

Abstract
The functional significance of mono-, di-, and tri-methylation of lysine residues within histone proteins is under investigation. Evidence from several model organisms suggests that different methylated states of H3 Lys9 (H3K9) are generated by specific histone methyltransferases (MTases) to mark distinct types of silent chromatin. Sequence alignment of all histone lysine MTases with known product specificity suggested that a key residue in the active site determines how many methyl groups they add. We examined this possibility both in vitro and in vivo and found that a Phe at the position equivalent to Phe281 of Neurospora crassa DIM-5 or Phe1205 of human G9a allows the enzyme to perform di and tri-methylation, whereas a Tyr at this position is restrictive, inhibiting tri-methylation and thus yielding a mono- or di-MTase. Phe to Tyr mutants of both DIM-5 and G9a restrict product specificity in vitro and in vivo without compromising overall catalysis. These mutants were employed to probe the biological significance of mono-, di-, and tri-methylation of H3K9 in both mouse embryonic stem cells and N. crassa. G9a F1205Y, when expressed in G9a (−/−) embryonic stem cells, rescued only H3K9 mono-methylation, but not di-methylation, to wild-type levels yet silenced Mage-a gene expression. When expressed in dim-5 strains, DIM-5 F281Y generated significant levels of mono- and di-H3K9 methylation (which are not observed in wild type Neurospora) as well as tri-methyl H3K9. The altered DIM-5 rescued the growth defect characteristic of dim-5 N. crassa but did not fully rescue the gross DNA hypomethylation of dim-5 strains.
Histones are subject to diverse post-translational modifications including acetylation, phosphorylation, ubiquitination, methylation, and sumoylation. Evidence accumulated over the past few years suggests that such modifications constitute a “histone code” that directs a variety of processes involving chromatin (1, 2). Considering just methylation of lysines, there are at least six modification sites (Lys4, Lys9, Lys27, Lys36, and Lys79 of histone H3 and Lys20 of histone H4), and in principle each site can have zero, one, two, or three methyl groups. It has been suggested that methylation at these sites, in combination with other nearby modifications, generates “modification cassettes” (3), yielding distinct patterns on chromatin for signaling downstream events (reviewed in Refs. 3 and 4).
With only one known exception, histone lysine methyltransferases (HKMTs)1 contain a SET domain of ~130 amino acids. SET proteins can be grouped into families according to the sequences surrounding this distinctive domain (5, 6) (see Table I). In this study we focus on two members of the SUV family that methylate Lys9 of histone H3, G9a characterized in mammals and DIM-5 characterized in the filamentous fungus Neurospora crassa. DIM-5 provided the first evidence that histone methylation can direct DNA methylation (7). DIM-5 normally generates primarily tri-methyl-Lys9 on histone H3 (8, 9). G9a is essential for early embryogenesis and is the predominant H3 Lys9 HKMT that directs euchromatic mono- and di-methylation (10–12). G9a apparently plays key roles in later development, because its activity is required for PRDI-BF1 (BLIMP-1) silencing in terminal B-cell differentiation (13), for neuron-restrictive silencing factor/REST-mediated silencing of neuronal genes in non-neuronal lineages (14), and for CCAAT displacement protein/cut-mediated silencing of many diverse genes involved in cell proliferation and differentiation (15). G9a and its close relative GLP-1 (G9a-like protein-1, also called EuHMT1) are also known to reside in CtBP co-repressor (16) and E2F-6.com-1 complexes (17). Like other members of the SUV family, DIM-5 of N. crassa (7), KYP of Arabidopsis (18), and Suv39h of mouse (19), G9a has been implicated in DNA methylation, because G9a (−/−) cells lack DNA methylation of the Prader-Willi syndrome imprinting center (20).
Table I.
The location of Phe/Tyr switch

|
We wished to investigate the mechanism and consequences of different product specificities (mono-, di-, or tri-methylation) of HKMTs. With the advent of antibodies specific for mono-, di-, or tri-methylation of various lysines, it became increasingly evident that product specificity can be important for generating distinct regulatory signals (11, 12). For example, in Saccharomyces cerevisiae, SET-1 can di- or tri-methylate H3 Lys4, but only tri-methylation is associated with the early stage of active transcription; di-methylation is globally distributed (21, 22). Human SET7/9 protein, on the other hand, generates mono-methyl Lys4 of H3 (9, 23). Mouse Suv39h specifically tri-methylates H3K9 at pericentric heterochromatin, whereas centro-meric regions display Suv39h-independent di-methylation (4, 19). Tri-methylated H3 Lys27 but not di-methylated Lys27 is associated with X inactivation (24). In contrast to DIM-5, which tri-methylates Lys9 of histone H3 to mark chromatin regions for DNA methylation (8), in Arabidopsis thaliana, the KRYP-TONITE HKMT dimethylates Lys9 (25).
By comparing the active sites of SET7/9 (a mono-MTase) and DIM-5 (a tri-MTase), we designed point mutations in both enzymes that profoundly altered their product specificities without affecting their catalytic activities (9). This led us to propose that a few key residues in the active site of HKMTs determine how many methyl groups they add, and the product specificity may be predictable from their primary sequences. We show that G9a forms mono- and di-methyl Lys9 as initial products and then slowly adds the third methyl group, consistent with reports that G9a is a global mono- and di-MTase (11, 12) but can generate trimethyl-H3K9 in some situations (26). We also demonstrate that a single point mutation, F1205Y, converts G9a to a mono-MTase. When expressed in G9a (−/−) ES cells, the F1205Y mutant rescues mono-, but not di-methyl H3K9 levels, yet represses Mage-a gene expression to wild-type levels. Similarly, the F281Y mutant of DIM-5, when expressed in a dim-5 null background, yields significant levels of mono- and di-methyl H3K9 that are not observed in the wild-type Neurospora. The presence of the mono- and di-methyl marks is incompatible with the function of DIM-5 in maintaining normal DNA cytosine methylation. In contrast, the remaining tri-methylation and/or the increased levels of mono- and di-methylation by the F281Y mutant is sufficient to rescue the characteristic growth defect of the dim-5 strain.
MATERIALS AND METHODS
Protein Expression and Purification
A fragment encoding the C-terminal 280 residues of human G9a was amplified from an expressed sequence tag clone BC002686 (Resgen) and subcloned between the BamHI and EcoRI sites of pGEx2T (Amersham Biosciences), yielding pXC428. BL21(DE3) Codon-Plus RIL (Stratagene) cells harboring pXC428 were grown in LB supplemented with 100 mg/liter ampicillin, 50 mg/liter chloramphenicol, and 25 μM ZnSO4 at 37 °C until A600 = 0.5. The culture was shifted to 16 °C and induced by the addition of 0.4 mM isopropyl β-D-thiogalactopyranoside for 16 h. The cells were collected by centrifugation, lysed with a French press, and applied to glutathione-Sepharose 4B (Amersham Biosciences) in a batch slurry. Thrombin cleavage of the glutathione S-transferase tag was performed on the beads, yielding the G9a catalytic domain following four residues: GSHM from the vector. Eluted protein was concentrated to 1 mg/ml, flash frozen, and stored at −80 °C in 25 mM Tris, pH 8.5, 50 mM NaCl, 2 mM dithiothreitol, and 5% glycerol. DIM-5 was expressed and purified as described (27).
Mutagenesis
Mutagenesis was performed with the QuikChange site-directed mutagenesis kit (Stratagene). The human G9a amino acid, corresponding to 1205 of the G9a mouse long isoform (10), in pXC428 was converted from Phe (TTT) to Tyr (TAT), yielding pXC444. The construct was fully sequenced to confirm the desired mutation and the lack of unintended mutations. Generation of DIM-5 mutants Y178V and F281Y has been described (9).
Mass Spectrometry Analysis
Methylation reactions for G9a were performed in 25 mM Tris, pH 8.5, 5 mM dithiothreitol, 250 μM S-adenosyl-L-methionine, 10 μM peptide (H3 amino acids 1–15, 1–24, or 21–34), and 0.1 mg/ml enzyme at room temperature. For DIM-5, 20 μM of peptide and 0.02 mg/ml of the enzyme were used. The reactions were stopped at various times by the addition of trifluoroacetic acid to 0.5%. MALDI-TOF mass spectrometry was performed on an Applied Biosystem Voyager System 4258 (Emory University Chemistry Department) with α-cyano-4-hydroxycinnamic acid as matrix. The final spectra are the averages of 200 shots/position at 10 different positions.
Construction of Neurospora Strains That Carry a dim-5 Nonsense Mutation at the dim-5 Locus and the dim-5WT, dim-5Y178V, or dim-5F281Y Allele at the his-3 Locus
A fragment containing 165 bp of 5′-noncoding sequence and the entire open reading frame of the dim-5 gene was amplified from a N. crassa wild-type strain (N1) by PCR with Herculase polymerase (Stratagene) and a pair of primers, oligo-nucleotide 1282 (5′-CGGAATTCTTACCACAGATAGCCTCTGCACTT-3′) and oligonucleotide 1283 (5′-CGGGATCCACGCTAAGCCATCTTTCTCTCTCA-3′). The resulting PCR product was digested with EcoRI and BamHI, gel-purified, and cloned into a his-3 targeting vector, pBM61 (28), yielding pHT15. A BglII fragment carrying the wild-type dim-5 sequence in pHT15 was replaced with the corresponding dim-5 fragment from pXC379F281Y or from pXC379Y178V (9), yielding pHT16 and pHT17, respectively. The dim-5 null strain N2264 (mat a his-3; leu-2 dim-5HT1pan-1) was transformed by electroporation (28) with NdeI-linearized pHT15, pHT16, or pHT17, and his-3+ transformants were isolated. Correctly targeted constructs at his-3 in the resulting transformant strains were verified by Southern hybridizations probed with a his-3 fragment.
PCR Primers
Pairs of PCR primers used to amplify fragments of pcn (470 bp), hH4 (425 bp), 1d21 (355 bp), 8A6 (302 bp), and 8F10 (316 bp) were: 560 pcn-FWD (ATGCTTGAAGCACGGTTGGAG), 561 pcn-REV (CCCAGGTGCTCCTGGTCAATG); 557 hH4-FWD (AACCACCGAAACCGTAGAGGGTAC), and 562 hH4-REV (ATCGCCGACACCGTGTGTTGTAAC); 1d21-FWD (5′-GTTCGGTTCCTCTGTCTTCCTTG-3′) and 1d21-REV-2 5′-CGC CGC ACA GAA TAT TAT AGC TAT C-3′; 8A6-FWD 5′-GGA TGG CGG ATC CTC AAA AAT A-3′and 8A6-REV 5′-TAA CCG CCG CTT TTT AAA ATT AGG A-3′; and 8F10-FWD 5′-GTA ACG CAA ATT CTA AAA TTG CAA TAC-3′and 8F10-REV 5′-CTT AGT AAT TAA TTT AAT ACG TGC GCC-3′.
Western Analysis of DIM-5 Variants
We germinated asexual spores (~5 × 106 spores/ml) of the Neurospora strain N150 (mat A), N2264 (mat a leu-2 dim-5HT1 pan-1), N2732 (mat a his-3+::dim-5WT; leu-2 dim-5HT1 pan-1) N2738 (mat a his-3+::dim-5F281Y; leu-2 dim-5HT1 pan-1), or N2744 (mat a his-3+::dim-5Y178V; leu-2 dim-5HT1 pan-1) for 16 h in 25 ml of Vogel’s minimal liquid medium supplemented with 1.5% sucrose, leucine, and pantothenic acid at 32 °C with shaking (150 rpm). 100 mg of tissue was quick frozen at −70 °C and then disrupted by bead beating in breaking buffer (20 mM HEPES-NaOH pH 7.5, 100 mM KCl, 2 mM EDTA, 10 mM dithiothreitol, 20% glycerol, 1 mM phenylmethylsulfonyl fluoride). The soluble fraction was separated by SDS-PAGE (12%) and transferred to polyvinylidene difluoride membrane in 10 mM 3-(cyclohexylamino)-1-propanesulfonic acid, pH 11, 20% methanol. Immunoblotting was performed using standard techniques. Rabbit polyclonal antibody against recombinant DIM-5 was affinity-purified using recombinant DIM-5 protein coupled to NHS-activated Sepharose (Amersham Biosciences).
Chromatin Immunoprecipitation
We performed chromatin immunoprecipitation as described previously (8), with minor modifications. Briefly, we germinated asexual spores (~5 × 106 spores/ml) of the Neurospora strain N2732 (mat a his-3+::dim-5WT; leu-2 dim-5HT1 pan-1) or N2738 (mat his-3+::dim-5F281Y; leu-2 dim-5HT1 pan-1) for 4.5 h in 50 ml of Vogel’s minimal liquid medium supplemented with 1.5% sucrose, leucine, and panthothenic acid at 32 °C with shaking (200 rpm). For chromatin fixation, paraformaldehyde was added to 2% and incubated at 32 °C for 30 min with shaking (100 rpm). After cell lysis, we sheared chromatin DNA to 0.5–0.8 kb and immunoprecipitated the soluble chromatin fraction using 2–8 μl of antibodies to mono-, di-, or tri-methylated Lys9 or dimethylated Lys4 of histone H3. Antibodies to methylated H3K9 were obtained from T. Jenuwein of University of Vienna (mono-methylated), Upstate Biotechnology Inc. (di-methylated), and P. B. Singh of Roslin Institute (tri-methylated) and the antibody to methylated H3K4 was obtained from Upstate Biotechnology Inc.
DNA was isolated from immunoprecipitated chromatin or total chromatin and subjected to PCR (95 °C for 30 s, 56 °C for 30 s, and 72 °C for 1 min for 24 cycles). PCRs (25 μl) included 50 mM KCl, 10 mM Tris-HCl, pH 9.0, 0.1% Triton X-100, 2.5 mM MgCl2, 0.2 mM dATP, 0.2 mM dTTP, 0.2 mM dCTP, 0.2 mM dGTP, 2.5 μCi of [γ-32P] dCTP, a 0.5-μl sample DNA, and 1.25 units of Taq polymerase (Promega). Under the chosen PCR conditions, a linear relationship was found between the amount of input DNA and the band intensities of PCR products (8).
Western Analysis of Neurospora Histone H3
Neurospora nuclei were isolated from N2732 (mat a his-3+::dim-5WT; leu-2 dim-5HT1 pan-1), N2744 (mat a his-3+::dim-5Y178V; leu-2 dim-5HT1 pan-1) or N2738 (mat his-3+::dim-5F281Y; leu-2 dim-5HT1 pan-1) strains and lysed in the buffer used for chromatin immunoprecipitation (ChIP) analysis (29). Nuclear lysates (~25 μg of nuclear proteins) were boiled in SDS gel loading buffer, clarified by centrifugation, fractionated by SDS-PAGE (18%), transferred to polyvinylidene difluoride membrane in 10 mM 3-(cyclohexylamino)-1-propanesulfonic acid, pH 11, containing 20% methanol at 70 V for 2 h, and immunoblotted as described previously (8). Calf thymus histones (Roche Applied Science) were included as control (2.5 μg). Obtained antibodies to methylated H3K9 were described in the previous section.
Analysis of DNA Methylation
Genomic DNA was isolated from liquid cultures grown for 2 days at 32 °C and analyzed for DNA methylation as described (30). The DNA (~1 μg) was digested with DpnII or Sau3AI and analyzed by Southern hybridization. Probes for methylated DNA regions 1d21, 8A6, and 8F10 used were PCR products generated with the primer pairs described above.
G9a ES Cells
Undifferentiated ES cells were maintained in 10% fetal calf serum and leukemia-inducing factor (500 units/ml)-containing medium. G9a long isoform (G9a-L) cDNA carrying the F1205Y mutation was generated by conventional double PCR mutagenesis and sub-cloned into the expression vector, pCAGGS. The G9a-L F1205Y expression vector was introduced into ES cells using Lipofectamine 2000 reagent (Invitrogen) according to the manual.
RESULTS
G9a Is a Fast Mono/di-MTase and a Slow Tri-MTase
We have previously used MALDI-TOF mass spectrometry to monitor the kinetic progression of methylation reactions and determined that DIM-5 is a tri-MTase and SET7/9 is a mono-MTase (8, 9) (Fig. 1A). Here we report a similar investigation of the product specificity of the G9a catalytic domain by monitoring the methylation of a synthetic histone H3 peptide, residues H3 1–24 (Fig. 1B) or H3 1–15 (Fig. 2, left panels), by mass spectrometry. G9a produced both mono- and di-methylated peptides early in the reaction. Di-methylated H3K9 was the dominant form after about 1 min on both the 1–24 and 1–15 peptides, shortly after a burst of mono-methyl product coincident with the rapid disappearance of unmethylated peptides. Notably, tri-methylated H3K9 was first detected only after the di-methyl product was maximal and both un-modified and mono-methylated peptides had disappeared. The conversion of di- to tri-methyl lysine was much slower, not becoming complete until after 30 min in both cases. These observations suggest that mono and di-methyl H3K9 are the initial products of G9a. This is in sharp contrast to DIM-5, which shows a direct progression to tri-methyl lysine without significant accumulation of di-methyl lysine (8, 9) (Fig. 1A) and SET7/9, which produces only mono-methyl lysine plus a trace of di-methyl lysine after overnight incubation (9). These biochemical findings are consistent with in vivo observations that G9a is a global mono- and di-HKMT; specifically, G9a (−/−) ES cells lose global euchromatic mono and di-methyl H3K9 immuno-staining (11, 12). Interestingly, our results suggest G9a could generate tri-methyl H3K9 at regions where it has high local concentrations. In fact, G9a tethered to a synthetic mini-locus as a Gal4DBD fusion generates tri-methyl lysine in vivo (26). Whether G9a makes trimethyl-H3K9 under normal circumstances in vivo remains to be determined.
Fig. 1. Mass spectrometry analysis of methylation kinetics.

A, DIM-5 on histone H3 peptide (amino acids 1–24). B, G9a on histone H3 peptide (amino acids 1–24). C, G9a on histone H3 peptide (amino acids 21–34). The average relative amounts of each peptide across the time course of the reaction as a percentage of the sum intensity of all of the peaks from an averaged (>200 shots) MALDI-TOF spectrum is reported.
Fig. 2. Generation of mono-methyl H3K9 (H3 amino acids 1–15) by G9a F1205Y variant without impaired catalytic ability.

A–C, representative mass spectra (from a single reaction) at various time points for WT G9a (left panels) and F1205Y (right panels). Unmodified, mono-, di-, and tri-methyl peptides are labeled. D, the relative amount of each peptide species over the full time courses of the reactions, expressed as a percentage of the sum of intensity of all related peaks. a.i., absolute intensity.
Considering that G9a has also been reported to have activity on H3K27 (10), we also tested G9a product specificity on a H3 peptide containing Lys27 (Fig. 1C). We found that G9a has very poor activity on this lysine, some 100-fold worse than on peptides of similar length that include Lys9. Nevertheless, G9a did produce mono- and di-methyl H3K27 simultaneously in the early stages of the reaction and very slowly generated tri-methyl Lys27 (~24 h). This low activity is consistent with the finding that methyl-H3K27 levels are unaffected in G9a (−/−) ES cells (11) and that G9a is a strong H3K9 MTase but has low or nondetectable H3K27 MTase activity, even when assayed as a member of a complex isolated by co-immunoprecipitation (13, 15). We conclude that G9a has poor activity on H3K27 in vitro, and it is unclear whether it has any significant effect on H3K27 methylation in vivo.
Replacement of Phe1205 with Tyr Converts G9a to a Mono-MTase
Comparison of the crystal structures for DIM-5 and SET7/9 led us to generate mutants (F281Y in DIM-5 and Y305F in SET7/9) with altered product specificity (9). To test the hypothesis that the corresponding residue Phe1205 in G9a (Table I) is the major determinant of product specificity, we replaced this residue with Tyr (F1205Y). The mutation did not significantly affect the catalytic activity of G9a, because the rate of loss of unmethylated peptide was roughly equal to that of wild-type (WT) enzyme (Fig. 2D, right panel). However, the reaction by F1205Y stalled at the mono-methyl stage (compare 1.5 and 10 min; Fig. 2, A and B), supporting our hypothesis. A trace amount of di-methylated product was observed only after prolonged incubation (45 min; Fig. 2C), which indicated that di-methylation by the altered enzyme was actually slower than the tri-methylation by the WT G9a. We conclude that the F1205Y mutation changed the product specificity of G9a from a fast mono/di-MTase with a slow tri-MTase activity to a predominantly mono-MTase without affecting overall catalytic activity, analogous to the F281Y mutation for DIM-5 (9).
Monomethyl G9a Silences Mage-a Gene Expression
G9a (−/−) ES cells lack global mono- and di-methyl H3K9 and show aberrant expression of Mage-a genes (11, 12, 31). A G9a trans-gene with the F1205Y mutation was expressed as a full-length G9a (long isoform) transgene in G9a (−/−) mouse ES cells. When expressed at WT levels (Fig. 3A), the F1205Y mutant rescued H3K9 mono- but not di-methylation to WT levels (Fig. 3B), consistent with our biochemical data. Interestingly, Northern hybridization results showed that this mono-methylation of Lys9 was sufficient to repress Mage-a (Fig. 3C). This result may reflect an unknown repressive lysine-binding module that recognizes mono- and/or di-H3K9, or it may be that mono- and di-H3K9 can both block H3K9 acetylation and neighboring marks required for transcription of the Mage genes.
Fig. 3. G9a F1205Y variant silences Mage-a gene expression.
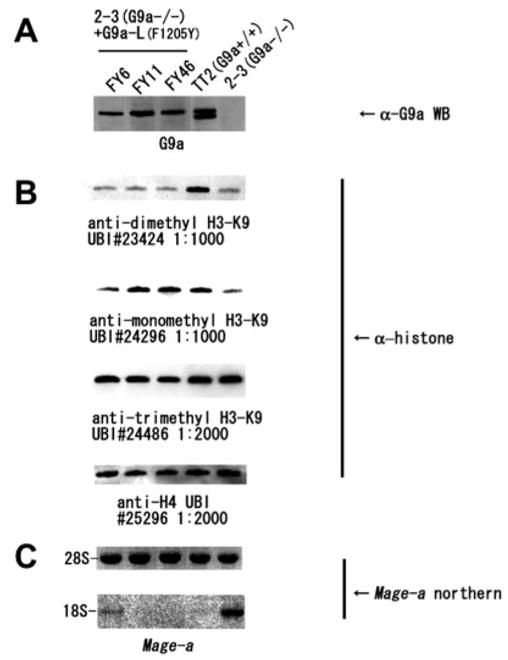
A, G9a long transgene expression level detected by Western blot. B, Western blot for mono-, di-, and tri-H3K9 levels with histone H4 loaded for control. C, Mage-a Northern blot. FY6, FY11, and FY46 indicate F1205Y transgene expression cell lines; TT2 indicates WT ES cells; and 2–3 indicates G9a(−/−) ES cells. UBI #23424, UBI #24296, UBI #24486, and UBI #25296 refer to the Upstate Biotechnology Inc. lot numbers for each product.
F281Y DIM-5 Partially Rescues Phenotypes of dim-5 Mutant
A lack of DIM-5 HKMT leads to gross hypomethylation of H3K9, which is a required mark for DNA methylation in Neurospora (7, 8). In addition, dim-5 strains are characterized by growth defects and poor fertility. Neurospora strains that lack all DNA methylation because of a null mutation in the DNA MTase gene dim-2 do not show these defects, suggesting that DIM-5, or its product trimethyl-H3K9, has functions beyond DNA methylation. To determine whether DNA methylation and other functions of DIM-5 activity rely on tri-methylation of H3K9, we introduced the F281Y substitution, which, in vitro, dramatically changed the product specificity of the enzyme from tri- to mono-H3K9 (with slight residual di-methyl activity) into a Neurospora strain bearing a nonsense mutation in dim-5. We then tested the transformed strains for mono-, di-, and tri-methylation and scored for physiological effects. As controls, we also transformed the dim-5 host strain with the WT dim-5 gene as well as with a second mutated gene (Y178V), resulting in a catalytically inactive variant (~0.5% WT activity) (9). All of the constructs were targeted to the his-3 locus. The ectopic WT and F281Y mutant expressed at similar levels as wild-type DIM-5, as determined by Western blot (Fig. 4A), but the Y178V mutant was not detected, presumably because of instability of the mutant protein.
Fig. 4. DIM-5 F281Y variant partially rescues defects of dim-5 mutant.

A, DIM-5 expression is similar from both the ectopic dim-5WT allele and the dim-5HT1 transformants carrying dim-5WT or dim-5F281Y alleles at his-3. The dim-5Y178V allele, however, is either not expressed or the resulting mutant protein is unstable. B, expression of DIM-5F281Y variant in N. crassa produces mono-, di-, and tri-methylated H3K9, whereas the WT DIM-5 produces exclusively trimethylated H3K9. Western blot analysis of N. crassa histone H3 using antibodies against mono-, di-, or tri-methylated H3K9. Coomassie Blue staining of a replica gel confirmed that the amount of histone H3 was approximately even between the WT, dim-5Y178V, and dim-5F281Y protein samples. C, chromatin immunoprecipitation experiments with chromatin from dim-5HT1 transformants carrying dim-5Y178V (inactive), dim-5WT, or dim-5F281Y alleles at his-3 were carried out using the indicated antibodies. Total DNA and immunoprecipitated DNA were subject to duplex PCR to amplify pairs of unmethylated (hH4 and pcn) and naturally methylated (1d21, 8A6, and 8F10; underlined) DNA regions of N. crassa. Note that the F281Y mutant, but not the inactive and wild-type alleles, show significant mono- and di-methyl-H3K9 signals in the regions that are subject to DNA methylation. The specificity mutant (F281Y) also shows a tri-methyl H3K9 signal, although it is somewhat weaker than in the ChIP with the wild-type allele (most obvious in the 1d21 region). In one experiment (right panel), regions were probed for dimethyl-H3K4, which is underrepresented at heterochromatic regions. D, DIM-5F281Y variant poorly complements DNA methylation defect in dim-5HT1 strain. Genomic DNA samples from two independently isolated dim-5HT1 transformants (1 and 2) carrying dim-5WT, dim-5F281Y, or dim-5Y178V alleles at his-3 were digested with DpnII (D) or Sau3AI (S) and analyzed by gel electrophoresis and Southern hybridization using probes for the indicated regions. A photograph of the ethidium bromide-stained gel (total) shows partial complementation on a global level. Note that Sau3AI and DpnII are isoschizomers, but only Sau3AI is sensitive to DNA cytosine methylation.
Western blots showed that the dim-5 strain transformed with the WT gene showed strong tri-methylation of H3K9, but no noticeable mono- or di-methylation of H3K9, as expected (Fig. 4B). The catalytically impaired enzyme also lacked mono-or di-methylated forms of H3K9 but showed a background of tri-methyl H3K9 (as does the dim-5 null strain (8)), possibly because of an unknown HKMT that is not preferentially targeted to regions that are subject to DNA methylation.
Interestingly, results of Western blot and ChIP experiments showed that the F281Y mutant produced mono-, di-, and tri-methyl H3K9 (Fig. 4, B and C). Although the anti-trimethyl antibody has been well characterized (8), we carried out an additional peptide competition experiment that demonstrated that the signal detected in the F281Y transformant is truly tri-methyl H3K9 (data not shown). Although the overall levels of trimethyl-H3K9 seem similar in the F281Y mutant and WT based on the semi-quantitative Western analysis, repeated duplex ChIP experiments, in which a region of the genome that is normally subject to DNA methylation (1d21, 8A6, or 8F10) was compared with a region not subject to DNA methylation (pcn or hH4), revealed modest reductions of tri-methyl Lys9, especially in the 1d21 and 8F10 regions (Fig. 4C). That the F281Y mutant produced substantial levels of tri-methyl H3K9, in addition to mono- and di-methyl H3K9, may be due to strong localization at heterochromatic loci, allowing its very slow residual di- and tri-MTase activity, detected in vitro (9), to act over a prolonged residence time. Another possibility is that an unknown HK-MT(s) can convert the mono- or di-H3K9 products generated by the mutant enzyme to tri-methyl H3K9. We have observed that WT DIM-5, for instance, has a much faster rate on modified (mono- or di-methyl peptides) than unmodified peptides (8). Nevertheless we cannot exclude the possibility that the F281Y mutant behaves principally as a tri-MTase in vivo, in contrast to the situation in vitro. Regardless of these possibilities, the presence of mono- and di-H3K9, which are not observed at significant levels in wild-type Neurospora, is clearly attributable to the reduced product specificity of F281Y DIM-5. The ChIP experiments showed that the mono- and di-methyl forms of H3K9 were specifically targeted to regions to be methylated, as is the case for DIM-5-dependent H3K9 tri-methylation in WT cells (Fig. 4C).
The Y178V mutant completely failed to complement the DNA methylation defect of dim-5 strains, as expected. More interestingly, F281Y poorly restored DNA methylation compared with ectopically expressed WT DIM-5 (Fig. 4D). We conclude that either the presence of mono- and di-methyl K9 or the slight reduction of tri-methyl H3K9 significantly compromised signals for DNA methylation. Curiously, although the F281Y mutant poorly restored DNA methylation, it rescued the dim-5 growth defect to the same degree as WT DIM-5 (Fig. 5). The catalytically impaired mutant (Y178V) did not complement the growth defect but because this mutant failed to produce stable protein (Fig. 4A), we could not determine whether the full rescue of the growth defect is due to some function of DIM-5 not dependent on its methyltransferase activity.
Fig. 5. DIM-5F281Y variant fully restores normal growth in dim-5HT1 strain.

Apical growth rates of six independent his-3-targeted dim-5HT1 transformants carrying dim-5WT, dim-5Y178V, or dim-5F281Y alleles at his-3 were measured at 32 °C using “race tubes” containing sucrose Vogel’s agar medium supplemented with leucine and pantothenic acid.
DISCUSSION
The multiplicity of methylation levels at a given residue is a newly recognized elaboration of the histone code. Each methylation mark (mono-, di-, or tri-) could serve to establish or mask unique binding sites of chromodomain proteins or associated proteins, thereby greatly expanding the complexity of the histone code. Variable degrees of methylation could also serve to tune the output of the “code” by modulating the affinity of lysine binding by chromodomains. The extent to which mono-, di-, and tri-methylation of a single lysine generates a unique mark or act to govern the signal strength of a single mark remains to be seen. The ability to predict the product of each HKMT will be useful in creating engineered mutants to test the biological impact of the variable methylation states of histone lysines. In this study, we found evidence that distinct processes may differ in their reading of a methyl mark. Specifically, in Neurospora, introduction of lower methylation states (mono-and di-methyl) at H3K9 appeared to preclude full complementation of the DNA methylation defect of dim-5 but did not hamper full complementation of the growth and fertility defects. The effect of mono- and/or di-methyl H3K9 on DNA methylation may reflect a requirement for a high local concentration of tri-methyl H3K9 and perhaps both H3 molecules in the nucleosome must be tri-methylated. This is consistent with the finding that the ectopic expression of histone H3 with K9R or K9L mutations in a wild-type background has the dominant effect of bringing about gross DNA hypomethylation (7). Additional work will be required to determine whether the striking difference in complementation reflects differences in the molecules that read the marks. It is conceivable, for example, that HP1, which is known to bind both di- and tri-methylated forms of H3K9 in other organisms (32, 33) and has been demonstrated to be essential for DNA methylation in Neurospora (34), forms distinct complexes that are differentially sensitive to the methylation state.
As previously reported, Phe281 of DIM-5 is critical in determining its product specificity (9). A Tyr at this position generates a mono/di-MTase. Sequence alignment including all HK-MTs with known product specificity suggests that this rule may be generalized (Table I). Arabidopsis KYP and SUVH6 both have a tyrosine and they are both mono/di-MTases (25). From a structural perspective, it appears the tyrosine hydroxyl can block substrate lysines with methyl group(s) attached from rotating into a position where they can be further methylated. In this work, we demonstrated that G9a, which has a Phe at this position, is a fast mono/di-MTase and slow tri-MTase. Interestingly, substituting the Phe of G9a with a Tyr also resulted in a mono-MTase, analogous to DIM-5. This mutant with “switched” product specificity rescued only mono-methyl H3K9 yet represses Mage-a gene expression. It is unknown whether di-methyl H3K9 is critical for other G9a-dependent processes (e.g. maintenance of DNA methylation at imprinting centers, PRDI-BF1 silencing in terminal B-cell differentiation, and neuron-restrictive silencing factor/REST-mediated silencing of neuronal genes).
At face value, S. cerevisiae SET1 protein appears to conflict with our Phe/Tyr switch hypothesis. It has a tyrosine at the position comparable with Phe281 of DIM-5, and yet results of ChIP experiments suggest that it can both di- and tri-methylate H3K4 (21, 22). It should be noted, however, that the published studies did not reveal whether SET1 also produces mono-methyl-Lys4, presumably because the mono-specific antibody was not yet available. Moreover, the product specificity of SET1 has not been determined in vitro because the enzyme appears to be only active as part of a large complex (COMPASS or SET1c), and an active recombinant protein complex has not been obtained (35, 36). It is possible that SET1 is intrinsically a mono/di-MTase but capable of di/tri-methylation when present in the large complex. Alternatively, SET1 may in fact make mono-methyl-K4 in vivo, but other HKMTs that can only utilize mono-methylated H3K4 as substrate carry the methylation to di- or tri-states. ESET, a di-HKMT, is stimulated and converted into a tri-MTase by binding mAM, a murine ATFa-associating modulator, both in vivo and in vitro (37). Curiously, this enzyme (SETDB1 in Table I) has a tryptophan at the key position, unlike other SUV SET domains. It was shown recently that ALL1/MLL1, another member of the SET1 family, is an H3K4 MTase and is required for assembly of a large complex on the promoter of target genes to activate their expression (38, 39). More importantly to this study, recombinant ALL1 fragments of C-terminal 224 residues (39) or 377 residues (38) have robust HKMT activity, but no activity on a di-methylated peptide, consistent with it being a mono/di-MTase. Our limited study of SET domain HKMTs suggests that a Phe/Tyr switch controls their product specificity and that alteration of this product specificity can have important biological consequences. It appears the histone code may show rigidity in some cases and surprising plasticity in others.
Acknowledgments
We are indebted to T. Jenuwein and P. B. Singh for the use of the H3K9 methyl-specific antibodies described in this manuscript.
Footnotes
This work was supported in part by United States Public Health Services Grants GM49245 and GM61355 (to X. Z. and X. C.) and GM35690 (to E. U. S.) and Training Grant T32 GM008367 (to R. E. C), and a Grant-in Aid (to Y. S.) from the Ministry of Education, Science, Technology, and Culture of Japan.
The abbreviations used are: HKMT, histone lysine methyltransferase; MTase, methyltransferase; H3K9, histone H3 lysine 9; ES, embryonic stem; WT, wild-type; MALDI-TOF, matrix-assisted laser desorption ionization time-of-flight; ChIP, chromatin immunoprecipitation.
References
- 1.Jenuwein T, Allis CD. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 2.Strahl BD, Allis CD. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 3.Fischle W, Wang Y, Allis CD. Nature. 2003;425:475–479. doi: 10.1038/nature02017. [DOI] [PubMed] [Google Scholar]
- 4.Lachner M, O’Sullivan RJ, Jenuwein T. J Cell Sci. 2003;116:2117–2124. doi: 10.1242/jcs.00493. [DOI] [PubMed] [Google Scholar]
- 5.Baumbusch LO, Thorstensen T, Krauss V, Fischer A, Naumann K, Assalkhou R, Schulz I, Reuter G, Aalen RB. Nucleic Acids Res. 2001;29:4319–4333. doi: 10.1093/nar/29.21.4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kouzarides T. Curr Opin Genet Dev. 2002;12:198–209. doi: 10.1016/s0959-437x(02)00287-3. [DOI] [PubMed] [Google Scholar]
- 7.Tamaru H, Selker EU. Nature. 2001;414:277–283. doi: 10.1038/35104508. [DOI] [PubMed] [Google Scholar]
- 8.Tamaru H, Zhang X, McMillen D, Singh PB, Nakayama J, Grewal SI, Allis CD, Cheng X, Selker EU. Nat Genet. 2003;34:75–79. doi: 10.1038/ng1143. [DOI] [PubMed] [Google Scholar]
- 9.Zhang X, Yang Z, Khan SI, Horton JR, Tamaru H, Selker EU, Cheng X. Mol Cell. 2003;12:177–185. doi: 10.1016/s1097-2765(03)00224-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tachibana M, Sugimoto K, Fukushima T, Shinkai Y. J Biol Chem. 2001;276:25309–25317. doi: 10.1074/jbc.M101914200. [DOI] [PubMed] [Google Scholar]
- 11.Peters AH, Kubicek S, Mechtler K, O’Sullivan RJ, Derijck AA, Perez-Burgos L, Kohlmaier A, Opravil S, Tachibana M, Shinkai Y, Martens JH, Jenuwein T. Mol Cell. 2003;12:1577–1589. doi: 10.1016/s1097-2765(03)00477-5. [DOI] [PubMed] [Google Scholar]
- 12.Rice JC, Briggs SD, Ueberheide B, Barber CM, Shabanowitz J, Hunt DF, Shinkai Y, Allis CD. Mol Cell. 2003;12:1591–1598. doi: 10.1016/s1097-2765(03)00479-9. [DOI] [PubMed] [Google Scholar]
- 13.Gyory I, Wu J, Fejer G, Seto E, Wright KL. Nat Immunol. 2004;5:299–308. doi: 10.1038/ni1046. [DOI] [PubMed] [Google Scholar]
- 14.Roopra A, Qazi R, Schoenike B, Daley TJ, Morrison JF. Mol Cell. 2004;14:727–738. doi: 10.1016/j.molcel.2004.05.026. [DOI] [PubMed] [Google Scholar]
- 15.Nishio H, Walsh MJ. Proc Natl Acad Sci U S A. 2004;101:11257–11262. doi: 10.1073/pnas.0401343101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi Y, Sawada J, Sui G, Affar el B, Whetstine JR, Lan F, Ogawa H, Luke MP, Nakatani Y. Nature. 2003;422:735–738. doi: 10.1038/nature01550. [DOI] [PubMed] [Google Scholar]
- 17.Ogawa H, Ishiguro K, Gaubatz S, Livingston DM, Nakatani Y. Science. 2002;296:1132–1136. doi: 10.1126/science.1069861. [DOI] [PubMed] [Google Scholar]
- 18.Jackson JP, Lindroth AM, Cao X, Jacobsen SE. Nature. 2002;416:556–560. doi: 10.1038/nature731. [DOI] [PubMed] [Google Scholar]
- 19.Lehnertz B, Ueda Y, Derijck AA, Braunschweig U, Perez-Burgos L, Kubicek S, Chen T, Li E, Jenuwein T, Peters AH. Curr Biol. 2003;13:1192–1200. doi: 10.1016/s0960-9822(03)00432-9. [DOI] [PubMed] [Google Scholar]
- 20.Xin Z, Tachibana M, Guggiari M, Heard E, Shinkai Y, Wagstaff J. J Biol Chem. 2003;278:14996–15000. doi: 10.1074/jbc.M211753200. [DOI] [PubMed] [Google Scholar]
- 21.Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, Schreiber SL, Mellor J, Kouzarides T. Nature. 2002;419:407–411. doi: 10.1038/nature01080. [DOI] [PubMed] [Google Scholar]
- 22.Ng HH, Robert F, Young RA, Struhl K. Mol Cell. 2003;11:709–719. doi: 10.1016/s1097-2765(03)00092-3. [DOI] [PubMed] [Google Scholar]
- 23.Xiao B, Jing C, Wilson JR, Walker PA, Vasisht N, Kelly G, Howell S, Taylor IA, Blackburn GM, Gamblin SJ. Nature. 2003;421:652–656. doi: 10.1038/nature01378. [DOI] [PubMed] [Google Scholar]
- 24.Plath K, Fang J, Mlynarczyk-Evans SK, Cao R, Worringer KA, Wang H, de la Cruz CC, Otte AP, Panning B, Zhang Y. Science. 2003;300:131–135. doi: 10.1126/science.1084274. [DOI] [PubMed] [Google Scholar]
- 25.Jackson JP, Johnson L, Jasencakova Z, Zhang X, PerezBurgos L, Singh PB, Cheng X, Schubert I, Jenuwein T, Jacobsen SE. Chromosoma. 2004;112:308–315. doi: 10.1007/s00412-004-0275-7. [DOI] [PubMed] [Google Scholar]
- 26.Osipovich O, Milley R, Meade A, Tachibana M, Shinkai Y, Krangel MS, Oltz EM. Nat Immunol. 2004;5:309–316. doi: 10.1038/ni1042. [DOI] [PubMed] [Google Scholar]
- 27.Zhang X, Tamaru H, Khan SI, Horton JR, Keefe LJ, Selker EU, Cheng X. Cell. 2002;111:117–127. doi: 10.1016/s0092-8674(02)00999-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Margolin BS, Garrett-Engele PW, Stevens JN, Fritz DY, Garrett-Engele C, Metzenberg RL, Selker EU. Genetics. 1998;149:1787–1797. doi: 10.1093/genetics/149.4.1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakayama J, Klar AJ, Grewal SI. Cell. 2000;101:307–317. doi: 10.1016/s0092-8674(00)80840-5. [DOI] [PubMed] [Google Scholar]
- 30.Foss HM, Roberts CJ, Claeys KM, Selker EU. Science. 1993;262:1737–1741. doi: 10.1126/science.7505062. [DOI] [PubMed] [Google Scholar]
- 31.Tachibana M, Sugimoto K, Nozaki M, Ueda J, Ohta T, Ohki M, Fukuda M, Takeda N, Niida H, Kato H, Shinkai Y. Genes Dev. 2002;16:1779–1791. doi: 10.1101/gad.989402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fischle W, Wang Y, Jacobs SA, Kim Y, Allis CD, Khorasanizadeh S. Genes Dev. 2003;17:1870–1881. doi: 10.1101/gad.1110503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jacobs SA, Khorasanizadeh S. Science. 2002;295:2080–2083. doi: 10.1126/science.1069473. [DOI] [PubMed] [Google Scholar]
- 34.Freitag M, Hickey PC, Khlafallah TK, Read ND, Selker EU. Mol Cell. 2004;13:427–434. doi: 10.1016/s1097-2765(04)00024-3. [DOI] [PubMed] [Google Scholar]
- 35.Roguev A, Schaft D, Shevchenko A, Pijnappel WW, Wilm M, Aasland R, Stewart AF. EMBO J. 2001;20:7137–7148. doi: 10.1093/emboj/20.24.7137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krogan NJ, Dover J, Khorrami S, Greenblatt JF, Schneider J, Johnston M, Shilatifard A. J Biol Chem. 2002;277:10753–10755. doi: 10.1074/jbc.C200023200. [DOI] [PubMed] [Google Scholar]
- 37.Wang H, An W, Cao R, Xia L, Erdjument-Bromage H, Chatton B, Tempst P, Roeder RG, Zhang Y. Mol Cell. 2003;12:475–487. doi: 10.1016/j.molcel.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 38.Milne TA, Briggs SD, Brock HW, Martin ME, Gibbs D, Allis CD, Hess JL. Mol Cell. 2002;10:1107–1117. doi: 10.1016/s1097-2765(02)00741-4. [DOI] [PubMed] [Google Scholar]
- 39.Nakamura T, Mori T, Tada S, Krajewski W, Rozovskaia T, Wassell R, Dubois G, Mazo A, Croce CM, Canaani E. Mol Cell. 2002;10:1119–1128. doi: 10.1016/s1097-2765(02)00740-2. [DOI] [PubMed] [Google Scholar]
- 40.Beisel C, Imhof A, Greene J, Kremmer E, Sauer F. Nature. 2002;419:857–862. doi: 10.1038/nature01126. [DOI] [PubMed] [Google Scholar]
- 41.Strahl BD, Grant PA, Briggs SD, Sun ZW, Bone JR, Caldwell JA, Mollah S, Cook RG, Shabanowitz J, Hunt DF, Allis CD. Mol Cell Biol. 2002;22:1298–1306. doi: 10.1128/mcb.22.5.1298-1306.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cao R, Zhang Y. Mol Cell. 2004;15:57–67. doi: 10.1016/j.molcel.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 43.Fang J, Feng Q, Ketel CS, Wang H, Cao R, Xia L, Erdjument-Bromage H, Tempst P, Simon JA, Zhang Y. Curr Biol. 2002;12:1086–1099. doi: 10.1016/s0960-9822(02)00924-7. [DOI] [PubMed] [Google Scholar]
- 44.Manzur KL, Farooq A, Zeng L, Plotnikova O, Koch AW, Sachchidanand, Zhou MM. Nat Struct Biol. 2003;10:187–196. doi: 10.1038/nsb898. [DOI] [PubMed] [Google Scholar]
- 45.Trievel RC, Flynn EM, Houtz RL, Hurley JH. Nat Struct Biol. 2003;10:545–552. doi: 10.1038/nsb946. [DOI] [PubMed] [Google Scholar]