

Abstract
Many organisms activate adaptive transcriptional programs to help them cope with decreased oxygen levels, or hypoxia, in their environment. These responses are triggered by various oxygen sensing systems in bacteria, yeast and metazoans. In metazoans, the hypoxia inducible factors (HIFs) mediate the adaptive transcriptional response to hypoxia by upregulating genes involved in maintaining bioenergetic homeostasis. The HIFs in turn are regulated by HIF-specific prolyl hydroxlase activity, which is sensitive to cellular oxygen levels and other factors such as tricarboxylic acid cycle metabolites and reactive oxygen species (ROS). Establishing a role for ROS in cellular oxygen sensing has been challenging since ROS are intrinsically unstable and difficult to measure. However, recent advances in fluorescence energy transfer resonance (FRET)-based methods for measuring ROS are alleviating some of the previous difficulties associated with dyes and luminescent chemicals. In addition, new genetic models have demonstrated that functional mitochondrial electron transport and associated ROS production during hypoxia are required for HIF stabilization in mammalian cells. Current efforts are directed at how ROS mediate prolyl hydroxylase activity and hypoxic HIF stabilization. Progress in understanding this process has been enhanced by the development of the FRET-based ROS probe, an vivo prolyl hydroxylase reporter and various genetic models harboring mutations in components of the mitochondrial electron transport chain.
Keywords: reactive oxygen species, prolyl hydroxylase, mitochondria, cellular oxygen sensing, hypoxia, hypoxia inducible factor
Introduction
Organisms from bacteria to metazoans have evolved oxygen (O2) sensing systems whichactivate adaptive transcriptional programs during times of O2 deprivation or hypoxia. Transcription factors known as hypoxia inducible factors (HIFs) are the major effectors of the hypoxic transcriptional response in metazoans. HIF activity is not only affected by cellular O2 levels but also by reactive oxygen species (ROS) produced during hypoxia. This review will discuss technologies employed to measure ROS, how hypoxia affects cellular ROS levels, evidence supporting a role for ROS in cellular O2 sensing and potential mechanisms by which ROS regulate HIF activity.
Cellular Oxygen Sensing
Aerobic life depends on O2 to generate ATP. Since organisms often encounter decreased O2 levels, or “hypoxia” in their environment, they have evolved various O2-sensing systems that trigger adaptive responses to help them maintain cellular and systemic energy homeostasis. Several O2 sensors have been described in bacteria, most of which respond to the O2-dependent redox status of the cell. For example, decreased O2 levels in E. Coli results in activation of the fumarate and nitrate reduction (FNR) transcription factor, via effects on the bound iron-sulfur cluster, which activates an adaptive transcriptional program [1]. In parallel with the FNR system in E. Coli, the two-component Arc A/B system induces transcription of adaptive genes in response to the redox state of electron carriers during hypoxic stress [2,3]. During O2 deprivation in Rhizobia and Bradyrhizobia, O2 is released from a heme moiety bound to the histidine kinase FixL which subsequently undergoes autophosphorylation and activates the transcription factor FixJ [4,5].
Many O2 sensing mechanisms have also been described in eukaryotes. In yeast, decreased de novo synthesis of sterol and heme due to O2 limitation affects the activities of multiple transcription factors, such as Hap1 and Sre1 [6,7]. In metazoans, O2 sensing systems are more complicated. Unlike unicellular organisms, whose O2 sensors are most sensitive to anoxic conditions, metazoans have evolved a more graded response to O2 levels due to the heterogeneity of O2 concentrations throughout tissues. For instance, in mammals normal O2 tensions in the lung are around 16% while other tissues are approximately 2–5%. In addition, metazoans must also elicit systemic adaptations to hypoxia which require multiple and more complex O2 sensing mechanisms beyond simple metabolic adaptations.
In metazoans, the major hypoxic transcriptional response is mediated by hypoxia inducible factors (HIFs) [8,9]. Standard and conditional knock-out mouse models have demonstrated a role for HIFs in embryonic vasculogenesis, as well as adult erythropoesis and the inflammatory response [10–13]. HIF is a heterodimeric transcription factor complex consisting of a constitutively stabilized β subunit and a more labile α subunit [14]. HIF transcriptional activity dramatically increases during hypoxia and leads to enhanced expression of hundreds of genes involved in maintaining homeostasis at the cellular, tissue and systemic level [15]. HIF complex activity is mainly mediated by protein stability of the α subunit [14,16–18]. Under normoxic conditions (21% O2), HIFα is continuously degraded by the 26S proteasome which is dependent on the hydroxylation of two key proline residues within the oxygen-dependent-degradation domain (ODDD) [19–22]. This hydroxylation allows an E3 ubiquitin ligase complex containing the von Hippel-Lindau protein (pVHL) to bind HIFα, earmarking it for degradation by the proteasome [18]. Under hypoxic conditions, HIFα is no longer hydroxylated and therefore no longer binds pVHL, allowing it to escape recognition by the proteasome. It can then translocate to the nucleus, dimerize with the β subunit and activate the transcription of target genes such as phosphoglycerate kinase and vascular endothelial growth factor [19,21–23]. O2-sensitive hydroxylation of HIFα is regulated by HIF-specific prolyl hydroxylases (PHDs), dioxygenases that use O2 and 2-oxoglutarate as co-substrates to carry out the hydroxylation reaction. In addition, PHDs require iron in the ferrous state for their activity and ascorbate to recycle that iron when oxidized in uncoupled hydroxylation reactions (Figure 1) [19,23].
Figure 1. The hydroxylase reaction.
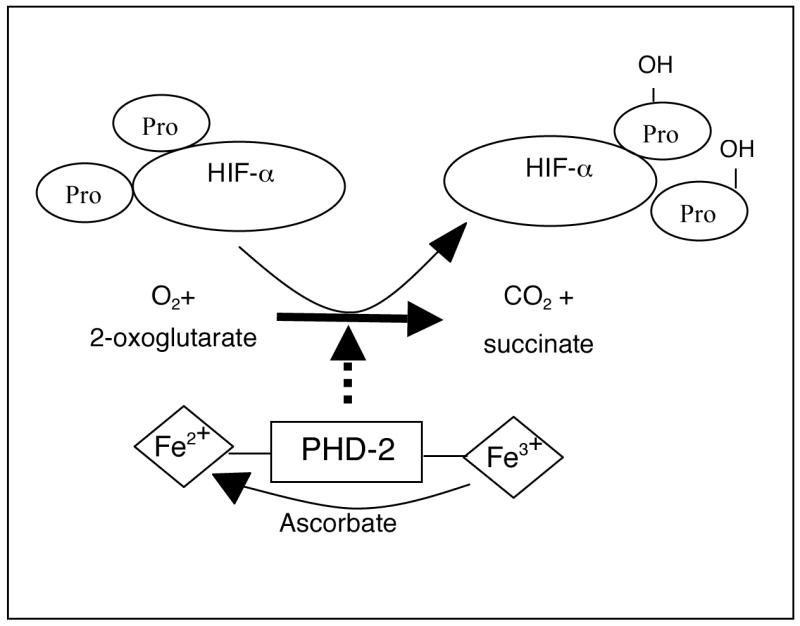
PHD2 utilizes cellular O2 and 2-oxoglutarate as co-substrates to hydroxylate two proline residues on HIFα. PHD2 also requires ferrous iron for its activity and ascorbate to recycle oxidized iron during uncoupled reactions.
Mammalian cells express three HIF-specific PHDs, PHD1, PHD2 and PHD3, though studies have shown PHD2 to be the most important regulator of HIF stabilization [24]. Since PHDs have a high Km for O2 in vitro, which presumably makes their activity sensitive to O2 tensions found in tissues, a simple regulatory model has been proposed whereby PHDs’ ability to hydroxylate HIFα, and thereby “sense O2,” is dictated by O2 availability. However, modes of regulation other than mere O2 availability have been reported. Some data suggest that byproducts of the tricarboxylic acid cycle such as succinate, malate and fumarate can inhibit PHD2 activity and stabilize HIFα when allowed to accumulate in cells or when added to in vitro hydroxylation reactions [25–27]. In addition, extensive data from multiple groups have suggested a role for mitochondrial reactive oxygen species (ROS) in regulating HIFα stabilization [28–31] The importance of ROS in cellular O2 sensing has been hotly debated and will be a focus of the latter part of this review. We will first focus on cellular sources of ROS, the methodology employed in measuring ROS and the relationship between ROS and mitochondria.
Types and Sources of Cellular ROS
Diatomic oxygen is a relatively non-reactive molecule that becomes highly reactive by gaining one electron to form superoxide anion (O2.−), two electrons to form hydrogen peroxide (H2O2), or three electrons to form hydroxyl radical (.OH). Cellular O2.−can dismutate into H2O2 spontaneously or by the enzymatic activites of cellular superoxide dismutates (SODs), the latter contributing to the majority of intracellular H2O2. Compared to O2.−, H2O2 is much more stable and can diffuse through biological membranes giving it the potential to act as a long-range signaling molecule [32–34]. Recent work has shown that ROS can serve as a regulated and specific second- messengers to propagate signals in multiple settings [35].
The main site of cellular ROS production is the mitochondrial electron transfer chain (ETC). Though it is commonly reported that 1–2% of electron flow on the ETC leads to ROS production, more recent reports show this value to be as low as 0.15%, depending on the carbon source being oxidized [36,37]. A high concentration of mitochondrial SOD ensures no significant O2.− accumulation beyond its initial site of production and allows for the generation of H2O2 which has second messenger effects in the cytosol [31,38–40]. The endoplasmic reticulum (ER) is another cellular source of ROS where resident cytochrome P-450 and b5 family members oxidize unsaturated fatty acids and xenobiotics to generate O2.− and H2O2 [41–43]. In addition, plasma membrane-associated oxidases such as NADPH oxidases generate ROS by oxidizing intracellular NADPH to reduce O2 into O2.− in order to achieve localized microbicidal function in phagosomes [44–47]. Peroxisomes contain multiple H2O2-generating enzymes including peroxisomal catalase which metabolizes toxic molecules such as ethanol. However, only a small percentage of peroxisomal H2O2 may escape this organelle [48]. Some soluble oxidases, dehydrogenases and dioxygenases in the cytoplasm can also generate ROS during catalytic cycling [49]. In summary, while many sources of intracellular ROS exist, the most significant contribution comes from the mitochondrion, which has also been shown to play the most important role in cellular O2 sensing.
Measurement of Cellular ROS
Compared to other biological metabolites, intracellular ROS are highly reactive and very short-lived, making their measurement a challenge. Currently there are no direct methods for measuring intracellular ROS levels. Some existing methods assay the downstream effects of ROS on cellular macromolecules such as lipid oxidation, protein carbonylation or the formation of 8-oxoguanine in DNA as a readout for ROS production [49,50]. More commonly used assays for assessing intracellular ROS levels involve loading cells with luminescent chemicals and fluorescent dyes which cross-react with different types of ROS to emit light. One example is lucigenin (Luc2+) which, after being reduced to LucH.+ within the cell, can react with O2.− to form an unstable product that chemiluminesces. The intensity of the light emitted is then taken as an indirect measure of the intracellular O2−. level [51]. For measuring H2O2, 2′,7′-dichlorfluorescein-diacetate (DCFH-DA) is the most extensively used chemical detector, given its simple and direct chemistry with ROS [52,53]. Esterized DCFH-DA enters cells via diffusion, becomes deacetylated and converted to DCFH. Nonfluorescent DCFH is subsequently oxidized by H2O2 to yield flurorescent DCF. Though more direct than cellular downstream readouts of cellular ROS levels, methodology employing fluorescent dyes also has drawbacks. For instance, DCFH has been shown to react with agents other than H2O2 making it more of a measure of total redox status rather than cellular H2O2 [54,55]. Moreover, both lucigenin and DCFH have been shown to yield a fluorescent signal in the absence of ROS, giving rise to the potential for high background and false positive readings [56–58]. In addition to these limitations, one must also assume that cellular uptake of DCFH-DA is equivalent under different conditions if a direct comparison is to be made. Finally, indirect effects of the experimental manipulation itself must also be considered. For instance, with the DCF fluorescence probe, an excitation violet-blue light is required, a wavelength which has been linked to cellular phototoxicity and H2O2 generation [59].
A recently developed fluorescence resonance energy transfer (FRET)-based assay holds potential to circumvent the limitations of chemical-based methods. In this assay, cyan and yellow-fluorescent proteins are bridged by part of a bacterial heat shock protein [29,60] which contains redox-sensitive cysteine thiols [61,62]. The thiol groups are reduced at normal conditions but upon oxidation, a disulfide bond is formed that changes the conformation of the hinge domain and separates the YFP and CFP. This decreases the energy transfer from CFP to YFP enhancing the CFP signal while decreasing the YFP signal during oxidation [63]. Such a probe can be overexpressed in cells and the ratio of CFP to YFP fluorescence taken as a measure of cellular ROS levels in real time, a readout independent of probe concentration. Though this technology eliminates concerns of equal probe distribution across samples and indirect effects from excessive experimental manipulation, it will be of interest to determine exactly which types of ROS affect the activity of this FRET probe.
ROS and Hypoxia
Though it is generally accepted that intracellular ROS levels change during hypoxia, in which direction this change occurs is still hotly debated. From an intuitive standpoint, one might assume that ROS levels drop during hypoxia since ROS require O2 as a substrate for their production and several studies support this prediction. For instance, it has been shown that endothelial cell plasma membranes release less extracellular H2O2 under hypoxia compared to normoxia [64,65]. In addition, perinuclear endoplasmic reticulum (ER) generation of OH, presumably derived from the “Fenton reaction” of H2O2 with transition metals, is higher under normoxic conditions compared to hypoxia [66]. Presumably, these studies are assessing the specific ROS-generating capacity of NADPH oxidase, the major ROS producer at the plasma membrane and in the ER. Other studies which assess total intracellular ROS content have made opposite observations. Using either DCFH and FRET technology discussed above, several groups have found an increase in total intracellular ROS upon exposure to hypoxia [29–31, 67]. In addition, it has been shown that protein cabonylation and formation of 8-hydroxy-2′-deoxyguanosine (8-OH-dG) in DNA both increase in yeast exposed to anoxia, an indirect indication that ROS increase during this stress [68]. In this same study, expression of SOD1 also increased, a further indirect confirmation that ROS increase during O2 limitation. In all, inconsistencies in reports on the direction and degree of hypoxic ROS production likely stem from differences in cell types and subcellular compartments examined as well as the inadequacy of current technology to accurately measure ROS levels. Establishing a reliable, quantitative and sensitive method for measuring ROS will be an important step in validating the role of ROS in the hypoxic response.
Role of ROS in Cellular O2 Sensing
The role of ROS in regulating the hypoxic response and HIF activity has remained controversial for over a decade. Early theories on the subject suggested that NADPH oxidase might be an important ROS-generating cellular O2 sensor since this multi-subunit membrane-bound enzyme is expressed in tissues implicated in systemic hypoxic responses and affects cellular redox status depending on cellular O2 concentrations [69,70]. However more in-depth studies did not support a requirement for NADPH in the hypoxic adaptive response [71]. Chandel et al later suggested that mitochondria are the source of ROS involved in the hypoxic response by showing that ρ° cells (which lack functional mitochondria) or treatment of cells with either inhibitors of the mitochondrial electron transport chain or antioxidants impede HIFα activation, though others failed to observe any effect of antioxidants on HIFα stabilization [31,72]. Experiments that utilized broader panels of specific inhibitors for each of the electron transport chain complex components later suggested that an important source of hypoxic ROS is complex III [39,73–75]. More recent studies further strengthen a role for mitochondrial ROS in hypoxic responses by utilizing genetic models showing that deficiencies in various mitochondrial electron transport chain components compromise hypoxic HIFα stabilization. These genetic models are improved systems for studying the effects of mitochondrial electron transport inhibition, as they elminate the effects of non-specificity encountered with chemical inhibitors. For example, genetic ablation of cytochrome c by targeted mutagenesis in murine embryonic cells or shRNA-mediated knockdown of the Rieske iron-sulfur protein of Complex III in HEK293 cells and 143B osteosarcoma cells attenuates hypoxic, but not anoxic, HIFα stabilization suggesting severe O2 limitation is sensed differently from more moderate hypoxia [29,30]. Interestingly, in the same studies, it was also shown that human fibroblasts with a deficiency in electron transport chain Complex IV, though defective in oxidative phosphorylation, maintained HIFα stabilization under hypoxia [28]. These results suggest that Complex III of the mitochondrial electron transport chain is the ROS-generating O2 sensor responsible for HIFα stabilization during hypoxia.
The idea that ROS might contribute to O2 sensing has been hotly contested, especially following the discovery of the HIF-specific prolyl hydoxylases, after which many assumed intracellular O2 must be the only determinant of hydroxylase and HIFα activity. In direct contrast to previous findings, some groups found that ρ° cell lines lacking functional mitochondria still stabilize HIFα under hypoxia [76,77]. Other studies suggested that cells with impaired mitochondrial function fail to stabilize HIFα under hypoxia, not because of defective ROS-generation, but because of intracellular O2 re-distribution which leads to increased availability of O2 as a substrate for the hydroxylation reaction [72,78]. Although these reports challenge a role for mitochondrial ROS in cellular O2 sensing, they do not explain why antioxidants, including the recently described mitochondrially-targeted compound MitoQ, block hypoxic HIFα activity [79]. In addition, they do not explain why defects which specifically compromise Complex III function prevent hypoxic HIFα activity, while Complex IV defects, that impair cellular respiration and presumably O2 distribution, leave the hypoxic response intact [28]. Complete validation of the importance of mitochondrial ROS most likely awaits the elucidation of the mechanisms by which ROS regulate HIFα stability during hypoxia.
Potential mechanisms for ROS in cellular O2 sensing
There are many possible mechanisms by which mitochondrial ROS regulate HIFα stability during hypoxia. These most likely involve the PHDs, since recent work employing a cellular prolyl hydroxylase reporter, which utilizes hydroxylation-specific mobility shifts on SDS-PAGE gels to monitor the hydroxylation of HIFα in vivo, showed that mitochondrial inhibitors and the anti-oxidant Mito-Q affect cellular hydroxylase activity [26]. One report has shown that elevated ROS levels due to genetic ablation of the transcription factor JunD and consequent downregulation of its anti-oxidant target genes, leads to PHD2 inactivation via oxidation of ferrous iron in the catalytic domain as determined by electron paramagnetic resonance spectroscopy [80]. However, it remains to be determined whether physiological levels of ROS produced during hypoxia can have the same effect on PHD2. Another plausible mechanism is that ROS alter the disulfide bond structure of PHD2 to inhibit its activity, a mode of regulation that has been well described for protein tyrosine phosphatases [66,81]. The recent discovery of the crystal structure of PHD2 may be an important step in assessing this possibility [82]. A final plausible mechanism by which ROS might affect the hypoxic response is the initiation of a signal transduction cascade resulting in inhibitory post-translational modifications on PHD2 (Figure 2). In support of this mechanism, recent work has shown that the p38 stress-activated MAPK cascade is involved in ROS-dependent hypoxic responses as mouse embryonic fibroblasts lacking either p38 MAPK or its upstream effectors MKK3/6 fail to stabilize HIFα under hypoxia and oxidant stress [83].
Figure 2. Plausible mechanisms by which ROS regulate PHD2 activity.
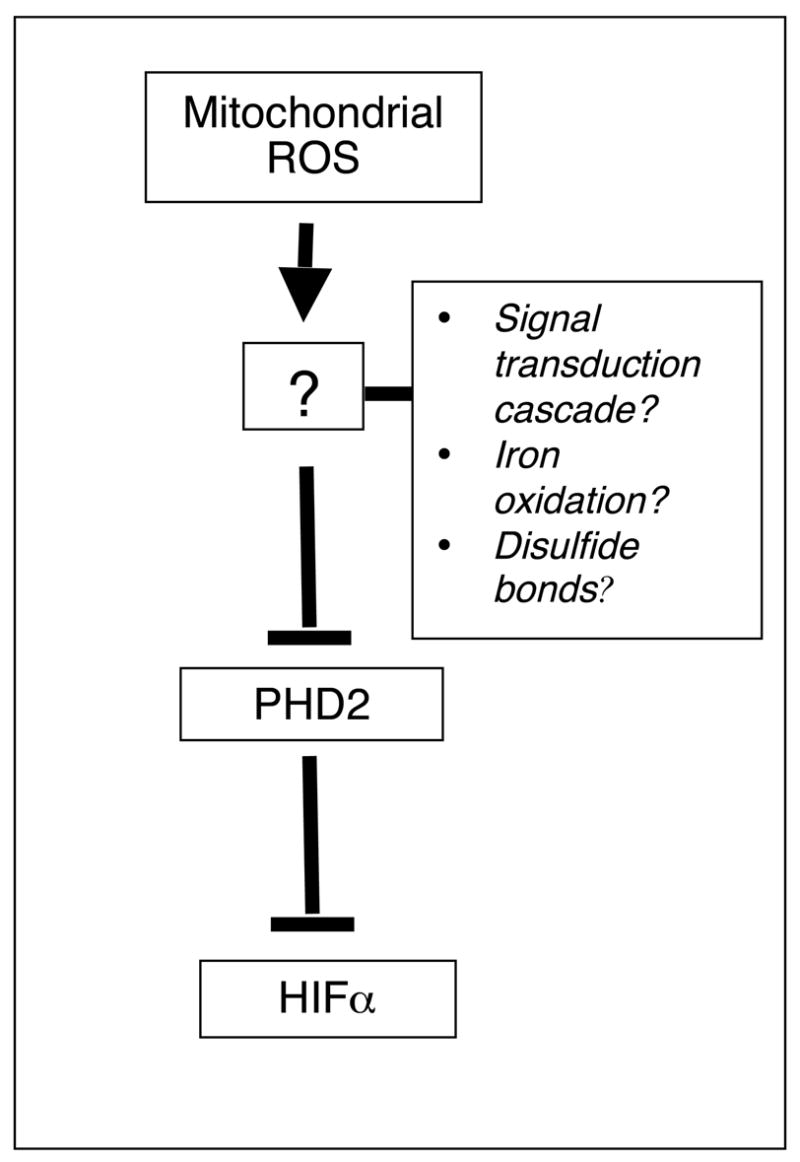
ROS may trigger a signal transduction cascade which results in post-translational modifications on PHD2, may oxidize bound iron, or may alter PHD2 disulfide bond structure to regulate PHD2 activity.
Summary
While there is no doubt that O2 substrate limitation can regulate PHD2 activity, it seems unlikely that this is the only determinant of HIFα stimulation in the complex environment of the cell. There is much evidence to suggest that other cellular factors, especially mitochondrial ROS, play a significant role in the hypoxic response. The development of reagents such as cellular genetic models like the cytochrome c null cells, the ROS-sensitive FRET probe and the in vivo hydroxylase reporter will most likely aid in uncovering the precise mechanism by which ROS participates in cellular O2 sensing.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kiley PJ, Beinert H. The role of Fe-S proteins in sensing and regulation in bacteria. Curr Opin Microbiol. 2003;6:181–185. doi: 10.1016/s1369-5274(03)00039-0. [DOI] [PubMed] [Google Scholar]
- 2.Unden G, Schirawski J. The oxygen-responsive transcriptional regulator FNR of Escherichia coli: the search for signals and reactions. Mol Microbiol. 1997;25:205–210. doi: 10.1046/j.1365-2958.1997.4731841.x. [DOI] [PubMed] [Google Scholar]
- 3.Alexeeva S, Hellingwerf KJ, Teixeira de Mattos MJ. Requirement of ArcA for redox regulation in Escherichia coli under microaerobic but not anaerobic or aerobic conditions. J Bacteriol. 2003;185:204–209. doi: 10.1128/JB.185.1.204-209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fischer HM. Genetic regulation of nitrogen fixation in rhizobia. Microbiol Rev. 1994;58:352–386. doi: 10.1128/mr.58.3.352-386.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gong W, Hao B, Chan MK. New mechanistic insights from structural studies of the oxygen-sensing domain of Bradyrhizobium japonicum FixL. Biochemistry. 2000;39:3955–3962. doi: 10.1021/bi992346w. [DOI] [PubMed] [Google Scholar]
- 6.Hon T, Dodd A, Dirmeier R, Gorman N, Sinclair PR, Zhang L, Poyton RO. A mechanism of oxygen sensing in yeast. Multiple oxygen-responsive steps in the heme biosynthetic pathway affect Hap1 activity. J Biol Chem. 2003;278:50771–50780. doi: 10.1074/jbc.M303677200. [DOI] [PubMed] [Google Scholar]
- 7.Hughes AL, Todd BL, Espenshade PJ. SREBP pathway responds to sterols and functions as an oxygen sensor in fission yeast. Cell. 2005;120:831–842. doi: 10.1016/j.cell.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 8.Kaelin WG., Jr The von Hippel-Lindau protein, HIF hydroxylation, and oxygen sensing. Biochem Biophys Res Commun. 2005;338:627–638. doi: 10.1016/j.bbrc.2005.08.165. [DOI] [PubMed] [Google Scholar]
- 9.Wang GL, Semenza GL. General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc Natl Acad Sci U S A. 1993;90:4304–4308. doi: 10.1073/pnas.90.9.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ryan HE, Lo J, Johnson RS. HIF-1 alpha is required for solid tumor formation and embryonic vascularization. Embo J. 1998;17:3005–3015. doi: 10.1093/emboj/17.11.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maltepe E, Schmidt JV, Baunoch D, Bradfield CA, Simon MC. Abnormal angiogenesis and responses to glucose and oxygen deprivation in mice lacking the protein ARNT. Nature. 1997;386:403–407. doi: 10.1038/386403a0. [DOI] [PubMed] [Google Scholar]
- 12.Gruber M, Hu CJ, Johnson RS, Brown EJ, Keith B, Simon MC. Acute postnatal ablation of Hif-2alpha results in anemia. Proc Natl Acad Sci U S A. 2007;104:2301–2306. doi: 10.1073/pnas.0608382104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–657. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med. 2003;9:677–684. doi: 10.1038/nm0603-677. [DOI] [PubMed] [Google Scholar]
- 16.Huang LE, Gu J, Schau M, Bunn HF. Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci U S A. 1998;95:7987–7992. doi: 10.1073/pnas.95.14.7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang LE, Arany Z, Livingston DM, Bunn HF. Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its alpha subunit. J Biol Chem. 1996;271:32253–32259. doi: 10.1074/jbc.271.50.32253. [DOI] [PubMed] [Google Scholar]
- 18.Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE, Pavletich N, Chau V, Kaelin WG. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol. 2000;2:423–427. doi: 10.1038/35017054. [DOI] [PubMed] [Google Scholar]
- 19.Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- 20.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 21.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 22.Yu F, White SB, Zhao Q, Lee FS. HIF-1alpha binding to VHL is regulated by stimulus-sensitive proline hydroxylation. Proc Natl Acad Sci U S A. 2001;98:9630–9635. doi: 10.1073/pnas.181341498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294:1337–1340. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- 24.Berra E, Benizri E, Ginouves A, Volmat V, Roux D, Pouyssegur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. Embo J. 2003;22:4082–4090. doi: 10.1093/emboj/cdg392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, Gottlieb E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7:77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 26.Pan Y, Mansfield KD, Bertozzi CC, Rudenko V, Chan DA, Giaccia AJ, Simon MC. Multiple factors affecting cellular redox status and energy metabolism modulate hypoxia-inducible factor prolyl hydroxylase activity in vivo and in vitro. Mol Cell Biol. 2007;27:912–925. doi: 10.1128/MCB.01223-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Isaacs JS, Jung YJ, Mole DR, Lee S, Torres-Cabala C, Chung YL, Merino M, Trepel J, Zbar B, Toro J, et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell. 2005;8:143–153. doi: 10.1016/j.ccr.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 28.Brunelle JK, Bell EL, Quesada NM, Vercauteren K, Tiranti V, Zeviani M, Scarpulla RC, Chandel NS. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005;1:409–414. doi: 10.1016/j.cmet.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 29.Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, Simon MC, Hammerling U, Schumacker PT. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005;1:401–408. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 30.Mansfield KD, Guzy RD, Pan Y, Young RM, Cash TP, Schumacker PT, Simon MC. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-alpha activation. Cell Metab. 2005;1:393–399. doi: 10.1016/j.cmet.2005.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A. 1998;95:11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev. 1979;59:527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- 33.Halliwell B, Gutteridge JM, Cross CE. Free radicals, antioxidants, and human disease: where are we now? J Lab Clin Med. 1992;119:598–620. [PubMed] [Google Scholar]
- 34.Cross CE, Halliwell B, Borish ET, Pryor WA, Ames BN, Saul RL, McCord JM, Harman D. Oxygen radicals and human disease. Ann Intern Med. 1987;107:526–545. doi: 10.7326/0003-4819-107-4-526. [DOI] [PubMed] [Google Scholar]
- 35.Rhee SG. Cell signaling. H2O2, a necessary evil for cell signaling. Science. 2006;312:1882–1883. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- 36.Freeman BA, Topolosky MK, Crapo JD. Hyperoxia increases oxygen radical production in rat lung homogenates. Arch Biochem Biophys. 1982;216:477–484. doi: 10.1016/0003-9861(82)90236-3. [DOI] [PubMed] [Google Scholar]
- 37.St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem. 2002;277:44784–44790. doi: 10.1074/jbc.M207217200. [DOI] [PubMed] [Google Scholar]
- 38.Lakshminarayanan V, Beno DW, Costa RH, Roebuck KA. Differential regulation of interleukin-8 and intercellular adhesion molecule-1 by H2O2 and tumor necrosis factor-alpha in endothelial and epithelial cells. J Biol Chem. 1997;272:32910–32918. doi: 10.1074/jbc.272.52.32910. [DOI] [PubMed] [Google Scholar]
- 39.Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, Schumacker PT. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem. 2000;275:25130–25138. doi: 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- 40.Banki K, Hutter E, Gonchoroff NJ, Perl A. Elevation of mitochondrial transmembrane potential and reactive oxygen intermediate levels are early events and occur independently from activation of caspases in Fas signaling. J Immunol. 1999;162:1466–1479. [PMC free article] [PubMed] [Google Scholar]
- 41.Aust SD, Roerig DL, Pederson TC. Evidence for superoxide generation by NADPH-cytochrome c reductase of rat liver microsomes. Biochem Biophys Res Commun. 1972;47:1133–1137. doi: 10.1016/0006-291x(72)90952-7. [DOI] [PubMed] [Google Scholar]
- 42.Capdevila J, Chacos N, Werringloer J, Prough RA, Estabrook RW. Liver microsomal cytochrome P-450 and the oxidative metabolism of arachidonic acid. Proc Natl Acad Sci U S A. 1981;78:5362–5366. doi: 10.1073/pnas.78.9.5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Capdevila J, Parkhill L, Chacos N, Okita R, Masters BS, Estabrook RW. The oxidative metabolism of arachidonic acid by purified cytochromes P-450. Biochem Biophys Res Commun. 1981;101:1357–1363. doi: 10.1016/0006-291x(81)91597-7. [DOI] [PubMed] [Google Scholar]
- 44.Babior BM. NADPH oxidase: an update. Blood. 1999;93:1464–1476. [PubMed] [Google Scholar]
- 45.Jones RD, Thompson JS, Morice AH. The NADPH oxidase inhibitors iodonium diphenyl and cadmium sulphate inhibit hypoxic pulmonary vasoconstriction in isolated rat pulmonary arteries. Physiol Res. 2000;49:587–596. [PubMed] [Google Scholar]
- 46.Jones RD, Morice AH. Hydrogen peroxide--an intracellular signal in the pulmonary circulation: involvement in hypoxic pulmonary vasoconstriction. Pharmacol Ther. 2000;88:153–161. doi: 10.1016/s0163-7258(00)00089-9. [DOI] [PubMed] [Google Scholar]
- 47.Segal AW, Shatwell KP. The NADPH oxidase of phagocytic leukocytes. Ann N Y Acad Sci. 1997;832:215–222. doi: 10.1111/j.1749-6632.1997.tb46249.x. [DOI] [PubMed] [Google Scholar]
- 48.Boveris A, Oshino N, Chance B. The cellular production of hydrogen peroxide. Biochem J. 1972;128:617–630. doi: 10.1042/bj1280617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Freeman BA, Crapo JD. Biology of disease: free radicals and tissue injury. Lab Invest. 1982;47:412–426. [PubMed] [Google Scholar]
- 50.Pryor WA, Godber SS. Noninvasive measures of oxidative stress status in humans. Free Radic Biol Med. 1991;10:177–184. doi: 10.1016/0891-5849(91)90073-c. [DOI] [PubMed] [Google Scholar]
- 51.Faulkner K, Fridovich I. Luminol and lucigenin as detectors for O2. Free Radic Biol Med. 1993;15:447–451. doi: 10.1016/0891-5849(93)90044-u. [DOI] [PubMed] [Google Scholar]
- 52.Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- 53.Frank J, Pompella A, Biesalski HK. Histochemical visualization of oxidant stress. Free Radic Biol Med. 2000;29:1096–1105. doi: 10.1016/s0891-5849(00)00395-6. [DOI] [PubMed] [Google Scholar]
- 54.Ischiropoulos H, Gow A, Thom SR, Kooy NW, Royall JA, Crow JP. Detection of reactive nitrogen species using 2,7-dichlorodihydrofluorescein and dihydrorhodamine 123. Methods Enzymol. 1999;301:367–373. doi: 10.1016/s0076-6879(99)01100-3. [DOI] [PubMed] [Google Scholar]
- 55.Royall JA, Ischiropoulos H. Evaluation of 2′,7′-dichlorofluorescin and dihydrorhodamine 123 as fluorescent probes for intracellular H2O2 in cultured endothelial cells. Arch Biochem Biophys. 1993;302:348–355. doi: 10.1006/abbi.1993.1222. [DOI] [PubMed] [Google Scholar]
- 56.Rota C, Chignell CF, Mason RP. Evidence for free radical formation during the oxidation of 2′-7′-dichlorofluorescin to the fluorescent dye 2′-7′-dichlorofluorescein by horseradish peroxidase: possible implications for oxidative stress measurements. Free Radic Biol Med. 1999;27:873–881. doi: 10.1016/s0891-5849(99)00137-9. [DOI] [PubMed] [Google Scholar]
- 57.Rota C, Fann YC, Mason RP. Phenoxyl free radical formation during the oxidation of the fluorescent dye 2′,7′-dichlorofluorescein by horseradish peroxidase. Possible consequences for oxidative stress measurements. J Biol Chem. 1999;274:28161–28168. doi: 10.1074/jbc.274.40.28161. [DOI] [PubMed] [Google Scholar]
- 58.Liochev SI, Fridovich I. Lucigenin (bis-N-methylacridinium) as a mediator of superoxide anion production. Arch Biochem Biophys. 1997;337:115–120. doi: 10.1006/abbi.1997.9766. [DOI] [PubMed] [Google Scholar]
- 59.Hockberger PE, Skimina TA, Centonze VE, Lavin C, Chu S, Dadras S, Reddy JK, White JG. Activation of flavin-containing oxidases underlies light-induced production of H2O2 in mammalian cells. Proc Natl Acad Sci U S A. 1999;96:6255–6260. doi: 10.1073/pnas.96.11.6255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Waypa GB, Guzy R, Mungai PT, Mack MM, Marks JD, Roe MW, Schumacker PT. Increases in mitochondrial reactive oxygen species trigger hypoxia-induced calcium responses in pulmonary artery smooth muscle cells. Circ Res. 2006;99:970–978. doi: 10.1161/01.RES.0000247068.75808.3f. [DOI] [PubMed] [Google Scholar]
- 61.Jakob U, Muse W, Eser M, Bardwell JC. Chaperone activity with a redox switch. Cell. 1999;96:341–352. doi: 10.1016/s0092-8674(00)80547-4. [DOI] [PubMed] [Google Scholar]
- 62.Graf PC, Martinez-Yamout M, VanHaerents S, Lilie H, Dyson HJ, Jakob U. Activation of the redox-regulated chaperone Hsp33 by domain unfolding. J Biol Chem. 2004;279:20529–20538. doi: 10.1074/jbc.M401764200. [DOI] [PubMed] [Google Scholar]
- 63.Guzy RD, Schumacker PT. Oxygen sensing by mitochondria at complex III: the paradox of increased reactive oxygen species during hypoxia. Exp Physiol. 2006;91:807–819. doi: 10.1113/expphysiol.2006.033506. [DOI] [PubMed] [Google Scholar]
- 64.Lopez-Barneo J, Pardal R, Ortega-Saenz P. Cellular mechanism of oxygen sensing. Annu Rev Physiol. 2001;63:259–287. doi: 10.1146/annurev.physiol.63.1.259. [DOI] [PubMed] [Google Scholar]
- 65.Zulueta JJ, Yu FS, Hertig IA, Thannickal VJ, Hassoun PM. Release of hydrogen peroxide in response to hypoxia-reoxygenation: role of an NAD(P)H oxidase-like enzyme in endothelial cell plasma membrane. Am J Respir Cell Mol Biol. 1995;12:41–49. doi: 10.1165/ajrcmb.12.1.7529030. [DOI] [PubMed] [Google Scholar]
- 66.Liu Q, Berchner-Pfannschmidt U, Moller U, Brecht M, Wotzlaw C, Acker H, Jungermann K, Kietzmann T. A Fenton reaction at the endoplasmic reticulum is involved in the redox control of hypoxia-inducible gene expression. Proc Natl Acad Sci U S A. 2004;101:4302–4307. doi: 10.1073/pnas.0400265101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mansfield KD, Simon MC, Keith B. Hypoxic reduction in cellular glutathione levels requires mitochondrial reactive oxygen species. J Appl Physiol. 2004;97:1358–1366. doi: 10.1152/japplphysiol.00449.2004. [DOI] [PubMed] [Google Scholar]
- 68.Dirmeier R, O’Brien KM, Engle M, Dodd A, Spears E, Poyton RO. Exposure of yeast cells to anoxia induces transient oxidative stress. Implications for the induction of hypoxic genes. J Biol Chem. 2002;277:34773–34784. doi: 10.1074/jbc.M203902200. [DOI] [PubMed] [Google Scholar]
- 69.Kummer W, Acker H. Immunohistochemical demonstration of four subunits of neutrophil NAD(P)H oxidase in type I cells of carotid body. J Appl Physiol. 1995;78:1904–1909. doi: 10.1152/jappl.1995.78.5.1904. [DOI] [PubMed] [Google Scholar]
- 70.Marshall C, Mamary AJ, Verhoeven AJ, Marshall BE. Pulmonary artery NADPH-oxidase is activated in hypoxic pulmonary vasoconstriction. Am J Respir Cell Mol Biol. 1996;15:633–644. doi: 10.1165/ajrcmb.15.5.8918370. [DOI] [PubMed] [Google Scholar]
- 71.Wenger RH, Marti HH, Schuerer-Maly CC, Kvietikova I, Bauer C, Gassmann M, Maly FE. Hypoxic induction of gene expression in chronic granulomatous disease-derived B-cell lines: oxygen sensing is independent of the cytochrome b558-containing nicotinamide adenine dinucleotide phosphate oxidase. Blood. 1996;87:756–761. [PubMed] [Google Scholar]
- 72.Hagen T, Taylor CT, Lam F, Moncada S. Redistribution of intracellular oxygen in hypoxia by nitric oxide: effect on HIF1alpha. Science. 2003;302:1975–1978. doi: 10.1126/science.1088805. [DOI] [PubMed] [Google Scholar]
- 73.Duranteau J, Chandel NS, Kulisz A, Shao Z, Schumacker PT. Intracellular signaling by reactive oxygen species during hypoxia in cardiomyocytes. J Biol Chem. 1998;273:11619–11624. doi: 10.1074/jbc.273.19.11619. [DOI] [PubMed] [Google Scholar]
- 74.Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem. 2003;278:36027–36031. doi: 10.1074/jbc.M304854200. [DOI] [PubMed] [Google Scholar]
- 75.Schroedl C, McClintock DS, Budinger GR, Chandel NS. Hypoxic but not anoxic stabilization of HIF-1alpha requires mitochondrial reactive oxygen species. Am J Physiol Lung Cell Mol Physiol. 2002;283:L922–931. doi: 10.1152/ajplung.00014.2002. [DOI] [PubMed] [Google Scholar]
- 76.Doege K, Heine S, Jensen I, Jelkmann W, Metzen E. Inhibition of mitochondrial respiration elevates oxygen concentration but leaves regulation of hypoxia-inducible factor (HIF) intact. Blood. 2005;106:2311–2317. doi: 10.1182/blood-2005-03-1138. [DOI] [PubMed] [Google Scholar]
- 77.Srinivas V, Leshchinsky I, Sang N, King MP, Minchenko A, Caro J. Oxygen sensing and HIF-1 activation does not require an active mitochondrial respiratory chain electron-transfer pathway. J Biol Chem. 2001;276:21995–21998. doi: 10.1074/jbc.C100177200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Metzen E, Zhou J, Jelkmann W, Fandrey J, Brune B. Nitric oxide impairs normoxic degradation of HIF-1alpha by inhibition of prolyl hydroxylases. Mol Biol Cell. 2003;14:3470–3481. doi: 10.1091/mbc.E02-12-0791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sanjuan-Pla A, Cervera AM, Apostolova N, Garcia-Bou R, Victor VM, Murphy MP, McCreath KJ. A targeted antioxidant reveals the importance of mitochondrial reactive oxygen species in the hypoxic signaling of HIF-1alpha. FEBS Lett. 2005;579:2669–2674. doi: 10.1016/j.febslet.2005.03.088. [DOI] [PubMed] [Google Scholar]
- 80.Gerald D, Berra E, Frapart YM, Chan DA, Giaccia AJ, Mansuy D, Pouyssegur J, Yaniv M, Mechta-Grigoriou F. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell. 2004;118:781–794. doi: 10.1016/j.cell.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 81.Salmeen A, Barford D. Functions and mechanisms of redox regulation of cysteine-based phosphatases. Antioxid Redox Signal. 2005;7:560–577. doi: 10.1089/ars.2005.7.560. [DOI] [PubMed] [Google Scholar]
- 82.McDonough MA, Li V, Flashman E, Chowdhury R, Mohr C, Lienard BM, Zondlo J, Oldham NJ, Clifton IJ, Lewis J, et al. Cellular oxygen sensing: Crystal structure of hypoxia-inducible factor prolyl hydroxylase (PHD2) Proc Natl Acad Sci U S A. 2006;103:9814–9819. doi: 10.1073/pnas.0601283103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Emerling BM, Platanias LC, Black E, Nebreda AR, Davis RJ, Chandel NS. Mitochondrial reactive oxygen species activation of p38 mitogen-activated protein kinase is required for hypoxia signaling. Mol Cell Biol. 2005;25:4853–4862. doi: 10.1128/MCB.25.12.4853-4862.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]