

Summary
Δ41-52 hPRL (human prolactin with residues 41-52 removed) is a lead compound for a new class of hPRL antagonists. The deleted sequence contains residues that functionally couple sites 1 and 2, the two hormone surfaces that each bind receptors. Δ41-52 hPRL retains 0.03% agonist activity in FDC-1 cell bioassays, a 3,054-fold reduction in activity, and displays approximately 100-fold less agonist activity than G129R hPRL, an antagonist that reduces the binding of hPRL receptor at site 2 during the formation of the heterotrimeric hormone/receptor complex. Replacement of various numbers and types of residues into the gap created by the deletion of residues 41 through 52 created hPRLs with varying agonist activities, suggested that manipulation of the sequence connecting the C-terminal of helix 1 with the disulfide bond (cysteines 58 with 174) linking helices 1 and 4 modulates articulation of these helices and influences agonist activity. We have compared the antagonist activities of G129R and Δ41-52 hPRLs to induce apoptosis in Jurkat cells, a human lymphoid cell line displaying an autocrine/paracrine hPRL/receptor system. Δ41-52 hPRL induces apoptosis in a time and dose-dependent fashion. Under these same conditions G129R hPRL fails to induce apoptosis. We conclude Δ41-52 hPRL is a lead compound of a new class of hPRL antagonists capable at low concentrations of inducing apoptosis in human cells expressing an autocrine/paracrine hPRL/receptor system.
Keywords: prolactin, human, antagonist
Introduction
Prolactin (PRL) is a hormone regulating mammary growth, development, and lactation in mammals [1]. It has been described as a mitogen, a hormone regulating differentiation, or, more recently, a viability factor [2]. In mammals prolactin has been proposed to play a role in the development and/or growth of breast and mammary tumors [3,4,5]. Human prolactin (hPRL) is produced as an endocrine hormone by the pituitary and placenta; in addition hPRL is produced by mammary epithelial cells, breast tumor cells, prostate epithelial cells, lymphoid cells, and other cell types [6] where it appears to function as an autocrine/paracrine system. With hPRL produced by these multiple sources and functioning in autocrine, paracrine, and endocrine roles, it is difficult to dissect the functions of this hormone using classic ablation and addition strategies. Additional tools are required to elucidate the paracrine or autocrine roles of hPRL. Development of hPRL antagonists offers a tool to identify specific functions of hPRL autocrine and paracrine systems. The development of hPRL antagonists is particularly of interest because these agents may provide a strategy for reducing the initiation or growth of cancers by blocking the actions of hPRL [7].
hPRL activates target cells by binding protein receptors that are primarily located within the plasma membranes of target cells [8]. These class 1 receptors are composed of extracellular and intercellular domains connected by a single transmembrane domain. Several isoforms of the hPRL receptor have been identified [9-14]. The functions of these various isoforms vary and are incompletely described [15]. The most commonly described mechanism activating the hPRL receptor is for the ligand to bind two receptors creating a heterotrimeric extracellular complex that also creates a properly oriented dimer of the receptor’s intercellular domains. The proper orientation of dimeric intercellular domains induced by hPRL binding is associated with the activation of intercellular signaling pathways. But, receptor dimerization appears to be somewhat promiscuous; in humans three lactogenic hormones (PRL, growth hormone, and placental lactogen) and certain divalent antibodies can bind and activate receptors. Recently, dimeric hPRL receptors have been reported in the absence of hormone [16,17]. The activity of these ligand-independent dimeric forms has not been reported.
We have recently demonstrated the mechanism by which hPRL binds the extracellular domains of the hPRL receptor. hPRL binds receptors in a strictly sequential fashion, where the ligand binds the first receptor inducing a change of the ligand’s conformation that organizes the hormone surface to create the binding site for the second receptor [18]. Without receptor bound at the first binding surface of hPRL, the second receptor binding surface does not bind hPRL receptor. In both human growth hormone (hGH)[19] and hPRL [20] we have identified a contiguous collection of amino acids that are required to propagate the binding-induced conformation change between the first and second hPRLr-binding surface of the ligand. This coupling motif is required for activity as a lactogenic hormone. Interestingly, this coupling motif is required for the lactogenic activity of hGH, but is not required for the somatotrophic activity, demonstrating that hGH uses different mechanisms to activate either the hGH or hPRL receptors. The motif that couples the binding sites of hGH when binding hGH receptors has recently been described Walsh et al. [21].
Most hPRL antagonists rely on steric hindrance within site 2. The best-documented hPRL antagonist (G129R) was created by replacing a glycine at position 129 with a more bulky and charged arginine [22,23]; this residue is believed to be in a pocket of the hormone that binds the second hPRL receptor. Thus, G129R hPRL binds a single hPRL receptor but is impeded by steric hindrance from binding a second receptor and forming active heterotrimeric complexes. Thus, at modest concentrations, it is believed that G129R hPRL binds receptors with a one to one stoichiometry, sequestering the receptor from functionally productive binding and activation by endogenous wild-type hPRL. Unfortunately, there are limitations to this approach. If structural changes, such as G129R, in site 2 of hPRL are not sufficient to eliminate affinity for the second receptor, the remaining agonist activity may be sufficient to overcome antagonism at concentrations required to bind and block all receptors. This is the case with G129R hPRL where between 1 and 10% of the agonist activity remains. Improvements to the G129R hPRL antagonist have recently been described [24] where several residues also have been removed from the N-terminus of the mature protein. An additional hPRL antagonist, a selective antagonist, has been created by Walker and colleagues [25], but the mechanism by which this antagonist selectively modulates the activities of the hPRL receptor remains to be fully described.
Despite its remaining agonist activity, G129R hPRL has been shown to provide significant antagonist activities in biological assays. Treatment of cells and animals with G129R hPRL can inhibit the growth of breast cancer cells, mammary development, and mammary tumor development. Treatment with G129R hPRL reduces cell viability in several human breast cancer cell lines, including MCF-7 and T47D [26]. G129R hPRL reduces viability of T47D cells by inducing apoptosis and increasing the ratio of BAX to bcl-2 in both in vitro [27] and in vivo [28] model systems. Transgenic mice expressing G129R hPRL had a reduced ductal branching [29]. After treatment with DMBA (dimethylbenzanthracene, a potent carcinogen) these mice displayed a reduced rate of mammary tumor development and a delayed the appearance of tumors. These studies suggest that exposure to hPRL antagonists may reduce the development of breast tumors and may reduce the viability of tumor cells expressing the hPRL/receptor autocrine/paracrine system. Therefore, hPRL antagonists have been shown to be an effective prophylactic tool in models for mammary cancer.
hGH and hPRL share a common four helix bundle structure [30-33] and both activate the hPRL receptor. hGH is found in 22 kDa and a 20 kDa isoforms in the human pituitary, the larger isoform has 190 residues, while the smaller isoform has only 175 residues [34]. The smaller form is produced by alternative splicing of the mRNA that removes the codons for residues 32 through 46 [35]. These residues are located within the C-terminal of helix 1 and extend into the non-helical sequence connecting helix 1 and the disulfide bond that covalently links this sequence most directly with helix 4 (cysteines 53 and 165). The structure of 20 kDa hGH is not known, but based on the structures of 22 kDa hGH [36,37], the loss of the 15 residues reduces the length of the backbone connecting helices 1 and 4, constraining the possible spatial relationships between these two helices. Tsunekawa et al. have shown that although 22 kDa hGH has both somatotrophic and lactogenic activities; 20 kDa hGH retains only somatotrophic activity [38]. We have confirmed the requirement for residues 32 through 46 for hGH lactogenic activity and expanded this work into hPRL [39]. We aligned the sequences of hGH and hPRL and determined that residues 41-52 of hPRL correlate with residues 32 through 46 that are absent from 20kDa hGH. These two segments are not highly conserved (Figure 1). We have prepared a recombinant hPRL missing these residues (Δ41-52 hPRL, Figure 1) and have observed that this change provides a significantly larger loss of lactogenic activity than that observed in 20 kDa hGH (approximately 10,000 fold vs. 200 fold for hPRL and hGH, respectively)(39) . Interestingly, several of the residues that are removed in the 20 kDa isoform of hGH and Δ41-52 hPRL are required for lactogenic activity and are part of the motif that is required to functionally couple this hormone’s lactogenic receptor-binding sites [18-20].
Figure 1. Structural Modifications for the Creation of Δ41-52 Human Prolactin.
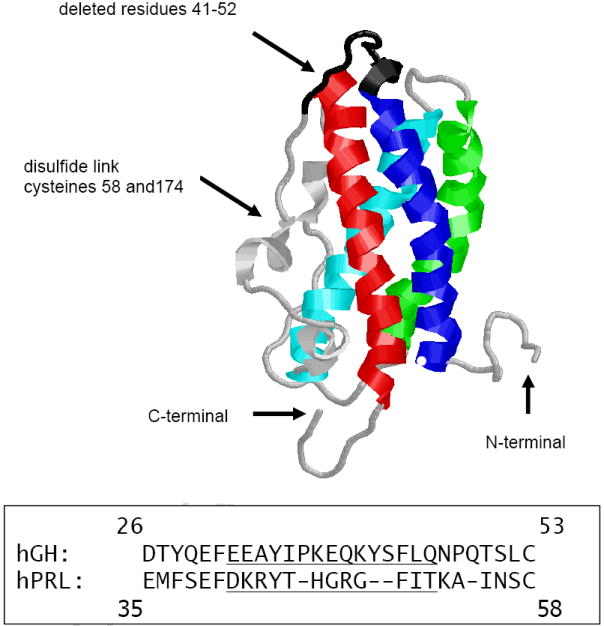
Three dimensional structure of hPRL with helices 1 through 4 shown in blue, cyan, green, and red, respectively. The deleted residues (41-52) are indicated by the black backbone. The lower panel shows a section of the aligned sequences of human growth hormone and prolactin. The underlined residues are not present in the 20 kDa form of hGH and have been deleted in Δ41-52 hPRL.
If several of the removed residues are required for the functional coupling of hPRL’s receptor binding sites, then their removal may not allow the binding-mediated conformational activation of the second receptor binding site. This fulfills the definition of an antagonist for hPRL, and constitutes a new class of hPRL antagonists that does not rely on steric blockage of the protein’s binding surface for a second hPRL receptor, but rather does not allow the formation of the second receptor-binding surface by uncoupling the structural connection between the first and second receptor binding surfaces. This appears to be the case for Δ41-52 hPRL where the remaining agonist activity is reduced greater than 500-fold below that of G129R hPRL, 10,000-fold below that of wild-type hPRL [39], and the protein functions as an effective antagonist for hPRL. Thus, the goal of this work is to characterize this new class of hPRL antagonists; including preparation, structural/function studies, remaining agonist activity, and documenting an antagonist activity.
Materials and Methods
Protein Expression and Characterization
Recombinant methionyl proteins, including wild-type, G129R, Δ41-52, and other mutant hPRLs, were prepared essentially as described by Peterson et al. [40] and Sivaprasad and Brooks [18]. In this study the residue numbering system starts with the N-terminal methionyl as residue 0, so that the subsequent residues in wild-type and G129R hPRLs correspond to those of the native protein. Folding of wild-type and G129R hPRLs was performed as described in Peterson et al. [40] where, following air oxidation, the pH was brought from 10.5 to 7.5 and the urea was removed simultaneously by dialysis, followed by purification on DEAE-Sepharose (GE Healthcare). These proteins were well-folded monomers. Δ41-52 hPRL required a more involved folding process where the denatured alkaline extract from inclusion bodies was diluted to reduce dimer formation and subjected to more controlled dialysis to achieve proper folding. Δ41-52 hPRL was folded by delaying neutralization of the pH from 11 to 7.5 until the concentration of urea had been reduced. It is our belief that by first reducing the urea concentration allows folding to occur prior to neutralization and the associated stabilization of the disulfide bonds. Δ41-52 hPRL was purified by DEAE-Sepharose followed by separation of the monomeric and remaining dimeric proteins were removed by gel filtration on a 5 × 60 cm column of Superdex 75 (GE Healthcare) in 10 mM NH4HCO3. Δ41-52 hPRL containing >90% monomer are commonly produced by this folding and purification procedure. Purified proteins were lyophilized and stored at -30°C.
Each batch of hormone was characterized for purity by SDS-containing polyacrylamide gel electrophoresis under reducing and non-reducing conditions. In addition, the molecular weights of each hormone were determined by liquid chromatography/mass spectroscopy (Micromass Q-TOF II). The spectroscopic signals of the mutant hPRLs were compared with wild-type hPRL to determine if the folds of mutant hPRLs were similar to the wild-type hormone. Samples were prepared in 10 mM NH4HCO3 and 150 mM NaCl, pH 8.0 at appropriate concentrations as determined by the bicinchroninic acid method [41]. Fluorescence emission was determined with a Perkin-Elmer Model 55 luminometer at room temperature. An excitation wavelength of 285 nm was used and the emission spectrum was recorded from 300 to 400 nm. Near-UV circular dichroism was measured from 200 to 260 nm using an Aviv Model 202 circular dichroism spectrophotometer. Absorbance was measured from 240 to 330 nm by a Perkin-Elmer Lambda 45 UV/VIS spectrometer. The thermal stabilities of wild-type and Δ41-52 hPRL structures were determined by absorbance, fluorescence, and circular dichroism spectroscopy recorded between 25°C and 95°C.
Mass Spectrometry and Disulfide Mapping
Disulfide bonds in hormones were determined by peptide mapping using trypsin digestion followed by reverse phase liquid chromatography and mass spectroscopy. Proteins were suspended in 2 M urea, 10 mM NH4HCO3 and 20 mM iodoacetamide and incubated for 15 minutes in the dark to complete the alkylation of free cysteines [42]. Digestion was accomplished with trypsin (Pierce) added at a 1:25 ratio and incubated at 37°C for either 4 or 24 hours, peptides were separated by capillary reverse phase liquid chromatography and injected for mass analysis on a Micromass Q-TOF II or a Bruker reflex III MALDI-TOF (Campus Chemical Instrumentation Center). MALDI-generated peak lists and MS/MS generated ion fragment data were analyzed by in-house-software, MASCOT [43], or Protein Prospector [44] to deduce sequence information.
Engineering the Deleted Sequence
Exploration of the sequence deleted from Δ41-52 hPRL was examined by replacing various amino acids into the deleted sequence. Deletion of residues 41-52 removed residues from C-terminus of helix 1 and the residues that link helix 1 and the disulfide bond, leaving only residues 52 through 57 to link the C-terminus of helix 1 to this disulfide bond (Figure 1). This deletion is likely to reduce the mobility of helices 1 and 4 relative to each other. Replacement of various residues into this deleted sequence can either enhance or reduce the effects of these structural restrictions. In addition, re-engineering of the deletion may provide a more effective hPRL antagonist or a protein that provides higher yields during folding. Additions of various residues within this deletion were performed by site-directed mutagenesis by the method of Kunkel et al. [45] and are shown in Table 1. Each of these hPRL variants was prepared, characterized, and tested for biological activity.
Table 1.
Structural Changes in the Sequence of Residues 41-52 of Human Prolactin.
| Structural Change |
Yield |
Agonist Activity |
|||
|---|---|---|---|---|---|
| Protein | Residues 35-55 with deletions or replacements underlined | Monomer yield | Monomer percentage | ED50 (nM) | Fold decrease |
| WT hPRL | EMFSEFDKRYTHGRGFITKAI | 15.4 mg | 90% | 0.23 | 1 |
| Δ41-52 hPRL | EMFSEFKAI | 9.4 mg | 42% | 702.42 | 3041 |
| G2-ΔhPRL | EMFSEFGGKAI | 2.7 mg | 39% | 5.25 | 23 |
| G3-ΔhPRL | EMFSEFGGGKAI | 7.9 mg | 40% | 18.91 | 82 |
| G4-ΔhPRL | EMFSEFGGGGKAI | 13.5 mg | 57% | 1277.16 | 5312 |
| G5-ΔhPRL | EMFSEFGGGGGKAI | 11.4 mg | 67% | 1.36 | 6 |
| A2-ΔhPRL | EMFSEFAAKAI | 6.2 mg | 34% | 163.38 | 707 |
| SPGG-ΔhPRL | EMFSEFSPGGKAI | 8.7 mg | 49% | 0.44 | 2 |
| DGFIT-ΔhPRL | EMFSEFDGFITKAI | 17.5 mg | 52% | 45.94 | 207 |
| DIT-ΔhPRL | EMFSEFDITKAI | 13.4 mg | 70% | 47.93 | 199 |
| G129R hPRL | ------- | 18.3 mg | 90% | 5.99 | 26 |
Biological Assay to Determine Agonist Activities
Various mutant hPRLs were tested in several cellular assays to determine their remaining agonist activity or to determine their ability to function as antagonists. FDC-P1 is a mouse leukemia cell line that had been transformed with the full-length hPRL receptor (a generous gift from Genentech Inc., South San Francisco). FDC-P1 cells expressing the hPRL receptor were maintained in RPMI-1640, 10% fetal bovine serum, 400 μg/mL G418, 10 μM 2-mercaptoethanol, and 2 nM mouse IL-3. Twenty four hours prior to the assay actively growing cells were starved in RPMI 1640 without phenol red but supplemented with 10% equine gelding serum, G418 sulfate and 10 μM 2-mercaptoethanol. Twenty thousand cells were seeded in wells of 96-well plates in starvation media and received increasing concentrations of wild-type, G129R, Δ41-52, or other hPRLs in triplicate wells. The cells were returned to the incubation for 48 hours and subsequently evaluated for relative cell numbers by a vital dye method (10 μL Alamar Blue per well, Accumed International)(46). The reduction of the vital dye was monitored between 2 and 4 hours of incubation at 570 and 600 nm and the percent of reduced dye calculated as a marker for cell number. This method correlates well with viable cell counts performed after Trypan Blue staining. The ED50 was calculated for each hormone tested and the ED50s were expressed as a ratio to that for wild-type hPRL.
Biological Assay to Determine Antagonist Activity
Jurkat cells are a human leukemia cell line [47] and express both hPRL and hPRL receptor [48]. Jurkat cells are available from the American Tissue Culture Collection (http://www.atcc.org/). Jurkat cells were grown in RPMI-1640, 15% fetal bovine serum, and penicillin/streptomycin. Cells were suspended in media at 106 cells/mL and placed in 6-well or 12-well plates and supplemented in triplicate wells with 0 to 1000 nM of either wild-type, G129R, or Δ41-52 hPRL. After 24, 48, 72, and 96 hours of incubation the Jurkat cells were analyzed by flow cytometry for the induction of apoptosis or the presence of necrosis by the method of Zamzami et al. and Castedo et al. [49,50]. Briefly, documentation of apoptosis used 20 nM 3,3’-dihexyloxacarbocyanine (DiOC6, Sigma) to assess the metabolic state of the mitochondria by monitoring the mitochondrial transmembrane potential in individual cells by flow cytometry (Coulter EPICS Elite). The loss of DiOC6 fluorescence is associated with ongoing apoptosis. In addition the cells were labeled with propidium iodide (PI) to assess cellular integrity. Increased PI fluorescence in conjunction with reduced DiOC6 fluorescence is an indication of necrotic cells. Individual cells were excited at 488 nm with a 15mW air-cooled argon ion laser (Cytonics, Uniphase). DiOC6 fluorescence emission was measured at 525 nm while PI fluorescence emission was measured at 635 nm. In addition, controls were run at each time point. Viable Jurkat cells, both stained and unstained were used to adjust gating, forward and side scattering. Jurkat cells not receiving hormone treatment were treated with either 100 μM carbamoyl cyanide m-chlorophenylhydrazone (m-CICCP, Sigma) to disrupt the mitochondrial membrane potential or with camptothecin (Sigma) to induce apoptosis. Only studies where the control cells responded to m-CICCP or camptothecin treatments were accepted. Fortunately, Jurkat cells are frequently used as control cells in the DiOC6 assay and in our assays the control cells responded similarly to those in previous studies. At the conclusion of cytometric fluorescence measurements, cells were graphed according to their DiOC6 and PI fluorescence. Gates were set using the control cells and the cells within in each of four quadrants were enumerated and the expressed as a percent of the total cells.
Results
Expression, Purification, and Characterization of Recombinant Human Prolactin Antagonists
Multiple batches of eleven recombinant hPRLs were prepared for this study. For wild-type and G129R hPRL SDS-containing acrylamide gel electrophoresis under reducing conditions demonstrated monomeric proteins with purities greater than 95%, and predominately monomeric protein when examined under non-reducing conditions (data not shown). Under reducing conditions the small amounts of dimeric proteins were entirely converted to monomers. Mass spectrometry of the proteins provided masses within ±2 Daltons of the molecular weight calculated from the amino acid sequence at a defined pH.
Thermal denaturation studies of wild-type and Δ41-52 hPRLs, as well as the extracellular domain of the hPRL receptor provided sharp thermal denaturations indicating a folded protein and a stable structure under physiological conditions when monitored by absorbance, circular dichroism, or fluorescence. Monitoring thermal denaturation by absorbance, circular dichroism (molar elipticity at 222 nm), or fluorescence (λmax) provided similar TMs, suggesting denaturation occurred in a single-step thermal transition. The α-helix dominated wild-type and Δ41-52 hPRLs provided TMs of 65°C and 80°C, respectively, indicating that deletion of residues 41 through 52 provided a more thermally stable molecule (Figure 2). The β-sheet-dominated extracellular domain of the hPRL receptor had a TM of 45°C.
Figure 2. Thermal Denaturation of Wild-type and Δ41-52 Human Prolactin.
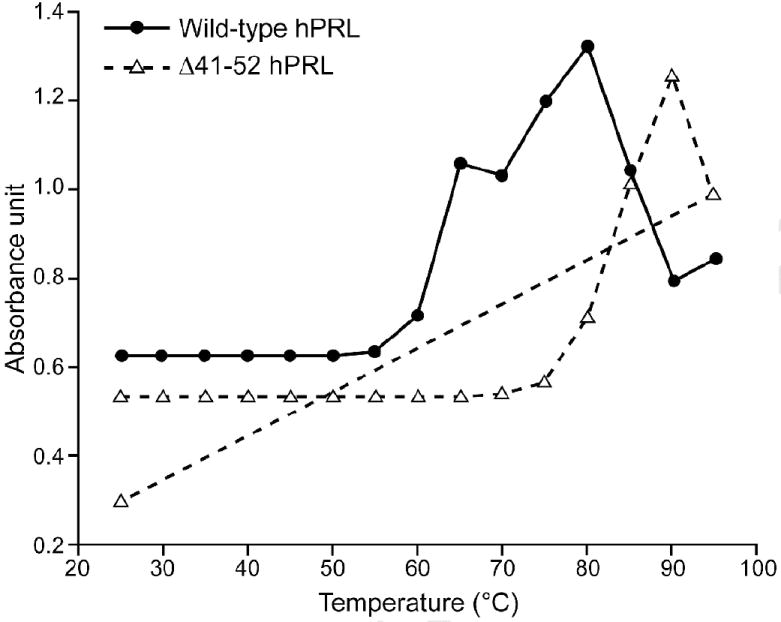
1.0 μM solution of either wild-type hPRL or Δ41-52 hPRL was placed into heated cuvets in a LS55 Perkin/Elmer spectrophotometer in 10 mM Tris pH 7.5 and 150 mM NaCl. Temperatures were increased at a rate of 1C° per minute from 25C° to 95C°. The absorbance was measured at 280 nm at 5C° increments. When 95C° was achieved the temperature of Δ41-52 hPRL was returned to 25C°; the reduction in absorbance observed between 90 and 95C° appears to be a result of precipitation of the denatured protein. This same reduction in absorbance is retained when the temperature is returned to 25C° suggesting that the protein precipitate does not return to a solution. A basal thermal-induced rate of absorbance change was determined at low temperatures and subtracted from the measured data.
Folding Δ41-52 hPRL by our standard method [40] followed by analysis of the products by non-reducing 15% acrylamide gel electrophoresis showed that Δ41-52 hPRL was largely a dimer. Mass spectroscopy (Nanospray Q-TOF) of mixed monomer and dimer of Δ41-52 hPRL demonstrated that the dimeric form had a molecular weight of 43,170 Daltons, twice that of the monomer (21,585 Daltons). Reduction of these Δ41-52 hPRL preparations eliminated the 43 kDa band while enhancing the 21 kDa band in SDS-containing polyacrylamide gels under reducing conditions. Extensive dimerization of wild-type hPRL was not observed when folded under identical conditions. Based on these data we conclude that elimination of residues 41 through 52 does not promote native folding of Δ41-52 hPRL under standard conditions and produces significant amounts of dimeric Δ41-52 hPRL. Inter-molecular pairing of cysteines to form disulfide bonds indicates that under standard folding conditions one or more cysteines have not been juxtaposed and oxidized in native form during the reduction of urea by dialysis, allowing the cysteines to seek inter-molecular partners for disulfide bond formation.
Based on these finding we made three changes in our preparation of Δ41-52 hPRL. We have delayed returning the pH from 11 to 7 until a significant reduction of urea has been accomplished. We also diluted the Δ41-52 hPRL to perform folding at a lower protein concentration, reducing the likelihood of inter-molecular disulfide bond formation. Finally, we separated monomeric Δ41-52 hPRLs from the remaining dimers by size exclusion chromatography. These three additions to our folding protocol significantly improved yields of well folded Δ41-52 hPRL monomers.
Mapping Intra-molecular Disulfide Bonds
We also sought to identify which cysteines were available for formation of Δ41-52 hPRL dimers. We blocked free cysteines with iodoacetamide [42], cleaved dimeric Δ41-52 hPRL with trypsin in the presence of 2 M urea, and performed mass spectrometry to determine the disulfide bonding pattern. Mapping the resolved peptides provided 80% sequence coverage. Tandem mass spectrometry identified the following sequence of amino acids: RLTVQCCQ. This peptide had a calculated molecular mass of 1,435.77 Daltons and the precursor ion had a mass of 1,435.81 Daltons (479.27 × 3) for the triple charged species (Figure 3). Inspection of the sequence of hPRL indicated that this peptide constituted a disulfide-linked pair of peptides that are residues 11 through 16 of hPRL. We suggest that cysteine 11 had formed inter-molecular dimers during the process of folding and oxidation. If cysteine 11 was available, then its normal intra-molecular disulfide partner (cysteine 4) also would have been available. Unfortunately, the N-terminal peptide (residues 1-10) was not recovered during peptide mapping. Thus, we were not able to confirm that cysteine 4 also participated in inter-molecular disulfide bond formation; but, without being reduced by its normal partner (cysteine 11) it was reasonable to assume that cysteine 4 would be available for inter-molecular disulfide bond formation.
Figure 3. Mass Spectrum for a Peptide Isolated from the Trypsin Digest of Δ41-52 hPRL.
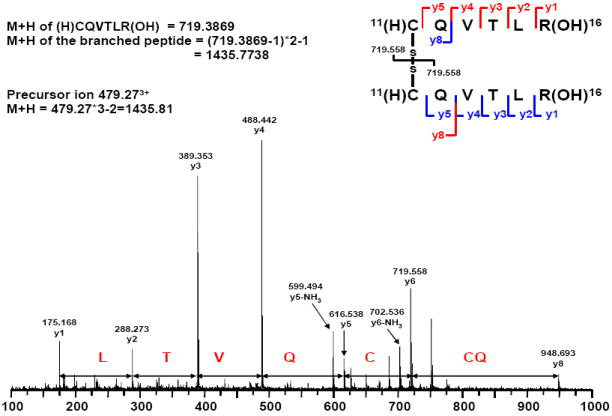
Dimeric Δ41-52 hPRL was isolated by size exclusion chromatography, digested with trypsin, and submitted to LC/MS/MS as described in the Materials and Methods section. The disulfide-linked peptides constituting residues 10-16 was identified by both the mass of the peptide fragment and the sequence information.
Engineering the Deleted Sequence of Human Prolactin Antagonists
Eight hPRLs each containing one or more amino acids added into the deletion site of Δ41-52 hPRL were successfully prepared and characterized. DNA sequencing and mass spectroscopy confirmed the addition of each amino acid addition (Table 1). The proteins’ molecular masses were all within 0.08% of the calculated values. The difference in molecular weights between Δ41-52 hPRL and the proteins with residues added to the gap were within 1 Dalton of the calculated values.
The absorbance spectra of wild-type and Δ41-52 hPRL closely overlay (Figure 4a) indicating that the proteins had similar folds. The fluorescence spectra of wild-type hPRL showed a maximum at 340 nm (Figure 4b). Δ41-52 hPRL showed an additional peak at 318 nm and a broadening of the signal toward the red end of the spectrum. All the hPRL mutants that added residues in the 41 through 52 deletion showed varying increases in the red end of the spectrum (340 to 400 nm). Some of these proteins also showed modest peaks at 318 nm (3G, A2, and DGFIT hPRLs, data not shown). The circular dichroism spectra of wild-type and Δ41-52 hPRLs indicated that the helical structure of the wild-type hormone was largely retained by Δ41-52 hPRL (Figure 4c). The only difference was that the mutant proteins showed increased negative deflections at 208 nm relative to the 222 nm signal. These spectral data indicate that the proteins are generally well folded with features intermediate to wild-type and Δ41-52 hPRLs.
Figure 4. Absorbance, Fluorescence and Circular Dichroism Spectrascopic Comparisons of Wild-type and Δ41-52 Human Prolactins.
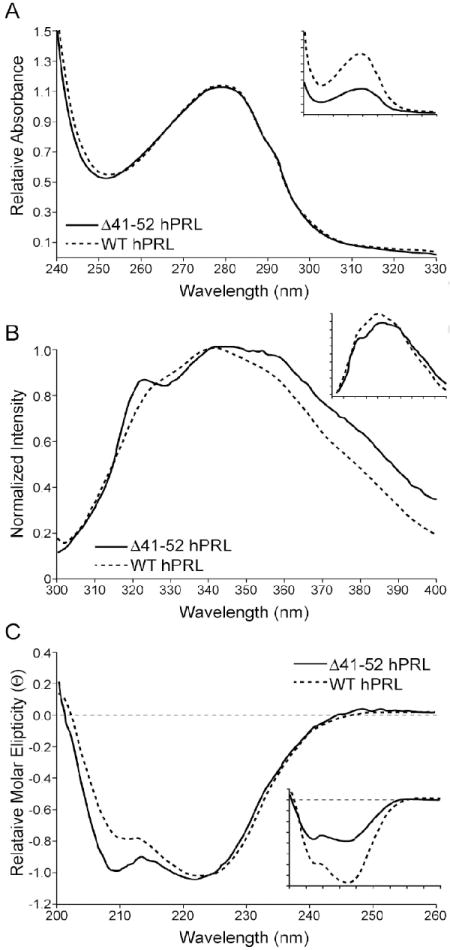
Samples were prepared in 10 mM NH4HCO3, 150 mM NaCl, pH 8.0 at appropriate concentrations as determined by the bicinchoninic acid method [41]. Fluorescence emission was determined on a Perkin-Elmer Model 55 luminometer with an excitation wavelength of 285 nm and the emission spectrum recorded from 300 to 400 nm. Near-UV circular dichroism was measured from 200 to 260 nm using an Aviv Model 202 circular dichroism spectrophotometer. Absorbance was measured from 240 to 330 nm by a Perkin-Elmer Lamda 45 UV/VIS spectrophotometer. Raw data is presented in the inset panel and the normalized data is presented in the main panel.
One of the goals of this study was to find hPRL antagonists in this class that display a highly reduced agonist activity and provide high yields during preparation. In these studies wild-type hPRL averages a yield of 15.4 mg per liter of bacterial broth (Table 1). The hormone recovered from the anion exchange column was judged to be 90% monomer on the basis of peak areas from the subsequent sizing column. Replacing glycine 129 with arginine (G129R hPRL, an antagonist that uses steric inhibition of site 2) produced average yields of 18.3 mg and 90% monomer, similar to that of wild-type hPRL. Following our modified folding and purification scheme Δ41-52 hPRL only produced an average of 9.4 mg of monomeric hormone with only 42% of the total hormone recovered from ion exchange chromatography as monomer. Clearly reducing the formation of dimeric species would dramatically increase yields of Δ41-52 hPRL.
Determination of Yields of Engineered Human Prolactins
Having optimized our folding and purification procedures, we next sought to engineer a protein that retained antagonist activities and provided high yields of monomeric hormone. Four replacement strategies were evaluated (Table 1). First, we added between 2 and 5 glycine residues (G2 through G5-ΔhPRL). As the number of glycine additions increased from 2 to 5 the percentage of monomeric hPRL recovered from the ion exchange column increased from 39 to 67%, the yields also increased with an average of 13.5 mg for the G4-ΔhPRL. The second strategy to influence the yield of monomeric hPRL antagonist was to add back two alanine residues into the position between residues 41 through 52. We observed that both the percentage of monomeric A2-ΔhPRL (34%) and the yield of A2-ΔhPRL (6.2 mg) were reduced when compared to Δ41-52 hPRL. The addition of two alanine residues in this position showed the lowest yields of purified protein and the lowest percentage of monomeric hormone. The third strategy was to add a β-turn (SPGG-ΔhPRL) at the top of helix 1. The addition of the ß-turn did not increase the yield (8.7 mg) and only modestly increased the percentage of monomer (49%). The forth strategy was to replace several of the original residues into Δ41-52 hPRL. Two proteins were prepared, the first included D41, G49, F50, I51, and T52 (DGFIT-ΔhPRL) while the second protein included D41, I51, and T52 (DIT-ΔhPRL). DIT-ΔhPRL, containing the smaller replacement, increased the yield of monomer to 13.4 mg with 70% of the folded protein being a monomer. The larger insert (DGFIT-ΔhPRL) produced the highest yield of monomeric hormone (17.5 mg) with 52% of the total protein being found as a monomeric species.
Determining the Agonist Activities of Engineered Human Prolactins
In addition to the yield of monomeric hormones, we also sought variants of Δ41-52 hPRL with minimal agonist activities. To this end we compared the agonist activities of wild-type hPRL and G129R hPRL to Δ41-52 hPRL and each of the hPRL variants. Each of these assays used increasing hormone concentrations to stimulate the growth of FDC-P1 cells that had been transformed with the hPRL receptor. The hPRL agonist activity was discerned following a 24 hour serum starvation and 48 hour stimulation by increasing hormone concentrations. ED50s were calculated and the fold reduction in agonist activity compared to that of wild-type hPRL (Table 1). The precision of these assays indicate that ED50s within approximately two-fold should be regarded as similar. Wild-type hPRL displayed a typical ED50 of 0.23 nM. The activity of G129R hPRL (5.99 nM) was significantly reduced, retaining approximately 4% of the agonist activity of wild-type hPRL. Our lead compound, Δ41-52 hPRL, displayed an agonist activity of 702.42 nM, a 3,054-fold reduction in agonist activity from that of wild-type hPRL. Δ41-52 hPRL retains approximately 100-fold less agonist activity than G129R hPRL. Disruption of the structure coupling of sites 1 and 2, the basis for this new class of hPRL antagonists, reduces the agonist activity to a much greater extent than antagonists that provide steric inhibition of site 2 (G129R hPRL).
In situations where residues were added between residues 40 and 53 reductions of agonist activity greatly varied (Table 1). Addition of 2, 3, and 4 glycine residues (G2 through G4-ΔhPRL) had progressively reduced agonist activity (between 23 and 5312-fold less activity than wild-type hPRL). G4 hPRL provided the greatest reduction of agonist activity (1277.16 nM, a reduction of 5,553-fold reduction from the agonist activity of wild-type hPRL). In contrast, addition of 5 glycines (G5 hPRL) produced a hormone with only a 6-fold reduction in agonist activity. The addition of two alanine residues (A2 hPRL) produced an agonist with intermediate activity 163.38 nM. These results indicate that extending helix 1 back to that of the wild-type hormone, a manipulation that we believe increases the stress within the molecule not only reduced the yield of hormone but provided agonist activity between those of wild-type and Δ41-52 hPRLs. Similarly, addition of a ß-turn into Δ41-52 hPRL also appeared to reduce the stress in Δ41-52 hPRL producing a hormone with activities approaching that of wild-type hPRL. Finally, addition of three or five original residues (DIT and DGFIT-ΔhPRLs, 199 and 207-fold decrease in agonist activity) produced hormones with agonist activities between wild-type and Δ41-52 hPRLs. These last two hormones have approximately 10-fold less agonist activity than G129R hPRL. In summary, few of the re-engineered hPRL antagonists had reductions in their remaining against activities similar to that of Δ41-52 hPRL.
Determination of the Antagonist Activities of Engineered Human Prolactins
The final objective of this study was to identify hPRLs that served as potent antagonists. These studies compared the activities of wild-type hPRL, G129R hPRL and Δ41-52 hPRL on the viability of Jurkat cells. We chose to use Jurkat cells for these studies, because hPRL promotes viability in these human lymphoma cells. Growth of these cells in 10% fetal bovine serum provided sufficient stimulus to insure viability and the synthesis and secretion of hPRL from the cells should increase the lactogenic stimulus during the 96 hour course of the experiment. We used flow cytometry to measure the influence of hPRLs on the proportions of viable cells, cells undergoing apoptosis, and necrotic cells. The exogenous hPRL concentrations and time of treatment were variables in these experiments. Thus, the ability of the two hPRL antagonists (G129R and Δ41-52 hPRL) to induce apoptosis can be directly compared to each other and to wild-type hPRL.
Treatment of Jurkat cells under the influence of 10% fetal bovine serum with Δ41-52 hPRL showed a time- and dose-dependent decrease in live cells, an increase in cells undergoing apoptosis, and an increased proportion of cells that are necrotic (Table 2 and Figure 5). For Δ41-52 hPRL, increasing antagonist concentrations produced a biphasic response where 1 nM Δ41-32 hPRL produced the smallest percentage in viable cells (27% viable) and the greatest percentage in necrotic cells (61% dead) after 72 hours of treatment. At both lower and higher concentrations of Δ41-52 hPRL (0.1,10, 100 and 1000 nM) the effects were smaller, but each dose at each time reduced percentages of viable cells, increased percentages of necrotic cells, and increased percentages of cells undergoing apoptosis. Apoptosis appeared to depend on the time of treatment; after 24 hours of treatment the maximal percentage of cells undergoing apoptosis was observed. At lower Δ41-52 hPRL concentrations (0.1 and 1.0 nM) the percentage of live cells decreased and the percentage of dead cells increased with time (up to 96 hours). At higher Δ41-52 hPRL concentrations (10, 100, and 1000 nM) the percentage of live cells first decreased and subsequently slowly increased during the 96 hours of treatment. This may be a result of the Jurkat cells increasing endogenous hPRL concentrations and resisting the antagonist effect of Δ41-52 hPRL.
Table 2.
Flow Cytometry Data for Hormone-treatment of Human Jurkat Cells (representative of three studies)*.
| Wild-type human prolactin | G129R human prolactin | Δ41-52 human prolactin | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dose | Time | % Live | % Apop. | % Necro. | % Total | % Live | % Apop. | % Necro. | % Total | % Live | % Apop. | % Necro. | % Total |
| 0.0 nM | 24 hrs | 93.9 | 1.9 | 3.8 | 99.6 | 93.9 | 1.9 | 3.8 | 99.6 | 93.9 | 1.9 | 3.8 | 99.6 |
| 48 hrs | 94.6 | 0.9 | 4.2 | 99.7 | 94.6 | 0.9 | 4.2 | 99.7 | 94.6 | 0.9 | 4.2 | 99.7 | |
| 72 hrs | 95.8 | 0.8 | 3.2 | 99.8 | 95.8 | 0.9 | 3.2 | 99.9 | 95.8 | 0.8 | 3.2 | 99.8 | |
| 96 hrs | 94.1 | 2.6 | 3.2 | 99.9 | 94.1 | 2.6 | 3.2 | 99.9 | 94.1 | 2.6 | 3.2 | 99.9 | |
| 0.1 nM | 24 hrs | 92.7 | 2.0 | 5.0 | 99.7 | 90.2 | 1.9 | 7.8 | 99.9 | 88.2 | 2.4 | 9.2 | 99.8 |
| 48 hrs | 92.8 | 1.3 | 5.3 | 99.4 | 93.6 | 1.0 | 5.2 | 99.8 | 77.1 | 9.4 | 12.4 | 98.9 | |
| 72 hrs | 95.2 | 1.0 | 3.7 | 99.9 | 95.9 | 1.1 | 2.8 | 99.8 | 58.7 | 7.1 | 33.3 | 99.1 | |
| 96 hrs | 91.0 | 4.1 | 4.8 | 99.9 | 92.9 | 3.1 | 3.6 | 99.6 | 54.5 | 5.5 | 38.6 | 98.6 | |
| 1.0 nM | 24 hrs | 90.7 | 2.0 | 7.1 | 99.8 | 89.7 | 1.9 | 8.2 | 99.8 | 85.6 | 2.4 | 11.7 | 99.7 |
| 48 hrs | 90.7 | 1.6 | 7.2 | 99.5 | 94.9 | 1.2 | 3.7 | 99.8 | 75.1 | 9.5 | 13.5 | 98.1 | |
| 72 hrs | 92.5 | 1.2 | 6.2 | 99.9 | 95.6 | 1.0 | 3.3 | 99.9 | 63.2 | 6.0 | 30.7 | 99.9 | |
| 96 hrs | 91.9 | 2.5 | 5.4 | 99.8 | 92.6 | 2.5 | 4.4 | 99.5 | 27.0 | 7.2 | 61.3 | 95.5 | |
| 10 nM | 24 hrs | 91.3 | 1.5 | 6.9 | 99.7 | 87.9 | 1.9 | 10.2 | 100 | 83.4 | 2.5 | 14.0 | 99.9 |
| 48 hrs | 92.7 | 1.4 | 5.5 | 99.6 | 94.3 | 1.3 | 4.2 | 99.8 | 74.2 | 11.1 | 13.9 | 99.2 | |
| 72 hrs | 95.1 | 0.8 | 4.0 | 99.9 | 95.4 | 0.9 | 3.6 | 99.9 | 49.5 | 7.1 | 42.8 | 99.4 | |
| 96 hrs | 91.0 | 2.0 | 6.8 | 99.8 | 93.6 | 2.5 | 3.6 | 99.7 | 57.7 | 4.9 | 37.6 | 100.2 | |
| 100 nM | 24 hrs | 92.0 | 1.6 | 6.2 | 99.8 | 91.3 | 1.6 | 6.9 | 99.8 | 88.8 | 1.7 | 9.4 | 99.9 |
| 48 hrs | 91.3 | 1.3 | 7.2 | 99.8 | 92.5 | 1.2 | 6.2 | 99.9 | 79.1 | 7.7 | 12.6 | 99.4 | |
| 72 hrs | 94.1 | 1.3 | 4.3 | 99.7 | 95.7 | 1.3 | 2.8 | 99.8 | 58.3 | 6.7 | 34.5 | 99.5 | |
| 96 hrs | 93.4 | 1.8 | 4.5 | 99.7 | 93.9 | 2.6 | 3.3 | 99.8 | 68.6 | 5.1 | 25.5 | 99.2 | |
| 0.5 μM | 24 hrs | 91.6 | 2.1 | 6.1 | 99.8 | 89.5 | 1.9 | 8.4 | 99.8 | - | - | - | - |
| 48 hrs | 90.8 | 1.7 | 7.3 | 99.8 | 92.2 | 2.3 | 6.2 | 100.7 | - | - | - | - | |
| 72 hrs | 89.6 | 2.9 | 7.3 | 99.8 | 95.2 | 0.6 | 4.0 | 99.8 | - | - | - | - | |
| 96 hrs | 88.6 | 2.6 | 8.6 | 99.8 | 93.8 | 2.3 | 3.6 | 99.7 | - | - | - | - | |
| 1.0 μM | 24 hrs | 88.5 | 2.4 | 9.0 | 99.9 | 91.6 | 1.8 | 6.5 | 99.9 | 85.1 | 2.1 | 12.6 | 99.8 |
| 48 hrs | 92.0 | 1.8 | 5.8 | 99.6 | 94.1 | 3.2 | 4.6 | 101.9 | 73.3 | 10.3 | 15.6 | 99.2 | |
| 72 hrs | 93.0 | 1.2 | 5.6 | 99.8 | 95.7 | 1.0 | 3.3 | 100 | 65.7 | 5.0 | 28.6 | 99.3 | |
| 96 hrs | 92.0 | 2.4 | 5.6 | 100 | 93.0 | 3.2 | 3.7 | 100 | 83.3 | 2.3 | 13.7 | 99.3 | |
Percent of the total cells recorded in quadrants that represent living cells, loss of mitochondrial membrane potential (apoptosis), loss of mitochondrial potential and increased PI permeability (necrosis), and total percentage of cells in the above three quadrants.
Figure 5. Time Course for the Induction of Apoptosis and Necrosis by 1 nM Δ41-52 hPRL.
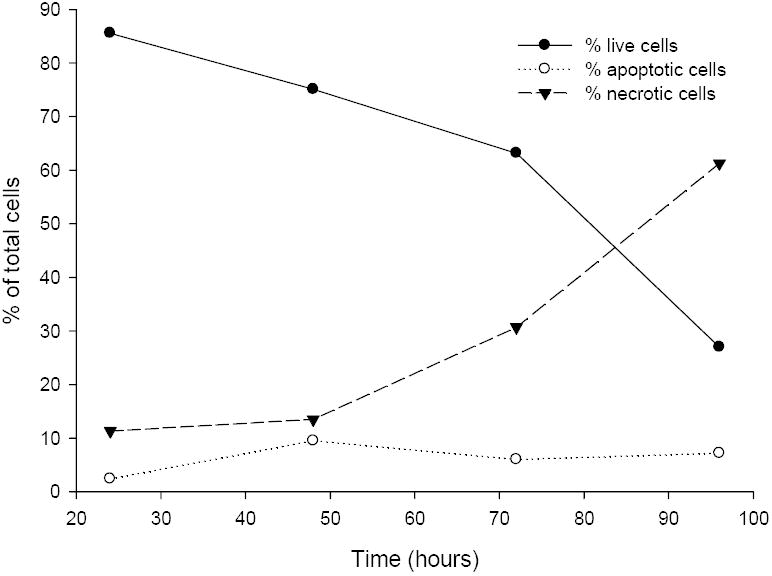
Jurkat cells were grown in RPMI-1640, 15% fetal bovine serum, and penicillin/streptomycin. Cells were suspended in media (106 cells/mL) and supplemented in triplicate wells with 1 nM Δ41-52 hPRL. After 24, 48, 72, and 96 hours of incubation the Jurkat cells were analyzed for the induction of apoptosis or the presence of necrosis by the method of Zamzami et al. and Castedo et al. [49,50]. This method used 20 nM 3,3’-dihexyloxacarbocyanine (DiOC6, Sigma) to assess induction of apoptosis by measuring the mitochondrial transmembrane potential in individual cells by flow cytometry (Coulter EPICS elite, Miami, FL). In addition the cells were labeled with propidium iodide (PI) to assess cellular integrity. Increased PI fluorescence in conjunction with reduced DiOC6 fluorescence is an indication of necrotic cells. Individual cells were excited at 488 nm with a 15mW air-cooled argon ion laser (Cytonics, Uniphase, San Jose, CA). PI fluorescence emission was measured at 635 nm. In addition, controls were run at each time point as described in Materials and Methods. Gates were set using the control cells and the cells within in each of four quadrants were enumerated and the expressed as a percent of the total cells counted. This data is representative of the three experiments performed.
Identical treatment with wild-type hPRL did not induce changes in the percentage of live, apoptotic, and necrotic Jurkat cells regardless of dose or time. Typically live cells represented between 89 to 95%, apoptotic cells represented between 0.8 to 2.9%, and necrotic cells represent 3.2 to 9.0% of the total cells (Table 2). Treatment with wild-type hPRL concentrations between 0.1 and 1000 nM did not change the percentages when compared to controls not receiving exogenous hPRL. These data indicate that a 15% fetal bovine serum stimulus is sufficient to fully stimulate viability of the Jurkat cells and addition of wild-type hPRL does not change the distribution of live, apoptotic, or necrotic cells.
Finally, Treatment of Jurkat cells with G129R hPRL at concentrations between 0.1 and 1000 nM for up to 96 hours produced results that are similar to the results of no treatment or treatment with wild-type hPRL. Living cells represented 88 to 95%, apoptotic cells represented 0.6 to 3.2%, and necrotic cells represented 2.8 to 10.2% of the total cells (data not shown). Treatment with G129R hPRL did not provide an effective antagonism in the presence of 15% fetal bovine serum and production of hPRL by Jurkat cells. Similar observations of Δ41-52 hPRL-induced apoptosis-mediated cell death were observed in the murine lymphoid cell lines Nb-2 cells and FDC-P1 that express the hPRL receptor (data not shown).
Discussion
The mechanism by which hPRL binds and activates the hPRL receptor is not fully understood. We have recently shown that sites 1 and 2 for both hPRL and hGH are functionally coupled [18-20]. Receptor binding to site 1 of hPRL initiates a conformation change across the structure of the hormone. This provides a second receptor binding site on the hormone with a biologically relevant affinity. We have performed numerous mutagenic studies that have identified various residues that are not among the functional binding epitopes of sites 1 or 2 but are required for lactogenic actions of these hormones; we believe that these residues constitute a motif that transmits the hPRL receptor binding-induced conformation change from site 1 to site 2 of the hormone.
Based on the work of Tsunekawa et al. [38] in the 20 kDa form of hGH, we prepared a similar deletion mutant in hPRL (Δ41-52 hPRL) [39] and were surprised to observe that this deletion provided a substantially greater reduction of lactogenic activity than that provided by the 20 kDa hGH. We also realized that this deletion contained some of the residues that we had identified as being critical for activity and necessary to functionally couple site 1 with site 2 in hPRL. Based on this information, we hypothesized that removal of residues contained within this motif might provide a new class of hPRL antagonists, where site 1 receptor binding would fail to initiate the organization of the hormone’s second binding site. This new class of hPRL antagonists could occupy but would fail to activate hPRL receptors. The results of our current work support this hypothesis where Δ41-52 hPRL has been shown to be a potent antagonist. The residues comprising site 2 of Δ41-52 hPRL are unchanged, yet no apparent result of site 2 binding is observed. This observation supports the notion that sites 1 and 2 are functionally coupled and the formation of site 2 is dependent on hPRLr binding at site 1 and the transmission of structural changes that organize a functional site 2.
In this work we have characterized the preparation of Δ41-52 hPRL and demonstrate that it represents a lead compound for this new class of hPRL antagonists. Further, we explored structural variants of the deleted sequence, characterizing the folding, yields, and remaining agonist activities. Finally, we demonstrated that Δ41-52 hPRL is a potent antagonist capable of inducing cell death through apoptosis in a cell line derived from a human T-cell leukemia [48].
Δ41-52 hPRL was more difficult to prepare than either wild-type or G129R hPRLs; dimeric Δ41-51 hPRL was the major product from our standard folding procedure. Analysis of dimeric Δ41-52 hPRL by trypsin cleavage and mass spectrometry indicated that the dimers were formed through the N-terminal cysteines of hPRL. Dimeric Δ41-52 hPRL was inactive as either an agonist or antagonist (Brooks and Almgren, unpublished) and thus represented a major inactive side product of protein folding. Thus, we developed a folding procedure that more carefully controlled disulfide bond formation. By delaying the neutralization of pH during the dialysis-mediated removal of urea, we reduced the production of this mis-folded hormone. Maintaining a higher pH delayed the formation of stable disulfide bonds allowing a longer opportunity for the folding process to juxtapose the proper cysteines for wild-type disulfide bond formation. The addition of size exclusion chromatography was an effective method to remove remaining dimeric hormone. Thus, we have developed a folding and purification scheme that produced a properly folded homologous monomeric Δ41-52 hPRL with high antagonist activity.
Thermal denaturation studies indicated that Δ41-52 hPRL was more stable than wild-type hPRL. The removal twelve residues decreased the length connecting helix 1 to helix 4 through the 58/174 disulfide bond; this shorter connector must reduce the possible movement between these two helices and stabilize the protein from thermal motion and denaturation. The relative movement between these helices may be required for the action of hPRL; this restriction of movement may provide the observed increase in thermal stability. The reduction in the potential conformers available to Δ41-52 hPRL also inhibits the site 1 receptor binding-induced conformation change within hPRL and thus inhibits the structuring of site 2 required for receptor binding. Residues in helices 3 and 4 are involved in the second binding site of hPRL for the receptor. Thus, the mechanism by which Δ41-52 hPRL has reduced agonist activity (Table 1) may be that binding of receptor at site 1 can no longer position helix 4 in an appropriate position to promote site 2 binding.
Replacement with native or other residues in the gap created by deletion of residues 41-52 was explored to determine their effects on yields, protein folding, and agonist activity. Four strategies for replacements were pursued. The first was to place between one and five glycines into the deletion. This approach was designed to relieve stress within the folded molecule that was induced by removal of the dozen residue sequence (residues 41 through 52). Glycine additions do not provide side chain chemistries, secondary structures, or backbone rigidity. Addition of increasing numbers of glycines tended to increase the yields of monomeric proteins. Glycine additions also changed the ED50s of agonist activities; this reduction in agonist activity was maximal with the addition of four glycines. Glycine additions provided a non-structured linker between residues 41 and 52. We believe these additions relieved the tension between the C-terminus of helix 1 and helix 4 (coupled by the disulfide bond between cysteines 58 and 174) without addition of structural features within the linker. This suggests that fine tuning of this spatial relationship was an effective means of regulating agonist activity. We have previously shown that residues between positions 41 and 52 are required for coupling sites 1 and 2 within hPRL [20]. But the spatial relationship between helices 1 and 4 is also important for site 1 binding. Thus, glycine addition may optimize site 1 binding while not providing efficient coupling between sites 1 and 2 of hPRL. These results are currently being evaluated by binding studies where site 1 and 2 binding events can be independently evaluated. The second approach was to add 2 alanines into the gap A2-Δ42-52 hPRL. These two alanines would extend helix 1 to a length described in structural studies [31,32] and increase the tension between helices 1 and 4. This structural change reduced the yields as well as the percentage monomer. The agonist activity of was intermediate between wild-type and Δ41-52 hPRL. A third structural study added a ß-turn (SPGG-hPRL) to the gap created by the deletion of residues 41 through 52. We believed that this addition would relieve some of the structural constraints associated with the deletion. Yields of SPGG-hPRL remained low and less than 50% folded into monomeric protein. Surprisingly, SPGG-hPRL regained much of its biological activity. This demonstrates that relief of the tension by addition of an engineered turn between the C-terminus of helix 1 and the central C58-C174 disulfide bond does not increase the efficiency of folding. Finally, selected sequences from wild-type hPRL, including DGFIT and DIT, were replaced in Δ41-52 hPRL. These additions increased both the yield of and percent of monomer. The addition of sequences that reduced the stress within the various hPRLs generally increased the yields of hPRLs; while the addition of alanines that increase the stress (A2-hPRL) reduced the yields of protein.
These same additions did not produce parallel results when considering their effects on agonist activities. The serial addition of glycines was illustrative, where only replacement with four glycines provided highly reduced agonist activity (a 5,312-fold reduction), while additions of two, three, or five glycines increased the remaining agonist activity from that of Δ41-52 hPRL (from 3041 to 23, 82, and 6-fold reductions in activity of wild-type hPRL, respectively). In contrast, the additions of 5 glycines (G5-ΔhPRL, 2-fold reduction in activity) or addition of four amino acids as a ß-turn (SPGG-hPRL, 6-fold reduction in activity) produced hormones with agonist activities similar to wild-type hPRL. Finally, addition of either three or five residues from the original sequence (DGFIT- and DIT-hPRL) produced hormones retaining some structure when compared to glycine additions and a modestly higher agonist activity (207 and 199-fold reduction of activity, respectively) then their glycine counterparts. Thus, it appeared that the nature of the linkage between residues 41 and 52 was important in the loss or retention of agonist activity. Both the number and nature of residues inserted into the deletion appeared to influence the loss of agonist activity.
Comparison of the remaining agonist activities of Δ41-52 and G129R hPRLs showed a clear advantage to our new class of antagonists when creating an ideal hPRL antagonist. The agonist activity of Δ41-52 was greatly reduced as we have previously reported [39] and was significantly less than G129R hPRL when tested in FDC-P1 cells expressing hPRL receptors. In these experiments Δ41-52 hPRL retained approximately 0.03% agonist activity while G129R hPRL retained approximately 3.8%. This greater than 100-fold reduction of agonist activity of Δ41-52 hPRL when compared to G129R hPRL should allow higher concentrations of this antagonist to be present before this protein affects significant agonist activities. Thus, when it is used in either in vitro or in vivo assays we anticipate that Δ41-52 hPRL will provide better antagonist activities than G129R hPRL.
Finally, low concentrations of Δ41-52 hPRL induced a substantial level of apoptosis in human Jurkat cells in a dose- and time-dependent fashion (Table 2, Figure 5). In parallel studies G129R hPRL did not induce apoptosis at doses up to 1000 ng/mL over the 96 hour time course of this study. Jurkat cells contain a paracrine/autocrine system, expressing both the hPRL receptor and hPRL [48]. Cells treated with no exogenous hPRL and those treated with up to 1000 nM wild-type hPRL displayed similar percentages of viable cells (94 to 96% vs. 89 to 95%, respectively), apoptotic cells (0.9 to 2.6% vs. 0.8 to 2.9%, respectively), and necrotic cells (3.2 to 4.2% vs. 3.2 to 9.0%, respectively), suggesting that addition of exogenous wild-type hPRL does not provide a greater stimulus for cell viability than that provided from endogenous sources and showing that Δ41-52 hPRL can effectively antagonize Jurkat cells under conditions that fully support cell viability.
Acknowledgments
This work was supported in part by Grant # R01-DK56117 form the National Institutes of Health. Patent # 6,995,244, Antagonists for Human Prolactin, was awarded for the preparation and use of this class of compounds.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Buhimschi C. Endocrinology of lactation. Obstet Gynecol Clinics North America. 2004;31:963–979. doi: 10.1016/j.ogc.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 2.Krishnan N, Thellin O, Buckley DJ, Horseman ND, Buckley AR. Prolactin suppresses glucocorticoid-induced thymocyte apoptiosis in vivo. Endocrinology. 2003;144:2102–2110. doi: 10.1210/en.2003-0053. [DOI] [PubMed] [Google Scholar]
- 3.Welsch CW, Nagasawa H. Prolactin and murine mammary tumorigenesis: a review. Cancer Research. 1977;37:951–963. [PubMed] [Google Scholar]
- 4.Welsch CW. Prolactin and the development and progression of early neoplastic mammary gland lesions. Cancer Research. 1978;38:4054–4058. [PubMed] [Google Scholar]
- 5.Clevenger CV, Furth PA, Hankinson SE, Schuler LA. The role of prolactin in mammary carcinoma. Endocrine Reviews. 2003;24:1–27. doi: 10.1210/er.2001-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ben-Jonathan N, Liby K, McFarland M, Zinger M. Prolactin as an autocrine/paracrine factor in human cancers. Trends Endocrinol Metab. 2002;13:245–250. doi: 10.1016/s1043-2760(02)00603-3. [DOI] [PubMed] [Google Scholar]
- 7.Goffin V, Bernichtein S, Touraine P, Kelly PA. Development and potential clinical uses of human prolactin receptor antagonists. Endocrine Reviews. 2005;26:400–422. doi: 10.1210/er.2004-0016. [DOI] [PubMed] [Google Scholar]
- 8.Kelly PA, Ali S, Rozakis M, Goujon L, Nagano M, Pellegrini I, Gould D, Djiane J, Edery M, Finidori J. The growth hormone/prolactin receptor family. Recent Progress in Hormone Research. 1993;48:123–164. doi: 10.1016/b978-0-12-571148-7.50009-9. [DOI] [PubMed] [Google Scholar]
- 9.Boutin JM, Edery M, Shirota M, Joliceur C, Lesueur L, Ali S, Gould D, Djiane J, Kelly PA. Identification of a cDNA encoding a long form of prolactin receptor in human hepatoma and breast cancer cells. Mol Endocrinol. 1989;3:1455–1461. doi: 10.1210/mend-3-9-1455. [DOI] [PubMed] [Google Scholar]
- 10.Kline JB, Roehrs H, Clevenger CV. Functional characterization of the intermediate isoform of the human prolactin receptor. J Biol Chem. 1999;274:35461–35468. doi: 10.1074/jbc.274.50.35461. [DOI] [PubMed] [Google Scholar]
- 11.Kline JB, Rycyzyn MA, Clevenger CV. Characterization of a novel and functional human prolactin receptor isoform (delta S1 PRLr) containing only one extracellular fibronectin-like domain. Mol Endocrinol. 2002;16:2310–22. doi: 10.1210/me.2001-0033. [DOI] [PubMed] [Google Scholar]
- 12.Kline JB, Clevenger CV. Identification and characterization of the prolactin-binding protein (PRLBP) in human serum and milk. J Biol Chem. 2001;276:24760–24766. doi: 10.1074/jbc.M011786200. [DOI] [PubMed] [Google Scholar]
- 13.Hu Z, Meng J, Dufau ML. Isolation and characterization of two novel forms of the human prolactin receptor generated by alternative splicing of a newly identified exon 11. J Biol Chem. 2001;276:41086–41094. doi: 10.1074/jbc.M102109200. [DOI] [PubMed] [Google Scholar]
- 14.Trott JF, Hovey RC, Koduri S, Vonderhaar BK. Alternative splicing of exon 11 of human prolactin receptor gene results in multiple isoforms including a secreted prolactin-binding protein. J Mol Endocrinol. 2003;30:31–47. doi: 10.1677/jme.0.0300031. [DOI] [PubMed] [Google Scholar]
- 15.Trott JF, Hovey RC, Koduri S, Vonderhaar BK. Multiple new isoforms of the human prolactin receptor gene. Adv Exptl Med Biol. 2004;254:495–499. doi: 10.1007/978-1-4757-4242-8_71. [DOI] [PubMed] [Google Scholar]
- 16.Qazi AM, Tsai-Morris C-H, Dufau ML. Ligand-independent homo- and heterodimerization of human prolactin receptor variants: inhibitory action of the short forms by heterodimerization. Mol Endocrinol. 2006;20:1912–1923. doi: 10.1210/me.2005-0291. [DOI] [PubMed] [Google Scholar]
- 17.Gadd S, Clevenger CV. Ligand-independent dimerization of the human prolactin receptor isoforms: functional implications. Mol Endocrinol. 2006;20:2734–2746. doi: 10.1210/me.2006-0114. [DOI] [PubMed] [Google Scholar]
- 18.Sivaprasad U, Canfield JM, Brooks CL. Mechanism of ordered binding by human prolactin. Biochem. 2004;43:13755–13765. doi: 10.1021/bi049333p. [DOI] [PubMed] [Google Scholar]
- 19.Duda KM, Brooks CL. Identification of residues outside the two binding sites that are critical for activation of the lactogenic activity of human growth hormone. J Biol Chem. 2003;278:22734–22739. doi: 10.1074/jbc.M212550200. [DOI] [PubMed] [Google Scholar]
- 20.Sivaprasad U. Thesis at The Ohio State University Libraries. 2003. The mechanism of lactogen receptor binding by human prolactin. call number THE:BIO2003PHDS582. [Google Scholar]
- 21.Walsh STR, Sylvester JE, Kossikoff AA. The high and low affinity of growth hormone are allosterically coupled. Proc Natl Acad Sci. 2004;101:17078–17083. doi: 10.1073/pnas.0403336101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goffin V, Kinet S, Ferrag F, Binart N, Martial JA, Kelly PA. Antagonistic properties of human prolactin analogs that show paradoxical agonistic activity in the Nb2 bioassay. J Biol Chem. 1996;271:16573–16579. doi: 10.1074/jbc.271.28.16573. [DOI] [PubMed] [Google Scholar]
- 23.Ramamoorthy P, Sticca R, Wagner TE, Chen WY. In vitro studies of a prolactin antagonist, hPRL-G129R, in human breast cancer cells. Internatl J Oncol. 2001;18:25–32. doi: 10.3892/ijo.18.1.25. [DOI] [PubMed] [Google Scholar]
- 24.Bernichtein S, Kayser C, Dillner K, Moulin S, Kopchick JJ, Martial JA, Norstedt G, Isaksson O, Kelly PA, Goffin V. Development of pure prolactin receptor antagonists. J Biol Chem. 2003;278:35988–35999. doi: 10.1074/jbc.M305687200. [DOI] [PubMed] [Google Scholar]
- 25.Walker AM. Theraputic potential for S179D prolactin-from prostate cancer to angioproliferative disorders: the first selective prolactin receptor modulator. Expert Opin Invest Drugs. 2006;15:1257–1267. doi: 10.1517/13543784.15.10.1257. [DOI] [PubMed] [Google Scholar]
- 26.Chen WY, Ramamoorthy P, Chen N-Y, Sticca R, Wagner TE. A human prolactin antagonist, hPRL-G129R, inhibits breast cancer cell proliferation through induction of apoptosis. Clin Cancer Res. 1999;5:3583–3593. [PubMed] [Google Scholar]
- 27.Beck MT, Pierce SK, Chen WY. Regulation of bcl-2 gene expression in human breast cancer cells by prolactin and its antagonist, hPRL-G129R. Oncogene. 2002;21:5047–5055. doi: 10.1038/sj.onc.1205637. [DOI] [PubMed] [Google Scholar]
- 28.Pierce SK, Chen WY. Human prolactin and its antagonist, hPRL-G129R, regulate bax and bxcl-2 gene expression in human cancer cells and transgenic mice. Oncogene. 2004;23:1248–1255. doi: 10.1038/sj.onc.1207245. [DOI] [PubMed] [Google Scholar]
- 29.Tomblyn S, Langenheim JF, Jacquemart IC, Holle E, Chen WY. The role of human prolactin and its antagonist, G129R, in mammary gland development and DMBA-initiated tumorigenesis in transgenic mice. Intl J Oncology. 2005;27:1381–1389. [PubMed] [Google Scholar]
- 30.Somers W, Ultsch M, De Vos AM, Kossiakoff AA. The X-ray structure of a growth hormone-prolactin receptor complex. Nature. 1994;372:478–481. doi: 10.1038/372478a0. [DOI] [PubMed] [Google Scholar]
- 31.Keeler C, Dannies PS, Hodsdon ME. The tertiary structure and backbone dynamics of human prolactin. J Mol Biol. 2003;328:1105–1121. doi: 10.1016/s0022-2836(03)00367-x. [DOI] [PubMed] [Google Scholar]
- 32.Teilum K, Hoch JC, Goffin V, Kinet S, Martial JA, Kragelund BB. Solution structure of human prolactin. J Mol Biol. 2005;351:810–823. doi: 10.1016/j.jmb.2005.06.042. [DOI] [PubMed] [Google Scholar]
- 33.Jomain J-B, Tallet E, Broutin I, Hoos S, van Agthoven J, Ducruix A, Kelly PA, Kragelund BB, England P, Goffin V. Structural and thermodynamic bases for the unique properties of del1-9-G129R-hPRL, a pure prolactin receptor antagonist. J Biol Chem. 2007;282:33118–33131. doi: 10.1074/jbc.M704364200. [DOI] [PubMed] [Google Scholar]
- 34.Lewis UJ, Dunn JT, Bonewald LF, Seavey BK, VanderLaan WP. A naturally occurring structural variant of human growth hormone. J Biol Chem. 1978;253:2679–2687. [PubMed] [Google Scholar]
- 35.DeNoto FM, Moore DD, Goodman HM. Human growth hormone DNA sequence and mRNA structure: possible alternative splicing. Nucleic Acids Res. 1981;9:3719–3730. doi: 10.1093/nar/9.15.3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de Vos AM, Ultsch M, Kossiakoff AA. Human growth hormone and extracellular domain of its receptor: crystal structure of the complex. Science. 1992;255:306–312. doi: 10.1126/science.1549776. [DOI] [PubMed] [Google Scholar]
- 37.Chantalat L, Jones ND, Korber F, Navaza J, Pavlovsky AG. The Crystal-Structure of wild-type growth-hormone at 2.5 Angstrom resolution. Prot Pep Letters. 1995;2:333–340. [Google Scholar]
- 38.Tsunekawa B, Wada M, Ikeda M, Uchida H, Naito N, Honjo M. The 20-kilodalton (kDa) human growth hormone (hGH) differs from the 22-kDa hGH in the effect on the human prolactin receptor. Endocrinology. 1999;140:3909–3918. doi: 10.1210/endo.140.9.6959. [DOI] [PubMed] [Google Scholar]
- 39.Peterson FC, Brooks CL. Different elements of mini-helix 1 are required for human growth hormone or prolactin action via the prolactin receptor. Prot Eng Design Select. 2004;17:417–424. doi: 10.1093/protein/gzh051. [DOI] [PubMed] [Google Scholar]
- 40.Peterson FC, Anderson PJ, Berliner LJ, Brooks CL. Expression, folding, and characterization of small proteins with increasing disulfide complexity by a pT7-7-derived phagemid. Prot Exp Purif. 1999;15:16–23. doi: 10.1006/prep.1998.0984. [DOI] [PubMed] [Google Scholar]
- 41.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 42.Tarr GE. Manual Edman Sequencing System. In: Shively JE, editor. Methods of Protein Microcharacterization. Clifton NJ: Humana Press; 1986. pp. 155–194. [Google Scholar]
- 43.Perkins DN, Pappin DJC, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 44.Chalkley RJ, Baker PR, Huang L, Hansen KC, Allen NP, Rexach M, Burlingame AL. Comprehensive analysis of a multidimensional liquid chromatography mass spectrometry dataset acquired on a quadrupole selecting quadrupole collision cell, time- of-flight mass spectrometer. II. new developments in protein prospector allow for reliable and comprehensive automatic analysis of large datasets. Mol Cell Proteomics. 2005;4:1194–1204. doi: 10.1074/mcp.D500002-MCP200. [DOI] [PubMed] [Google Scholar]
- 45.Kunkel TA, Bebenek K, McClary J. Efficient site-directed mutagenesis using uracil-containing DNA. Meth Enzymol. 1991;204:125–139. doi: 10.1016/0076-6879(91)04008-c. [DOI] [PubMed] [Google Scholar]
- 46.O’Brian J, Wilson I, Orton T, Pognan F. Investigation of the Alamar Blue (resazurin) Fluorescent Dye for the Assessment of Mammalian Cell Cytotoxicity. Europ J Biochem. 2000;267:5421–5426. doi: 10.1046/j.1432-1327.2000.01606.x. [DOI] [PubMed] [Google Scholar]
- 47.Schneider U, Schwenk H, Bornkamm G. Characterization of EBV-genome negative “null” and “T” cell lines derived from children with acute lymphoblastic leukemia and leukemic transformed non-Hodgkin lymphoma. Intl J Cancer. 1977;19:621–626. doi: 10.1002/ijc.2910190505. [DOI] [PubMed] [Google Scholar]
- 48.Matera L, Cutufia M, Geuna M, Contarini M, Buttiglieri S, Galin S, Fazzari A, Cavaliere C. Prolactin is an autocrine growth factor for jurkat human T-leukemic cell line. J Neuroimmunol. 1997;79:12–21. doi: 10.1016/s0165-5728(97)00096-9. [DOI] [PubMed] [Google Scholar]
- 49.Zamzami N, Marchetti P, Castedo M, Zanin C, Vayssiere J-L, Petit PX, Kroemer G. Reduction in mitochondrial potential constitutes an early irreversible step of programmed lymphocyte death in vivo. J Exper Med. 1995;181:1661–1672. doi: 10.1084/jem.181.5.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Castedo M, Ferri K, Roumier T, Metivier D, Zamzami N, Kroemer G. Quantitation of mitochondrial alterations associated with apoptosis. J Immunol Meth. 2002;265:39–47. doi: 10.1016/s0022-1759(02)00069-8. [DOI] [PubMed] [Google Scholar]