

Abstract
Diets deficient in an indispensable amino acid are known to suppress food intake in rats. Few studies were focused at understanding how amino acid-deficient diets may elicit biochemical changes at the mitochondrial level. The goal of this study was to evaluate mitochondrial function in rats fed diets with 0.00, 0.18, 0.36, and 0.88% threonine (Thr) (set at 0, 30, 60, and 140% of Thr requirement for growth). Here, it is described for the first time that Thr-deficient diets induce a specific uncoupling of mitochondria in liver, especially with NADH-linked substrates, not observed in heart (except for Thr-devoid diet). The advantage of this situation would be to provide ATP to support growth and maintenance when high-quality protein food (or wealth of high-quality food in general) is available, whereas Thr-deficient diets (or deficient-quality protein food) promote the opposite, increasing mitochondrial uncoupling in liver. The uncoupling with NADH substrates would favor the use of nutrients as energy sources with higher FADH-to-NADH ratios, such as fat, minimizing the first irreversible NADH-dependent catabolism of many amino acids, including Thr, thus enhancing the use of the limiting amino acid for protein synthesis when a low quality protein source is available.
Keywords: mitochondria, oxidative stress, oxygen consumption, oxidative phosphorylation, amino acid imbalance, amino acid deficiency
mammals require an adequate supply of essential amino acids in their diets to grow and thrive. Deficiency in one or more essential amino acids in the diet of rodents results in anorexia and loss of body protein (25, 26). It has been reported that detection of dietary deficiency takes place in the anterior pyriform cortex (3, 33). In this area, it has been found that during feeding of an essential amino acid-deficient diet, a translation inhibition by tRNA activation of the eukaryotic initiation factor-2α kinase 4 (eIF2αk4) occurs (22). This kinase, termed general control nonderepressible 2 (GCN2) in yeast where it was described first, is activated under conditions of nutrient deprivation or UV irradiation (54). In yeast, starvation of any single amino acid results in intracellular accumulation of uncharged tRNA, which binds to GCN2 on a domain homologous to His-tRNA synthetases. This binding event enhances the phosphorylation of eIF2α. Whereas eIF2α phosphorylation serves to slow global rates of translation, at the same time, it enhances translation of the transcriptional activator, GCN4. Increased levels of GCN4 leads to induction of genes that encode amino acid biosynthetic enzymes and other related metabolic proteins in an attempt to correct for the nutritional deficiency (54).
Animals fed these amino acid-deficient diets showed a significant depression in growth (25, 34, 42). Growth is a complex process driven by nutrient availability and hormonal status and, at the molecular level, is essentially based on macromolecule synthesis. Anabolic reactions are expensive processes supported by cellular ATP. Cellular ATP is mainly obtained through oxidative phosphorylation at the mitochondrial level and secondarily through substrate level phosphorylation. Recent studies showed that the response to amino acid deficiency takes 15 min to develop [if previously fed a low threonine (Thr) diet; Refs. 22, 28], but few data exist to understand biochemical changes related to bioenergetics at longer periods and in organs other than brain.
The goal of this study was to test whether the depression in body weight gain observed in animals fed diets deficient in one essential amino acid was associated with mitochondrial dysfunction. Diets deficient in one essential amino acid might result in a defective oxidative phosphorylation that could provide support for few cellular processes but not for all those required for an adequate body growth. To test this hypothesis, rats were fed diets deficient in one essential amino acid, Thr, and the functionality of mitochondria from two key tissues, liver (organ after intestine that is exposed to a dietary change) and heart (highly aerobic organ for which function depends on oxidative phosphorylation) were evaluated.
The novel aspects of this study are the following: 1) the effect of amino acid deficiency was tested in the whole animal as opposed to in vitro studies (i.e., cells in culture; e.g., Refs. 23, 41), which abrogate organ cross talk and hormonal status; 2) the deficiency of an essential amino acid was tested using diets with 0.00, 0.18, 0.36, and 0.88% Thr (set at 0, 30, 60, and 140% of Thr requirement for growth). The 0.18 and 0.36% Thr diets were designed to minimize the severe anorectic effect of essential amino acid-devoid diets, thus overcoming the caloric restriction issue that has confounded most of the results obtained under certain experimental settings; 3) the diets supplied at least 80% or higher of all nutrients required for growth or maintenance (except Thr); and 4) mitochondria biochemistry was studied in two tissues (heart and liver) to evaluate whether a dietary restriction affected oxidative phosphorylation (the main source for cellular ATP).
MATERIALS AND METHODS
Chemicals and biochemicals.
EDTA, EGTA, sodium succinate, mannitol, sucrose, HEPES, and BSA (fatty-acid free) were all purchased from Sigma (St. Louis, MO). Tris, glycine, sodium chloride, and potassium chloride were purchased from Fisher (Pittsburgh, PA). All other reagents were of analytical grade.
Animals and diets.
The University of California Davis Animal Care and Use Committee approved the experiments described below. Male Sprague-Dawley rats (6-wk-old) weighing 180–200 g or 14-wk-old weighing 360–370 g were purchased from a commercial supplier (Harlan Sprague Dawley, Indianapolis, IN, except as noted). All cages were kept in a vivarium maintained at 22 ± 1°C, with a 12:12-h light-dark cycle, following National Institutes of Health (NIH) guidelines for animal care and use. The lights were set to go out at 1900. Cage maintenance was conducted during the light cycle to avoid interference with behavioral measurements.
Briefly, after arrival in our laboratory, 5–8 rats were fed a commercially available chow (Purina cat. no. 5001) or purified, amino acid-based, Thr-deficient diets. All diets, except chow, were manufactured in our laboratory using crystalline l-amino acids (Tables 1 and 2). The Thr-deficient diets provided other essential amino acids at ratios based on egg albumin (Table 1; Ref. 45). Thr was set at 0, 30, 60, or 140% of the reported growth requirement in the 0.00, 0.18, 0.36, and 0.88% Thr diets, respectively (Table 2; Ref. 27a). Thr-deficient diets were formulated with appropriate energy density for growth and to provide protein and nitrogen at ±25% of the requirement for growth (Table 3). Over the testing period, the rats had free access to their food 24 h/day except when cage maintenance was conducted. Every day the rats were weighted to record body growth. At days 1, 3, 6, 9, and 15, the rats were removed from their cages and taken to the surgery room, where they were euthanized with CO2. The liver and heart were quickly removed from the rats, wet weights recorded, and rapidly homogenized for mitochondria isolation or amino acid analyses.
Table 1.
Amino acid mix compositions of diets used in this study
| Amino Acid | Amino Acid Mix 1 | Amino Acid Mix 2 |
|---|---|---|
| Alanine | 0.73 | 1.03 (0.78) |
| Arginine | 0.75 | 1.06 (0.81) |
| Asparagine-H2O | 1.34 | 1.88 (1.43) |
| Cystine | 0.3 | 0.42 (0.32) |
| Glutamic acid | 1.58 | 2.21 (1.68) |
| Glycine | 0.41 | 0.57 (0.43) |
| Histidine-HCl-H2O | 0.41 | 0.57 (0.43) |
| Isoleucine | 0.78 | 1.09 (0.83) |
| Leucine | 1.09 | 1.53 (1.17) |
| Lysine-HCl | 1.08 | 1.51 (1.15) |
| Methionine | 0.42 | 0.58 (0.44) |
| Phenylalanine | 0.71 | 0.99 (0.75) |
| Serine | 0.95 | 1.32 (1.00) |
| Tyrosine | 0.17 | 0.24 (0.18) |
| Tryptophan | 0.52 | 0.72 (0.55) |
| Valine | 0.85 | 0.19 (0.14) |
| Total | 12.09 | 15.91 (12.12) |
Both amino acid mixes were expressed in g/100 g diet. All amino acids were purchased from Ajinomoto, Tokyo, Japan. Numbers in parentheses represent an Amino Acid Mix 2 diet formulated to comply with the nitrogen and protein requirements for growth. See Thr-deficient diets depress body weight under results.
Table 2.
Composition of diets used in this study
| Component | 0.00% Thra | 0.18% Thr | 0.36% Thr | 0.88% Thr |
|---|---|---|---|---|
| Amino acid mix 1 | 12.09 | 12.09 | 12.09 | |
| Amino acid mix 2 | 12.12 (15.91) | |||
| Thrb | 0 | 0.18 | 0.36 | 0.88 (0.88) |
| Prolinec | 0.82 | 0.67 | 0.52 | 0.00 (0.72) |
| Vitamin mixd | 1 | 1 | 1 | 1 (1) |
| Mineral mixe | 5 | 5 | 5 | 5 (5) |
| Corn oilf | 5 | 5 | 5 | 5 (5) |
| Choline chlorideg | 0.1 | 0.1 | 0.1 | 0.1 (0.1) |
| Sucroseh | 25.27 | 25.26 | 25.25 | 23.47 (23.47) |
| Corn starchi | 50.72 | 50.7 | 50.68 | 47.94 (47.94) |
| Total | 100 | 100 | 100 | 100 |
Numbers in parentheses represent a 0.88% Thr diet formulated to comply with the nitrogen and protein requirements for growth. See Thr-deficient diets depress body weight under results.
Threonine (Thr) content expressed as g/100 g diet.
Thr (Ajinomoto): levels of Thr were formulated at 0, 30, 60, or 140% of the nutritional requirements for growth (NRG) 1995 requirement for growth.
Proline (Ajinomoto): concentration was adjusted to keep the 0, 0.18, 0.36, and 0.88% Thr diets isonitrogenous.
Vitamin supplement (TD USB Vitamin Supplement; Purina Test Diets) composition per kilogram of supplement: α-tocopherol, 1,000 IU/g, 5.0 g; l-ascorbic acid, 45 g; choline chloride, 75.0 g; d-calcium pantothenate, 3.0 g; inositol, 5.0 g; menadione, 2.25 g; niacin, 4.5 g; p-aminobenzoic acid, 5.0 g; pyridoxine HCl, 1.0 g; riboflavin, 1.0 g; thiamin HCl, 1.0 g; retinyl acetate, 900,000 units; ergocalciferol, 100,000 units; biotin, 20 mg; folic acid, 90 mg; vitamin B12, 1.35 mg.
Minerals (g/kg; AIN-76 Mineral Mixture, ICN Biochemicals): calcium phosphate dibasic, 500 g; sodium chloride, 74 g; potassium citrate monohydrate, 220 g; potassium sulfate, 52 g; magnesium oxide, 24 g; manganese carbonate (43–48% Mn), 3.5 g; ferric citrate (16–17% Fe), 6 g; zinc carbonate (70% ZnO), 1.6 g; cupric carbonate (53–55% Cu), 0.3 g; potassium iodate, 0.01 g; sodium selenite, 0.01 g; chromium potassium sulfate, 0.55 g, sucrose, finely powdered, 118.0 g.
Mazola corn oil (Best Foods, Englewood Cliffs, NJ).
Choline chloride (Fisher): 50% in water.
Sucrose (Spreckles, San Francisco, CA).
Corn starch (National Starch and Chemical, Bridgewater, NJ): Melojel food grade corn starch.
Table 3.
Nitrogen, total protein, and caloric content comparisons among diets
| 0.00% Thr | 0.18% Thr | 0.36% Thr | 0.88% Thr | Chow | NRG 1995 | |
|---|---|---|---|---|---|---|
| Nitrogen | 1.8 | 1.8 | 1.8 | 1.8 (2.4) | 3.2 | 2.4 |
| Protein | 13 | 13 | 13 | 13 (17.5) | 23 | 15 |
| Caloric content | 4.0 | 4.0 | 4.0 | 3.8 (4.0) | 3.3 | 3.8–4.1 |
Nitrogen (calculated) and protein contents were expressed in g/100 g diet. Caloric content was expressed in kcal/g diet. Numbers in parentheses represent a 0.88% Thr diet formulated to comply with the nitrogen and protein requirements for growth. See Thr-deficient diets depress body weight under results.
Food intake recordings.
Rats were housed individually in hanging wire cages so that food intake could be recorded. Preweighed food cups containing the appropriate diet were placed in each cage. Rats housed in this way were fed the diets from start to euthanasia. Food intake was recorded on a digital balance (corrected for spillage) each day for the duration of the experiment.
Mitochondria isolation.
Rat liver and heart mitochondria were isolated as previously described (14, 18). Briefly, mitochondria were isolated by differential centrifugation followed by Percoll gradient purification. This procedure yielded intact mitochondria minimally contaminated with other subcellular compartments (17, 18). Rats were anesthetized using a CO2 chamber. The livers (and hearts) were quickly removed, washed with 0.22 M mannitol, 70 mM sucrose, 0.5 mM EGTA, 2 mM HEPES, 0.1% fatty acid-free BSA, pH 7.4 (buffer A), and homogenized in a 10:1 buffer to liver wet weight ratio. Large cell debris and nuclei were pelleted by centrifuging at 600 g for 5 min in a Sorvall refrigerated centrifuge. This supernatant was filtered through two layers of cheesecloth to remove fat. Mitochondria were pelleted by centrifuging the supernatant for 10 min at 10,300 g in the same centrifuge. After suspending the pellet in 5 ml of the previous buffer, the mitochondrial fraction was loaded on 20 ml of 30% (vol/vol) Percoll, 0.225 M mannitol, 1 mM EGTA, 25 mM HEPES, 0.1% defatted BSA, pH 7.4, and spun for 30 min at 95,000 g in a Beckman Ti-60 rotor. Mitochondria were collected from the lowest band and washed twice with buffer A by centrifuging 10 min at 6,300 g and finally washed using 0.15 M KCl. Mitochondrial pellets were gently suspended in a small volume of ice-cold buffer A. Purified mitochondria from homogenized livers or hearts were collected by differential centrifugation followed by Percoll gradient purification and multiple washes in MSHE buffer (220 mM d-mannitol, 70 mM sucrose, 0.5 mM EGTA, 2 mM HEPES, 0.1% fatty acid-free BSA). The resulting mitochondria were washed twice with 150 mM KCl. The final pellet was resuspended in MSHE. The KCl step is essential for the removal of catalase and other unspecifically bound proteins, which comes not only from tissue homogenization, but also from the lysis of red blood cells (14, 18). Average yields of heart and liver mitochondria were 1.6 ± 0.1 mg protein/g hearts and 13 ± 1 mg/g livers, independently of the diets.
Oxygen consumption.
The oxygen uptake of mitochondria was measured using a Clark-type O2 electrode from Hansatech (King's Lynn, United Kingdom) at 22°C (14, 18). Intact, purified mitochondria were suspended in 1 ml (liver) or 0.5 ml (heart) of reaction buffer containing 0.225 M sucrose, 5 mM MgCl2, 20 mM KCl, 10 mM potassium phosphate, and 20 mM HEPES/KOH, pH 7.4, in the presence of 1 mM malate-10 mM glutamate (malate-glutamate), 2.5 mM malate-5 mM pyruvate (pyruvate), or 10 mM succinate as substrates and 0.45 mM ADP. The respiratory control ratio (RCR) was obtained by dividing the rate of oxygen consumption in State 3 (expressed as nanomoles of oxygen per minute per milligram of protein, obtained with a substrate and ADP) by that of State 4 (in the presence of a substrate without ADP; Refs. 8, 9).
Amino acid analyses.
Tissue samples (liver and liver mitochondria) were homogenized in 200 μl of aqueous sulfosalicylic acid (3 g/100 g deionized water) spiked with 2.45 μM l-γ-amino-n-butyric acid as the internal standard. Each sample was then centrifuged at 14,000 g for 30 min at 4°C. All samples were processed for amino acid analysis using a commercially available kit [AccQ-Tag, Waters (10, 52)]. Amino acids were visualized using HPLC with fluorescence detection. Serum samples were processed directly without the precipitation procedure.
Protein, lipid, and glycogen analyses.
In all samples, protein concentrations were determined by use of the BCA protein assay kit. BSA was used to generate standard curves. For total liver lipid, a frozen piece of liver was homogenized with Folch reagent (32). The organic layer was dried, and the lipid remaining on evaporation was weighted. Glycogen was evaluated as described in Ref. 37.
Core body temperature.
Core body temperature was measured in all animals between 9 and 10 AM using a digital rectal probe.
Mitochondria number per cell.
The relative amounts of liver mitochondria in rats were determined using a quantitative real-time PCR (QRTPCR) method described by Wong and Cortopassi (56) and modified for our experiments as follows. The targeted genes were the single-copy nuclear pyruvate kinase (PK) and mitochondrial cytochrome b (CYTB). Species-specific primers were selected using the Oligo 6.0 software (MedProbe). Rat primers for PK were: forward 5′-act ggc cgg tgt cat agt ga-3′; reverse 5′-tgt tga cca gcc gta tgg ata-3′. Primers for CYTB were: forward 5′-cac cca cat ctg ccg aga c-3′; reverse 5′-aat gcg aag aag cgt gtt a-3′. Genomic DNA was extracted from 10 mg of liver with the QIAamp DNA Micro Kit (Qiagen). Each DNA concentration was determined by duplicate measurements of the absorbance at 260 nm. DNA was diluted to 50 ng/μl and served as stock DNA template for QRTPCR. This assay was performed in a Mastercycler ep realplex thermocycler (Eppendorf, Westbury, NY) with 9 μl of master mixture (2.5× RealMasterMix; Eppendorf) containing: 20 milliunits (mU)/μl HotMaster Taq DNA Polymerase; 4 mM magnesium acetate; 0.4 mM dNTPs with dUTP, and SYBR Green I; 0.3 μM each of the forward and reverse primers; 5 μl of the DNA template (50, 5, 0.5, 0.05, and 0.005 ng/μl); and water in a final volume of 20 μl. The same DNA template dilutions were used for both nuclear (nDNA) and mitochondrial (mtDNA) DNA amplification. The reaction was conducted as follows: 1 cycle of initial template denaturation for 2 min at 94°C, followed by 15 s of denaturation at 94°C, 30 cycles of annealing at 60°C, and 30 s of extension at 68°C for 40 cycles. At the end of the amplification process, melting curves were analyzed between 60 and 95°C (temperature transition of 0.1°C/s) with continuous fluorescence monitoring to control for the absence of nonspecific products. The cycle threshold was designated as Ct. Each sample was analyzed in duplicate. The corresponding real-time PCR efficiencies for each mitochondrial and nuclear gene amplification were calculated according to the equation E = 10(−1/slope) − 1 according to the Mastercycler ep manual.
Relative mitochondrial gene copy number to nuclear gene copy number was assessed by a comparative Ct method using the equation ΔCtmitochondria/nuclear = Ctmitochondria − Ctnuclear. The fold change relative to control animals (4-wk-old on 0.88% Thr) was calculated using the equation 2^(−ΔΔCtmitochondria/nuclear), where ΔΔCtmitochondria/nuclear = mean ΔCtmitochondria/nuclear of the control animals − ΔCtmitochondria/nuclear of each animal from different dietary groups.
RNA quantification.
Total RNA was isolated from liver with TRIzol (Invitrogen) and treated with RNase-free DNase (Ambion) to remove any contaminating genomic DNA. First-strand cDNA synthesis was performed using oligo(dT) primers (Invitrogen). Real-time PCR reactions were performed on the Eppendorf MasterCycler real-time detection system using SYBR Green. Each assay included (in triplicates) a standard curve of five serial dilution points of control cDNA (ranging from 100 ng to 100 pg), a no template control, and 25–50 ng of each sample cDNA. The relative concentrations of the endogenous controls hypoxanthine phosphoribosyltransferase (HPRT) and uncoupling protein-1 (UCP-1) were determined by plotting the cycle threshold (Ct) vs. the log of the serial dilution points, and the relative expression of gene of interest was determined after normalizing to endogenous controls. Primers used for real-time PCR were as follows: HPRT (forward 5′-gctggtgaaaaggacctct-3′; reverse 5′-cacaggactagaacacctgc-3′); UCP-1 (forward 5′-gggcccttgtaaacaacaaa; reverse 5′-gtcggtccttccttggtgta-3′).
Statistical analyses.
Data were analyzed using ANOVA, and results were expressed as means ± SE except as noted.
RESULTS
Thr-deficient diets depress body weight.
Rats (6 wk old) were fed diets with 0, 0.18, 0.36, and 0.88% Thr to provide 0, 30, 60, and 140% of the Thr requirement for growth (Table 2). For comparison purposes, a group of rats was also fed a commercially available chow (Purina cat. no. 5001; Table 3). The body weight of rats fed the various diets was assessed daily (Fig. 1A). The average daily food intake and weight gain of rats fed a commercial chow (0.91% Thr) or the 0.88% Thr diet was not significantly different (7–8 g weight gain per day; Fig. 1A), indicating that the differences in protein and caloric content between these diets did not affect food intake behavior or growth response (1.3-fold higher protein content and 0.8-fold of the caloric content in chow vs. 0.88% Thr diet; Table 3). Furthermore, a 0.88% Thr diet formulated to comply with the nitrogen and protein requirements for growth (numbers between parentheses in Tables 1–3) did not differ significantly from chow or 0.88% Thr (data not shown). It could be argued that parameters obtained with amino acid-based diets could not be compared with those obtained with chow because of the differences in amino acid bioavailability (free amino acids vs. amino acids from corn or soy protein as in chow), digestibility, fiber contents, physical form of the diets (powder vs. chow pellets), or other potential factors (including ingredients inherently present in chow, e.g., plant pigments). However, parameters related to mitochondrial function (see below) from 0.88% Thr and chow were also not significantly different, indicating that a comparison between the diets is appropriate under our experimental conditions.
Fig. 1.
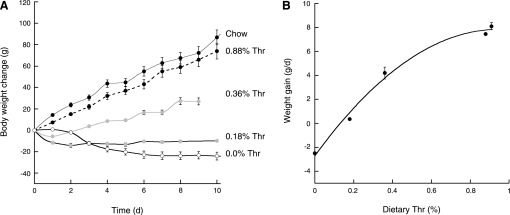
Changes in body weight of rats fed ad libitum commercial chow or purified diets providing varying concentrations of threonine (Thr). A: the rats (6 wk old) were weighed on a daily basis throughout the period tested (n = 4 per group; repeated in 3 separate experiments). Chow (black circles and line), 0.88% Thr (black circles, dotted line), 0.36% Thr (gray circles and line), 0.18% Thr (gray circles, black line), and 0.00% Thr (white circles, black line) are shown. B: daily gain (in grams) of rats fed diets varying in Thr concentration. The daily weight gains were taken as the slopes of the linear part of the plots indicated in the main panel. d, Day.
The daily weight gain of rats fed the 0.36 and 0.18% Thr diets was significantly depressed (weight gain, 4.2 and 0.35 g/day respectively; Fig. 1B) compared with that of rats fed 0.88% Thr. Rats fed a Thr-devoid diet showed more profound weight gain depression (−2.5 g/day; Fig. 1B). Daily weight gain increased proportionally with dietary Thr concentration, reaching a plateau at concentrations >0.6% Thr (Fig. 1B). Of note, the inflexion point of this curve was at 0.57% Thr, value between the nutritional requirements for growth (NRG) from 1978 (0.50% Thr) and 1995 (0.62% Thr; Refs. 2, 27a). 1
Thr-deficient diets decrease food intake.
Rats fed chow or 0.88% Thr diet had comparable daily food intake (26 ± 1 and 27.4 ± 0.9 g/day), whereas rats fed the 0.18% Thr diet had, on average, an 84% (84 ± 4%, mean ± SD) of the food intake of rats fed the 0.36% Thr diet (Fig. 2). The food intake of rats fed a 0.00% Thr diet was significantly depressed compared with all groups (in average 12 g/day vs. 18.4, 21.9, and 26–27 g/day for the 0.18%, 0.36%, and 0.88% Thr or chow).
Fig. 2.

Daily average food intake of rats fed ad libitum Thr-deficient diets. The procedure to record food intake was described in detail under materials and methods. *P < 0.05. All values obtained with 0.00% Thr were significantly different from 0.36 and 0.18% Thr. Chow (black circles and line), 0.88% Thr (black circles, dotted line), 0.36% Thr (gray circles and line), 0.18% Thr (gray circles, black line), and 0.00% Thr (white circles, black line) are shown.
The lack of weight gain in rats fed Thr-deficient diets can be explained by the anorectic response, given that food ingestion is depressed in rats offered an amino acid-deficient diet (3, 26). It has been shown that the rapid rejection of amino acid-decent diets corresponds to decreases in the level of the limiting indispensable amino acid in the anterior pyriform cortex of rats (3, 28). The smaller depression in both food intake (see below) and weight gain values obtained in this study compared with those previously published can be explained by essentially three factors: 1) the age of the rats; 2) the protein selected to design the diet; and 3) the nutritional requirements for rat growth. We used 6-wk-old rats (180 ± 20 g body wt) compared with other studies in which larger changes (30%) in food intake were observed with weanling (12) or 4-wk-old rats (100 ± 10 g body wt) exposed to amino acid-imbalanced diets (3, 28). The amino acid concentrations and ratios used in this study were based on egg protein, whereas those performed before were based on the casein amino acid composition and/or previous nutritional requirements (28, 34, 35). Finally, the design of our diets was based on the current nutritional growth requirements in which the quantitative contribution of each amino acid is generally larger (23% in average) than those based on previous ones.
Thr-deficient diets affect significantly liver but not heart weights.
Weights of liver and heart were obtained from rats fed the Thr-deficient diets. Liver wet weights, normalized per 100 g of body wt, showed a linear correlation with the daily weight gain (Fig. 3, closed circles; r2 = 0.71). The daily body weight gain was significantly influenced by the liver weight, whereas the heart weight had minimal impact. Similar results were obtained from Sprague-Dawley rats in which liver and gut weights were significantly affected by the daily weight gain when fed different planes of nutrition, whereas heart and kidney showed almost no change (15). In another study, liver and gastrocnemius muscle weights along with body weights were also significantly decreased in rats fed ad libitum Thr-devoid diets (50).
Fig. 3.

Relationship between liver and heart weights and average daily weight gains in rats fed diets varying in Thr concentration. Rats were fed ad libitum the 0, 0.18, 0.36, and 0.88% Thr and chow diets. Organ wet weights were taken at day 10. Daily weight gain was obtained from Fig. 1B.
The correlation between weight gain and liver weight did not change significantly when the liver weights were corrected by the weights of hydrated glycogen (assuming that water binds to dry glycogen weight per weight at a 2:1 ratio), indicating that the changes in liver weights were not significantly affected by different amounts of glycogen. In agreement with these observations, when the total protein, lipid, and glycogen contents of livers from rats fed the Thr-deficient diets were evaluated, protein and glycogen contents were not significantly different (when normalized per 100 g of liver dry weight; Table 4), whereas the lipid content decreased by 27%. This could be the result of an increased lipolysis, a decreased fatty acid synthesis or decreased formation of triacylglyceride deposits, or a combination of both processes. However, given that the diets provided appropriate amounts of fat to sustain adequate growth, it seems more likely that an increased use of fat has been developed in the livers of rats fed Thr-deficient diets.
Table 4.
Protein, lipid, and glycogen composition of livers from rats fed Thr-deficient diets
| Dietary Thr | Liver Protein, % liver dry wt | Liver Lipid, % liver dry wt | Liver Glycogen, % liver dry wt | Liver Mitochondrial Protein, mg/100 g body wt | Heart Mitochondrial Protein, mg/100 g body wt |
|---|---|---|---|---|---|
| Chow | 61.3 | 24.1 | 14.6 | 106 | 1.17 |
| 0.88% | 62.1 | 23.5 | 14.4 | 103 | ND |
| 0.36% | 63.1 | 22.3 | 14.6 | 89* | 0.93 |
| 0.18% | 62.9 | 20.1* | 17.0 | 75* | 0.87 |
| 0.00% | 63.9 | 17.5* | 18.6 | 64* | ND |
These parameters were evaluated at day 9 of the diet. The values were the mean of at least 3 experiments with SD ≤10%.
Significantly different from the values obtained with either chow or 0.88% Thr. ND, not determined.
The total liver protein concentration did not parallel the changes in mitochondrial protein (39% decrease) indicating that a general protein loss was not observed, in contrast to a loss in a specific compartment. Conversely, heart mitochondrial protein followed the lack of heart weight change with the daily body weight gain (Fig. 3 and last column Table 4).
Thr-deficient diets decrease liver mitochondrial coupling and alters mitochondria number.
Surprisingly, if a simple calculation is made to determine the true energy intake (obtained from the food intake data) and compared with the “expected” energy intake to sustain growth or maintenance, it is evident that the energy intake is in excess of the need for growth or maintenance as Thr becomes limiting. 2
We reasoned that the excess of energy intake observed in rats fed the Thr-deficient diets could be the result of a less efficient use of energy, accompanied or not, by a decrease in the mitochondria number. To this end, we evaluated the ratio of mtDNA to nDNA and mitochondrial protein per gram of liver to estimate the mitochondria number per cell. These parameters indicated that the mitochondria number almost doubled on going from the growth to the maintenance phase (6-wk-old exhibited 47% of 14-wk-old for both parameters when compared with diets with 100% of the requirements for each phase; Fig. 4A). During the same transition, the RCR increased by 3-fold (Fig. 4B). This is indicative that under maintenance conditions there is a need for more OXPHOS capacity supported by more mitochondria with higher efficiency. On feeding the rats of either age group with the Thr-deficient diets, the mitochondria number decreased significantly, 4 and 1.6 times lower for younger and older rats fed the 0.00% Thr diet compared with the ones with 100% requirements (Fig. 4A). These results indicated that younger rats under active growth rate are more susceptible to exhibit mitochondria changes with dietary changes than older rats. Probably the difference is based on the ability of older rats to access a larger pool of nonessential (because they are of bigger size and have less stringent nutrient requirements) proteins and replenish the endogenous Thr pool.
Fig. 4.
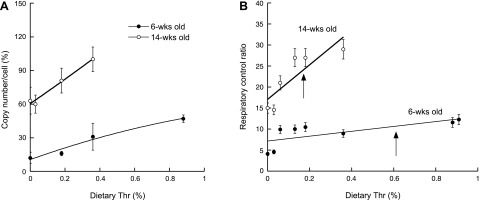
Changes of mitochondria number and respiratory control ratio (RCR) of liver mitochondria with dietary Thr in 6- and 14-wk-old rats. A: the mitochondria number per cell was evaluated as described in materials and methods. The numbers were expressed as percentage of the value obtained with the 14-wk-old rats fed the 0.36% Thr diet, which has 100% of the nutritional requirements for maintenance sustenance. B: the RCR of purified liver mitochondria was obtained with malate-glutamate. Six-week-old rats were fed on the 0.00, 0.03, 0.06, 0.13, 0.18, 0.36, and 0.88% Thr for 10 days; 14-wk-old rats were fed the same diets except the 0.88%.
It could be hypothesized that the excess of energy intake (not used to increase body weight) observed with Thr-deficient diets could be dissipated by increasing mitochondrial uncoupling and releasing heat. If this excess of energy were diverted from oxidative phosphorylation to support heat production, then it can be calculated that the body temperature would have increased ∼1°C. The body temperatures were experimentally evaluated in rats fed for 5 days the 0.18 and 0.89% Thr diets. Rats fed 0.89% Thr diet had a core body temperature of 97.1 ± 0.2°F (n =16), whereas those fed the 0.18% diet had a significantly different temperature (98.0 ± 0.3°F; n = 14; P < 0.05), accounting for an increase of 0.90 ± 0.09°F. This could not be accounted for by increased thermogenesis due to higher levels of UCP-1 mRNA in Thr-deficient animals, which were similar to controls. The increase in body temperature is not surprising given that liver is one of the internal organs with the highest resting energy expenditure and, after adipose tissue, shows the greatest weight change with caloric restriction or with amino acid-imbalanced diets (Refs. 19, 20) and that 60–70% of the resting energy expenditure are either mitochondrial processes or dependent on mitochondrial function (mitochondrial proton leak and Na+-K+-ATPase; Refs. 1, 43).
The RCRs of purified liver mitochondria from each experimental group were evaluated to test for coupling between electron transport and oxidative phosphorylation. The RCR is usually used as an index of mitochondria coupling and functionality, spanning from a value of 1 for uncoupled mitochondria to values of 10 or higher (depending on substrates, tissues, and species) for highly coupled mitochondria. The rates were tested by using an NADH-linked substrate, malate-glutamate (Fig. 4B), and an FADH-linked substrate, succinate (data not shown). Given that growing rats (i.e., 6 wk old) would be more prone to show this effect based on the higher need of ATP required to sustain the costly processes of macromolecule biosynthesis, we also obtained the RCR of liver mitochondria from 14-wk-old rats fed the same Thr-deficient diets (Fig. 4B) because these rats were considered to be under maintenance conditions since their original body weight was already 360 ± 10 g, and their daily average weight gain was 1.6 ± 0.3 g/day, significantly lower than that obtained with the 4-wk-old (7–8 g/day; see before).
The RCR of liver mitochondria from 6- and 14-wk-old rats fed the Thr-deficient diets increased with dietary Thr (Fig. 4B). This indicated that this correlation was not solely related to body weight gain but an adaptation to Thr deficiency that takes place regardless of the condition (growth or maintenance). Of note, under optimal supply of Thr for growth or maintenance, the RCR of 14-wk-old rats was threefold higher than that of 6-wk-old, suggesting that under maintenance conditions there is a need for a more efficient OXPHOS supported by mitochondria with higher RCR. From Fig. 4B, it can be concluded that as dietary Thr increased, liver mitochondria were more coupled and more efficient at producing ATP.
Thr-deficient diets significantly decrease State 3 respiration in liver.
A decrease in RCR could be the result of a decrease in State 3 (oxygen consumption with available substrate, oxygen, and ADP), increase in State 4 (oxygen consumption when ADP is limiting), or a combination of both. To evaluate these different scenarios, the individual rates of State 3 and 4 of rat liver mitochondria from rats fed the various Thr-deficient diets were evaluated with malate-glutamate, pyruvate, and succinate (Table 5). Our results indicated that the increase in uncoupling observed when going from 0.88% Thr (or chow) to 0.18% Thr was not the result of a significant change in State 4 respiration, succinate oxidase (Complex II → III → IV), or cytochrome oxidase (Complex IV) activities but a decline in NADH oxidase (47%; Table 5). Considering that NADH oxidase is constituted by Complexes I, III, and IV and that no changes were obtained with succinate oxidase, then it is surmised that the decrease in RCR was mainly attributed to a lower Complex I activity. Similar results were obtained with pyruvate-malate, another NAD-linked substrate, indicating that the decline in NADH oxidase activity did not show a substrate dependency.
Table 5.
States 3 and 4 oxygen uptake of liver mitochondria from rats fed Thr-deficient diets
| Thr, % | State 3 |
State 4 | ||
|---|---|---|---|---|
| NADH Oxidase | Succinate Oxidase | Cytochrome Oxidase | ||
| 0.00 | 29±1* | 70.9±0.9* | 44.8±0.7* | 5.2±0.2* |
| 0.18 | 22.8±0.4* | 92±3 | ND | 4.38±0.08 |
| 0.36 | 38±2* | 93±2 | 54±2 | 3.7±0.2 |
| 0.88 | 42.7±0.3 | 110±9 | 55±1 | 3.6±0.2 |
Values represent means ± SE (n ≥ 5). States 3 and 4 were expressed as nmol oxygen·min−1·mg mitochondrial protein−1. NADH oxidase was assessed with malate-glutamate, succinate oxidase with succinate in the presence of rotenone, and cytochrome oxidase with N,N,N′,N′-tetramethylphenylenediamine and ascorbate in the presence of antimycin; State 4 was taken after State 3 was obtained with malate-glutamate.
Significantly different from 0.88% Thr (P < 0.05).
When the diet was devoid of Thr, changes in all Complexes were observed (similar decline in NADH oxidase, 35% decline in succinate oxidase, and 18.5% decline in cytochrome oxidase; Table 5) in addition to an increase in State 4 respiration (1.44-fold; Table 5). Given that the hepatic mitochondrial proton leak was not altered (1–12 mo) or decreased (18 mo) in rats on a 40% caloric restriction diet (20), it is suggested that the increase in State 4 and time course (5–9 days vs. mo) elicited by the 0.00% Thr diet cannot be attributed to the similar (40–50%; Fig. 1) putative caloric restriction developed by the anorectic effect, in turn, triggered by the lack of this amino acid in the diet.
Thr-deficient diets do not induce changes in heart mitochondria.
No changes (except at 0.00% Thr) in NADH oxidase, succinate oxidase, or cytochrome oxidase were observed in heart mitochondria. A 20–30% decrease in NADH oxidase and succinate oxidase was observed when rats were fed the 0.00% Thr diet, decreases smaller than those observed with liver mitochondria (Table 5).
Thr-deficient diets increase the excess energy.
The association between RCR and energy efficiency was tested by plotting RCR values for both age groups at all Thr with the excess energy intake (expressed per body surface; Fig. 5). 3 These energy values were obtained as the difference between the energy intake calculated with the experimental daily food intake and that calculated using the experimental daily weight gain (Ref. 27a and references therein). The excess energy intake values had a reciprocal correlation with RCR values (Fig. 5; r2 = 0.97) indicating that more mitochondrial uncoupling leads to an apparent excess of energy not destined to support weight gain.
Fig. 5.

Correlation between liver mitochondrial RCR and energy intake. All RCRs were taken from Fig. 4. Energy intake was calculated from body weights, food intake, and weight gain as indicated in the text. A linear correlation was found between RCR and energy intake (r2 = 0.93).
DISCUSSION
When supplies of high-quality protein foods are insufficient, omnivores are at risk for essential amino acid deficiency. The choice of these foods has played an important role in human evolution (38). Protein malnutrition is a worldwide problem still common today, for example, in the form of kwashiorkor in developed and underdeveloped regions (36, 40). Combining protein sources with complementary essential amino acid patterns to maintain appropriate essential amino acid balance is a longstanding cultural practice that implies the existence of instinctive detection of essential amino acid deficiency in humans (6, 7) as well as in other mammals and species (16, 16a, 39).
It is known that rats given a diet deficient or devoid of one essential amino acid will eat less of it than they will of a protein-free diet (26). It is also known that if rats are force-fed a diet that lacks one essential amino acid they will develop pathological lesions and their survival will be shorter, whereas if they are allowed to eat ad libitum, their food intake will be depressed, no pathological lesions will develop, and they will survive longer (26, 48–50). Harper and Rogers (26) indicated that the depression of food intake that ensues from an amino acid-imbalanced diet is a protective mechanism designed not to exacerbate further the amino acid imbalance created by the deficiency.
In this study, we explored novel aspects of an essential amino acid deficiency: this effect was tested at various levels of Thr deficiency and in the whole animal allowing to evaluate organ cross talk and hormonal status, minimizing the anorectic effect due to essential amino acid deficiency (0.18 and 0.36% Thr diets). In addition, mitochondria biochemistry was studied in two tissues (heart and liver) to evaluate whether the dietary restriction affected oxidative phosphorylation, the main source for cellular ATP. In our study, we confirmed previous findings (depression of food intake and body weight gain) using Thr-deficient diets, except that a different, novel protective mechanism, triggered by a deficiency in one essential amino acid, was observed. Hepatic mitochondrial bioenergetics was affected by the Thr deficiency. The NADH oxidase activity declined with the severity of Thr deficiency, whereas succinate or cytochrome c oxidases were not affected unless no Thr was present in the diet. This situation suggests that a specific metabolic adaptation at the level of Complex I had occurred. In addition, the mitochondrial changes were specific for liver mitochondria, not observed in heart (unless Thr-devoid diets were used). Thus this response elicited a specific metabolic change (or adaptation) as opposed to a generalized response such as anorexia, and it is specific in terms of the targeted proteins within mitochondria and organs.
The availability of dietary Thr increased the efficiency of oxidative phosphorylation in liver. The advantage of this situation is that more ATP is provided to support growth and maintenance when high-quality protein food (or wealth of high-quality food in general) is available. Conversely, Thr-deficient diets (or low quality of protein food) promoted mitochondrial uncoupling in liver. Considering organs with a high rate of energy expenditure relative to their size, changes in size and metabolic rate of these tissues may have a substantial impact on maintenance requirements. In support of this observation, a significant decrease in liver weight (not heart) was found in rats exposed to Thr-deficient diets to decrease the energy expenditure when the anorectic response triggered by the amino acid deficiency has been established.
The introduction of hepatic uncoupling with an amino acid-deficient diet might seem counterintuitive, for it might be assumed that decreasing the food intake and the energy expenditure to match the lower caloric intake (and at the same time, minimizing the amino acid imbalance) might suffice without expecting changes in mitochondrial biochemistry. However, there are three clear advantages to the liver mitochondrial uncoupling: 1) it restricts the growth of liver (Fig. 3), minimizing energy expenditure. Diets influence energy use through adaptation of high energy-expending internal organs [especially liver (29, 30)]; 2) heat production increases with uncoupling in an attempt to overcome a lower body insulation (less fat deposits, increased lipolysis; see below), resulting in higher core body temperatures; and 3) it forces a dietary switch. With Thr-deficient diets, the use of NADH-linked substrates (e.g., malate-glutamate or pyruvate) was limited compared with that of FADH ones (e.g., succinate), even though the yield of oxidative phosphorylation is lower with the latter ones (P/O ratios of 2.5 vs. 1.5; Ref. 27). Evidently, fatty acid catabolism through β-oxidation falls in this category (glucose provides a 5:1 NADH-to-FADH ratio, essential amino acids in average 4:1, fatty acids 2:1; these numbers were calculated by using data from Ref. 53 assuming full oxidation to CO2 and H2O). This switch in the diet would favor the use of fat as energy source, as evidenced by the lower lipid content of liver, producing ATP at a lower yield even when mild uncoupling conditions had been established. In addition, the use of fatty acids would spare the use of the limiting amino acid for protein synthesis when a low quality protein source is available and possibly decreases the first irreversible NADH-dependent step in the catabolism of amino acids in an attempt to minimize the deficiency. In this regard, most amino acid dehydrogenases exhibit product inhibition by NADH. 4
In rats fed diets ad libitum (no preconditioning), the anorectic response seemed to be the first event in the cascade of adaptations to diets devoid of one essential amino acid. However, in situations in which an intermediate amino acid deficiency is encountered (e.g., 0.18–0.36% Thr), an almost simultaneous response (1–2 days) in hepatic bioenergetics and food intake changes seem to develop. It would be interesting to evaluate the exact kinetics of these processes to ascertain whether there is a cause-consequence link or whether they are triggered as independent, protective mechanisms. Although at the present moment we do not know the exact molecular mechanism responsible for the activity decline and changes in mitochondria number per cell, our laboratory is actively working to identify the signal transduction pathway involved in this effect. In this regard, the integrated stress response (ISR; Ref. 13), the pathway highly conserved from yeast to mammals that integrates signaling from multiple stress pathways including amino acid starvation, seems to be playing a major role. The ISR acts downstream of eIF2α phosphorylation (13, 24, 46). In mammals, distinct stress signals activate four eIF2α kinases: PKR (activated by double-stranded RNA during viral infection; Ref. 47), GCN2 (activated by uncharged tRNAs, adapts cells to amino acid starvation; Refs. 23, 57), HRI (activated by heme deficiency; Ref. 21), and PERK (activated by protein load in the endoplasmic reticulum stress; Ref. 44). The reversible phosphorylation on Ser51 of eIF2α by any of these kinases inhibits initiation of mRNA translation. Although global protein synthesis is inhibited, the translation of specific mRNAs such as ATF4 is strongly induced (46). This transcription factor plays a crucial role for the adaptation to stresses by regulating the expression of many genes involved in metabolism and transport of amino acid among others (24). Ongoing studies from our laboratory suggest that several nuclear-encoded genes for mitochondrial proteins are repressed, whereas others are induced by the Thr-deficient diets. The observed mtDNA depletion and changes in the electron transport chain could be explained by the specific repression and induction of genes that encode for proteins involved (directly or not) in mtDNA replication and maintenance and protein synthesis folding and targeting to mitochondria, respectively. One of the mechanisms that could trigger these events could be the increase in uncharged tRNAs ensuing from the Thr deficiency and subsequent activation of the GCN2; however, the fact that the concentrations of hepatic Thr (even under the most deficient diets) were well above the Km for the tRNA aminoacyl synthetases argues against the occurrence of this pathway. Instead, an adaptation to Thr deficiency was observed that involves the participation of several genes that attenuate the ISR, preventing an uncontrolled response to stress and its subsequent deleterious effects (Ross-Inta CM, Almendares A, Fujisawa Y, and Giulivi C, unpublished observations).
GRANTS
This study was supported by National Institute of Environmental Health Sciences Grants ES-012691 and ES-005707.
Acknowledgments
We thank Prof. Quinton Rogers for excellent comments, helpful discussions, and advice throughout this study and during the preparation of this manuscript. We are grateful to Dr. Jennifer Larsen for useful comments. We thank the technical assistance of Chern-Yi Tsai.
Footnotes
The reason for the difference in the requirements is based on the methods used to estimate them. Estimates reported earlier were based on significant differences between means by use of a multiple-range test, which produced values similar to results obtained with broken-stick models. Estimates calculated in this way will be lower than those made by the four-parameter logistical model and, as it was observed in the guinea pig, underestimated the requirement for indispensable amino acids by ∼20% (2).
For the 0.36% diet: 3.8 kcal/g diet × 21.9 g diet/day ÷ 4.2 g weight gain/day = 19.8 kcal/g weight gain; for the 0.88% Thr diet: 3.8 kcal/g × 21.9 g/day ÷ 7.45 g/day = 11 kcal/g weight gain; for chow: 3.8 kcal/g × 21.9 g/day ÷ 8.1 g/day = 10.3 kcal/g weight gain.
Energy intake for growth based on food intake (kcal/day) = food intake (g/day) × 3.8 kcal/g diet; energy intake (kcal/dm2) = energy intake (kcal/day) ÷ body surface (dm2); body surface (dm2) = 11.36 × (body weight plus weight gain in kg)0.67.
Energy intake for growth based on weight gain = 128/(body weight plus weight gain in kg)0.75 × (body weight plus gain in g)/1,000, divided by the body surface calculated as indicated above.
Energy intake for maintenance based on body weights: use 64 instead of 128 in the equations indicated above and replace with corresponding experimental body weights.
REFERENCES
- 1.Baldwin RL, Smith NE, Taylor J, Sharp M. Manipulating metabolic parameters to improve growth rate and milk secretion. J Anim Sci 51: 1416–1428, 1980. [DOI] [PubMed] [Google Scholar]
- 2.Benevenga NJ, Gahl MJ, Crenshaw TD, Finke MD. Protein and amino acid requirements for maintenance and amino acid requirements for growth of laboratory rats. J Nutr 124: 451–453, 1994. [DOI] [PubMed] [Google Scholar]
- 3.Beverly JL, Gietzen DW, Rogers QR. Effect of dietary limiting amino acid in prepyriform cortex on food intake. Am J Physiol Regul Integr Comp Physiol 259: R709–R715, 1990. [DOI] [PubMed] [Google Scholar]
- 4.Bird MI, Nunn PB. Metabolic homoeostasis of l-threonine in the normally-fed rat. Importance of liver threonine dehydrogenase activity. Biochem J 214: 687–694, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carpenter KJ A short history of nutritional science: part 1 (1785–1885). J Nutr 133: 638–645, 2003. [DOI] [PubMed] [Google Scholar]
- 7.Carpenter KJ A short history of nutritional science: part 2 (1885–1912). J Nutr 133: 975–984, 2003. [DOI] [PubMed] [Google Scholar]
- 8.Chance B, Williams GR. A simple and rapid assay of oxidative phosphorylation. Nature 175: 1120–1121, 1955. [DOI] [PubMed] [Google Scholar]
- 9.Chance B, Williams GR. The respiratory chain and oxidative phosphorylation. Adv Enzymol Relat Subj Biochem 17: 65–134, 1956. [DOI] [PubMed] [Google Scholar]
- 10.Cohen SA, De Antonis KM. Applications of amino acid derivatization with 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate. Analysis of feed grains, intravenous solutions and glycoproteins. J Chromatogr A 661: 25–34, 1994. [DOI] [PubMed] [Google Scholar]
- 12.Deshpande PD, Harper AE, Elvehjem CA. Amino acid imbalance on low fibrin diets. J Biol Chem 230: 327–333, 1958. [PubMed] [Google Scholar]
- 13.Dever TE Gene-specific regulation by general translation factors. Cell 108: 545–556, 2002. [DOI] [PubMed] [Google Scholar]
- 14.Elfering SL, Haynes VL, Traaseth NJ, Ettl A, Giulivi C. Aspects, mechanism, and biological relevance of mitochondrial protein nitration sustained by mitochondrial nitric oxide synthase. Am J Physiol Heart Circ Physiol 286: H22–H29, 2004. [DOI] [PubMed] [Google Scholar]
- 15.Ferrell CL, Koong KJ. Influence of plane of nutrition on body composition, organ size and energy utilization of Sprague-Dawley rats. J Nutr 116: 2525–2535, 1986. [DOI] [PubMed] [Google Scholar]
- 16.Fromentin G, Nicolaidis S. Rebalancing essential amino acids intake by self-selection in the rat. Br J Nutr 75: 669–682, 1996. [DOI] [PubMed] [Google Scholar]
- 16a.Gietzen DW Amino acid recognition in the central nervous system. In: Neural and Metabolic Control of Macronutrient Intake, edited by Berthoud HR and Seeley RJ. Boca Raton, FL: CRC, 2000, p. 339–357.
- 17.Giulivi C Functional implications of nitric oxide produced by mitochondria in mitochondrial metabolism. Biochem J 332: 673–679, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giulivi C, Poderoso JJ, Boveris A. Production of nitric oxide by mitochondria. J Biol Chem 273: 11038–11043, 1998. [DOI] [PubMed] [Google Scholar]
- 19.Greenberg JA, Boozer CN. Metabolic mass, metabolic rate, caloric restriction, and aging in male Fischer 344 rats. Mech Ageing Dev 113: 37–48, 2000. [DOI] [PubMed] [Google Scholar]
- 20.Hagopian K, Harper ME, Ram JJ, Humble SJ, Weindruch R, Ramsey JJ. Long-term calorie restriction reduces proton leak and hydrogen peroxide production in liver mitochondria. Am J Physiol Endocrinol Metab 288: E674–E684, 2005. [DOI] [PubMed] [Google Scholar]
- 21.Han AP, Yu C, Lu L, Fujiwara Y, Browne C, Chin G, Fleming M, Leboulch P, Orkin SH, Chen JJ. Heme-regulated eIF2alpha kinase (HRI) is required for translational regulation and survival of erythroid precursors in iron deficiency. EMBO J 20: 6909–6918, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hao S, Sharp JW, Ross-Inta CM, McDaniel BJ, Anthony TG, Wek RC, Cavener DR, McGrath BC, Rudell JB, Koehnle TJ, Gietzen DW. Uncharged tRNA and sensing of amino acid deficiency in mammalian piriform cortex. Science 307: 1776–1778, 2005. [DOI] [PubMed] [Google Scholar]
- 23.Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell 6: 1099–1108, 2000. [DOI] [PubMed] [Google Scholar]
- 24.Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11: 619–633, 2003. [DOI] [PubMed] [Google Scholar]
- 25.Harper AE, Benevenga NJ, Wohlhueter RM. Effects of ingestion of disproportionate amounts of amino acids. Physiol Rev 50: 428–558, 1970. [DOI] [PubMed] [Google Scholar]
- 26.Harper AE, Rogers QR. Amino acid imbalance. Proc Nutr Soc 24: 173–190, 1965. [DOI] [PubMed] [Google Scholar]
- 27.Hinkle PC P/O ratios of mitochondrial oxidative phosphorylation. Biochim Biophys Acta 1706: 1–11, 2005. [DOI] [PubMed] [Google Scholar]
- 27a.Institute for Laboratory Animal Research. Nutrient requirements of laboratory animals (4th ed.). Washington, DC: National Academy Press, 1995, p. 11–59.
- 28.Koehnle TJ, Russell MC, Gietzen DW. Rats rapidly reject diets deficient in essential amino acids. J Nutr 133: 2331–2335, 2003. [DOI] [PubMed] [Google Scholar]
- 29.Koong LJ, Ferrell CL. Effects of short term nutritional manipulation on organ size and fasting heat production. Eur J Clin Nutr 44, Suppl 1: 73–77, 1990. [PubMed] [Google Scholar]
- 30.Koong LJ, Ferrell CL, Nienaber JA. Assessment of interrelationships among levels of intake and production, organ size and fasting heat production in growing animals. J Nutr 115: 1383–1390, 1985. [DOI] [PubMed] [Google Scholar]
- 31.Krebs HA Pyridine nucleotides and rate control. Symp Soc Exp Biol 27: 299–318, 1973. [PubMed] [Google Scholar]
- 32.Lebaron FN, Folch J. The effect of pH and salt concentration on aqueous extraction of brain proteins and lipoproteins. J Neurochem 4: 1–8, 1959. [DOI] [PubMed] [Google Scholar]
- 33.Leung PM, Rogers QR. Importance of prepyriform cortex in food-intake response of rats to amino acids. Am J Physiol 221: 929–935, 1971. [DOI] [PubMed] [Google Scholar]
- 34.Leung PM, Rogers QR, Harper AE. Effect of amino acid imbalance in rats fed ad libitum, interval-fed or force-fed. J Nutr 95: 474–482, 1968. [DOI] [PubMed] [Google Scholar]
- 35.Leung PM, Rogers QR, Harper AE. Effect of amino acid imbalance on dietary choice in the rat. J Nutr 95: 483–492, 1968. [DOI] [PubMed] [Google Scholar]
- 36.Liu T, HR, Mancini AJ, Weston WL, Paller AS, Drolet BA, Esterly NB, Levy ML, SL, Frieden IJ. Kwashiorkor in the United States: fad diets, perceived and true milk allergy, and nutritional ignorance. Arch Dermatol 137: 630–636, 2002. [PubMed] [Google Scholar]
- 37.Lo S, Russell JC, Taylor AW. Determination of glycogen in small tissue samples. J Appl Physiol 28: 234–236, 1970. [DOI] [PubMed] [Google Scholar]
- 38.Milton K The critical role played by animal source foods in human (Homo) evolution. J Nutr 133: 3886S–3892S, 2003. [DOI] [PubMed] [Google Scholar]
- 39.Murphy ME, Pearcy SD. Dietary amino acid complementation as a foraging strategy for wild birds. Physiol Behav 53: 689–698, 1993. [DOI] [PubMed] [Google Scholar]
- 40.Oyelami OA, Ogunlesi TA. Kwashiorkor–is it a dying disease? S Afr Med J 97: 65–68, 2007. [PubMed] [Google Scholar]
- 41.Pham PT, Heydrick SJ, Fox HL, Kimball SR, Jefferson LS Jr, Lynch CJ. Assessment of cell-signaling pathways in the regulation of mammalian target of rapamycin (mTOR) by amino acids in rat adipocytes. J Cell Biochem 79: 427–441, 2000. [DOI] [PubMed] [Google Scholar]
- 42.Rogers QR, Harper AE. Amino acid diets and maximal growth in the rat. J Nutr 87: 267–273, 1965. [DOI] [PubMed] [Google Scholar]
- 43.Rolfe DF, Brown GC. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol Rev 77: 731–758, 1997. [DOI] [PubMed] [Google Scholar]
- 44.Ron D Translational control in the endoplasmic reticulum stress response. J Clin Invest 110: 1383–1388, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ross-Inta C, Tsai CY, Giulivi C. The mitochondrial pool of free amino acids reflects the composition of mitochondrial-DNA encoded proteins: indication for a post-translational quality control for protein synthesis. Biosci Rep 28: 239–249, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rutkowski DT, Kaufman RJ. All roads lead to ATF4. Dev Cell 4: 442–444, 2003. [DOI] [PubMed] [Google Scholar]
- 47.Scheuner D, Patel R, Wang F, Lee K, Kumar K, Wu J, Nilsson A, Karin M, Kaufman RJ. Double-stranded RNA-dependent protein kinase phosphorylation of the alpha-subunit of eukaryotic translation initiation factor 2 mediates apoptosis. J Biol Chem 281: 21458–21468, 2006. [DOI] [PubMed] [Google Scholar]
- 48.Sidransky H, Baba T. Chemical pathology of acute amino acid deficiencies. III. Morphologic and biochemical changes in young rats fed valine- or lysine-devoid diets. J Nutr 70: 463–483, 1960. [DOI] [PubMed] [Google Scholar]
- 49.Sidransky H, Farber E. Chemical pathology of acute amino acid deficiencies. I. Morphologic changes in immature rats fed threonine-, methionine-, or histidine-devoid diets. AMA Arch Pathol 66: 119–134, 1958. [PubMed] [Google Scholar]
- 50.Sidransky H, Farber E. Chemical pathology of acute amino acid deficiencies. II. Biochemical changes in rats fed threonine-or methionine-devoid diets. AMA Arch Pathol 66: 135–149, 1958. [PubMed] [Google Scholar]
- 51.Siess EA, Brocks DG, Wieland OH. Subcellular distribution of key metabolites in isolated liver cells from fasted rats. FEBS Lett 69: 265–271, 1976. [DOI] [PubMed] [Google Scholar]
- 52.Strydom DJ, Cohen SA. Comparison of amino acid analyses by phenylisothiocyanate and 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate precolumn derivatization. Anal Biochem 222: 19–28, 1994. [DOI] [PubMed] [Google Scholar]
- 53.van Milgen J Modeling biochemical aspects of energy metabolism in mammals. J Nutr 132: 3195–3202, 2002. [DOI] [PubMed] [Google Scholar]
- 54.Wek SA, Zhu S, Wek RC. The histidyl-tRNA synthetase-related sequence in the eIF-2 alpha protein kinase GCN2 interacts with tRNA and is required for activation in response to starvation for different amino acids. Mol Cell Biol 15: 4497–4506, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Williamson JR, Scholz R, Browning ET. Control mechanisms of gluconeogenesis and ketogenesis. II. Interactions between fatty acid oxidation and the citric acid cycle in perfused rat liver. J Biol Chem 244: 4617–4627, 1969. [PubMed] [Google Scholar]
- 56.Wong A, Cortopassi G. Reproducible quantitative PCR of mitochondrial and nuclear DNA copy number using the LightCycler. Methods Mol Biol 197: 129–137, 2002. [DOI] [PubMed] [Google Scholar]
- 57.Zhang P, McGrath BC, Reinert J, Olsen DS, Lei L, Gill S, Wek SA, Vattem KM, Wek RC, Kimball SR, Jefferson LS, Cavener DR. The GCN2 eIF2α kinase is required for adaptation to amino acid deprivation in mice. Mol Cell Biol 22: 6681–6688, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]