

Abstract
Mature enzymes encoded within the human immunodeficiency virus type 1 (HIV-1) genome [protease (PR), reverse transcriptase (RT) and integrase (IN)] derive from proteolytic processing of a large polyprotein (Gag-Pol). Gag-Pol processing is catalyzed by the viral PR, which is active as a homodimer. The HIV-1 RT functions as a heterodimer (p66/p51) composed of subunits of 560 and 440 amino acids, respectively. Both subunits have identical amino acid sequence, but p51 lacks 120 residues which are removed by the HIV-1 PR during viral maturation. While p66 is the catalytic subunit, p51 has a primarily structural role. Amino acid substitutions affecting the stability of p66/p51 (i.e. F130W) have a deleterious effect on viral fitness. Previously we showed that the effects of F130W are mediated by p51 and can be compensated by mutation T58S. While studying the dynamics of emergence of the compensatory mutation, we observed that mutations in viral PR-coding region were selected in HIV clones containing the RT substitution F130W, before the imposition of T58S/F130W mutations. The identified PR mutations (G94S and T96S) improved the replication capacity of the F130W mutant virus. By using a trans-complementation assay, we demonstrate that the loss of p66/p51 heterodimer stability caused by Trp-130 can be attributed to an increased susceptibility of RT to viral PR degradation. Recombinant HIV-1 PRs bearing mutations G94S or T96S showed decreased dimer stability and reduced catalytic efficiency. These results were consistent with crystallographic data showing the location of both residues in the PR dimerization interface.
Keywords: HIV, reverse transcriptase, protease, retrovirus, fitness, Gag-Pol
Introduction
The genome of all replication-competent retroviruses consists of at least three genes required for viral infection and replication, arranged in the order 5′-gag-pol-env-3′. In the human immunodeficiency virus type 1 (HIV-1), the expression of those retroviral genes leads to the formation of three polyproteins Gag (Pr55Gag), Gag-Pol (Pr160Gag-Pol), and Env (Pr160Env). The structural proteins of the virus [matrix protein (MA), capsid protein (CA), p2 and nucleocapsid protein (NC)] arise from the proteolytic processing of Gag by the retroviral protease (PR).1 A ribosomal frameshift between the gag and pol genes leads to the synthesis of the Pr160Gag-Pol precursor. The three retroviral enzymes: PR, reverse transcriptase (RT) and integrase (IN) are encoded in the Pol open reading frame, and arise from the proteolytic processing of Gag-Pol by the viral PR.
The HIV-1 PR is a homodimeric enzyme, composed of two identical subunits of 99 amino acids. Each subunit contains the conserved sequence Asp-Thr-Gly, which provides the aspartyl group necessary for catalysis. Crystal structures and biochemical studies have shown that major interactions that stabilize the mature dimer appear in a four-stranded antiparallel β-sheet, where the N-terminal residues of each of the PR monomers form the outer strands of the β-sheet, and the C-terminal residues of each monomer form the two inner strands2 (review 3). PR dimerization and autocatalytic release from Gag-Pol are critical steps in the viral life cycle.4–6
The HIV-1 RT is a heterodimer composed of subunits of 66 kDa (p66) and 51 kDa (p51).7,8 The p66 subunit has 560 amino acids and contains DNA polymerase and endonuclease (RNase H) domains. The p51 subunit has the same amino acid sequence as p66, but lacks the 120 residues of the C-terminal region forming the RNase H domain. Crystallographic studies have shown that the polymerase domains of both p66 and p51 contain four subdomains: fingers, palm, thumb and connection.9 In the p51 subunit, these subdomains adopt a “close” conformation, with its catalytic residues occupying an internal position in the molecule.9–11 The HIV-1 RT heterodimer arises from the viral PR-mediated cleavage of the Pr160Gag-Pol precursor, although the precise pathway for the formation of RT heterodimers in HIV-infected cells is not fully understood. The p51 subunit plays a largely structural role in the heterodimer.12–14 Structural studies have allowed the identification of major contacts between p66 and p51. In p51, residues 52–55 and 135–140 in the fingers subdomain, 255–265 and 286–290 in the thumb subdomain, and 393–402 and 420–423 in the connection subdomain generate the largely hydrophobic surface that interacts with p66.15 Several non-conservative mutations in p51 that affect residues of the β7-β8 loop (positions 134–139)16–19 or the so-called “Trp motif” (residues 398, 401, 402, 406, 410 and 414)20,21 are known to impair RT dimerization, while producing a loss of viral infectivity.
Previously, we showed that the substitution of Trp for Phe-130 in the HIV-1 RT rendered a virus with diminished replication capacity.22 The deleterious effects of F130W were attributed to the loss of stability of p51, based on the characterization of chimeric recombinant RTs with mutated or wild-type subunits. A compensatory mutation in the RT-coding region (T58S) was selected upon passaging the virus in cell culture.22 A detailed study of the dynamics of imposition of double-mutant T58S/F130W has now revealed the early presence of viral subpopulations containing mutations in the dimerization region of the viral PR. Here, we provide further evidence on the deleterious effects of F130W on the stability of p51 using a trans-complementation assay.23 The presence of F130W renders p51 susceptible to the viral PR. The increased viral replication capacity conferred by the compensatory PR mutations G94S (and less frequently, T96S) appears to be a consequence of their destabilizing effect on the PR dimer, which renders an enzyme with reduced proteolytic activity.
Results
Analysis of compensatory mutations emerging with the replication-deficient mutant F130W
Our previous studies showed that cultures transfected with a mutant proviral DNA of HIV-1 subtype B strain 89ES061, carrying a F130W substitution in the RT-coding region rendered virus with impaired replication.22 However, successive passages of the mutant virus in cell culture led to selection of T58S in the viral RT. This compensatory mutation restored viral replication capacity, in the absence of additional mutations in the gag and pol genes of mutant HIV-1 containing F130W (Figure 1(a),(b)).22 Viral protein expression analysis revealed that all three viruses (i.e. wild-type HIV-1 and mutants F130W and T58S/F130W) produced significant amounts of p24 and IN, although mutant F130W showed delayed maturation. As shown in Figure 1(c), the ratio of p24 to its Gag precursor is similar for the wild-type HIV-1 and the double mutant T58S/F130W, but significantly lower for the single-mutant F130W. Interestingly, Western blot analysis with monoclonal antibodies against the RNase H domain of the RT (p66 subunit) (Figure 1(e)) or the RT heterodimer (p66/p51) (Figure 1(f)) revealed undetectable levels of the viral polymerase in the cultures infected with the F130W mutant. These results were consistent with the lower RT activity detected in those culture supernatants (Figure 1(a)). In addition, the similar levels of IN observed with the three variants analyzed suggested that the lower RT levels were not related to a defect in Gag-Pol expression.
Figure 1.

Replication kinetics of wild-type HIV-1 and mutants F130W and T58S/F130W in MT-4 cells. MT-4 cells (6 × 106) were infected at a multiplicity of infection of 0.0001 TCID50 per cell. Infections were monitored by measuring RT activity (a) and p24 production (b). Cell lysates collected when the p24 levels reached the highest values (day 4 for wild-type HIV-1, day 6 for mutant T58S/F130W, and day 9 for mutant F130W) were analyzed by Western blot (c – f). Immunoblots were probed with monoclonal antibodies against p24 (c), IN (d), the RNase H domain of the RT (e), and the RT heterodimer (f). Samples were normalized using a β-actin-specific antibody.
In an attempt to study the population dynamics leading to the imposition of the double mutant T58S/F130W, we carried out a series of transfection experiments with mutant HIV-189ES061 provirus containing the F130W mutation alone. The viral stocks obtained after infecting MT-4 cells with supernatants from transfection experiments were analyzed by DNA sequencing. The nucleotide sequence of the viral stock of mutant F130W used in the experiments shown in Figure 1 was rather homogeneous, since it had only the relevant mutation, with potential heterogeneities representing less than 10 % of the total virus population. However, other stocks of the F130W mutant contained nucleotide sequence heterogeneities in the PR-coding region (Figure 2(a)). In one of them, the compensatory mutation in the RT-coding region (T58S) was present in about 20 % of the viral population. Clonal analysis of that viral stock showed that 40 % of the clones contained the F130W mutation alone. A number of clones had F130W in combination with the PR mutation G94S and others contained both RT mutations (T58S and F130W) (Figure 2(b)). After two passages of this virus in MT-4 cells, the double-mutant T58S/F130W became predominant.
Figure 2.

Emergence of compensatory mutations in the PR-coding region of HIV-189ES061 containing a F130W mutation in its RT-coding region. (a) Sequence heterogeneities in viral stocks derived from transfections of COS-1 cell monolayers, obtained with mutant HIV-1 carrying the amino acid change F130W in the RT-coding region. Numbers indicate the estimated relative percentage of wild-type and mutant virus, as determined after DNA sequencing of the whole viral population. (b) Clonal analysis of HIV variants found in stock 1, before and after two serial passages in MT-4 cells, at a multiplicity of infection of 0.001 TCID50 per cell. The ratios shown on the right indicate the number of clones containing the mutations studied, relative to the total number of clones analyzed. (c) Kinetics of imposition of Ser over Gly-94 in an infection of MT-4 cells (106), using the virus obtained from stock 2, at a multiplicity of infection of 0.0001 TCID50 per cell. RT-PCR was carried out with culture supernatants collected at various days after infection, using primers 198U5U (5′-GTC TGT TGT GTG ACT CTG GT-3′) and 292GD (5′-GAT TGA CTG CGA ATC GTT C-3′). Numbers in the lower panel indicate the relative percentage of virus carrying a mutated (G94S) or a wild-type PR, while maintaining the RT mutation F130W, as determined by DNA sequencing.
Additional transfection experiments with mutant HIV-1 having the RT mutation F130W revealed the presence of HIV variants carrying mutations in the C-terminal region of the PR. In four out of five viral stocks, G94S-containing variants represented 10 to 60 % of the total population found in the corresponding viral stock. In one case, the transfection supernatant contained virus which had acquired the PR mutation T96S (Figure 2(a)).
Although the PR mutation G94S was usually found as a relatively minor component of the quasispecies recovered from viral stocks, infection of MT-4 cells with stock no. 2 (which contained the lowest proportion of G94S-containing virus) showed the rapid imposition of the double mutant [PR (G94S) + RT (F130W)] over the mutant containing the F130W mutation alone (Figure 2(c)). Taken together, these data suggest that G94S has a positive effect on viral fitness, although the clonal analysis of the viral populations passaged in MT-4 cells showed that the double-mutant T58S/F130W was even fitter and became predominant after two passages in cell culture (Figure 2(b)).
The compensatory effect of G94S was confirmed with infectious HIV-189ES061 clones, after cotransfection of 293T cell monolayers. In those cells, the emergence of compensatory mutations is minimized because cell culture supernatants are used directly in replication kinetics and infectivity assays, without further viral replication in MT-4 cells, as required for transfections of COS-1 cells. The amount of p24 antigen in 293T cell supernatants was roughly similar for wild-type and mutant HIV-189ES061 clones, although we detected large differences in viral infectivity (Figure 3(a)). The wild-type was about 42 times more infectious than the double mutant [PR (G94S) + RT (F130W)], and >2900 times more infectious than the virus collected from cell supernatants of transfections carried out with mutant F130W. Although both mutant viruses showed diminished replication capacity in comparison with the wild-type HIV-1, the detection of RT activity at days 17 - 21 in MT-4 cell culture supernatants infected with the double mutant [PR (G94S) + RT (F130W)] (Figure 3(b)) helps to further confirm the compensatory effect of the G94S mutation.
Figure 3.

Compensatory effects of the PR mutation G94S on the replication capacity of HIV-1 clones carrying the F130W substitution in their RT. (a) Characterization of virus recovered from culture supernatants of 293T cell monolayers transfected with HIV-1 cDNA clones carrying the indicated mutations. The p24 antigen levels were determined by ELISA. Culture supernatants were titrated in MT-2 cells. Infectivity values (expressed as TCID50 units per pg of p24) are averages of two independent determinations. (b) Replication kinetics of wild-type and mutant HIV-1 clones in MT-4 cells. MT-4 cells (106) were infected with a volume of culture supernatants of 293T cell monolayers transfected with the corresponding proviral DNA, containing 1 ng of p24 antigen. Samples were withdrawn from the infected cultures at the indicated times and virion-associated RT activity was determined as described.22
By themselves, the PR mutations G94S and T96S had a relatively minor effect on viral infectivity in comparison with the wild-type HIV-189ES061 clones, with titers in the range of 0.8 – 1.4 and 0.6 – 1.4 TCID50 per pg of p24, for G94S and T96S, respectively. However, competition assays carried out in MT-2 cells at a multiplicity of 0.001 showed the higher fitness of the wild-type clones over the mutant HIV carrying mutation G94S (data not shown).
Effects of F130W on the stability of p51
The constructs M7 and vpr-IN, together with wild-type or mutated vpr-p51/p66 (Table 1) were used to cotransfect 293T cell monolayers. M7 is a provirus lacking the RT- and IN-coding regions.23 In this trans-complementation assay, Vpr-51 incorporates p66 through interaction and stable association of the two RT subunits (Vpr-51 and p66) within the cellular cytoplasm. Specific interaction between Vpr and the Gag precursor polyprotein leads to the incorporation of the Vpr-p51/p66 complex into the progeny virus, and subsequent cleavage by the viral PR generates wild-type RT heterodimer.
Table 1.
Abbreviations for plasmids
| Plasmid | Abbreviation |
|---|---|
| pSG3M7 | M7 |
| pLR2P-vpr-45Prop51-IRES-p66 | vpr-p51/p66 |
| pLR2P-vprΔp51-IRES-p66 | vprΔp51/p66 |
| pLR2P-vpr-p51(F130W)-IRES-p66 | vpr-p51(F130W)/p66 |
| pLR2P-vpr-p51(T58S/F130W)-IRES-p66 | vpr-p51(T58S/F130W)/p66 |
| pLR2P-vpr-IN | vpr-IN |
Immunoblot analysis of the transfection-derived virions showed that the trans-p66/p51F130W mutant had significantly reduced amounts of both p66 and p51 RT subunits compared to the wild-type trans-p66/p51 (Figure 4(a)). Interestingly, the presence of the T58S mutation in combination with F130W in the p51 subunit restored the levels of virion-associated RT subunits (lane 4). As expected, M7 virions trans-complemented with vpr-Δp51/p66 did not show RT subunit packaging (lane 2). We confirmed that approximately the same amount of virus was analyzed in each sample by probing the blot with a monoclonal antibody recognizing HIV-1 CA (Figure 4(a), lower panel). Western blot analysis of the corresponding cell lysates using a monoclonal antibody specific for the 66-kDa subunit of HIV-1 RT (Figure 4(b)), or antisera against Vpr (not shown) revealed that the expression of both proteins was not impaired for the mutant clones. The amount of cellular protein analyzed in each sample was similar, as demonstrated with a monoclonal antibody specific for human α-tubulin.
Figure 4.

Analysis of p51 mutants containing F130W. M7 proviral DNA was transfected into 293T cells alone or together with wild-type or mutant vpr-p51/p66 and vpr-IN expression plasmid DNAs. (a) Virion incorporation and proteolytic processing of trans-heterodimeric RT. Immunoblot analysis of transfection-derived virions obtained with M7 plus vpr-p51/p66, vprΔp51/p66, vpr-p51(F130W)/p66, and vpr-p51(T58S/F130W)/p66, using the RT-specific monoclonal antibody 8C4 (upper panel) and a CA-specific monoclonal antibody 183-H12-5C (lower panel). (b) Immunoblot analysis of the expression of p66 and α-tubulin in the transfected 293T cells. Samples were analyzed with a mixture of monoclonal antibodies against the RNase H domain of HIV-1 RT (7E5) and α-tubulin.
The results shown in Figure 4 suggest that the mutant p51 is degraded during viral packaging or maturation. We studied the effects of p51F130W after cotransfecting the 293T cell monolayers with an HIV construct lacking an active PR (designated as PM-3),4 in order to discriminate between viral or cellular PR-mediated processing. The PM-3 virus contains Asn instead of Asp-25 in the catalytic site of the viral PR. The immunoblot analysis of virions generated by cotransfection with PM-3, vpr-p51/p66 and vpr-IN showed significant amounts of Vpr-p51 and p66 in the released viral particles (Figure 5, lane 1). As expected, the p66 subunit was not incorporated into virions obtained with PM-3 and vpr-Δp51/p66 (lane 2). Interestingly, the PM-3 virions obtained with vpr-p51(F130W)/p66 contained wild-type levels of Vpr-p51, suggesting that the lower stability of p51 due to the presence of F130W could be a consequence of its higher susceptibility to viral PR degradation (lane 3). Also, two bands related to p66 were observed in the vpr-p51(F130W)/p66 virions. Similar p66-derived double bands have been described for RT mutations that are in the vicinity of the dimer interface and likely result from cleavage by cellular proteases.19,21 The simultaneous presence of both mutations T58S and F130W in the p51 subunit also prevented processing of the p66 subunit by non-viral PRs (lane 4), further emphasizing the importance of the T58S mutation in increasing p66/p51F130W heterodimer stability and decreasing sensitivity to PR degradation.
Figure 5.

Immunoblot analysis of mutant heterodimers in PM-3 virions (containing catalytically inactive viral PR). Virus obtained by cotransfection of PM-3 together with vpr-p51/p66 expression plasmids were analyzed by immunoblot using a monoclonal antibody against the RT.
Effects of G94S and T96S on the proteolytic activity and dimerization properties of the viral PR
Since G94S and T96S emerged as mutations that compensate for the replication defect caused by F130W in the viral RT, we analyzed the effects of substituting Ser for Gly-94 or Thr-96 in the viral PR. Wild-type and mutant PRs were expressed in E. coli and purified from inclusion bodies. The specific activities of the three enzymes were compared in an HPLC assay, using a synthetic nonapeptide (Val-Ser-Gln-Asn-Tyr*Pro-Ile-Val-Gln, asterisk indicates the cleavage site) as a substrate of the proteolytic reaction. The peptide mimics the naturally occurring cleavage site between the HIV-1 proteins MA and CA. As shown in Table 2, wild-type and mutant PRs are able to cleave the oligopeptide substrate. However, in comparison with the wild-type enzyme, both mutants G94S and T96S showed decreased catalytic efficiency. The largest effects were observed with G94S. Its catalytic constant (kcat) was about 4.4 times reduced in comparison with the wild-type PR, while its Km was increased by about 5-fold.
Table 2.
Kinetic parameters for the cleavage of peptide VSQNYPIVQ by wild-type and mutant HIV-1 PRs.
| Enzyme | kcat (s−1)a | Km (mM) | kcat/Km (mM s−1) |
|---|---|---|---|
| WT | 6.14 ± 0.32 | 0.20 ± 0.03 | 30.70 ± 4.88 |
| G94S | 1.40 ± 0.08 | 1.01 ± 0.12 | 1.39 ± 0.18 |
| T96S | 4.57 ± 0.61 | 1.14 ± 0.13 | 4.01 ± 0.70 |
Catalytic constants were referred to the amount of active PR used in the assays (7.5 nM for WT PR, 63.5 nM for mutant G94S, and 23.5 nM for mutant T96S). PR active site titrations were carried out with DMP 323. Inhibition constants (Ki) obtained for this compound were 0.5 nM (for wild-type PR), 8.5 nM (for G94S), and 4.3 nM (for T96S).
Gly-94 and Thr-96 map within the dimerization region of the PR homodimer, and away from the substrate binding site.2 Since dimerization properties and the proteolytic activity of the viral PR are related to each other, we measured the effects of the mutations in PR stability by urea-denaturation. As shown in Figure 6(a), both mutants G94S and T96S showed increased susceptibility to the denaturing agent in comparison with the wild-type PR. In agreement with the lower catalytic activity and susceptibility to urea denaturation, the dissociation constants (Kd) for G94S and T96S were estimated at 26.3 and 4.94 nM, respectively (Figure 6(b)). In contrast, we did not observe any significant decrease in the specific proteolytic activity at the lowest measured concentration of active wild-type PR (< 0.37 nM). These data reveal a correlation between the lower specific activity of both PR mutants, and particularly of G94S, and their reduced dimer stability.
Figure 6.
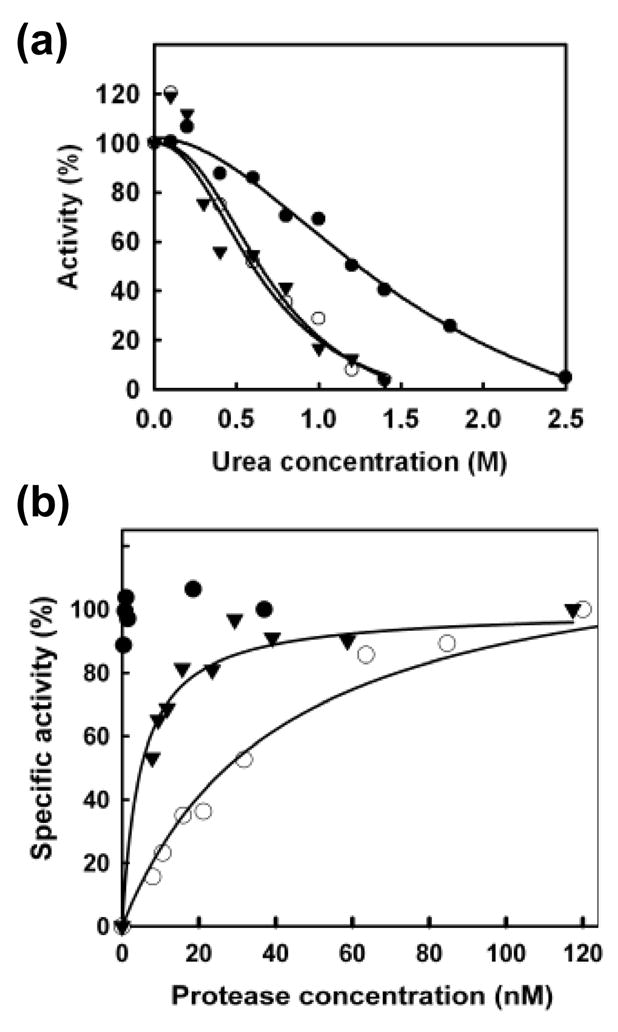
PR stability. (a) Sensitivity to urea: wild-type HIV-1 PR (filled circles) (UC50 = 1.22 M), G94S (open circles) (UC50 = 0.63 M), and T96S (filled triangles) (UC50 = 0.74 M). (b) Dimer dissociation: wild-type HIV-1 PR (filled circles) (Kd < 0.37 nM, no dissociation observed), G94S (open circles) (Kd = 26.3 ± 11.7 nM), and T96S (filled triangles) (Kd = 4.94 ± 1.07 nM).
Discussion
Both HIV-1 PR and RT are synthesized as part of large precursor polyproteins which are used in the assembly of immature virions. The PR-mediated cleavage of these precursors leads to the formation of infectious virus. The active form of HIV-1 RT is a p66/p51 heterodimer, which can be obtained in vitro by treatment of purified recombinant p66/p66 RT homodimers with HIV-1 PR.24 In addition, coexpression of p66 RT and HIV-1 PR provides good yields of stable p66/p51 RT heterodimers,25 an observation that has been also interpreted as evidence suggesting that RT heterodimer formation proceeds through a p66/p66 RT homodimer intermediate. Recent data based on a model 90-kDa Pol polyprotein containing the transframe protein, PR, RT and the N-terminus of IN appears to support this proposal.26 However, the precise pathway for the formation of RT heterodimers in HIV-infected cells is still poorly understood. Biochemical studies have shown that both p66/p66 homodimers and p66/p51 heterodimers retain significant DNA polymerase activity in vitro,12,13,27 although heterodimers are more stable than p66/p66 homodimers.28 Available evidence suggest that the stabilizing role of p51 is critical to maintain RT function during the viral life cycle.
Our previous studies showed that substituting Trp for Phe-130 had a deleterious effect on viral replication. In addition, we showed that the F130W substitution compromised the stability of p51, probably by making the polypeptide more susceptible to proteases.22 However, these studies were carried out with recombinant HIV-1 RT expressed in E. coli, and extrapolating these results to conditions found in HIV-infected cultures may not be correct. In the context of a normal proviral genome, the F130W substitution had a significant impact on RT stability. Western blot analysis showed that the expression levels of p24 and IN were normal, in contrast with the RT which remained almost undetectable (Figure 1).
The results of the trans-complementation system, developed to study the impact of mutations in specific subunits of HIV-1 RT in the context of a viral infection,29 were in agreement with the data obtained with recombinant chimeric RTs. Interestingly, the presence of F130W in p51 produced a large reduction in the amount of packaged p66/p51 in mature virions, although the proportion of p66 to p51 was relatively close to 1:1. These results were rather different from those obtained with mutants impairing RT dimerization, whose levels of packed Vpr-p51 (and processed p51) were roughly similar to those obtained with the wild-type constructs.21,23 The reduction in RT subunit content was consistent with a reduced viral infectivity (data not shown). The absence of both RT subunits in virions could not be attributed to reduced expression of either p66 or p51. In contrast to the results described above, the analysis of virions recovered after cotransfection with an HIV construct containing a catalytic mutant PR showed similar levels of packed Vpr-p51, thereby suggesting that the viral PR could be responsible for the loss in RT heterodimer stability produced by F130W when present in p51. The presence of the T58S mutation along with F130W in the 51-kDa subunit produced a significant increase in the levels of virion-associated RT subunits, in comparison with mutant F130W, while protecting the RT subunits from cellular PR-mediated degradation within the viral particles.
Although the compensatory mutation T58S in the viral RT restores the replication capacity of virus harboring F130W, a detailed study of the viral populations present in transfection supernatants and early passages of the F130W mutant revealed that the first second-site mutations that appear in the viral population occur within the PR-coding region. G94S appeared in 4 out of 5 viral stocks, while T96S emerged as a compensatory mutation in the fifth experiment. The appearance of G94S in the viral PR improved the viral replication capacity of mutant virus carrying the substitution F130W, but the viral fitness of the double-mutant was relatively low in comparison with the wild-type enzyme. The biochemical characterization of the mutant PRs carrying mutations G94S or T96S showed that both mutations produced a significant reduction in proteolytic activity, which was more pronounced in the case of G94S. These data were consistent with published rough estimates of proteolytic activity obtained with PR mutants such as G94A, T96A, or T96S.30,31 Further evidence on the effects of G94S in viral maturation was obtained from immunoblot analysis of vesicular stomatitis virus glycoprotein-pseudotyped HIV-1 virions. Virus carrying the PR mutation G94S and the RT substitution F130W showed higher amounts of unprocessed Gag than those virions containing only F130W (A. Mulky, unpublished observations).
The effects on the proteolytic activity can be attributed to impaired dimerization, as shown by the results of urea denaturation assays and Kd determinations. In agreement with this proposal, the analysis of published crystal structures shows that residues 94 and 96 are part of the four-stranded β-sheet responsible for a large portion of the PR dimerization interactions (Figure 7). Hydrogen bonds between the CO and NH groups of Thr-96 of one of the PR subunits and the NH and CO groups of Asn-98 in the other subunit contribute to the stabilization of the β-sheet. In addition, hydrogen bonds involving hydroxyl and amido groups in the side chains of Thr-96 and Asn-98, respectively contribute to stabilize each PR subunit. The substitution of Ser for Thr-96 could affect the hydrogen bond network. Gly-94 is not involved in hydrogen bonding interactions within the β-sheet. However, it is located next to a loop and at the N-terminus of a β-strand comprising residues 94–99. The substitution of Ser for Gly-94 is expected to destabilize the β-sheet that forms the dimerization interface of the PR.
Figure 7.
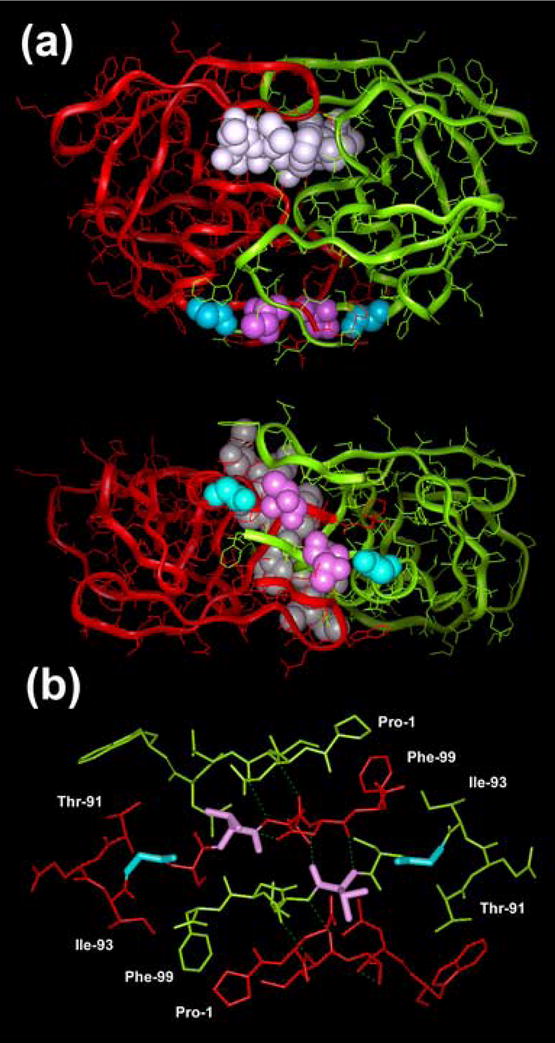
Location of Gly-94 and Thr-96 in the HIV-1 PR homodimer. (a) Crystallographic structure of HIV-1 PR complexed with an inhibitor (Protein Data Bank File 7HVP) showing the location of the relevant residues. The dimerization interface of the enzyme is shown below. PR subunits are represented with solid ribbon diagrams (red and green for subunits A and B, respectively). CPK models are used to represent a substrate analogue bound to the PR (white), and the side chains of Gly-94 and Thr-96 (blue and magenta, respectively). (b) Spatial arrangement and hydrogen bonding interactions observed in the four-stranded antiparallel β-sheet that stabilizes the dimeric structure of the viral PR. As above, Gly-94 and Thr-96 are shown in blue and magenta, respectively.
Although the identified PR mutations may cause a delay in viral maturation, their lower proteolytic activity is expected to increase the stability of the mutated p51. It has been reported that mutations that impair RT heterodimerization such as N136A, W401A or W401L lead to the formation of noninfectious viruses with reduced levels of virion-associated and intracellular RT activity in comparison with the wild-type variants.19,21,32 Using the trans-complementation assay, Mulky et al. have shown that substituting Ala for Asn-136 in the β7-β8 loop of p51 destabilizes the heterodimeric RT, while rendering the 51-kDa subunit susceptible to degradation by the viral PR.19 In addition, Wapling et al. showed that the HIV-1 PR contributed to the degradation of virion-associated subunits when mutations at position 401 were present.32 Authors argued that the loss of a proper RT dimer interface led to inefficient Gag-Pol dimerization, a step that is required for autocatalytic activation of the viral PR.32 Although NC binding to viral RNA as well as other dimerization domains in Gag and Pol also have an important role in viral assembly,33 minimizing at least in part the effects of RT mutations, we cannot rule out the possibility of F130W having a deleterious effect on Gag-Pol polyprotein conformation. Crystal structures of HIV-1 RT show that the side-chain of Phe-130 in the p51 subunit contacts residues such as Asn-57 and Arg-143, which are involved in interactions stabilizing the RT heterodimer.9–11,22 These interactions might be required for the proper temporal cleavage of Gag-Pol, and their loss could be compensated in part by mutations in the PR dimer interface.
Recently published data suggest that the proteolytic cleavage rendering the p66/p51 heterodimer plays an important role in the stabilization of the viral RT during viral maturation. Extensive mutagenesis studies have shown that amino acid substitutions affecting residues in the vicinity of the p51/RNase H cleavage site (RT positions 440–441) often lead to the loss of viral infectivity due to diminished virion RT levels.34 Although those experiments show no correlation between the type of mutation, its location and the obtained RT levels, it was clear that certain mutations rendered RTs highly susceptible to PR degradation. Taking together, those data suggest that proteolytic removal of one of the RNase H domains in the p66/p66 homodimers may be essential for providing a stable conformation of the viral RT, resistant to further proteolytic degradation within the virion. Further evidence suggesting that mutations affecting RT conformation have an impact on viral maturation was obtained from experiments carried out with virus carrying RT mutations L234D and W239A. Those residues are not involved in dimer interactions, but produce a loss of infectivity, which has been attributed to the premature cleavage of the Gag-Pol precursor.35 Although Phe-130 does not participate in intersubunit interactions, this residue could have an influence on the conformation of the β7-β8 loop of p51.22 Several amino acid substitutions within the β7-β8 loop of p51 are known to impair heterodimer formation, while decreasing the DNA polymerase activity of the viral RT in enzymatic assays.16–18
Molecular mechanisms leading to fitness recovery play an important role in antiretroviral therapy. Evolution of drug resistance is characterized by severe fitness losses, which can be partially overcome by compensatory mutations or other adaptive changes that restore viral replication capacity. This process involves structurally relevant amino acid substitutions in target enzymes (i.e. PR, RT or IN) (review36), changes in substrates of those enzymes (i.e. substitutions in the cleavage sites of Gag and Gag-Pol polyproteins),37–39 or modifications elsewhere in the genome that could affect viral protein expression (i.e. changes in the frameshift signal40) or packaging of Pol proteins.41 In the case of HIV-1 RT, all second-site reversions described so far involve mutations within the RT-coding region.42,43 The identification of PR mutations that affect viral maturation, while compensating for viral replication defects caused by mutations affecting the stability of the viral RT emerges as a novel strategy to increase viral fitness. It is also anticipated that compensatory mutations in the PR dimer interface could play a role in the acquisition of resistance to inhibitors of RT dimerization, whose development still remains at a pre-clinical stage.
Materials and Methods
Cell lines, antibodies and molecular clones
COS-1, MT-4 and MT-2 cells were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum and 2 mM glutamine. The 293T cell cultures were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10 % fetal bovine serum, penicillin (100 units/ml) and streptomycin (0.1 mg/ml).
Antibodies used included polyclonal anti-Vpr serum44 and monoclonal antibodies to human α-tubulin and β-actin (Sigma), HIV-1 CA (183-H12-C5, contributed by B. Chesebro and H. Chen), HIV-1 IN (8G4, contributed by D. E. Helland), HIV-1 RT (p66/p51 heterodimer) (8C4 and 5B2B2, contributed by D. E. Helland and A. M. Szilvay), and the RNase H domain (p66 subunit) of HIV-1 RT (7E5, contributed by D. E. Helland). Antibodies against HIV proteins were obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH.
Plasmids p61F A and p61F B containing the 5′ and 3′ ends of the proviral DNA of the HIV-1 subtype B isolate 89ES061 and a mutant derivative of p61F A containing the F130W mutation in the RT-coding region were previously described.22,45 Cotransfection with plasmids p61F A and p61F B renders infectious virus after in vivo ligation.45 Mutant HIV-189ES061 clones were obtained by site-directed mutagenesis using the QuikChange Site-Directed Mutagenesis Kit (Stratagene) by following the manufacturer’s instructions, using the plasmid p61F A as template. The mutagenic primers 5′-CTG TTG ACT CAG ATT AGT TGC ACT TTA AAT TTT CCC-3′ and 5′-GGG AAA TTT AAA GTG CAA CTA ATC TGA GTC AAC AG-3′ were used to obtain virus harboring the PR mutation G94S. Primers 5′-CTG TTG ACT CAG ATT GGT TGC TCT TTA AAT TTT CCC-3′ and 5′-GGG AAA TTT AAA GAG CAA CCA ATC TGA GTC AAC AG-3′ were used to introduce T96S in the viral PR. All mutations were confirmed by DNA sequencing.
The HIV-1 pSG3 (SG3) proviral clone (GenBank accession number L02317)46 was used to construct all proviral and recombinant RT and IN expression plasmids, used in the trans-complementation assay (construct names are listed in Table 1).21 The PM-3 proviral plasmid contains a mutation in the PR-coding region that renders PR inactive due to the presence of Asn instead of Asp-25 in its catalytic site.4 The RT-IN-minus pSG3M7 (M7) proviral construct described previously23 was used for trans-complementation analysis with all the pLR2P-based RT and IN expression plasmids. The RT subunits were expressed in trans using the previously described pLR2P-vpr-45Prop51-IRES-p66 (vpr-p51/p66) plasmid.29 Derivatives of vpr-p51/p66 having mutations in p51 were obtained by cloning the RT-coding region from the previously described pRT6 plasmid containing mutations F130W or T58S/F130W,22 into vpr-p51/p66. All clones were confirmed by nucleotide sequencing.
Transfections and analysis of virus infectivity
DNA transfections were performed on monolayer cultures of COS-1 cells, by electroporation as previously described22,45 or 293T cells using the calcium phosphate precipitation method. Usually, 293T cells were seeded at 4 × 106 in 10-cm-diameter tissue culture plates one day prior to transfection. Monolayers were then transfected with 10 μg of proviral HIV-189ES061 DNA. Culture supernatants from the 293T cells were collected 72 h post-transfection, clarified by low-speed centrifugation (1,000 × g, 10 min), and filtered through 0.45 μm pore-size sterile filters. The clarified supernatants were assayed for HIV-1 p24 concentration by ELISA. In transfections carried out with COS-1 cell monolayers, MT-4 cells were added 48 h after transfection to increase the relatively low viral titers.
To obtain a virus stock, supernatants from transfection experiments on COS–1 cells, collected at days when maximum levels of RT activity were detected were used to infect 5 × 106 fresh MT-4 cells to increase viral production. Viral stocks were titrated in MT-2 cells and titers were calculated by the Spearman-Karber method, and expressed as TCID50.47 Infections were then carried out at a multiplicity of infection of 10−3 to 10−4 TCID50 per cell. Viral replication was monitored by measuring RT activity and p24 concentration in culture supernatants as previously described,22 and estimating cell viability using the trypan blue staining method. Detection of viral proteins by Western blot was carried out as described.22 Supernatants from 293T cell transfections were used without further co-cultivation in infectivity assays.
RNA isolation, amplification, and nucleotide sequence analysis
Culture supernatants were passed through 0.45-μl pore size filters and treated with DNase A for 30 min to eliminate input DNA. Total RNA was isolated from 20 μl-aliquots of culture supernatants obtained from transfections (or infections).48 RNA amplification was done with the Qiagen OneStep RT-PCR kit (Qiagen) using primers PRF1 (5′-CAG CCC CAC CAG AAG AGA GC-3′) and 59RD (5′-ATG ATT CCT AAT GCA TAT TGT GAG T-3′), which correspond to positions 2156–2175 and 4042–4066, respectively, according to the numbering of the HIV-1 HXB2CG strain (GenBank accession no. K03455). For quasispecies analysis, the polymerase chain reaction products were cloned using the TA cloning kit (Invitrogen, Carlsbad, California), and sequenced with the Big Dye Terminator Cycle Sequencing kit (Applied Biosystems), using an ABI-PRISM 377 DNA sequencer.
Immunoblot analysis
For trans-complementation studies, DNA transfections of 293T cells were performed in six-well plates. Monolayers were transfected with 6 μg of proviral DNA (M7 or PM-3), 3 μg of the vpr-p51/p66 constructs and 1 μg of the vpr-IN construct. Culture supernatants from the 293T cells were collected 60 h posttransfection, clarified by low-speed centrifugation (1000 × g, 10 min), and filtered through 0.45 μm pore-size sterile filters. Transfection-derived virions were concentrated by ultracentrifugation through 20% (w/v) sucrose cushion (125,000 × g, 2 h, 4 °C) using a SW41 rotor (Beckman Inc.). Pellets were solubilized in Laemmli loading buffer (62.5 mM Tris, pH 6.8, 0.2 % SDS, 5 % β-mercaptoethanol, 10 % glycerol), boiled, and proteins were separated on 12 % polyacrylamide gels containing SDS. Following electrophoresis, proteins were transferred to nitrocellulose (0.2 μm pore size) by electroblotting and incubated for 1 h at room temperature in blocking buffer (5 % non-fat dry milk in phosphate-buffered saline). The blocked blots were exposed to antibody for 1 h in blocking buffer with constant mixing. After extensive washing, bound antibodies were detected by chemiluminiscence using horseradish peroxidase-conjugated species-specific secondary antibodies (Southern Biotechnology Associates, Inc.) as described by the manufacturer (GE Healthcare).
Mutagenesis, expression and purification of recombinant HIV-1 PRs
The E. coli strain BL12(DE3)pLysS harboring the plasmid pET-HIVPR49 was kindly provided by Dr. Jordan Tang, Oklahoma Medical Research Foundation. The amino acid sequence of the PR encoded within pET-HIVPR contains Met-36, Ser-37 and Ile-64, instead of Ile-36, Asn-37 and Val-64, as found in the HIV-1 89ES061 isolate.45 Mutations M36I, S37N and I64V were introduced in pET-HIVPR with the QuikChange Site-Directed Mutagenesis Kit (Stratagene), using the mutagenic primers: 5′-GTT CTG GAA GAA ATC AAT CTG CCG GGT CGT TGG AAG CCG-3′ and 5′-CGG CTT CCA ACG ACC CGG CAG ATT GAT TTC TTC CAG AAC-3′ for M36I and S37N; and 5′-CAG TAT GAT CAG ATC CTG GTT GAA ATC TGC GGT CAC-3′ and 5′-GTG ACC GCA GAT TTC AAC CAG GAT CTG ATC ATA CTG-3′ for I64V. Then, we introduced a TAA termination codon and a HindIII restriction site, using primers 5′-CGG TTG CAC TTT GAA CTT CTA AGC TTC CCC GAT TGA AAC TGT TCC GG-3′ and 5′-CCG GAA CAG TTT CAA TCG GGG AAG CTT AGA AGT TCA AAG TGC AAC CG-3′ to obtain a modified pET-HIVPR construct, designated as pET-HIVPR61. Plasmids for expression of mutant PRs carrying mutations G94S or T96S were obtained with the QuikChange mutagenesis kit using pET-HIVPR61 as template and the mutagenic primers: 5′-CCT GCT GAC TCA GAT CTC TTG CAC TTT GAA CTT CTA AGC-3′ and 5′-GCT TAG AAG TTC AAA GTG CAA GAG ATC TGA GTC AGC AGG-3′ for G94S, and 5′-CTG ACT CAG ATC GGT TGC TCT TTG AAC TTC TAA GCT TTC-3′ and 5′-GAA AGC TTA GAA GTT CAA AGA GCA ACC GAT CTG AGT CAG-3′ for T96S. In all cases, mutations were confirmed by DNA sequencing.
Expression and purification of mutant and wild-type PRs was carried out essentially as described previously.49 Briefly, freshly prepared E. coli BL12(DE3)pLysS containing the plasmid pET-HIVPR61, or the mutated PRs were grown in 1 liter of Luria broth medium containing 75 μg/ml ampicillin and 25 μg/ml chloramphenicol, to an A600 of 0.6–0.8. After induction with 0.5 mM isopropyl-β-D-thiogalactopyranoside for 2.5 h, cells were harvested by centrifugation at 5,000 × g and stored at −20 °C. The bacteria (8 g of cell paste) were processed as described to obtain a pellet, which contains the majority of the proteins expressed (as inclusion bodies).49 The inclusion bodies were solubilized in 1 ml of 10 mM Tris, pH 7.5, 8 M urea and 10 mM dithiothreitol, and centrifuged at 100,000 × g. The clear supernatant was transferred to a 2-ml DEAE Sephadex A-25 column (previously equilibrated in the dilution buffer) and permitted to flow at room temperature. Fractions of 0.3 ml were collected and those containing the PR were pooled and stored at 4 °C. The denatured protein solution in 8 M urea was then acidified by mixing with 0.02 volume of 10 % (v/v) trifluoroacetic acid to make a final pH of 3.5, and then refolded by dialysis against 1 mM dithiothreitol, 10 mM sodium acetate buffer, pH 3.5. After centrifugation at 100,000 × g, the supernatants were collected, and after addition of glycerol to a concentration of 10 % (v/v), samples were stored at −70 °C.
Oligopeptide substrates and proteolytic assays
The synthetic nonapeptide substrate Val-Ser-Gln-Asn-Tyr-Pro-Ile-Val-Gln, corresponding to the MA/CA cleavage site in the natural Gag precursor and the products of cleavage were analyzed by reverse-phase high-pressure liquid chromatography (RP-HPLC).50 The PR assays were initiated by mixing 5 μl of enzyme, 10 μl of 2x incubation buffer [0.5 M sodium phosphate buffer (pH 5.6), containing 4 M NaCl, 10 mM dithiothreitol, 2 mM EDTA and 10 % glycerol] and 5 μl 0.05 – 10 mM substrate. The reaction mixture was incubated at 37 °C for 1 h and terminated by the addition of 180 μl of 1 % trifluoroacetic acid. The hydrolysis products were separated by RP-HPLC and analyzed as previously described.51 Kinetic parameters were determined by fitting the data obtained at <20 % substrate hydrolysis to the Michaelis-Menten equation by using SigmaPlot 8.02 (Systat Software Inc., Richmond, CA) software. The active enzyme concentrations of the wild-type and mutant PRs were determined with the HPLC assay in the presence of a substrate concentration of 0.4 mM and 1.25 mM, respectively, and using DMP 323 as a tight-binding inhibitor of HIV-1 PR.52 DMP 323 was a king gift from Dr. Bruce Korant (DuPont Experimental Station).
Urea-denaturation assay
The HPLC assay with concentrations of 50–200 nM (wild-type PR) or 250–900 nM (mutant PRs), and 400 μM substrate was used to determine PR activity in the presence of 0–3.2 M urea.53 Before adding the substrate, the enzyme was incubated in the presence of urea (at appropriate concentrations) for 5 min at 37 °C. The UC50 values for half-maximal velocity were obtained by plotting the initial velocities against concentration of urea and fitting to a curve for solvent-denaturation of protein using SigmaPlot 8.02 software.
Kd determination
Specific activity was measured as a function of dimeric enzyme concentration in 0.25 M sodium phosphate buffer (pH 5.6), containing 2 M NaCl, 5 mM dithiothreitol, 1 mM EDTA and 5 % glycerol, using a 1.25 mM substrate concentration. Samples were incubated for 1 h at 37 °C, and processed for HPLC analysis as described above.53,54
Acknowledgments
We thank Fernando Barahona, Beatriz Delgado Marcos and Beatriz López Merino for technical assistance. This work was supported in part by Fondo de Investigación Sanitaria (through the “Red Temática de Investigación Cooperativa en SIDA” RD06/006). In addition, work in the CBMSO (Madrid) was supported by grant BIO2003/01175 (Spanish Ministery of Education and Science) and an institutional grant of Fundación Ramón Areces. Work in the CNM (Majadahonda) was supported by grants SAF2002/626, SAF2003/4987 and SAF2005/3833 (Spanish Ministery of Education and Science), and by the Plan Nacional sobre el SIDA. Work in the UAB was supported by National Institutes of Health grants CA73470 and AI47714 and core facilities of Birmingham Center for AIDS Research (P30-AI-27767). Support from the Spanish-Hungarian Intergovernmental Science and Technology Cooperation Program (grant HH2005-0020) is also acknowledged.
Abbreviations used
- HIV-1
human immunodeficiency virus type-1
- MA
matrix protein
- CA
capsid protein
- NC
nucleocapsid protein
- PR
protease
- RT
reverse transcriptase
- IN
integrase
- DMEM
Dulbecco’s modified Eagle’s medium
- TCID50
tissue culture infectious dose 50
- RP-HPLC
reverse-phase high-pressure liquid chromatography
Footnotes
J.C.K. has a stock interest in a company that licensed technology utilized in this study from the UAB Research Foundation, and UAB Patent Policy controls the consideration of and/or revenues received arising from the commercialization of said technology.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Oroszlan S, Luftig TB. Retroviral proteinases. Curr Top Microbiol Immunol. 1990;157:153–185. doi: 10.1007/978-3-642-75218-6_6. [DOI] [PubMed] [Google Scholar]
- 2.Weber IT. Comparison of the crystal structures and intersubunit interactions of human immunodeficiency and Rous sarcoma virus proteases. J Biol Chem. 1990;265:10492–10496. [PubMed] [Google Scholar]
- 3.Dunn BM, Rao M. Human immunodeficiency virus 1 retropepsin. In: Barrett AJ, Rawlings ND, Woessner JF, editors. Handbook of Proteolytic Enzymes. 2. Elsevier; London: 2004. pp. 144–154. [Google Scholar]
- 4.Kohl NE, Emini EA, Schleif WA, Davis LJ, Heimbach JC, Dixon RAF, Scolnick EM, Sigal IS. Active human immunodeficiency virus protease is required for viral infectivity. Proc Natl Acad Sci USA. 1988;85:4686–4690. doi: 10.1073/pnas.85.13.4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Louis JM, McDonald RA, Nashed NT, Wondrak EM, Jerina DM, Oroszlan S, Mora PT. Autoprocessing of the HIV-1 protease using purified wild-type and mutated fusion proteins expressed at high levels in Escherichia coli. Eur J Biochem. 1991;199:361–369. doi: 10.1111/j.1432-1033.1991.tb16132.x. [DOI] [PubMed] [Google Scholar]
- 6.Tessmer U, Kräusslich HG. Cleavage of human immunodeficiency virus type 1 proteinase from the N-terminally adjacent p6* protein is essential for efficient Gag polyprotein processing and viral infectivity. J Virol. 1998;72:3459–3463. doi: 10.1128/jvi.72.4.3459-3463.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Di Marzo Veronese F, Copeland TD, DeVico AL, Rahman R, Oroszlan S, Gallo RC, Sarngadharan MG. Characterization of highly immunogenic p66/p51 as the reverse transcriptase of HTLV-III/LAV. Science. 1986;231:1289–1291. doi: 10.1126/science.2418504. [DOI] [PubMed] [Google Scholar]
- 8.Lightfoote MM, Coligan JE, Folks TM, Fauci AS, Martin MA, Venkatesan S. Structural characterization of reverse transcriptase and endonuclease polypeptides of the acquired immunodeficiency syndrome retrovirus. J Virol. 1986;60:771–775. doi: 10.1128/jvi.60.2.771-775.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kohlstaedt LA, Wang J, Friedman JM, Rice PA, Steitz TA. Crystal structure at 3.5 Å resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science. 1992;256:1783–1790. doi: 10.1126/science.1377403. [DOI] [PubMed] [Google Scholar]
- 10.Ding J, Das K, Hsiou Y, Sarafianos SG, Clark AD, Jr, Jacobo-Molina A, Tantillo C, Hughes SH, Arnold E. Structural and functional implications of the polymerase active site region in a complex of HIV-1 RT with a double-stranded DNA template-primer and an antibody Fab fragment at 2.8 Å resolution. J Mol Biol. 1998;284:1095–1111. doi: 10.1006/jmbi.1998.2208. [DOI] [PubMed] [Google Scholar]
- 11.Huang H, Chopra R, Verdine GL, Harrison SC. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science. 1998;282:1669–1675. doi: 10.1126/science.282.5394.1669. [DOI] [PubMed] [Google Scholar]
- 12.Le Grice SF, Naas T, Wohlgensinger B, Schatz O. Subunit-selective mutagenesis indicates minimal polymerase activity in heterodimer-associated p51 HIV-1 reverse transcriptase. EMBO J. 1991;10:3905–3911. doi: 10.1002/j.1460-2075.1991.tb04960.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hostomsky Z, Hostomska Z, Fu TB, Taylor J. Reverse transcriptase of human immunodeficiency virus type 1: functionality of subunits of the heterodimer in DNA synthesis. J Virol. 1992;66:3179–3182. doi: 10.1128/jvi.66.5.3179-3182.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amacker M, Hubscher U. Chimeric HIV-1 and feline immunodeficiency virus reverse transcriptases: critical role of the p51 subunit in the structural integrity of heterodimeric lentiviral DNA polymerases. J Mol Biol. 1998;278:757–765. doi: 10.1006/jmbi.1998.1739. [DOI] [PubMed] [Google Scholar]
- 15.Menéndez-Arias L, Abraha A, Quiñones-Mateu ME, Mas A, Camarasa MJ, Arts EJ. Functional characterization of chimeric reverse transcriptases with polypeptide subunits of highly divergent HIV-1 group M and O strains. J Biol Chem. 2001;276:27470–27479. doi: 10.1074/jbc.M104342200. [DOI] [PubMed] [Google Scholar]
- 16.Pandey PK, Kaushik N, Talele TT, Yadav PNS, Pandey VN. The β7-β8 loop of the p51 subunit in the heterodimeric (p66/p51) human immunodeficiency virus type 1 reverse transcriptase is essential for the catalytic function of the p66 subunit. Biochemistry. 2001;40:9505–9512. doi: 10.1021/bi002872j. [DOI] [PubMed] [Google Scholar]
- 17.Auwerx J, van Nieuwenhove J, Rodríguez-Barrios F, de Castro S, Velázquez S, Ceccherini-Silberstein F, De Clercq E, Camarasa MJ, Perno CF, Gago F, Balzarini J. The N137 and P140 amino acids in the p51 and the P95 amino acid in the p66 subunit of human immunodeficiency virus type 1 (HIV-1) reverse transcriptase are instrumental to maintain catalytic activity and to design new classes of anti-HIV-1 drugs. FEBS Lett. 2005;579:2294–2300. doi: 10.1016/j.febslet.2005.02.077. [DOI] [PubMed] [Google Scholar]
- 18.Balzarini J, Auwerx J, Rodríguez-Barrios F, Chedad A, Farkas V, Ceccherini-Silberstein F, García-Aparicio C, Velázquez S, De Clercq E, Perno CF, Camarasa MJ, Gago F. The amino acid Asn136 in HIV-1 reverse transcriptase (RT) maintains efficient association of both subunits and enables the rational design of novel RT inhibitors. Mol Pharmacol. 2005;68:49–60. doi: 10.1124/mol.105.012435. [DOI] [PubMed] [Google Scholar]
- 19.Mulky A, Vu BC, Conway JA, Hughes SH, Kappes JC. Analysis of amino acids in the β7-β8 loop of human immunodeficiency virus type 1 reverse transcriptase for their role in virus replication. J Mol Biol. 2007;365:1368–1378. doi: 10.1016/j.jmb.2006.10.089. [DOI] [PubMed] [Google Scholar]
- 20.Tachedjian G, Aronson HE, de los Santos M, Seehra J, McCoy JM, Goff SP. Role of residues in the tryptophan repeat motif for HIV-1 reverse transcriptase dimerization. J Mol Biol. 2003;326:381–396. doi: 10.1016/s0022-2836(02)01433-x. [DOI] [PubMed] [Google Scholar]
- 21.Mulky A, Sarafianos SG, Jia Y, Arnold E, Kappes JC. Identification of amino acid residues in the human immunodeficiency virus type-1 reverse transcriptase tryptophan-repeat motif that are required for subunit interaction using infectious virions. J Mol Biol. 2005;349:673–684. doi: 10.1016/j.jmb.2005.03.057. [DOI] [PubMed] [Google Scholar]
- 22.Olivares I, Gutiérrez-Rivas M, López-Galíndez C, Menéndez-Arias L. Tryptophan scanning mutagenesis of aromatic residues within the polymerase domain of HIV-1 reverse transcriptase: critical role of Phe-130 for p51 function and second-site revertant restoring viral replication capacity. Virology. 2004;324:400–411. doi: 10.1016/j.virol.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 23.Mulky A, Sarafianos SG, Arnold E, Wu X, Kappes JC. Subunit-specific analysis of the human immunodeficiency virus type 1 reverse transcriptase in vivo. J Virol. 2004;78:7089–7096. doi: 10.1128/JVI.78.13.7089-7096.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chattopadhyay D, Evans DB, Deibel MR, Jr, Vosters AF, Eckenrode FM, Einspahr HM, Hui JO, Tomasselli AG, Zurcher-Neely HA, Heinrikson RL, Sharma SK. Purification and characterization of heterodimeric human immunodeficiency virus type 1 (HIV-1) reverse transcriptase produced by in vitro processing of p66 with recombinant HIV-1 protease. J Biol Chem. 1992;267:14227–14232. [PubMed] [Google Scholar]
- 25.Le Grice SF, Gruninger-Leitch F. Rapid purification of homodimer and heterodimer HIV-1 reverse transcriptase by metal chelate affinity chromatography. Eur J Biochem. 1990;187:307–314. doi: 10.1111/j.1432-1033.1990.tb15306.x. [DOI] [PubMed] [Google Scholar]
- 26.Sluis-Cremer N, Arion D, Abram ME, Parniak MA. Proteolytic processing of an HIV-1 pol polyprotein precursor: insights into the mechanism of reverse transcriptase p66/p51 heterodimer formation. Int J Biochem Cell Biol. 2004;36:1836–1847. doi: 10.1016/j.biocel.2004.02.020. [DOI] [PubMed] [Google Scholar]
- 27.Restle T, Muller B, Goody RS. Dimerization of human immunodeficiency virus type 1 reverse transcriptase. A target for chemotherapeutic intervention. J Biol Chem. 1990;265:8986–8988. [PubMed] [Google Scholar]
- 28.Sluis-Cremer N, Dmitrienko GI, Balzarini J, Camarasa MJ, Parniak MA. Human immunodeficiency virus type 1 reverse transcriptase dimer destabilization by 1-{spiro[4″-amino-2″,2″-dioxo-1″,2″-oxathiole-5″,3′-[2′,5′-bis-O-(tert-butyldimethylsilyl)-β-D-ribofuranosyl]]}-3-ethylthymine. Biochemistry. 2000;39:1427–1433. doi: 10.1021/bi991682+. [DOI] [PubMed] [Google Scholar]
- 29.Mulky A, Kappes JC. Analysis of human immunodeficiency virus type 1 reverse transcriptase subunit structure/function in the context of infectious virions and human target cells. Antimicrob Agents Chemother. 2005;49:3762–3769. doi: 10.1128/AAC.49.9.3762-3769.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Loeb DD, Swanstrom R, Everitt L, Manchester M, Stamper SE, Hutchison CA., III Complete mutagenesis of the HIV-1 protease. Nature. 1989;340:397–400. doi: 10.1038/340397a0. [DOI] [PubMed] [Google Scholar]
- 31.Pettit SC, Gulnik S, Everitt L, Kaplan AH. The dimer interfaces of protease and extra-protease domains influence the activation of protease and the specificity of GagPol cleavage. J Virol. 2003;77:366–374. doi: 10.1128/JVI.77.1.366-374.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wapling J, Moore KL, Sonza S, Mak J, Tachedjian G. Mutations that abrogate human immunodeficiency virus type 1 reverse transcriptase dimerization affect maturation of the reverse transcriptase heterodimer. J Virol. 2005;79:10247–10257. doi: 10.1128/JVI.79.16.10247-10257.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alfadhli A, Dhenub TC, Still A, Barklis E. Analysis of human immunodeficiency virus type 1 Gag dimerization-induced assembly. J Virol. 2005;79:14498–14506. doi: 10.1128/JVI.79.23.14498-14506.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abram ME, Parniak MA. Virion instability of human immunodeficiency virus type 1 reverse transcriptase (RT) mutated in the protease cleavage site between RT p51 and RT RNase H domain. J Virol. 2005;79:11952–11961. doi: 10.1128/JVI.79.18.11952-11961.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu Q, Ottmann M, Pechoux C, Le Grice S, Darlix JL. Mutations in the primer grip of human immunodeficiency virus type 1 reverse transcriptase impair proviral DNA synthesis and virion maturation. J Virol. 1998;72:7676–7680. doi: 10.1128/jvi.72.9.7676-7680.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Menéndez-Arias L, Martínez MA, Quiñones-Mateu ME, Martínez-Picado J. Fitness variations and their impact on the evolution of antiretroviral drug resistance. Curr Drug Targets – Infect Dis. 2003;3:355–371. doi: 10.2174/1568005033481033. [DOI] [PubMed] [Google Scholar]
- 37.Doyon L, Croteau G, Thibeault D, Poulin F, Pilote L, Lamarre D. Second locus involved in human immunodeficiency virus type 1 resistance to protease inhibitors. J Virol. 1996;70:3763–3769. doi: 10.1128/jvi.70.6.3763-3769.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang YM, Imamichi H, Imamichi T, Lane HC, Falloon J, Vasudevachari MB, Salzman NP. Drug resistance during indinavir therapy is caused by mutations in the protease gene and in its Gag substrate cleavage sites. J Virol. 1997;71:6662–6670. doi: 10.1128/jvi.71.9.6662-6670.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fehér A, Weber IT, Bagossi P, Boross P, Mahalingam B, Louis JM, Copeland TD, Torshin IY, Harrison RW, Tözsér J. Effect of sequence polymorphism and drug resistance on two HIV-1 Gag processing sites. Eur J Biochem. 2002;269:4114–4120. doi: 10.1046/j.1432-1033.2002.03105.x. [DOI] [PubMed] [Google Scholar]
- 40.Doyon L, Payant C, Brakier-Gingras L, Lamarre D. Novel Gag-Pol frameshift site in human immunodeficiency virus type 1 variants resistant to protease inhibitors. J Virol. 1998;72:6146–6150. doi: 10.1128/jvi.72.7.6146-6150.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bleiber G, Munoz M, Ciuffi A, Meylan P, Telenti A. Individual contributions of mutant protease and reverse transcriptase to viral infectivity, replication, and protein maturation of antiretroviral drug-resistant human immunodeficiency virus type 1. J Virol. 2001;75:3291–3300. doi: 10.1128/JVI.75.7.3291-3300.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olivares I, Sánchez-Merino V, Martínez MA, Domingo E, López-Galíndez C, Menéndez-Arias L. Second-site reversion of a human immunodeficiency virus type 1 reverse transcriptase mutant that restores enzyme function and replication capacity. J Virol. 1999;73:6293–6298. doi: 10.1128/jvi.73.8.6293-6298.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pelemans H, Esnouf R, Min KL, Parniak M, De Clercq E, Balzarini J. Mutations at amino acid positions 63, 189, and 396 of human immunodeficiency virus type 1 reverse transcriptase (RT) partially restore the DNA polymerase activity of a Trp229Tyr mutant RT. Virology. 2001;287:143–150. doi: 10.1006/viro.2001.1032. [DOI] [PubMed] [Google Scholar]
- 44.Wu X, Liu H, Xiao H, Kim J, Seshaiah P, Natsioulis G, Boeke JD, Hahn BH, Kappes JC. Targeting foreign proteins to human immunodeficiency virus particles via fusion with Vpr and Vpx. J Virol. 1995;69:3389–3398. doi: 10.1128/jvi.69.6.3389-3398.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Olivares I, Shaw G, López-Galíndez C. Phenotypic switch in a Spanish HIV type 1 isolate on serial passage on MT-4 cells. AIDS Res Human Retrovir. 1997;13:979–984. doi: 10.1089/aid.1997.13.979. [DOI] [PubMed] [Google Scholar]
- 46.Ghosh SK, Fultz PN, Keddie E, Saag MS, Sharp PM, Hahn BH, Shaw GM. A molecular clone of HIV-1 tropic and cytopathic for human and chimpanzee lymphocytes. Virology. 1993;194:858–864. doi: 10.1006/viro.1993.1331. [DOI] [PubMed] [Google Scholar]
- 47.Mascola JR. Neutralization of HIV-1 infection of human peripheral blood mononuclear cells (PBMC): antibody dilution method. In: Michael NL, Kim JH, editors. HIV Protocols. Methods in Molecular Medicine. Vol. 17. Humana Press; Totowa, NJ.: 1999. pp. 317–322. [DOI] [PubMed] [Google Scholar]
- 48.Boom R, Sol CJA, Salimans MMM, Jansen CL, Wertheim-van Dillen PME, van der Noordaa J. Rapid and simple method for purification of nucleic acids. J Clin Microbiol. 1990;28:495–503. doi: 10.1128/jcm.28.3.495-503.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ido E, Han H, Kezdy FJ, Tang J. Kinetic studies of human immunodeficiency virus type 1 protease and its active-site hydrogen bond mutant A28S. J Biol Chem. 1991;266:24359–24366. [PubMed] [Google Scholar]
- 50.Copeland TD, Oroszlan S. Genetic locus, primary structure and chemical synthesis of HIV protease. Gene Anal Tech. 1988;5:109–115. doi: 10.1016/0735-0651(88)90010-6. [DOI] [PubMed] [Google Scholar]
- 51.Tözsér J, Bláha I, Copeland TD, Wondrak EM, Oroszlan S. Comparison of the HIV-1 and HIV-2 proteinases using oligopeptide substrates representing cleavage sites in Gag and Gag-Pol polyproteins. FEBS Lett. 1991;281:77–80. doi: 10.1016/0014-5793(91)80362-7. [DOI] [PubMed] [Google Scholar]
- 52.Erickson-Viitanen S, Klabe RM, Cawood PG, O’Neal PL, Meek JL. Potency and selectivity of inhibition of human immunodeficiency virus protease by a small nonpeptide cyclic urea, DMP 323. Antimicrob Agents Chemother. 1994;38:1628–1634. doi: 10.1128/aac.38.7.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wondrak EM, Louis JM. Influence of flanking sequences on the dimer stability of human immunodeficiency virus type 1 protease. Biochemistry. 1996;35:12957–12962. doi: 10.1021/bi960984y. [DOI] [PubMed] [Google Scholar]
- 54.Louis JM, Clore GM, Gronenborn AM. Autoprocessing of HIV-1 protease is tightly coupled to protein folding. Nature Struct Biol. 1999;6:868–875. doi: 10.1038/12327. [DOI] [PubMed] [Google Scholar]