

Abstract
Using immunohistochemistry and mass spectrometry differentiation-dependent changes in the subcellular distribution and processing of aquaporin-0 (AQP0) have been mapped in the rat lens. Sections labelled with C-terminal tail AQP0 antibodies yielded two concentric rings of labelling with minimal signal in the lens core. The rings were separated by a transient zone of decreased labelling located prior to the transition of differentiating fiber (DF) cells into mature denucleated fiber (MF) cells. Mass spectrometry showed the loss of core labelling was due to AQP0 cleavage, while the transient loss of labelling was more likely caused by masking of the antibody epitope. AQP0 subcellular distribution changed with radial distance into the lens. In peripheral DF cells, AQP0 was found throughout both broad and narrow side membranes. In deeper lying DF cells, AQP0 aggregated into plaque-like structures located on the broad sides. This shift occurred prior to the transient loss of AQP0 signal, and coincided with formation of broad side membrane invaginations between adjacent fiber cells to which filensin, a known binding partner of AQP0, were also localized to. After nuclei loss, AQP0 was once again distributed throughout MF cell membranes. In the absence of protein synthesis the observed subcellular redistribution of AQP0 in DF and subsequent cleavage of AQP0 in MF is suggestive of a switch in the function of AQP0 from a water channel to a junctional protein.
Keywords: AQP0, membrane junctions, fiber cells, lens, confocal microscopy
INTRODUCTION
The ocular lens has been used extensively as a model system to study the general mechanisms involved in development, differentiation and growth (Lovicu and Robinson, 2004). Lens transparency is closely linked to the unique structure and function of its fiber cells. These highly differentiated cells are derived from equatorial epithelial cells, which exit the cell cycle and embark upon a differentiation process that produces extensive cellular elongation (Piatigorsky, 1981), loss of cellular organelles and nuclei (Bassnett, 2002), and expression of fiber-specific proteins (Wistow and Piatigorsky, 1988). Because this process continues throughout life, a gradient of fiber cells at different stages of differentiation is established around an internalized core of mature, anucleate fiber cells. To maintain its structural organisation the lens operates an internal microcirculation system that delivers nutrients to, and removes waste products from these internalized fiber cells (Mathias et al., 1997). This system is thought to be generated by spatial differences in ion transport processes that operate in peripheral and deeper regions of the lens. Since an inherent consequence of fiber cell differentiation is nuclear degradation and the removal of protein synthesis machinery, this raises the question of how, in the absence of de novo protein synthesis, the necessary spatial differences in membrane protein function are established as fiber cells differentiate.
In the absence of protein synthesis, changes to spatial location and function of membrane proteins in the inner cortex and core of the lens can only occur via the redistribution of, and/or post-translational modification to, existing proteins. Previously, we used immunohistochemistry and confocal microscopy to map the differentiation dependent changes in the distribution of key membrane proteins in the lens (Donaldson & Lim 2008). Using this approach we have shown that the subcellular distribution of the putative adhesion proteins MP20 and its binding partner galectin-3 undergo a sudden shift from a predominately cytoplasmic to a membranous location that coincided with the loss of cell nuclei and the formation of a barrier to extracellular space diffusion (Grey et al., 2003; Jacobs et al., 2004). Additionally, via the use of specific antibodies directed against the cytoplasmic loop or tail of the gap junction protein Cx46, we showed that Cx46 is abruptly cleaved at two distinct stages during fiber cell differentiation (Jacobs et al., 2004). This processing of Cx46 correlated with the dispersion of the broad side gap junction plaques that directed fluxes within peripheral fiber columns towards the lens equator, to a more uniform subcellular distribution which supported more isotropic fluxes in mature fiber cells (Jacobs et al., 2004). In the present study we have adopted similar methodological approaches to map the differentiation dependent changes in the processing and subcellular distribution of Aquaporin-0 (AQP0), another protein that is known to undergo extensive post translational modification in different regions of the lens.
AQP0, originally known as major intrinsic protein 26 (MIP26), is the most abundant lens membrane protein, in some species constituting over 60% of total fiber cell membrane protein (Broekhuyse et al., 1979; Broekhuyse et al., 1976; Bloemendal, 1982; Fitzgerald et al., 1983). It is interesting to note that AQP0 is known to undergo extensive post-translational modification (Ball et al., 2004; Schey et al., 1997; Schey et al., 1999), and has long been thought to play dual functional roles in the lens. AQP0 is mandatory for normal lens function, as lenses either lacking AQP0 (Shiels et al., 2001), or expressing a mutant form of AQP0 display distinct cataract phenotypes (Shiels and Bassnett, 1996; Shiels et al., 1999). When expressed in Xenopus oocytes, AQP0, like other members of the aquaporin family, increased membrane permeability for water, but relative to other aquaporins, AQP0 is a poor water channel (Chandy et al., 1997; Kushmerick et al., 1995; Mulders et al., 1995). However, its water permeability can be modulated by changes in pH and calcium concentration (Németh-Cahalan et al., 2004; Varadaraj et al., 2005). While it is now known that AQP0 functions as a water channel, an extensive literature exists that suggests AQP0(MIP) also plays a role in cell adhesion (Bok et al., 1982; Michea et al., 1995; Sas et al., 1985). More recently, high-resolution structural studies indicate that AQP0 exists as two separate structures with different functions (Gonen et al., 2004b; Harries et al., 2004; Palanivelu et al., 2006). Post-translational modifications are thought to play a role in modulation of the dual functionality of AQP0. Gonen et al showed that truncated AQP0 isolated from the lens core formed junctional structures in which the central water pore was closed (Gonen et al., 2004a), and post-translational phosphorylation of specific residues in the AQP0 tail region affect binding of calmodulin (Rose et al., 2008), which is thought to mediate the response of AQP0 water permeability to changes in Ca2+ concentration (Németh-Cahalan and Hall, 2000; Varadaraj et al., 2005). Although the functional consequences of other post-translational modifications to the AQP0 C terminus, such as glycation and deamidation, are not well understood, they suggest that this region of the protein is important for its regulation, and that regulation would be lost upon cleavage of the C terminus in the core of the lens.
To place these well characterised changes in AQP0 structure, post-translational processing and function into the context of the continuum that is lens fiber cell differentiation we have mapped the distribution of AQP0 throughout the lens using our immunohistochemical imaging approaches developed to map the distributions of other membrane proteins (Donaldson et al., 2004). Using an AQP0 antibody directed against its cytoplasmic tail we have identified areas in the lens where AQP0 is cleaved, and the subcellular locations associated with a potential shift in AQP0 from a non-junctional to a junctional configuration. Furthermore, to verify our immunohistochemical results we have used mass spectrometry to assess the extent of AQP0 cleavage in different regions of the lens. Our results show that in the core of the lens the regulatory C-terminal tail of AQP0 is abruptly removed, while in the outer cortex the homogenous distribution of AQP0 around the fiber cell membrane undergoes an extensive rearrangement that results in AQP0 aggregation on the broad sides of differentiating fiber cells. However, this aggregation of AQP0 is a transient phenomenon, since upon nuclear degradation a homogenous AQP0 labelling of the membranes of mature fiber cells returns. These results show that changes in the subcellular distribution and processing of AQP0 occur at discrete stages during fiber cell differentiation and suggest that these stages of differentiation are associated with a switch in the mode of operation of this multifunctional protein.
METHODS
Reagents
Anti-AQP0 C-terminal antibodies were obtained from Alpha Diagnostic International (ADI) (San Antonio, TX) and have been used previously by others (Shiels and Bassnett, 1996). An alternative anti-AQP0 C-terminal antibody (designated IIIp11) was kindly provided by Dr. Nalin Kumar (University of Illinois at Chicago, IL). Anti-filensin antibodies were a gift from Dr Roy Quinlan (University of Durham, Durham, UK). Anti-MP20 antibodies were gifted by Dr Charles Louis (University of Minnesota, St. Paul, MN). Goat anti-rabbit Alexa Fluor 488 secondary antibody and Texas red-dextran were obtained from Molecular Probes (Eugene, OR). TRITC-conjugated wheat germ agglutinin (WGA) was obtained from Sigma (Sigma Chemical Company, Australia). Phosphate buffered saline (PBS) was prepared fresh from tablets (Sigma Chemical Company, Australia). Unless otherwise stated all other chemicals were from Sigma.
Immunohistochemistry
Lenses were removed from 21 day-old Wistar rats immediately following death, fixed with 0.75% paraformaldehyde at room temperature (r.t.) for 24 hours, and prepared for cryosectioning using published protocols (Jacobs et al., 2003). For sectioning, whole lenses were mounted on, and encased in, optimal cutting temperature compound (Tissue-Tek; Sakura Finetek, Zoeterwoude, The Netherlands). Lenses were cryosectioned at −18°C using a cryostat (CM3050, Leica, Germany). Equatorial (10 μm) or axial (16 μm) sections were transferred onto microscope slides and incubated in blocking solution (3% bovine serum albumin, 3% fetal calf serum in PBS) for 1 hour (r.t.). After washing in PBS, sections were incubated in primary antibody in blocking solution (1:100) for 2 hours (r.t.). Slides were washed in PBS and incubated for 1.5 hours, in the dark (r.t.), with fluorescent secondary antibodies in blocking solution (1:200). Where necessary, sections were then incubated with WGA-TRITC in PBS (1:100) for 1 hour (r.t.) to label cell membranes. Alternatively, cell nuclei were stained with 100 μmol/l propidium iodide for 5 minutes (r.t.). Images of each fluorophore-staining pattern were acquired using a laser scanning confocal microscope (Leica TCS 4D or Leica TCS SP2, Heidelberg, Germany). Generally, a series of adjacent image stacks were collected for each protein target, allowing production of high-resolution image montages over large distances in the lens. For presentation, labelling patterns were pseudo-colored and combined using Adobe Photoshop software (Adobe Systems Inc., San Jose, CA).
En Face Preparations
Following fixation as above, lenses were washed three times for 10 minutes in PBS, then cut in two along the equator. Fixed lens hemispheres were further cut into quarters, yielding eight ‘en face’ preparations, allowing visualisation of broad sides of lens fiber cells in the x-y plane. Layers of cells from the center of the lens out were peeled iteratively from each en face preparation, divided into six separate wells, representing six layers of lens cells in order of depth in the lens, and immunolabelled as above. In this way a gradient of lens fiber cell age and differentiation could be translated from in situ to tissue preparations on slides for capture of antibody labelling patterns.
Image processing and quantification
Image processing and analysis were performed using Image J (National Institute of Health, Bethesda, MD). Extended focus images of AQP0, Cx46 tail and Cx46 loop-specific antibody staining were generated by maximum projection of image stacks over a depth of 10 μm. High-resolution extended focus image montages were assembled using Adobe Photoshop software, and rotated such that radial cell columns were horizontal and the lens periphery was to the left-hand side of the image. Image J was used to sum the pixel intensities for each antibody labelling pattern in each column of the two-dimensional images, and produce a one-dimensional vector that represented the total signal (cytoplasm and membrane) for a given protein target. The vector of normalized pixel intensity values was plotted as a function of normalized distance into the lens, expressed as r/a, where r is the radial distance of the signal from the center of the lens section, and a is the radius of the lens section.
Mass spectrometry
Lenses were extracted from 21 day-old Wistar rats immediately following death, and dissected in 37°C PBS into outer cortex, inner cortex, and core regions as described by Lim et al (Lim et al., 2005). Tissue was homogenised in buffer containing 10mM Tris, 1mM EDTA. Membrane and cytoplasmic fractions were separated via centrifugation at 16,000 g for 30 mins at 4°C in an Eppendorf 5402 centrifuge. The supernatant was discarded, and the pellet containing lens membrane proteins washed in 4M Urea in Tris buffer, 7M Urea in Tris buffer, and 100mM NaOH to remove bound crystallin proteins. Membrane proteins were then washed with water prior to delipidation in 95% EtOH at −20°C overnight. Finally, membrane proteins were washed in water, solubilised in 7:2 Formic Acid:Isopropanol, and spotted onto a MALDI plate using saturated 2,5-Dihydroxybenzoic acid in 70% ACN/0.1% TFA as matrix. Samples were analysed using a Voyager-DE™ PRO MALDI-TOF mass spectrometer (Applied Biosystems, Inc., California, USA). Prior to data collection, a linear external calibration was performed using the mass calibrants bovine insulin (Mr = 5734), E. coli thioredoxin (Mr = 11674), and equine apomyoglobin (Mr = 16952).
RESULTS
Differentiation-dependent changes in AQP0 distribution
The distribution of AQP0 in the lens was initially investigated using equatorial cryosections cut from whole rat lenses, and labelled with two different antibodies directed against the C-terminal tail of AQP0. In low power images, both antibodies yielded the same pattern of AQP0 protein expression, characterized by two concentric rings of AQP0 labelling in the outer and inner cortex, with minimal signal in the lens core (Fig. 1A and B, left panel). To ensure that the abrupt reduction of AQP0 signal in the lens core was not due to inadequate fixation, sections were prepared from rat lenses cut in half immediately prior to immersion in fixative. Although the morphology of the sections was somewhat compromised, AQP0 labelling was identical to that seen using conventional procedures (Fig. 1B, left panel). Additionally, labelling for another lens membrane protein, MP20, was detected in the lens core (Fig. 1B, right panel). This suggests that the AQP0 labelling pattern seen in Fig. 1A is not due to an artefact caused by an inadequate penetration of fixative into the lens core.
Figure 1. Overview of AQP0 immunolabelling pattern in rodent lenses.

Equatorial cryosections from rodent lenses labelled with antibodies directed against the C terminus of AQP0. (A) Expression pattern of AQP0 in a 21 day-old Wistar rat labelled with the IIIp11 antibody. The expression pattern changes radially from lens cortex to core. (B) Left panel: The concentric ring pattern is not an artefact, as seen by the cut lens fixation control of AQP0 labelling using the Alpha Diagnostic International (ADI) AQP0 antibody that produced an identical pattern to (A). Right panel: Labelling of the lens core with antibodies to MP20. (C) ADI AQP0 antibody labelling in a 21 day-old mouse lens, showing the same pattern as in the rat lens (A). Scale bar = 500 μm.
This conclusion is also supported by the observation of a similar AQP0 labelling pattern in sections prepared from the mouse lens, in which due to its smaller size one would expect greater rates of fixative penetration (Fig. 1C). Hence, the spatial distribution of AQP0 is conserved in rats and mice. Furthermore, the similarity of the pattern obtained in the two rodent lenses indicates that the changes in AQP0 labelling may reflect differentiation-dependent changes in the subcellular distribution of, and/or post-translational modifications to AQP0, as has been observed for other lens proteins (Grey et al., 2003; Jacobs et al., 2004). To investigate this, axial sections from the rat lens were double-labelled with AQP0 antibodies and propidium iodide, allowing changes in AQP0 labelling to be correlated to nuclear degradation, a marker of fiber cell differentiation (Fig. 2). Changes in labelling intensity of AQP0 were evident in high magnification images selected from areas of the lens cortex (Fig. 2A). In the outer cortex differentiating nucleated fiber cells initially exhibit extensive AQP0 labelling (Fig. 2B), but with depth into the lens these nucleated fiber cells exhibit a loss of AQP0 labelling (Fig. 2C), and upon complete loss of cell nuclei an increase in labelling intensity (Fig. 2D). The initial decrease in AQP0 labelling intensity occurs just prior to nuclear degradation, which reflects the transition from differentiating to mature fiber cells (Fig. 2B). In contrast, the second major decrease in AQP0 labelling intensity evident in Fig. 1A occurs distal to nuclear degradation. It is possible that the two zones in which the loss of labelling from anti-AQP0 antibodies we observe could be due to a masking of the antibody epitope located on the C terminus of AQP0. Alternatively, the observation that rat AQP0 is cleaved in vivo (Schey et al., 1999) suggests that the observed loss of labelling could also be due to truncation of the cytoplasmic tail of AQP0, and subsequent loss of the antibody epitope. To distinguish between these two possibilities we endeavoured to repeat our immunohistochemical mapping experiments using peptide specific AQP0 antibodies raised against regions other than the cytoplasmic tail. Two different antibodies raised against the E loop of AQP0 were trialled, and although they yielded satisfactory Western blotting data (data not shown), when used for immunohistochemistry they resulted in high levels of non-specific background labelling which made the mapping of changes in AQP0 signal intensity unfeasible. Thus an alternative technique is required to determine the mechanism(s) responsible for the two observed changes in AQP0 labelling intensity detected by the cytoplasmic tail antibody.
Figure 2. Changes in AQP0 immunolabelling as a function of fiber cell differentiation.
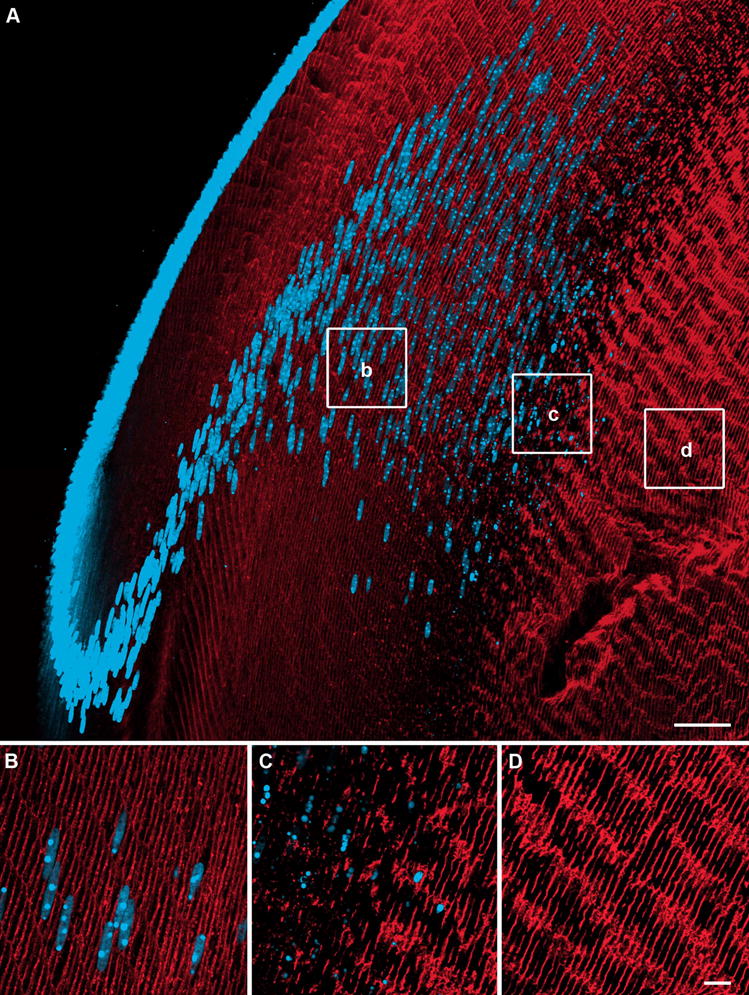
Axial cryosections of a rat lens double labelled with an AQP0 antibody (red) and the nuclear stain propidium iodide (blue). (A) Overview of AQP0 expression in the outer cortex, showing dispersal and degradation of cell nuclei. (B) In differentiating fiber cells AQP0 expression is predominantly membranous. (C) Just prior to nuclear degradation, a decrease in AQP0 labelling is seen. (D). Mature fiber cells in the inner cortex show a dramatic increase in AQP0 labelling following nuclei loss. Scale bars: A = 50 μm; B–D = 10 μm.
Mass spectrometry analysis of lens membrane proteins from distinct lens regions
To verify our immunohistochemical mapping and to provide information on specific sites of AQP0 truncation, mass spectrometry was used to analyse intact AQP0 protein from different areas of the lens (Fig. 3). Dissected lens membrane fractions were subjected to urea/alkali stripping to remove contaminating crystallins. The urea/alkali stripping of the membranes removed the majority of the crystallins, although some residual crystallins were detected as peaks at approximately m/z 20,000. The major peaks in the spectra occurred at approximately m/z 28,000. Similar spectra were obtained from the outer and inner lens cortex, whereas large differences were observed in spectra obtained from the lens core (Fig. 3, bottom). In the outer cortex, the most abundant ion signal represented full-length intact AQP0 at m/z 28,232 (predicted [M+H]+ = 28,210) (Fig. 3, top). Little difference was observed between the outer and inner cortex spectra, with full-length intact AQP0 detected at m/z 28,228 (Fig. 3, middle). In the lens core, multiple peaks were observed, representing several forms of AQP0 protein (Fig. 3, bottom). Major peaks for full-length AQP0 at m/z 28,261, AQP0 1-253 at m/z 27,151 (predicted [M+H]+ = 27,101), AQP0 1-238 at m/z 25,740 (predicted [M+H]+ = 25,689), and AQP0 1-234 at m/z 25,300 (predicted [M+H]+ = 25,248) were observed. Additionally, minor peaks corresponding to AQP0 1-260 at m/z 27,950 (predicted [M+H]+ = 27,898), AQP0 1-250 at m/z 26,943 (predicted [M+H]+ = 26,886), and AQP0 1-231 at m/z 24,910 (predicted [M+H]+ = 24,849) were also observed. There is a consistent discrepancy of ~50Da between predicted and observed masses of AQP0 in the lens core, probably due to the observed masses falling outside the range over which the instrument was calibrated (5–17 kDa). In a previous study on the rat lens we characterized the major cleavage sites using a more accurate mass determination that was obtained by first cleaving the protein and accurately measuring the masses of the smaller truncated pieces that were within the calibration range of the instrument (Schey et al., 1999). A summary of the observed and predicted masses for all AQP0 peaks in all lens regions is presented in Table 1.
Figure 3. Mass spectrometry of intact AQP0 protein from different lens regions.

Representative MALDI mass spectra of AQP0 obtained from different regions of a rat lens. In the outer cortex (top panel) and inner cortex (middle panel), AQP0 exists in its full-length form. In the lens core (bottom panel), a small amount of full-length AQP0 is detected, and multiple forms of truncated AQP0 are present, indicating extensive processing of full-length AQP0. Hashed areas of inset images represent approximate dissected regions of rat lens.
Table 1.
Predicted and observed m/z ratios for all forms of AQP0 detected in the rat lens
| Lens Region | AQP0 Form | Predicted m/z | Observed m/z | Relative Abundance |
|---|---|---|---|---|
| Outer Cortex | 1-263 | 28,210 | 28,232 | 1.0 |
| Inner Cortex | 1-263 | 28,210 | 28,228 | 1.0 |
| Core | 1-263 | 28,210 | 28,261 | 0.12 |
| 1-260 | 27,898 | 27,950 | 0.06 | |
| 1-253 | 27,101 | 27,151 | 0.09 | |
| 1-250 | 26,886 | 26,943 | 0.05 | |
| 1-238 | 25,689 | 25,740 | 0.32 | |
| 1-234 | 25,248 | 25,300 | 0.33 | |
| 1-231 | 24,849 | 24,910 | 0.03 |
Clearly the cytoplasmic tail of AQP0 is highly processed through numerous cleavage events in the lens core, consistent with a loss of antibody binding sites and the decrease in antibody labelling observed in the lens core. Since no truncation of AQP0 was detected in the outer or inner cortex, our mass spectrometry results suggest that the initial transient decrease in AQP0 antibody labelling intensity is due to a transient masking of the antibody interaction with its epitope on the cytoplasmic tail than to protein truncation.
Mapping of cleavage zones in the rat lens
From our mass spectrometry results, the most abundant form of AQP0 in the lens core is 1–238. Previous studies showed that AQP0 is cleaved in vitro at K238 by calpain (Schey et al., 1999; Ma et al., 2005). Additionally, accelerated cleavage of AQP0 at K238 is associated with formation of lens cataract (Schey et al., 1999). It is possible that calpain acts to cleave rat AQP0 in the lens core in vivo. Interestingly, the tail of the lens gap junction protein Cx50 is also cleaved by calpain, which occurs as cell nuclei degrade (Lin et al., 1997). The other lens fiber cell gap junction protein, Cx46, also undergoes truncation in vivo. While the protease(s) responsible for Cx46 cleavage in rat is not known, immunohistochemical experiments have mapped the zones in which cleavage occurs (Jacobs et al., 2004). Since our result shows a truncation of the AQP0 C-terminal tail, we have compared the relative locations of Cx46 and AQP0 cleavage zones.
To achieve this, intensity profiles were extracted from high-resolution maximum projection image montages that captured AQP0 labelling from the periphery to the lens core (Fig. 4). Results from a previous study mapping Cx46 cleavage zones in the rat lens (Jacobs et al., 2004) are presented to contrast AQP0 labelling. To facilitate comparison between different lens sections, labelling intensity was plotted against normalized distance from the lens center. While Cx46 cleavage occurred in two distinct zones in the lens cortex, the intensity profile for AQP0 C-terminal antibody labelling was completely different. A transient decrease in AQP0 signal preceded nuclear degradation, and the second Cx46 cleavage zone. The subsequent increase in AQP0 signal that occurred upon nuclear degradation suggests that the loss of labelling is not due to AQP0 cleavage, a result consistent with our mass spectrometry analysis of protein fractions isolated from this zone (Fig. 3). Thus, although Cx46 (Fig. 4) and Cx50 (Lin et al., 1997) undergo cleavage at the transition between differentiating fiber (DF) and mature fiber (MF) cells where cell nuclei are degraded, AQP0 does not. AQP0 cleavage occurs abruptly in a much deeper zone that is spatially distinct from the two cleavage zones observed for Cx46. These results indicate that at three different and distinct stages of fiber cell differentiation, proteases selectively modify existing membrane proteins. Furthermore, since AQP0 truncation occurs after nuclear degradation, it would appear that the differentiation of MF cells continues in the lens core with the truncation of the cytoplasmic tail of AQP0.
Figure 4. Comparison of AQP0 and Cx46 cleavage zones.
Normalized intensity profiles were extracted from image montages labelled with either loop specific anti-Cx46 (yellow), tail specific anti-Cx46 (blue), or tail specific anti-AQP0 (red), and plotted against normalized distance from the lens center (r/a). Signal from loop specific anti-Cx46, which detects the uncleaved form of Cx46, stays relatively constant. In contrast, two distinct drops in signal intensity were seen with the tail specific anti-Cx46, indicating two putative cleavage zones. The loss of signal from tail specific anti-AQP0 occurs deeper into the lens than both Cx46 cleavages.
Differentiation-dependent changes in AQP0 distribution in the lens cortex
Taken together, our immunolabelling and mass spectrometry data show that loss of AQP0 labelling in the lens core is due to cleavage of the protein, while the outer transient decrease in AQP0 labelling intensity is not. This suggests that the initial decrease in AQP0 labelling intensity is caused by a reversible loss of antibody access to its binding site located on the cytoplasmic tail. This could be due to a conformational change in the AQP0 protein itself, or binding of another protein to the antibody epitope. In an attempt to distinguish between these possibilities we mapped at high resolution the subcellular distribution of AQP0 throughout the lens cortex (Fig. 5). Changes in subcellular distribution were evident in high magnification images of AQP0 immunolabelling from the lens outer and inner cortex (Fig. 5A). In the outer cortex, regular arrays of hexagonal lens fiber cells were clearly identifiable (Fig. 5B). An overlay of AQP0 signal (red) and general lens membrane marker (WGA, green) gave a yellow color, indicating a homogenous distribution of AQP0 throughout the cell membrane in this region. Some AQP0 signal was also observed in the fiber cell cytoplasm of these lens fiber cells, consistent with AQP0 labelling of vesicles observed by others using immunoelectron microscopy (Lo et al., 2003). Deeper into the cortex, homogenous labelling of the cell membranes was replaced by an aggregation of AQP0 signal specifically on broad side fiber cell membranes (Fig. 5C). Further into the lens, where fiber cell nuclei degrade, the AQP0 signal decreased dramatically. The subsequent increase in AQP0 labelling intensity in mature lens fiber cells of the inner cortex was associated with a redistribution of AQP0 around the entire cell membrane (Fig. 5D). The changes in the subcellular distribution of AQP0 seem to be associated with changes in fiber cell morphology. In the outer cortex, fiber cells are aligned in columns, but during the course of fiber cell differentiation, the cross-sectional profile of cells within a column changes from hexagonal to more circular (Fig. 5B–C). This phenomenon was recently quantified as a function of fiber cell differentiation (Jacobs et al., 2004). Following nuclear degradation, there is a sudden loss of the columnar alignment of the fiber cells (Fig. 5D). Against this background of changing fiber cell morphology, the subcellular distribution of AQP0 changes from uniform, to fragmented plaques, and then following a transient loss of AQP0 labelling, back to a uniform membranous distribution. This sequence of events implies a redeployment of AQP0 from general membrane labelling to junction formation as fiber cells differentiate. AQP0 has previously been shown via immunoelectron microscopy to form junctional structures distinct from gap junctions (Zampighi et al., 1989), and to alter its association with gap junctional plaques as a function of fiber cell age (Dunia et al., 1998). If AQP0 redeployment is indeed the case, one would expect that the redistribution would be driven by changes in the interaction of AQP0 with the cytoskeleton, and furthermore this interaction could potentially result in the transient decrease in AQP0 labelling that precedes nuclei loss.
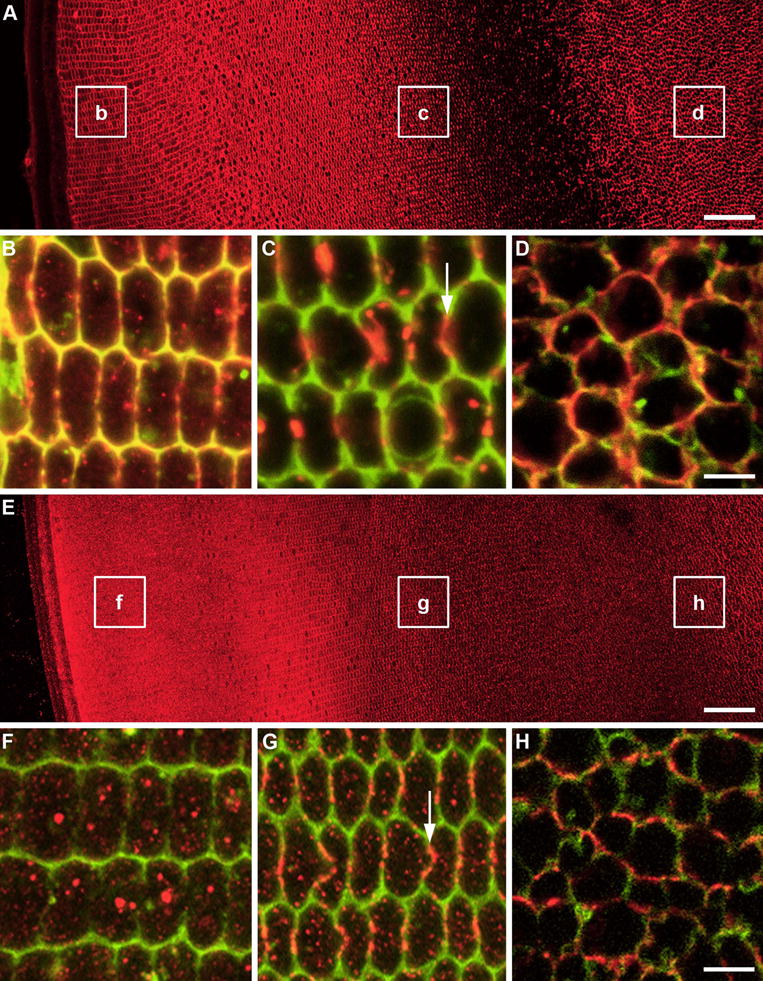
Figure 5. Mapping of AQP0 and filensin distribution in the lens cortex.

Equatorial sections from a 21 day-old Wistar rat double-labelled with either AQP0 or filensin antibodies (red), and the general membrane marker WGA (green). (A) Overview of AQP0 labelling pattern showing location of high power images (B-D). (B) Membranous AQP0 labelling in nucleated fiber cells of the lens cortex and WGA gives a yellow color. Some cytoplasmic labelling of AQP0 is also detectable. (C) Prior to cell nuclear degradation, AQP0 labelling is localized to plaque-like structures predominantly on the broad sides of fiber cells. (D) In non-nucleated fiber cells of the inner cortex, AQP0 labelling returns and is distributed throughout the cell membrane but now the hexagonal shape of the cells is lost. (E) Overview of filensin labelling pattern showing location of high power images (F–H). (F) In the peripheral outer cortex filensin labelling is predominately cytoplasmic with no colocalisation with the membrane label WGA. (G) In the deeper outer cortex filensin is still cytoplasmic but there is significant association of filensin with the broad sides of fiber cell membranes. (H) In the inner cortex filensin is localized exclusively to the membrane in mature fiber cells. Scale bars A,E = 50 μm; B–D & F–H = 5 μm.
Since the lens-specific cytoskeletal protein filensin has recently been shown to associate with the C terminus of AQP0 during lens fiber cell differentiation (Lindsey Rose et al., 2006), we hypothesized that this association may be responsible for the observed redistribution of AQP0 and/or the transient decrease in AQP0 labelling. To distinguish between these two possibilities, equatorial lens sections were immunolabelled for filensin (Fig. 5E–H), and compared to the labelling pattern obtained for the AQP0 C-terminal antibody (Fig. 5A–D). Unlike AQP0, filensin labelling intensity was relatively uniform throughout the lens cortex (Fig. 5E). At high resolution it is evident that in peripheral nucleated lens fiber cells, filensin was found predominantly in the cell cytoplasm, although faint membranous labelling was also detected (Fig. 5F). Deeper into the lens cortex, membranous labelling of filensin was more evident, particularly on the broad sides of fiber cells (Fig. 5G). The localisation of filensin to broad sides occurred in the same zone where AQP0 aggregation into broad side plaques and the bowing of the broadside membranes first occurred (Fig. 5C & G, arrows). In non-nucleated cells deeper in the rat lens, membranous labelling of filensin was more uniform, and cytoplasmic labelling had disappeared (Fig. 5H). Interestingly, the association of filensin with AQP0 plaque formation occurred before the observed transient decrease in AQP0 labelling, indicating that the interaction of filensin with AQP0 is not responsible for the decrease in AQP0 labelling. Thus, either side of the transient loss of AQP0 signal, there is a strong association between the subcellular distribution of filensin and AQP0 suggesting a role for filensin in mediating the differentiation-dependent subcellular redistribution of AQP0.
Redistribution of AQP0 is associated with the formation of a new membrane specialization in mature fiber cells
While our observations suggest a rearrangement of the subcellular distribution of AQP0 occurs as a function of fiber cell differentiation, it is difficult to determine what these changes are in images of fiber cells cut in cross section since there is limited z resolution in this orientation. To provide higher resolution views of the AQP0 labelling patterns in the three regions shown in Fig. 5, fiber cells were peeled off from fixed lenses, double-labelled with AQP0 and WGA, and viewed en face. In this orientation, membrane specializations, such as the ball-and-socket joints, and gap junction plaques are more evident in the x-y plane. Using this approach, it was possible to identify the three areas of AQP0 labelling observed in cross section in Fig. 5B–D. This approach revealed some interesting associations of AQP0 with junctional structures, which may indicate a change in the functional role of AQP0 in different lens regions. In the lens outer cortex, three distinct labelling patterns were observed for WGA (Fig. 6A, left panel). The narrow sides of fiber cells showed an intense punctate signal, consistent with the labelling of the ball-and-socket joints (Kistler et al., 1986). On the broad sides, WGA staining was less intense, and was interspersed with areas of no staining. Since WGA binds specifically to membrane proteins with N-acetylglucosamine-linked carbohydrates, these negatively stained membrane domains represent areas which do not contain glycosylated membrane proteins, and correspond to the location of gap junction plaques in the lens (Jacobs et al., 2004). In contrast, AQP0 was found throughout the fiber cell membrane, but was excluded from the WGA-negative domains (Fig. 6A, middle panel). By combining the two signals, it was apparent that AQP0 was also surrounding the WGA negative domains (Fig. 6A, right panel), suggesting that AQP0 associates with the periphery of gap junction plaques, a result consistent with previous ultrastructural studies (Dunia et al., 1998). Cells lying deeper in the lens cortex, where cell nuclei are dispersed, exhibit less distinct ball-and-socket joints, and smaller WGA-negative gap junction plaques (Fig. 6B, left panel). In these cells, while labelling for AQP0 was similar to that seen in more peripheral cells, a plaque-like AQP0 labelling was observed (Fig. 6B, middle panel), which was associated with some WGA-negative domains, indicating that AQP0 had infiltrated gap junction plaques (Fig. 6B, right panel). In MF cells isolated from the inner cortex, no WGA- (Fig. 6C, left panel) or AQP0- (Fig. 6C, middle panel) negative domains were observed, and both labels were uniformly distributed throughout the cell membrane (Fig. 6C, right panel). Thus in MF cells, the AQP0 plaques found in WGA-negative domains of nucleated fiber cells have disappeared, and AQP0 and WGA-labelled glycosylated membrane proteins colocalize throughout the entire cell membrane.
Figure 6. En face immunolabelling of AQP0 and WGA in isolated lens fiber cells.

Layers of fiber cells were sequentially peeled off the lens to yield isolated cells from the periphery (A), deeper cortex (B), and mature lens fiber cells from the inner cortex (C). Cells were labelled with the general membrane label WGA (green, left panel) and an AQP0 antibody (red, middle panel). Images shown are single optical sections taken to capture the broad side surface of the cell. Arrowheads indicate ball-and-socket joints located to the narrow sides of fiber cells, while asterisks highlight WGA-negative regions indicative of large broad side gap junction plaques. The two channels are merged in the right panel. Scale bar = 5 μm.
By collecting image stacks through individual fiber cells immunolabelled for AQP0, it is possible to visualize fiber cells in three dimensions at high resolution. Furthermore, the three-dimensional reconstruction of isolated fiber cells allowed a cross-sectional profile to be obtained, which could be compared to the AQP0 staining patterns observed in equatorial lens sections (Fig. 5), thereby confirming the relative locations of the isolated cells. The three-dimensional reconstructions revealed that in the deeper cortex, the emergence of broad side AQP0 plaques observed en face (Fig. 6B) was associated with changes in membrane morphology (Fig. 7). Two adjacent lens fiber cells isolated from the deeper lens cortex were volume-rendered and cross-sections were taken through areas where AQP0 plaques were observed to illustrate the diversity of plaque morphology in this region (Fig. 7A). The broad sides labelled more strongly for AQP0, and often featured flattened plaques (Fig. 7B). In addition, discontinuous AQP0 plaque labelling was observed in regions of bowed fiber cell broad side morphology (Fig. 7C, asterisk). Of particular interest was the formation of ‘domes’ of AQP0 labelling, observed folding into and out of adjacent fiber cells (Fig. 7D, arrows). Dome morphology was observed to change from in to out within a short distance along the fiber cell broad sides (Fig. 7E & F, dagger). Over the 40 μm length of the two fiber cells shown, ten outward and nine inward domes were observed. The broad side domes typically measured 1.9 μm wide, and ranged between 1.7 and 2.0 μm deep. Recently, the emergence of a new structural specialization in mouse lenses older than 2 weeks was reported (Blankenship et al., 2007). These paddlelike structures projected from the short sides of cells, were aligned with similar structures in adjacent cells, and were most obvious in MF cells. While the appearance of a new structural specialization that forms during lens fiber cell differentiation is interesting, it is clear that the paddlelike structures described by Blankenship et al are different to the ‘dome’ structures reported here.
Figure 7. AQP0 plaque morphologies in isolated lens fiber cells.

Labelling for AQP0 in two adjacent lens fiber cells isolated from the lens cortex region where AQP0 aggregates into plaque-like structures. (A) Broad side view of a three-dimensional volume rendering of two lens fiber cells. (B–F) A series of single cross-sections extracted from the volume rendered image obtained from the regions indicated by the dashed lines in (A). The cross-sections feature discontinuous AQP0 plaque labelling (asterisk), inward- and outward-folding AQP0 plaques in adjacent fiber cells (arrows), and transitions from inward- to outward-folding plaques in short distances in the same fiber cell (dagger). Scale bar A, B–F = 5 μm, 2.5 μm.
Taken together our data suggest that the interaction of AQP0 with the cytoskeletal protein filensin drives the subcellular aggregation of AQP0 on the broad sides of deeper fiber cells resulting in the formation of domes. These changes precede the loss of AQP0 signal intensity, which occurs as cell nuclei are lost. The subsequent recovery of AQP0 labelling in MF cells is associated with a loss of domes, suggesting that dome formation represents yet another phase in the differentiation-dependent subcellular redistribution of AQP0 that presumably results in a shift in the function of AQP0 from a water channel to a junctional protein. Consistent with this view is the observation that a barrier to the extracellular diffusion of fluorescent dyes is formed upon nuclear degradation, which correlates with the insertion of MP20 into the membrane from a cytoplasmic pool (Grey et al., 2003), and the redeployment of AQP0 from broad side plaques to a more homogenous membrane distribution (Fig. 5). Thus while our observations do not ‘prove cause and effect’ they do show that the dramatic reorganisation of membrane protein distribution that occurs as cell nuclei are lost has a functional consequence, namely a restriction of the extracellular space.
DISCUSSION
Using antibodies directed against the C-terminal tail of AQP0 we have produced the first comprehensive map of how the subcellular distribution and processing of AQP0 changes as fiber cells differentiate. In peripheral fiber cells, AQP0 was distributed homogenously throughout the cell membrane, but was excluded from large gap junction plaques. Deeper into the lens cortex, AQP0 aggregated into plaques, which were predominantly located on the broad sides of fiber cells, and often formed dome-like structures, a novel morphological feature that, to the authors’ knowledge, have not previously been reported. The cytoskeletal protein filensin labelled similar structures, suggesting it is involved in the observed aggregation of AQP0. As fiber cell nuclei degraded AQP0 antibody signal was transiently lost, presumably due to a masking of the antibody epitope, before labelling throughout the membranes of MF cells in the inner cortex. This redeployment of AQP0 coincided with nuclear degradation, loss of the hexagonal fiber cell shape, cleavage of Cx46 (Jacobs et al., 2004) and Cx50 (Lin et al., 1997), membrane insertion of MP20 and formation of a barrier to extracellular diffusion (Grey et al., 2003). Deeper into the lens AQP0 signal was abruptly lost due to AQP0 C-terminal tail truncation that removes peptide motifs involved in the regulation of AQP0 function. Thus AQP0 cleavage in the core of the lens appears to be the final step in a programme of changes that occur during fiber cell differentiation designed to alter the function of AQP0 in the absence of de novo protein synthesis.
AQP0 cleavage in the lens core is a well established finding in a variety of species (Ball et al., 2004; Thibault et al., 2008) and is therefore not a new finding. However in the present study we show at least in the rat lens that this cleavage of AQP0 occurs abruptly and completely over a distance of ~10–20 cell layers (Fig. 4). Furthermore, our mass spectrometric data indicates that this abrupt cleavage produces the major truncation products at residues L234 and K238 in the rat lens that are consistent with truncation products obtained in the presence of calpain. In contrast, major truncation sites observed for human AQP0 in the lens core are residues N259 and N246, with D243 as minor species (Ball et al., 2004). Since asparagine residues can undergo deamidation, Ball et al. have proposed that AQP0 cleavage in human lenses is due to the age-dependent deamidation of AQP0, which in turn induces a general non-enzymatic age-dependent truncation of the AQP0 peptide backbone. Differences in the sequence of rat and human AQP0, and higher levels of calpain (Nakajima et al., 2006) in the rat lens relative to the human lens may explain why we observe such a rapid and discrete zone of AQP0 cleavage in the rat lens. Thus in the rat lens it appears that the cleavage of the C-terminal tail of AQP0 is not a general age-dependent protein degradation but a programmed event that is presumably mediated by calpain.
In the lens outer cortex Cx46 also undergoes cleavage of its C-terminal tail but in two spatially distinct steps. Since calpain can cleave connexins (Lin et al., 1997) and AQP0 (Schey et al., 1999) in vitro, it suggests in vivo that calpain mediated cleavage of Cx46 and AQP0 is differentially regulated since they occur at spatially different locations. Calpains are intracellular cysteine proteases which require Ca2+ for activation. A variety of calpains, and the endogenous calpain inhibitor, calpastatin, are expressed in the lens (Azuma et al., 1997; Huang and Wang, 2001; Ma et al., 1998a; Ma et al., 2001; Ma et al., 1998b; Shearer et al., 2000; Yoshida et al., 1985). While the rules governing the specificity of calpain cleavage are not well understood, cleavage of lens connexins has been shown to alter their functional and structural properties (Lin et al., 1998; Lin et al., 1997). Cleavage of the C-terminal tail of Cx50 by calpain coincided with nuclear degradation, and resulted in the loss of pH sensitivity, presumably allowing the remaining gap junction channels to remain open in the acidified lens core. While cleavage of some Cx46 also occurred upon nuclear degradation, the majority of cleavage occurred earlier in fiber cell differentiation and was associated with the redistribution of gap junction plaques (Jacobs et al., 2004). Thus, the loss of cell nuclei and other organelles appears to be associated with an increase in calpain activity. However, our mass spectrometry data suggests that this localized increase in calpain activity does not result in cleavage of AQP0, suggesting that at this stage in fiber cell differentiation, AQP0 is calpain-resistant. In other cell types, phosphorylation has been shown to protect Cx32 against calpain cleavage (Elvira et al., 1993). Therefore it is possible that post-translational modifications (phosphorylation) to, and/or protein interactions (calmodulin binding) with the C-terminal tail of AQP0 may inhibit its cleavage by calpain in this region. This idea is supported by the observed transient decrease in AQP0 signal that precedes cell nuclei loss which we have attributed to the masking of the antibody epitope by such interactions.
Since in the lens core AQP0 is cleaved, it suggests that the protective effects of this interaction are lost and AQP0 becomes susceptible to cleavage. The major AQP0 truncation sites in the rat lens are L234 and K238, which lie within the putative calmodulin recognition/binding domain. Therefore, it is likely that rat lens core AQP0 cannot bind calmodulin. Since calmodulin is thought to mediate the response of AQP0 water permeability to changes in Ca2+ concentration (Németh-Cahalan and Hall, 2000; Varadaraj et al., 2005), it is conceivable that the water permeability of AQP0 in the rat lens core is not affected by changes in Ca2+ concentration. In contrast, pH regulation of AQP0 water permeability would not be expected to change in the rat lens core, since the regulatory histidine residues are not located in the C-terminal region (Németh-Cahalan et al., 2004).
The recently published structures for AQP0 (Gonen et al., 2004a; Harries et al., 2004; Palanivelu et al., 2006) confirm the previously held belief (Shiels and Bassnett, 1996; Shiels et al., 2001) that AQP0 is a multifunctional protein, acting both as a water channel and a structural protein in the lens. Our mapping of changes in the subcellular distribution and cleavage of AQP0 has localized stages in the continuum of fiber cell differentiation where a shift in the function of AQP0 may occur. In the outer cortex, AQP0 immunolabelling observed in this and other studies (Zampighi et al., 2002) is consistent with the function of a water channel (general membrane staining) to one more appropriate for junction formation (plaque-like distribution). In the cortex, these changes occur against a background in which cell nuclei and the cellular machinery required for de novo protein synthesis are lost. This indicates that the observed shift in the subcellular distribution of AQP0 towards the start of junction formation (dome formation) occurs via rearrangement of pre-existing membranous AQP0. The colocalisation of filensin and AQP0 in domes and the subsequent masking of the AQP0 epitope suggest that this rearrangement is mediated via protein interactions with the C-terminal tail of AQP0. The return of membranous labelling of AQP0 following nuclear degradation also appears to be related to junction formation since extracellular space diffusion becomes abruptly restricted in this region (Grey et al., 2003).
Based on the observation that insertion of MP20 into lens cell membranes correlates with formation of an extracellular diffusion barrier that is associated with formation of a central lens core syncytium (Grey et al., 2003), and reference to previous literature (Fotiadis et al., 2000; Gonen et al., 2004a; Gonen et al., 2001), we propose a working model to explain how the observed restriction of the extracellular space might occur in the absence of de novo protein synthesis (Fig. 8). In this schematic model, MP20 and galectin-3 are found in intracellular vesicles of DF cells, while AQP0 is located throughout the cell membrane, consistent with its role as a water channel (Fig. 8A). The extracellular space is relatively large and permits passage of large molecules between cells. Just prior to nuclear degradation, AQP0 interacts with the cytoskeletal protein filensin, and becomes aggregated on the broad sides of fiber cells, often in dome-like structures (Fig. 8B). Upon nuclear degradation, MP20 and galectin-3 associate with the cell membrane, while existing membranous AQP0 undergoes redistribution around the entire cell membrane, to form an adhesion complex that restricts extracellular space diffusion (Fig. 8C).
Figure 8. Model for interaction of MP20, galectin-3 and AQP0 in formation of the extracellular diffusion barrier.

(A) Nucleated cortical fiber cells contain intracellular vesicles of MP20 (red) and galectin-3 (green), while AQP0 (blue) is expressed predominantly in the cell membrane. The passage of large molecules (dashed arrow) through the extracellular space is possible. (B) Prior to degradation of cell nuclei, AQP0 redistributes in the membrane to broad side plaque-like structures, often in dome-like invaginations. (C) Upon nuclear degradation, MP20 and galectin-3 relocate to the cell membrane, and interact (i) to greatly attenuate the extracellular space. AQP0 molecules are brought into close apposition to form a junctional structure (ii). Alternatively, alternating aggregates of AQP0 in apposing cell membranes could lead to the formation of wavy junctions (iii). The passage of large molecules through the extracellular space is obliterated.
Based on studies of the protein-protein interactions of MP20/galectin-3 (Gonen et al., 2001), AQP0/filensin (Lindsey Rose et al., 2006), and AQP0/AQP0 (Fotiadis et al., 2000; Gonen et al., 2004a), it is possible to speculate on the processes involved in the formation of this adhesion complex. Galectin-3 forms homomers via its C and N terminus (Kuklinski and Probstmeier, 1998), contains carbohydrate and lectin binding domains, and is known to interact with MP20, with a possible stoichiometry of 2:2 (Gonen et al., 2001). Its interaction with MP20 is inhibited by lactose, suggesting that it interacts with MP20 via its carbohydrate binding domain. Recently, Ervin et al showed that W43 and W61 are glycosylated (Ervin et al., 2005). Therefore the concomitant insertion of MP20 and delivery of galectin-3 to the extracellular space may facilitate the linking of MP20 from adjacent cell membranes via the binding of galectin-3 to carbohydrate groups on the first extracellular loop of MP20 (Fig. 8Ci). It is conceivable that MP20/galectin-3 binding brings fiber cells closer together, facilitating interactions between AQP0 accumulated in the broad side membranes. Two types of AQP0/AQP0 interaction are conceivable. It is known from in vitro reconstitution studies that dialysis of detergent-purified AQP0 results in spontaneous formation of double-layered two-dimensional crystals. These crystals are formed by the hydrophilic interaction of the extracellular surfaces of AQP0 protein in apposing layers (Fig. 8Cii) (Fotiadis et al., 2000; Gonen et al., 2004a). These in vitro junctions appear to have a similar structure to thin 11–13nm junctions and square arrays that are a feature of cell-to-cell interactions in the lens core (Gonen et al., 2004a). Alternatively, alternating aggregates of AQP0 in apposing cell membranes could lead to formation of wavy junctions, the predominant junctional structure in the lens core (Fig. 8Ciii). Regardless of the actual protein-protein interactions involved, the sequence of events occurring at this transition zone appear to mediate a switch in the function of AQP0 from a water channel in the cortex, to its involvement in the different junctional structures seen in the lens core.
Validation of this model obviously requires further investigation. At the molecular level, the stoichiometry of the MP20/galectin-3 interaction and the precise protein domains that are interacting remain unclear. Additionally, while cleavage of AQP0 facilitates junction formation in vitro (Gonen et al., 2004a), it is unclear whether a similar cleavage is required to facilitate junction formation in vivo. Indeed, we have shown that cleavage of AQP0 C-terminal tail occurs some distance past the events summarized in Fig. 8. Furthermore, since the C-terminal tail of AQP0 contains many regulatory residues and sequences, it is likely that this domain is required for the protein-protein interactions that accompany junction formation. The answers to these, and many other questions, require further development of targeted proteomic approaches, such as Laser Capture Microdissection/Mass Spectrometry, or MALDI Imaging Mass Spectrometry that can utilize and build on the maps of AQP0 processing and redistribution presented in this study.
Acknowledgments
Supported by the Health Research Council of New Zealand, the New Zealand Lottery Grants Board, and the University of Auckland Research Committee.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Azuma M, Fukiage C, David LL, Shearer TR. Activation of calpain in the lens: A review and proposed mechanism. Experimental Eye Research. 1997;64:29–38. doi: 10.1006/exer.1996.0234. [DOI] [PubMed] [Google Scholar]
- Ball LE, Garland DL, Crouch RK, Schey KL. Post-translational modifications of aquaporin 0 (AQP0) in the normal human lens: Spatial and temporal occurrence. Biochemistry. 2004;43:9856–9865. doi: 10.1021/bi0496034. [DOI] [PubMed] [Google Scholar]
- Bassnett S. Lens organelle degradation. Experimental Eye Research. 2002;74:1–6. doi: 10.1006/exer.2001.1111. [DOI] [PubMed] [Google Scholar]
- Blankenship T, Bradshaw L, Shibata B, Fitzgerald P. Structural specializations emerging late in mouse lens fiber cell differentiation. Investigative Ophthalmology and Visual Science. 2007;48:3269–3276. doi: 10.1167/iovs.07-0109. [DOI] [PubMed] [Google Scholar]
- Bloemendal H. Lens proteins. CRC Critical Reviews in Biochemistry. 1982;12:1–38. doi: 10.3109/10409238209105849. [DOI] [PubMed] [Google Scholar]
- Bok D, Dockstader J, Horwitz J. Immunocytochemical localization of the lens main intrinsic polypeptide (MIP26) in communicating junctions. Journal of Cell Biology. 1982;92:213–220. doi: 10.1083/jcb.92.1.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broekhuyse RM, Kuhlmann ED, Stols AL. Lens membranes II. Isolation and characterization of the main intrinsic polypeptide (MIP) of bovine lens fiber membranes. Experimental Eye Research. 1976;23:365–371. doi: 10.1016/0014-4835(76)90135-4. [DOI] [PubMed] [Google Scholar]
- Broekhuyse RM, Kuhlmann ED, Winkens HJ. Lens membranes VII. MIP is an immunologically specific component of lens fiber membranes and is identical with 26K band protein. Experimental Eye Research. 1979;29:303–313. doi: 10.1016/0014-4835(79)90009-5. [DOI] [PubMed] [Google Scholar]
- Chandy G, Zampighi GA, Kreman M, Hall JE. Comparison of the water transporting properties of MIP and AQP1. Journal of Membrane Biology. 1997;159:29–39. doi: 10.1007/s002329900266. [DOI] [PubMed] [Google Scholar]
- Donaldson P, Grey AC, Merriman-Smith BR, Sisley AMG, Soeller C, Cannell MB, Jacobs MD. Functional imaging: New views on lens structure and function. Clinical and Experimental Pharmacology and Physiology. 2004;31:890–895. doi: 10.1111/j.1440-1681.2004.04099.x. [DOI] [PubMed] [Google Scholar]
- Dunia I, Recouvreur M, Nicolas P, Kumar N, Bloemendal H, Benedetti EL. Assembly of connexins and MP26 in lens fiber plasma membranes studied by SDS-fracture immunolabeling. Journal of Cell Science. 1998;111:2109–2120. doi: 10.1242/jcs.111.15.2109. [DOI] [PubMed] [Google Scholar]
- Elvira M, Diez JA, Wang KK, Villalobo A. Phosphorylation of connexin-32 by protein kinase C prevents its proteolysis by mu-calpain and m-calpain. Journal of Biological Chemistry. 1993;268:14294–14300. [PubMed] [Google Scholar]
- Ervin LA, Ball LE, Crouch RK, Schey KL. Phosphorylation and glycosylation of bovine lens MP20. Investigative Ophthalmology and Visual Science. 2005;46:627–635. doi: 10.1167/iovs.04-0894. [DOI] [PubMed] [Google Scholar]
- Fitzgerald PG, Bok D, Horwitz J. Immunocytochemical localization of the main intrinsic polypeptide (MIP) in ultrathin frozen sections of rat lens. Journal of Cell Biology. 1983;97:1491–1499. doi: 10.1083/jcb.97.5.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fotiadis D, Hasler L, Muller DJ, Stahlberg H, Kistler J, Engel A. Surface tongue-and-groove contours on lens MIP facilitate cell-to-cell adherence. Journal of Molecular Biology. 2000;300:779–789. doi: 10.1006/jmbi.2000.3920. [DOI] [PubMed] [Google Scholar]
- Gonen T, Cheng Y, Kistler J, Walz T. Aquaporin-0 membrane junctions form upon proteolytic cleavage. Journal of Molecular Biology. 2004a doi: 10.1016/j.jmb.2004.07.076. [DOI] [PubMed] [Google Scholar]
- Gonen T, Grey AC, Jacobs MD, Donaldson PJ, Kistler J. MP20, the second most abundant lens membrane protein and member of the tetraspanin superfamily, joins the list of ligands of galectin-3. BMC Cell Biology. 2001;2:17. doi: 10.1186/1471-2121-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonen T, Sliz P, Kistler J, Cheng Y, Walz T. Aquaporin-0 membrane junctions reveal the structure of a closed water pore. Nature. 2004b;2503:1–4. doi: 10.1038/nature02503. [DOI] [PubMed] [Google Scholar]
- Grey AC, Jacobs MD, Gonen T, Kistler J, Donaldson P. Insertion of MP20 into lens fibre cell plasma membranes correlates with the formation of an extracellular diffusion barrier. Experimental Eye Research. 2003;77:567–574. doi: 10.1016/s0014-4835(03)00192-1. [DOI] [PubMed] [Google Scholar]
- Harries WEC, Akhavan D, Miercke LJW, Khademi S, Stroud RM. The channel architecture of aquaporin 0 at a 2.2A resolution. Proceedings of the National Academy of Sciences. 2004;101:14045–14050. doi: 10.1073/pnas.0405274101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YH, Wang KKW. The calpain family and human disease. Trends in Molecular Medicine. 2001;7:355–362. doi: 10.1016/s1471-4914(01)02049-4. [DOI] [PubMed] [Google Scholar]
- Jacobs MD, Donaldson PJ, Cannell MB, Soeller C. Resolving morphology and antibody labeling over large distances in tissue sections. Microscopy Research and Techniques. 2003;62:83–91. doi: 10.1002/jemt.10360. [DOI] [PubMed] [Google Scholar]
- Jacobs MD, Soeller C, Sisley AM, Cannell MB, Donaldson PJ. Gap junction processing and redistribution revealed by quantitative optical measurements of connexin46 epitopes in the lens. Investigative Ophthalmology and Visual Science. 2004;45:191–199. doi: 10.1167/iovs.03-0148. [DOI] [PubMed] [Google Scholar]
- Kistler J, Gilbert K, Brooks HV, Jolly RD, Hopcroft DH, Bullivant S. Membrane interlocking domains in the lens. Investigative Ophthalmology and Visual Science. 1986;27:1527–1534. [PubMed] [Google Scholar]
- Kuklinski S, Probstmeier R. Homophilic binding properties of Galectin-3: Involvement of the carbohydrate recognition domain. Journal of Neurochemistry. 1998;70:814–823. doi: 10.1046/j.1471-4159.1998.70020814.x. [DOI] [PubMed] [Google Scholar]
- Kushmerick C, Rice SJ, Baldo GJ, Haspel HC, Mathias RT. Ion, water, neutral solute transport in Xenopus oocytes expressing frog lens MIP. Experimental Eye Research. 1995;61:351–362. doi: 10.1016/s0014-4835(05)80129-0. [DOI] [PubMed] [Google Scholar]
- Lim J, Lam YC, Kistler J, Donaldson PJ. Molecular characterization of the cystine/glutamate exchanger and the excitatory amino acid transporters in the rat lens. Investigative Ophthalmology and Visual Science. 2005;46:2869–2877. doi: 10.1167/iovs.05-0156. [DOI] [PubMed] [Google Scholar]
- Lin JS, Eckert R, Kistler J, Donaldson P. Spatial differences in gap junction gating in the lens are a consequence of connexin cleavage. European Journal of Cell Biology. 1998;76:246–250. doi: 10.1016/s0171-9335(98)80002-2. [DOI] [PubMed] [Google Scholar]
- Lin JS, Fitzgerald S, Dong Y, Knight C, Donaldson P, Kistler J. Processing of the gap junction protein connexin50 in the ocular lens is accomplished by calpain. European Journal of Cell Biology. 1997;73:141–149. [PubMed] [Google Scholar]
- Lindsey Rose KM, Gourdie RG, Prescott AR, Quinlan RA, Crouch RK, Schey KL. The C terminus of lens Aquaporin 0 interacts with the cytoskeletal proteins filensin and CP49. Investigative Ophthalmology and Visual Science. 2006;47:1562–1570. doi: 10.1167/iovs.05-1313. [DOI] [PubMed] [Google Scholar]
- Lo WK, Wen XJ, Zhou CJ. Microtubule configuration and membranous vesicle transport in elongating fiber cells of the rat lens. Experimental Eye Research. 2003;77:615–626. doi: 10.1016/s0014-4835(03)00176-3. [DOI] [PubMed] [Google Scholar]
- Lovicu FJ, Robinson ML, editors. Development of the Ocular Lens. Cambridge University Press; Cambridge: 2004. [Google Scholar]
- Ma H, Azuma M, Shearer TR. Degradation of human aquaporin 0 by m-calpain. FEBS Letters. 2005;579:6745–6748. doi: 10.1016/j.febslet.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Ma H, Fukiage C, Azuma M, Shearer TR. Cloning and expression of mRNA for calpain Lp82 from rat lens: Splice variant of p94. Investigative Ophthalmology and Visual Science. 1998a;39:454–461. [PubMed] [Google Scholar]
- Ma H, Fukiage C, Kim YH, Duncan MK, Reed NA, Shih M, Azuma M, Shearer TR. Characterization and expression of calpain 10 - A novel ubiquitous calpain with nuclear localization. Journal of Biological Chemistry. 2001;276:28525–28531. doi: 10.1074/jbc.M100603200. [DOI] [PubMed] [Google Scholar]
- Ma H, Shih M, Hata I, Fukiage C, Azuma M, Shearer TR. Protein for Lp82 calpain is expressed and enzymatically active in young rat lens. Experimental Eye Research. 1998b;67:221–229. doi: 10.1006/exer.1998.0515. [DOI] [PubMed] [Google Scholar]
- Mathias RT, Rae JL, Baldo GJ. Physiological properties of the normal lens. Physiological Reviews. 1997;77:21–50. doi: 10.1152/physrev.1997.77.1.21. [DOI] [PubMed] [Google Scholar]
- Michea LF, Andrinolo D, Ceppi H, Lagos N. Biochemical evidence for adhesion-promoting role of major intrinsic protein isolated from both normal and cataractous human lenses. Experimental Eye Research. 1995;61:293–301. doi: 10.1016/s0014-4835(05)80124-1. [DOI] [PubMed] [Google Scholar]
- Mulders SM, Preston GM, Deen PMT, Guggino WG, An Os CD, Agre P. Water channel properties of major intrinsic protein of lens. Journal of Biological Chemistry. 1995;270:9010–9016. doi: 10.1074/jbc.270.15.9010. [DOI] [PubMed] [Google Scholar]
- Nakajima E, Walkup RD, Ma H, Shearer TR, Azuma M. Low activity by the calpain system in primate lenses causes resistance to calcium-induced proteolysis. Experimental Eye Research. 2006;83:593–601. doi: 10.1016/j.exer.2006.02.014. [DOI] [PubMed] [Google Scholar]
- Németh-Cahalan KL, Hall JE. pH and calcium regulate the water permeability of aquaporin 0. Journal of Biological Chemistry. 2000;275:6777–6782. doi: 10.1074/jbc.275.10.6777. [DOI] [PubMed] [Google Scholar]
- Németh-Cahalan KL, Kalman K, Hall JE. Molecular basis of pH and Ca2+ regulation of aquaporin water permeability. The Journal of General Physiology. 2004;123:573–580. doi: 10.1085/jgp.200308990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palanivelu DV, Kozono DE, Engel A, Suda K, Lustig A, Agre P, Schirmer T. Co-axial association of recombinant eye lens aquaporin-0 observed in loosely packed 3D crystals. Journal of Molecular Biology. 2006;355:605–611. doi: 10.1016/j.jmb.2005.10.032. [DOI] [PubMed] [Google Scholar]
- Piatigorsky J. Lens differentiation in vertebrates: A review of cellular and molecular features. Differentiation. 1981;19:134–153. doi: 10.1111/j.1432-0436.1981.tb01141.x. [DOI] [PubMed] [Google Scholar]
- Rose KML, Wang Z, Magrath GN, Hazard EN, Hildebrandt JD, Schey KL. Aquaporin 0-calmodulin interaction and the effect of aquaporin 0 phosphorylation. Biochemistry. 2008;47:339–347. doi: 10.1021/bi701980t. [DOI] [PubMed] [Google Scholar]
- Sas DF, Sas J, Johnson KR, Menko AS, Johnson RG. Junctions between lens fiber cells are labeled with a monoclonal antibody shown to be specific for MP26. Journal of Cell Biology. 1985;100:216–225. doi: 10.1083/jcb.100.1.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schey KL, Fowler JG, Schwartz JC, Busman M, Dillon J, Crouch RK. Complete map and identification of the phosphorylation site of bovine lens major intrinsic protein. Investigative Ophthalmology and Visual Science. 1997;38:2508–2515. [PubMed] [Google Scholar]
- Schey KL, Fowler JG, Shearer TR, David L. Modifications of rat lens major intrinsic protein in selenite-induced cataract. Investigative Ophthalmology and Visual Science. 1999;40:657–667. [PubMed] [Google Scholar]
- Shearer TR, Ma H, Shih M, Fukiage C, Azuma M. Calpains in the lens and cataractogenesis. Methods in Molecular Biology. 2000;144:277–285. doi: 10.1385/1-59259-050-0:277. [DOI] [PubMed] [Google Scholar]
- Shiels A, Bassnett S. Mutations in the founder of the MIP gene family underlie cataract development in the mouse. Nature Genetics. 1996;12:212–215. doi: 10.1038/ng0296-212. [DOI] [PubMed] [Google Scholar]
- Shiels A, Bassnett S, Varadaraj K, Mathias RT, Al-Ghoul K, Kuszak JR, Donoviel D, Lilleberg S, Friedrich G, Zambrowicz B. Optical dysfunction of the crystalline lens in aquaporin-0-deficient mice. Physiological Genomics. 2001;7:179–186. doi: 10.1152/physiolgenomics.00078.2001. [DOI] [PubMed] [Google Scholar]
- Shiels A, Collis H, Bassnett S. Transposin mutagenesis and intracellular misrouting of lens Major Intrinsic Protein (MIP) in the cataract Fraser (CatFr) mouse. Investigative Ophthalmology and Visual Science. 1999;40:s878. [Google Scholar]
- Thibault DB, Gillam CJ, Grey AC, Han J, Schey KL. MALDI tissue profiling of integral membrane proteins from ocular tissues. Journal of the American Society for Mass Spectrometry. 2008;19:814–822. doi: 10.1016/j.jasms.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadaraj K, Kumari S, Shiels A, Mathias RT. Regulation of aquaporin water permeability in the lens. Investigative Ophthalmology and Visual Science. 2005;46:1393–1402. doi: 10.1167/iovs.04-1217. [DOI] [PubMed] [Google Scholar]
- Wistow G, Piatigorsky J. Lens crystallins: The evolution and expression of proteins for a highly specialized tissue. Annual Review of Biochemistry. 1988;57:479–504. doi: 10.1146/annurev.bi.57.070188.002403. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Murachi T, Tsukahara I. Distribution of calpain I, calpain II, and calpastatin in bovine lens. Investigative Ophthalmology and Visual Science. 1985;26:953–956. [PubMed] [Google Scholar]
- Zampighi GA, Eskandari S, Hall JE, Zampighi L, Kreman M. Micro-domains of AQP0 in lens equatorial fibers. Experimental Eye Research. 2002;75:505–519. doi: 10.1006/exer.2002.2041. [DOI] [PubMed] [Google Scholar]
- Zampighi GA, Hall JE, Ehring GR, Simon SA. The structural organization and protein composition of lens fiber junctions. Journal of Cell Biology. 1989;108:2255–2275. doi: 10.1083/jcb.108.6.2255. [DOI] [PMC free article] [PubMed] [Google Scholar]