrine pancreatic toxicity induced by cyanohydroxybutene. Toxicol Sci 96: 174–183, 2007. [DOI] [PubMed] [Google Scholar]
- 48.Weaver JG, Rouse MS, Steckelberg JM, Badley AD. Improved survival in experimental sepsis with an orally administered inhibitor of apoptosis. FASEB J 18: 1185–1191, 2004. [DOI] [PubMed] [Google Scholar]
- 49.Weaver JG, Tarze A, Moffat TC, Lebras M, Deniaud A, Brenner C, Bren GD, Morin MY, Phenix BN, Dong L, Jiang SX, Sim VL, Zurakowski B, Lallier J, Hardin H, Wettstein P, van Heeswijk RP, Douen A, Kroemer RT, Hou ST, Bennett SA, Lynch DH, Kroemer G, Badley AD. Inhibition of adenine nucleotide translocator pore function and protection against apoptosis in vivo by an HIV protease inhibitor. J Clin Invest 115: 1828–1838, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zaninovic V, Gukovskaya AS, Gukovsky I, Mouria M, Pandol SJ. Cerulein upregulates ICAM-1 in pancreatic acinar cells, which mediates neutrophil adhesion to these cells. Am J Physiol Gastrointest Liver Physiol 279: G666–G676, 2000. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.