

Abstract
Background & Aims
Intestinal dysmotility is a component of many neurodegenerative disorders, including some characterized by neuronal intranuclear inclusions. PrP-SCA7-92Q transgenic mice phenocopy many aspects of the human polyglutamine neurodegenerative disorder, spinocerebellar ataxia type 7 (SCA7). The enteric neuropathology of PrP-SCA7-92Q mice was investigated after observing that they develop signs of intestinal pseudo-obstruction.
Methods
Gastrointestinal transit of radio-opaque pellets through pre-symptomatic and symptomatic PrP-SCA7-92Q mice and non-transgenic littermates were compared. Gross, microscopic, and ultrastructural studies were conducted, along with histological and whole-mount immunohistochemistry, to identify intranuclear inclusions and quantify subsets of enteric neurons. Immunoblot analysis was performed to confirm selective loss of particular neuronal populations.
Results
A subset of cholinergic enteric ganglion cells in PrP-SCA7-92Q mice harbor nuclear inclusions comprised of transgene-derived ataxin-7, which contains a pathogenic polyglutamine expansion. These animals die between 15 and 20 weeks with intestinal distension and enterocolitis. Signs of disease are preceded by selective loss of nitric oxide synthase-positive neurons (which lack nuclear inclusions), loss of nerve fibers in the myenteric nerve plexus, and delayed gastrointestinal transit. Cholinergic neurons, including those with inclusions, are spared.
Conclusions
PrP-SCA7-92Q mice may be useful models for human intestinal pseudo-obstruction, particularly visceral neuropathies with neuronal intranuclear inclusions. Loss of inclusion-free inhibitory neurons supports the hypothesis that inclusions may be neuroprotective or coincidental, as opposed to harbingers of neuron death. Since enteric neuropathology in PrP-SCA7-92Q animals is easily missed by routine histopathology, quantitative immunohistochemical approaches may be required to recognize analogous forms of human enteric neuropathy.
Keywords: polyglutamine, nuclear inclusion, enteric neuropathy, pseudo-obstruction, mouse, spinal cerebellar ataxia, neuronal degeneration, transgenic model
INTRODUCTION
Neuronal intranuclear inclusion disease (NIID) and polyglutamine disorders refer to two groups of progressive neurodegenerative disorders that share clinical and pathological features. In each, affected individuals exhibit a variety of symptoms that correlate with neuropathological changes in their central and peripheral nervous systems, including intranuclear neural inclusions composed in part of glutamine-rich proteins and other shared components.
NIID usually presents in childhood or adolescence, but infantile and adult onsets are described1. Ataxia, tremor, lower motor neuron impairment, oculogyric crises, and autonomic findings are common. The underlying etiology of NIID is unknown and is likely to be heterogeneous. Familial recurrences suggest a genetic basis for most, if not all, cases. Large eosinophilic nuclear inclusions in NIID are readily apparent in routine histological sections.
Intestinal pseudo-obstruction is unusual, but can be a significant problem in NIID. Schuffler and colleagues reported a brother and sister with NIID, who exhibited signs of intestinal pseudo-obstruction since childhood and eventually required total parenteral nutrition.2 At autopsy, the density of myenteric ganglion cells was severely reduced and a third of the surviving neurons contained nuclear inclusions. Another family in which three siblings were affected with similar findings was reported by Barnett.3 In the latter instance, a rectal biopsy from the father also demonstrated neuronal inclusions, but he evidenced no signs of NIID other than mild abdominal pain. It is unclear why intestinal dysmotility is a serious problem for some patients with NIID, but not others.
Some of the clinical and pathological features of NIID resemble spinocerebellar ataxia type 7 (SCA7) and several other neurodegenerative disorders caused by intragenic expansions of polyglutamine-encoding tracts.4 Clinical features of SCA7 include cerebellar gait ataxia, dysarthria, and decreased visual acuity due to retinal degeneration. Dysphagia and/or esophageal or anal sphincter dysfunction affect as many as 50% of patients, but intestinal pseudo-obstruction has not been reported.
In neurons of patients with SCA7 or other polyglutamine diseases, expanded ataxin-7 and other proteins aggregate in nuclei as “inclusion bodies” (IBs).5 Although IBs are less obvious than the nuclear inclusions of NIID in routine histological sections, they share many immunohistochemical properties.6, 7 For example, non-expanded polyglutamine proteins (e.g., ataxin 3) are present in the nuclear inclusions of NIID and some, but not all, NIID inclusions are labeled by 1C2 antibody, which recognizes polyglutamine expansions.8–10 In addition, both types of nuclear inclusions contain SUMO-1, a ubiquitin-like peptide that covalently binds to other proteins and influences their localization and metabolism.11, 12 It remains to be determined whether NIID is caused by the same type of mutations as polyglutamine diseases. Animal models are needed to better understand the pathogenesis of pseudo-obstruction in NIID or related neurodegenerative diseases.
In an effort to model SCA7, transgenic mice that express human ataxin-7 with an expanded polyglutamine tract have been generated.13, 14 The PrP-SCA7-92Q mice recapitulate phenotypic features of SCA7, including retinal degeneration and cerebellar ataxia. Here, we investigated the premature demise of these mice, and found that enteric neuronal intranuclear inclusions, progressive neurodegeneration of myenteric inhibitory neurons, and intestinal pseudo-obstruction are consistent findings in this transgenic model.
METHODS
Mice
The PrP-SCA7-92Q transgenic mice were created as described elsewhere 13 and received standard laboratory chow ad libitum. Animals were handled in accordance with guidelines established by the National Institutes of Health and our institutional animal care and use committee. To harvest tissues, mice were asphyxiated with carbon dioxide.
Mapping the transgene insertion site by fluorescence in situ hybridization
Cells were teased out of the spleen from male PrP-SCA7-92Q transgene-positive mice, pelleted, and cultured in RPMI with 10% fetal bovine serum and 40 μg/ml lipopolysaccharide (Sigma L4391) for 48 hours. Cells were harvested using 0.1 μg/ml KaryoMAX Colcemid (Invitrogen Corp.) and 0.075 M KCl, and fixed in 3:1 methanol:acetic acid.
Plasmid DNA containing the PrP-SCA7-92Q transgene was labeled with Spectrum orange dUTP (Abbott Laboratories) by nick translation (Nick Translation kit, Abbott Laboratories) according to the manufacturer’s protocol. DNA from a plasmid containing the DXWas70 locus, which maps near the centromere of the mouse X chromosome (MGI accession ID: MGI:95244), was labeled with Spectrum green dUTP and used as a control probe to identify the mouse X chromosome. The PrP-SCA7-92Q- and DXWas70-labeled probes were co-hybridized to metaphase spreads using a HYBrite TM denaturation/hybridization system (Vysis, Inc.). Post-hybridization washes were performed according to the manufacturer protocol (Vysis, Inc.). Chromosomes were counterstained using DAPI and probe signals were visualized using a fluorescent microscope.
In vivo gastrointestinal transit assay
Gastrointestinal transit was evaluated as described previously.15 Mice were induced to swallow radioopaque pellets and progression of the pellets was assessed radiographically immediately after administration and 16–20 hours later.
Microscopic studies
Segments of gut were fixed for 2-4 hours on ice in 4% paraformaldehyde or 3% glutaraldehyde for light and electron microscopy, respectfully. Paraformaldehyde-fixed tissues were dehydrated, embedded in paraffin, and sectioned for staining with hematoxylin-and-eosin or immunohistochemistry. Standard methods and plastic resin were used to process for electron microscopy. Immunostaining was performed as described previously.16 Antibodies that specifically recognize Hu (Invitrogen), ataxin-7 (Affinity Bioreagents), vasoactive intestinal peptide (VIP, Chemicon), calretinin (Zymed) and SUMO-1 (Sigma) were diluted 1:1250, 1:1000, 1:1000, 1:500 and 1:200, respectively. VIP and calretinin immunolocalization was performed using an automated immunostainer (Benchmark XT, Ventana Medical Systems).
Whole-mount immunostaining
Segments of distal ileum and colon were placed in phosphate-buffered saline and the mucosa and submucosa were manually removed with fine forceps. The muscularis propria and enclosed myenteric plexus were fixed 10 minutes in ice-cold acetone and then immersed in 1X PBS before blocking the tissues in 1X PBST (0.1% Tween 20) with 1% BSA for 1-3 hours at room temperature with gentle rocking.17 The tissues were then incubated in dilutions of primary antibodies overnight at 4°C (nNOS, Santa Cruz Biotechnology, 1:50; SCA7, 1:200; Hu, 1:100; ChAT, Chemicon, 1:200). After primary antibody incubation, the tissues were washed and then incubated in fluorescently-labeled secondary antibodies (1:200) for one hour at room temperature (goat anti-mouse FITC, goat anti-rabbit Texas Red, donkey anti-goat Alexa 488, Invitrogen). After incubation in secondary antibodies, the tissues were treated as above, except 1X PBS was used in the final wash. Six representative fields from each tissue sample were captured at 40X magnification using a Deltavision SA3.1 Widefield Deconvolution Microscope (Applied Precision, Inc.). Digital images were colorized with Adobe Photoshop software, and the neurons in each field were counted, and then averaged over the six fields.
For acetylcholinesterase and NADPH-diaphorase wholemount immunohistochemistry, neuromuscular preparations were fixed 1 hour in 4% paraformaldehyde and then handled as described elsewhere.15
Western blot
Blots were prepared from protein (10 μg/lane) extracted from small intestinal neuromuscular preparations as described previously 15, probed overnight with either mouse anti-nNOS (1:1000) or goat anti-ChAT (1:500), washed, and incubated in biotinylated secondary antibody with peroxidase-conjugated anti-biotin (Santa Cruz Biotechnology). Immunlabelling was visualized by chemiluminescence (Luminol, Santa Cruz Biotechnology).
RESULTS
To model the retinal and cerebellar degeneration of SCA7, a human ATAXIN-7 cDNA that encodes a mutant form of ataxin-7 with a pathogenic 92 glutamine (Q)-long expansion (≤ 35 glutamines is normal) was inserted into the murine prion promoter (PrP) expression construct.13 The PrP promoter directs expression to neurons and a subset of glial cells of the central and peripheral nervous systems.18 Four independent lines of PrP-SCA7-92Q transgenic mice were produced by micronuclear injection and exhibited differences in transgene expression levels that correlated with the onset and rate of progression of cerebellar and retinal degeneration.13, 14 For the present study, we focused on line 6529 PrP-SCA7-92Q mice; however, the gross intestinal pathology, enteric neural inclusions, and premature death reported for line 6529 transgene-positive individuals were also observed in hemizygous PrP-SCA7-92Q males and females from two other independent lines.
As the 6529 PrP-SCA7-92Q line was expanded, hemizygous transgenic males were observed to die prematurely between 12 and 21 weeks of age (mean = 17.2 ± 2.2 weeks, n= 40). With rare exception, hemizygous females survived over 70 weeks. This sexual dimorphism suggested that the transgene had integrated into the X-chromosome, which was confirmed by fluorescence-in-situ hybridization to metaphase chromosomes (data not shown). Hemizygous females presumably survive due to inactivation of the “transgenic” X-chromosome. Days prior to death, affected animals developed lethargy and abdominal distension, with marked dilatation of the proximal colon and distal, and sometimes proximal, small intestine (Figure 1A). A limited set of crosses between hemizygous animals produced only transgenic female offspring, half of which (presumed PrP-SCA7-92Q homozygotes) recapitulated the male PrP-SCA7-92Q phenotype.
Figure 1.

(A) A photograph of the gastrointestinal tracts from a symptomatic, 15-week, PrP-SCA7-92Q mouse and its non-transgenic littermate demonstrates marked distension of the proximal colon (co) and distal small intestine (di) of the transgenic animal. The stomach (s) and proximal small intestine (psi) are not affected. (B) A photomicrograph from an H&E-stained section demonstrates a rare eosinophilic intranuclear inclusion (arrowhead) in a myenteric neuron from a transgenic mouse. (C) Ultrastructurally, a subset of neurons in transgenic mice contained granular electron dense inclusions (arrowhead and inset), which appeared more heterogenous than neighboring nucleoli (n).
An in vivo intestinal transit assay using barium impregnated pellets was performed on symptomatic and asymptomatic line 6529 PrP-SCA7-92Q male mice and non-transgenic littermates at 7 and 17 weeks. After the radio-opaque pellets were administered, transit was allowed to proceed for 16 hours. Subsequently, the mice were sacrificed and the gastrointestinal tracts removed and radiographed. Sixteen-to-twenty hours after administration, no pellets were retained in control mice at either age. However, symptomatic and asymptomatic 17-week-old, Prp-SCA7-92Q transgenic mice retained pellets in the small or large intestine, indicating an underlying motility defect (Table 1). It is noteworthy that delayed transit precedes signs of intestinal distension in some PrP-SCA7-92Q animals and is not an agonal phenomenon. Impaired transit in older, but not younger, animals is consistent with a progressive disease process, and parallels the development of CNS and retinal neuropathies characteristic of the SCA7 mouse model.13, 14
H&E-stained histological sections of bowel wall from PrP-SCA7-92Q mice at different ages failed to reveal alterations in smooth muscle or obvious abnormalities in the density or distribution of enteric neurons or nerves. Enterocolitis was detected in some symptomatic animals, but ganglionitis was not. Inconspicuous, solitary, round intranuclear inclusions, more eosinophilic than nucleoli, were found in less than 5% of ganglion cells (Figure 1B). These were 1–2 μm in diameter and correlated ultrastructurally with heterogenous electron-dense inclusions, consisting of both filamentous and granular areas (Figure 1C). The light and electron microscopic appearance of these inclusions are identical to neuronal intranuclear inclusions seen in SCA7 patients and mice, but appear smaller and less filamentous than inclusions of human NIID.
Immunohistochemistry typically reveals many more inclusion bodies in neurons of patients with polyglutamine disorders than are apparent by light microscopy.1, 6 Immunolabelling of paraffin sections (Figures 2A) or whole-mount preparations (Figures 2C–D) with a human ataxin-7-specific antibody showed single or multiple ataxin-7-positive intranuclear inclusions in numerous myenteric and submucosal ganglion cells of PrP-SCA7-92Q mice, but not in non-transgenic controls. The inclusions also demonstrated SUMO-1 immunoreactivity (Figure 2B), a feature of human polyglutamine bodies and NIID.
Figure 2.
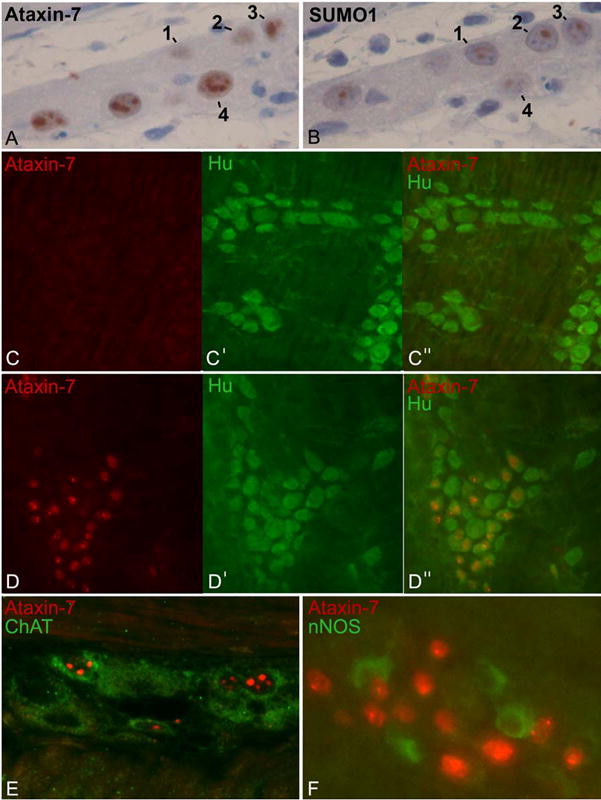
Ataxin-7 inclusions in enteric neurons of PrP-SCA7-92Q mice. (A) Human ataxin-7-specific antibodies demonstrate punctate brown inclusions in the nuclei of a subset of enteric ganglion cells from a PrP-SCA7-92Q mouse. (B) An immediately adjacent paraffin section immunostained with SUMO-1-specific antisera shows SUMO-1 immunoreactivity in the same nuclei (numbered 1 to 4). (C,D) Wholemount preparations or myenteric plexus immunostained with antibodies that recognize ataxin-7 (red) and the neural marker, Hu (green), demonstrate no intranuclear inclusions in a non-transgenic sample (C), but abundant intranuclear inclusions in a subset of neurons from a transgenic mouse (D). (E) In a paraffin section, ataxin-7 (red) immunoreactivity is present in the nuclei of a subset of myenteric neurons that co-express the cytoplasmic marker, choline acetyltransferase (ChAT, green). (F) In contrast, a wholemount preparation of a myenteric ganglion does not show cellular colocalization of ataxin-7 (red) with neuronal nitric oxide synthase (nNOS, green).
Immunohistochemistry was performed on sections and wholemount preparations of distal ileum from 4-week and 15-week asymptomatic PrP-SCA7-92Q male mice to localize ataxin-7 and either Hu (all neurons; Figures 2D), choline acetyltransferase (ChAT; excitatory motor and interneurons; Figure 2E), or neuronal nitric oxide synthetase (nNOS; inhibitory motor neurons; Figure 2F). As expected, all cells in the gut wall that contained ataxin-7-positive nuclear inclusions co-expressed the neural marker Hu. Approximately 50% of neurons (Hu-positive cells) contained inclusions, and this percentage remained relatively stable between 4 and 15 weeks (Figure 3A).
Figure 3.
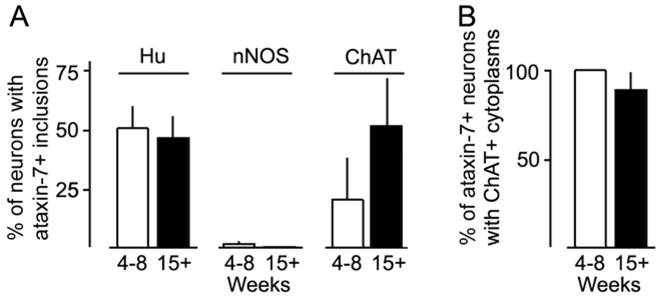
(A) Immunofluorescent colocalization studies of ataxin-7 intranuclear inclusions with various cytoplasmic markers in intestinal samples shows no age-related differences in the fraction of total neurons (Hu+) or cholinergic neurons (ChAT+) with inclusions at either age. The mean value of ChAT neurons with inclusions was greater at 15 weeks than at 4–8 weeks, but this difference was not statistically significant due to animal-to-animal variability at both time points. Virtually no nNOS+ neurons contained inclusions at either age. (B) Conversely, nearly all of the neurons with ataxin-7-immunoreactive inclusions co-expressed ChAT at either age.
In the small intestines of adult male mice, ChAT-positive cell bodies represent about one-third of the myenteric ganglion cell population.19 At every age examined, ataxin-7-positive intranuclear inclusions were only detected in a subset of these cholinergic neurons (Figure 3B). However, wide variability was observed between mice in the fraction of ataxin-7-positive ChAT neurons (Figure 3A). Significant differences between 4-to-8-week versus 15-week mice were not resolved, despite a trend towards a greater percentage of dual labeled cells in older mice (Figure 3A). In contrast, ataxin-7-immunoreactive inclusions were not detected at any age in the nuclei of nNOS immunoreactive neurons, which represent a different third of the myenteric ganglion cell population (Figure 3A).19
As PrP-SCA7-92Q mice aged, myenteric ganglion cells were lost (Figure 4). At 4 and 8 weeks of age, the densities of Hu-positive ganglion cells in the small intestines of transgenic male mice and their littermates were identical. At 15 weeks, the average ganglion cell density in transgenic mice was only 40–60% of the control average (Figure 4A, B). This reduction in total ganglion cell number correlated with a dramatic loss of nNOS neurons, which declined from control values to only 20% of control values between 4 and 15 weeks (Figure 4C). In contrast, the density of ChAT neurons did not differ significantly from controls at both time points (Figure 4D). Similar to nNOS neurons, the density of calretinin-immunoreactive myenteric neurons (putative intrinsic primary afferent neurons) was significantly less in transgenic mice versus controls at 15 weeks (Figure 4E). Immunoblot analyses of nNOS and ChAT in protein extracts from intestinal neuromuscular tissue also produced results consistent with loss of nNOS neurons, but not ChAT neurons, in PrP-SCA7-92Q mice between 4 and 15 weeks (Figure 5).
Figure 4.
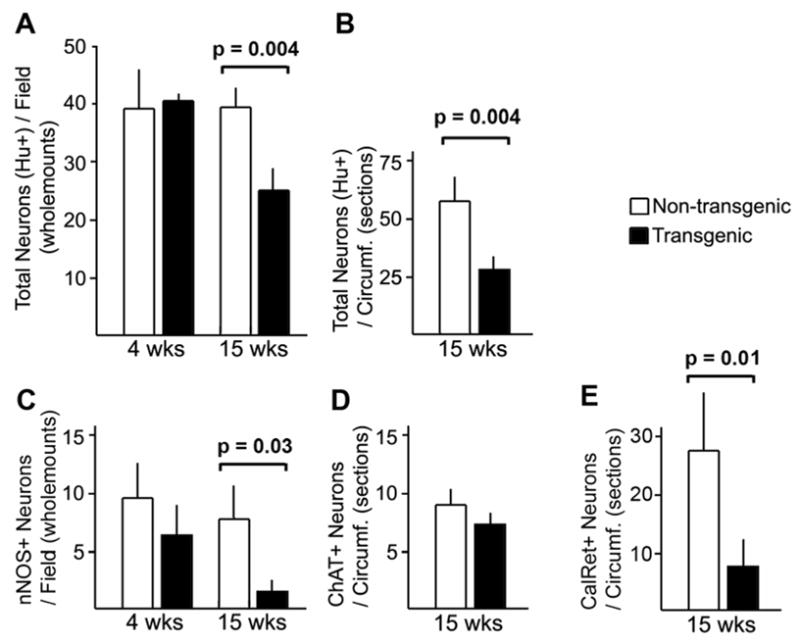
(A, B) Counts of myenteric neurons, identified by Hu immunostaining in wholemount preparations (A) or paraffin sections (B) of transgenic vs. non-transgenic mice demonstrate equivalent numbers at 4 weeks of age and a significant reduction at 15 weeks of age. (C) The density of nNOS-immunoreactive neurons was not signficaintly between the two genotypes at 4 weeks, but a marked loss of nNOS-immunoreactive neurons is evident at 15 weeks. (D) No significant difference existed in the density of ChAT-immunoreactive neurons at 15 weeks. (E) The density of calretinin-immunoreactive myenteric ganglion cells was significantly less in transgenic mice at 15 weeks.
Figure 5.
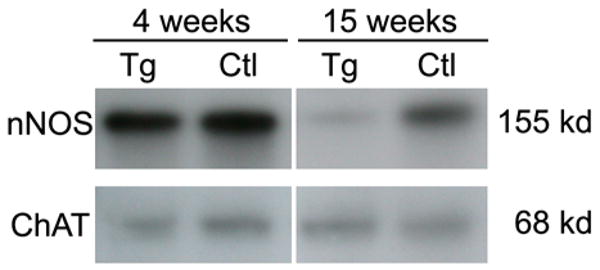
Western blot analysis of equivalent amounts of total protein from transgenic (Tg) and non-transgenic (Ctl), enteric neuromuscular samples shows no differences in the amount of nNOS or ChAT at 4 weeks of age. In extracts from 15-week mice, the amount of nNOS in the Tg samples is dramatically reduced relative to Ctl samples. However, the equivalent amounts of ChAT are detected in extracts from each genotype.
Loss of nNOS ganglion cell bodies was accompanied by qualitative changes in the network of nerves in the myenteric plexus. Wholemount preparations stained histochemically for acetylcholinesterase (all nerve fibers) or nNOS (inhibitory motor fibers) demonstrated reductions in thickness of tertiary branches to the muscularis propria (Figure 6). The majority of nNOS neurons also express VIP.19 Light microscopic analysis of anti-VIP-immunostain sections did not provide sufficient resolution to quantitate VIP-positive ganglion cell bodies, but showed a striking reduction in the density of VIP-immunoreactive nerve fibers in the muscularis propria (Supplemental Figure 1). The residual nNOS-positive neurons had enlarged cell bodies and some of their axons were unusually thick and tortuous with abnormal varicosities, not observed in non-transgenic animals.
Figure 6.

(A, B) Wholemount preparations of distal ileum from non-transgenic (A) and PrP-SCA7-92Q transgenic, 15-week-old mice (B) mice stained histochemically for NADPH-diaphorase (nNOS) activity demonstrate reduction in the density of nNOS-cell bodies in the transgenic animals. Those nNOS neurons that remain are enlarged, but the network of nNOS-positive nerve fibers is simplified with fewer and thinner cell processes. (C, D) Acetylcholinesterase histochemical staining of similar preparations also demonstrates reduction of the density and number of nerve processes in the myenteric plexus of a PrP-SCA7-92Q transgenic mouse (D) compared to its non-transgenic littermate (C).
DISCUSSION
PrP-SCA7-92Q mice show line-to-line phenotypic variability, but a consistent finding in at least three lines is premature death that is associated with intestinal dilatation and enteric neuronal intranuclear inclusions. Our detailed investigation of the 6529 line suggests that distension and death are the consequences of progressive loss of enteric neurons and delayed intestinal transit. In these respects, PrP-SCA7-92Q mice may be a useful model for humans with visceral neuropathies, intranuclear ganglion cell inclusions, and intestinal pseudo-obstruction.
Apart from viral infections, intranuclear inclusions in enteric neurons have been reported in two contexts, polyglutamine diseases and NIID. Polyglutamine diseases and NIID share many clinical and pathological features, but potentially important differences include: (1) inclusions in polyglutamine disorders are typically smaller, fewer, and less conspicuous; (2) etiologies (expanded glutamine repeats) of polyglutamine disorders are known; (3) only normal, as opposed to expanded, polyglutamine proteins, have been detected in the inclusions of NIID; and (4) intestinal pseudo-obstruction has been reported in NIID patients, but not patients with SCA7 or other polyglutamine diseases. In the first three respects, the properties of PrP-SCA7-92Q mice are more like those of polyglutamine disorders than NIID. However, the fact that PrP-SCA7-92Q mice lose enteric neurons and develop intestinal pseudo-obstruction more closely models some humans with NIID.
It has been hypothesized that NIID may either represent a polyglutamine disease for which the affected gene has not been determined or may comprise a genetic disorder that impacts overlapping molecular pathways. The fact that PrP-SCA7-92Q mice develop the intestinal pseudo-obstruction like some NIID patients lends support to this hypothesis. Our observations also suggest that intestinal dysmotility may be an under-reported phenomenon in some SCA7 patients, the majority of whom suffer from dysphagia of unknown etiology. Recently, we learned of a 20-year-old female SCA7 patient whose clinical presentation has been confirmed by molecular testing (71-glutamine expansion). Interestingly, this SCA7 patient carries a diagnosis of pseudo-obstruction, but no gastrointestinal pathology specimens have been obtained (J. Garbern, personal communication).
Immunohistochemical staining and formal neuronal counts demonstrate a significant reduction in enteric neurons of PrP-SCA7-92Q mice, which precedes gross anatomical signs of pseudo-obstruction. Inhibitory motor neurons that express nNOS appear to be affected disproportionately, despite the fact that ataxin-7 inclusions are not found in this subpopulation of ganglion cells. The only cells with ataxin-7 intranuclear inclusions also expressed ChAT. In contrast to nNOS+ neurons, ChAT-expressing ganglion cells were relatively spared. These data support mounting evidence that large neuronal inclusions (i.e. visible at the light microscope level) in polyglutamine diseases can be “protective”, and that it is neuronal subpopulations that lack such visible inclusions, which are most vulnerable to the toxicity pathway.20, 21 The proportion of ataxin-7-inclusion positive ChAT cells actually increased with age, either because new inclusions form de novo in this cellular population, or because ChAT-positive cells lacking inclusions degenerate. If nuclear inclusions are neuroprotective, other populations of neurons that do not express ChAT or nNOS might be also vulnerable to PrP-SCA7-92Q expression. Consistent with this hypothesis, calretinin-immunoreactive neurons in myenteric ganglia (putative intrinsic primary afferent neurons) were also significantly reduced in density in PrP-SCA7-92Q mice. Although a severe loss of myenteric ganglion cells has been reported in patients with intestinal pseudo-obstruction and NIID, no published data exists regarding degeneration or survival of specific types of neurons.
Our results indicate that further careful analysis of intestinal pseudo-obstruction in PrP-SCA7-92Q mice may yield clue to the pathogenic nature of this condition. In addition to documenting the fates of other neuronal subtypes in the gut wall, changes in these populations will need to be correlated with physiological studies of peristaltic activity and neuromuscular electrophysiology. Furthermore, a subset of ChAT-positive ganglion cells form inclusions and survive, whereas nNOS neurons fail to form inclusions and degenerate in this disease process, raising the question of cell-type selective vulnerability. The architecture of the myenteric plexus may be an advantageous mammalian system for addressing this important question, because the neuronal subtypes can be easily distinguished in wholemount preparations, as opposed to the complex three-dimensional organization of the brain. Finally, our findings with this mouse model highlight challenges that exist in the evaluation of biopsies from humans with motility disorders. In routine H&E-stained sections, the loss of approximately 50% of enteric neurons in older PrP-SCA7-92Q animals was not readily apparent. Formal counts with appropriate controls were necessary to appreciate a significant difference. The fact that nNOS+ and calretinin-immunoreactive neurons were so profoundly affected could not be recognized without immunohistochemistry. Therefore, similarly subtle alterations in the enteric nervous system may characterize many human forms of intestinal dysmotility, suggesting that their discovery and characterization will require specialized approaches that exceed standard techniques in most clinical laboratories.
Supplementary Material
VIP-immunostained sections from the proximal colons of a transgenic (A) and non-transgenic (B) demonstrate reduced density of myenteric nerve fibers, particularly the intramuscular fibers of the muscularis propria (arrowheads).
Acknowledgments
Grant support: UW NIEHS-sponsored Center for Ecogenetics and Environmental Health (NIEHS P30ES07033); CHRMC Research Administration Award (RPK); R01 EY14061 (NIH; ARL)
Abbreviations
- NIID
neuronal intranuclear inclusion disease
- SCA
spinocerebellar ataxia
Footnotes
Conflicts of Interest: No conflicts of interest exist
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Takahashi-Fujigasaki J. Neuronal intranuclear hyaline inclusion disease. Neuropathology. 2003;23:351–9. doi: 10.1046/j.1440-1789.2003.00524.x. [DOI] [PubMed] [Google Scholar]
- 2.Schuffler MD, Bird TD, Sumi M, Cook A. A familial neuronal disease presenting as intestinal pseudoobstruction. Gastroenterology. 1978;75:889–98. [PubMed] [Google Scholar]
- 3.Barnett JL, McDonnell WM, Appelman HD, Dobbins WO. Familial visceral neuropathy with neuronal intranuclear inclusions: diagnosis by rectal biopsy. Gastroenterology. 1992;102:684–91. doi: 10.1016/0016-5085(92)90121-e. [DOI] [PubMed] [Google Scholar]
- 4.David G, Durr A, Stevanin G, Cancel G, Abbas N, Benomar A, Belal S, Lebre AS, Abada-Bendib M, Grid D, Holmberg M, Yahyaoui M, Hentati F, Chkili T, Agid Y, Brice A. Molecular and clinical correlations in autosomal dominant cerebellar ataxia with progressive macular dystrophy (SCA7) Hum Mol Genet. 1998;7:165–70. doi: 10.1093/hmg/7.2.165. [DOI] [PubMed] [Google Scholar]
- 5.Koeppen AH. The pathogenesis of spinocerebellar ataxia. Cerebellum. 2005;4:62–73. doi: 10.1080/14734220510007950. [DOI] [PubMed] [Google Scholar]
- 6.Ross CA. Intranuclear neuronal inclusions: a common pathogenic mechanism for glutamine-repeat neurodegenerative diseases? Neuron. 1997;19:1147–50. doi: 10.1016/s0896-6273(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 7.Takahashi J, Tanaka J, Arai K, Funata N, Hattori T, Fukuda T, Fujigasaki H, Uchihara T. Recruitment of nonexpanded polyglutamine proteins to intranuclear aggregates in neuronal intranuclear hyaline inclusion disease. J Neuropathol Exp Neurol. 2001;60:369–76. doi: 10.1093/jnen/60.4.369. [DOI] [PubMed] [Google Scholar]
- 8.Lieberman AP, Robitaille Y, Trojanowski JQ, Dickson DW, Fischbeck KH. Polyglutamine-containing aggregates in neuronal intranuclear inclusion disease. Lancet. 1998;351:884. doi: 10.1016/S0140-6736(05)70296-8. [DOI] [PubMed] [Google Scholar]
- 9.Takahashi J, Fukuda T, Tanaka J, Minamitani M, Fujigasaki H, Uchihara T. Neuronal intranuclear hyaline inclusion disease with polyglutamine-immunoreactive inclusions. Acta Neuropathol (Berl) 2000;99:589–94. doi: 10.1007/s004010051166. [DOI] [PubMed] [Google Scholar]
- 10.McFadden K, Hamilton RL, Insalaco SJ, Lavine L, Al-Mateen M, Wang G, Wiley CA. Neuronal intranuclear inclusion disease without polyglutamine inclusions in a child. J Neuropathol Exp Neurol. 2005;64:545–52. doi: 10.1093/jnen/64.6.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pountney DL, Huang Y, Burns RJ, Haan E, Thompson PD, Blumbergs PC, Gai WP. SUMO-1 marks the nuclear inclusions in familial neuronal intranuclear inclusion disease. Exp Neurol. 2003;184:436–46. doi: 10.1016/j.expneurol.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 12.Amiel J, Attie T, Jan D, Pelet A, Edery P, Bidaud C, Lacombe D, Tam P, Simeoni J, Flori E, Nihoul-Fekete C, Munich A, Lyonnet S. Heterozygous endothelin receptor B (EDNRB) mutations in isolated Hirschsprung disease. Hum Molec Genet. 1996;5:355–7. doi: 10.1093/hmg/5.3.355. [DOI] [PubMed] [Google Scholar]
- 13.La Spada AR, Fu YH, Sopher BL, Libby RT, Wang X, Li LY, Einum DD, Huang J, Possin DE, Smith AC, Martinez RA, Koszdin KL, Treuting PM, Ware CB, Hurley JB, Ptacek LJ, Chen S. Polyglutamine-expanded ataxin-7 antagonizes CRX function and induces cone-rod dystrophy in a mouse model of SCA7. Neuron. 2001;31:913–27. doi: 10.1016/s0896-6273(01)00422-6. [DOI] [PubMed] [Google Scholar]
- 14.Garden GA, Libby RT, Fu YH, Kinoshita Y, Huang J, Possin DE, Smith AC, Martinez RA, Fine GC, Grote SK, Ware CB, Einum DD, Morrison RS, Ptacek LJ, Sopher BL, La Spada AR. Polyglutamine-expanded ataxin-7 promotes non-cell-autonomous purkinje cell degeneration and displays proteolytic cleavage in ataxic transgenic mice. J Neurosci. 2002;22:4897–905. doi: 10.1523/JNEUROSCI.22-12-04897.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Howe DG, Clarke CM, Willis BS, Yan H, Schneider DA, McKnight GS, Kapur RP. Inhibition of protein kinase A in enteric neurons causes lethal intestinal pseudoobstruction. J Neurobiol. 2006;66:256–72. doi: 10.1002/neu.20217. [DOI] [PubMed] [Google Scholar]
- 16.Parisi MA, Baldessari AE, Iida MHK, Clarke CM, Doggett B, Shirasawa S, Kapur RP. Genetic background modifies intestinal pseudo-obstruction and the expression of a reporter gene in Hox11L1 −/− mice. Gastroenterology. 2003;125:1428–40. doi: 10.1016/j.gastro.2003.08.021. [DOI] [PubMed] [Google Scholar]
- 17.Wang XY, Vannucchi MG, Nieuwmeyer F, Ye J, Faussone-Pellegrini MS, Huizinga JD. Changes in interstitial cells of Cajal at the deep muscular plexus are associated with loss of distention-induced burst-type muscle activity in mice infected by Trichinella spiralis. Am J Pathol. 2005;167:437–53. doi: 10.1016/S0002-9440(10)62988-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borchelt DR, Davis J, Fischer M, Lee MK, Slunt HH, Ratovitsky T, Regard J, Copeland NG, Jenkins NA, Sisodia SS, Price DL. A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet Anal. 1996;13:159–63. doi: 10.1016/s1050-3862(96)00167-2. [DOI] [PubMed] [Google Scholar]
- 19.Sang Q, Young HM. Chemical coding of neurons in the myenteric plexus and external muscle of the small and large intestine of the mouse. Cell Tissue Res. 1996;284:39–53. doi: 10.1007/s004410050565. [DOI] [PubMed] [Google Scholar]
- 20.Woulfe JM. Abnormalities of the nucleus and nuclear inclusions in neurodegenerative disease: a work in progress. Neuropathol Appl Neurobiol. 2007;33:2–42. doi: 10.1111/j.1365-2990.2006.00819.x. [DOI] [PubMed] [Google Scholar]
- 21.Sisodia SS. Nuclear inclusions in glutamine repeat disorders: are they pernicious, coincidental, or beneficial? Cell. 1998;95:1–4. doi: 10.1016/s0092-8674(00)81743-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
VIP-immunostained sections from the proximal colons of a transgenic (A) and non-transgenic (B) demonstrate reduced density of myenteric nerve fibers, particularly the intramuscular fibers of the muscularis propria (arrowheads).