

Abstract
We have shown in two accompanying papers that TNF induces oscillations in (1) ~13% of the genome, and (2) the activation of MAP kinase and NF-κB signaling pathways. Here we aim to bridge oscillations in signal transduction activation to oscillations in genetic output. Specifically, we sought to study how these oscillations can combine in a ligand-specific manner at the level of the promoter to initiate gene transcription. We utilize the late onset gene CD38 as a model gene since it has previously been shown that TNF, but not the related cytokine RANK-L, induces its expression. We find that TNF-induced oscillations in p65 and p50 recruitment to the CD38 promoter correlated with recruitment of MAPK-induced AP-1 recruitment, as analyzed by quantitative ChIP analysis. Through re-ChIP analysis we show that a unique transcriptional complex is seen on the promoter at 3 hours post TNF addition, corresponding to the onset of CD38 transcription, which is not seen in the basal state. Moreover, we show that RANK-L was unable to combinatorially recruit AP-1 and NF-κB transcription factors to the CD38 promoter, despite inducing the activation of both signaling pathways. These results, in sum with the two accompanying papers, constitute a new paradigm through which cells dynamically orchestrate signaling molecules to coordinate time-resolved gene transcription by the formation of novel time-specific transcriptional complexes.
Keywords: CD38, TNF, RANK-L, NF-κB, AP-1, osteoclast, promoter, Brg-1, CBP, ChIP, re-ChIP
Introduction
One cell type where TNF superfamily cytokines and NF-κB proteins crucially regulate gene activation programs and cell differentiation is the monocytic precursor. These cells are capable of differentiating into macrophages, dendritic cells and osteoclasts depending on which cytokines they become exposed to [1]. Exposure to TNF up-regulates genes needed for dendritic cell differentiation and macrophage activation. For example, TNF induces pro-inflammatory immunomodulators such as interferons and enzymes involved in chemotaxis such as CD38 [2]. The profile of gene activation by TNF contrasts with that of RANK-L, a TNF superfamily member that up-regulates genes needed for attachment to and degradation of bone, such as αvβ3 integrins and TRAP, respectively [3]. Thus, although members of the TNF superfamily induce similar signaling pathways (NF-κB, JNK, MAPK) [4], each member induces genes needed to determine specific cellular fates.
We hypothesized that different gene induction profiles for members of the TNF superfamily may be related to differences in the temporal profile of activation of NF-κB and AP-1 pathways. In unstimulated cells, NF-κB regulated genes are repressed by p50 or p52 homodimers that tether co-repressor complexes [5]. For p50 homodimers, two repressive complexes are p50/SMRT/HDAC3 and p50/Bcl-3/NCoR/HDAC3 [6; 7]. The first complex undergoes de-repression when IKK-α phosphorylates SMRT leading to its export [7]. The later complex undergoes de-repression when MEKK1 phosphorylates TAB2 leading to nuclear export of NCoR and HDAC3 [7].
For the transcriptional induction phase of gene expression, p65 is believed to mediate recruitment of histone acetyltransferases (HATs), which include p300, CBP, p300 and SRC-1,-2,-3. For full recruitment of the HATs, p65 needs concomitant activation of MAPK1 and PKC [8]. Once activated, the HATs acetylate p65 further augmenting induction. Of note is that p65 binding is transient, and while other transcription factors appear to be present for a longer time, p65 is believed to be the key recruiter of RNA polymerase II (pol II) [8]. This notion however, does not hold up with extended stimulation times, where pol II recruitment is temporally dissociated from p65 binding, as shown for the IκB-α and MIP-2α genes [8].
Thus a key question arises: is there an ordered cyclical recruitment of transcription factors to promoters? To address this question, we have focused on the CD38 gene, which is regulated by NF-κB, AP-1 and PKC, thus fulfilling the three components of the p65-mediated HAT activation [4]. Moreover, to understand how these components bring about transcriptional specificity, we have chosen to study CD38 upregulation in the context of two related ligands, TNF and RANK-L. While both ligands are capable of activating NF-κB, AP-1 and PKC, only TNF causes CD38 gene induction [4]. Our hypothesis is that this differential upregulation occurs through temporal and/or combinatorial differences at the level of promoter activation. To investigate if this is indeed the case, we started by performing quantitative chromatin immunoprecipitation (ChIP) assays examining NF-κB and AP-1 recruitment to the CD38 promoter over time.
Results
In osteoclast precursors, the cytokines TNF and RANK-L induce similar downstream pathways and share some of the same adaptor molecules. We have previously shown that TNF and RANK-L differentially regulate ADP-ribosyl cyclases –enzymes that are crucial for immune function, but are detrimental to osteoclastogenesis [4]. We found that TNF induced sustained upregulation of CD38 via the signaling molecules JNK, PKC and NF-κB [4]. Here we analyze how these pathways downstream of TNF cooperate to induce CD38 expression.
The first time point where TNF can up-regulate CD38 but RANK-L fails to do so is at 3 hours following stimulation [4]. To analyze if signaling downstream of JNK and NF-κB lead to transcription factor recruitment on the CD38 promoter, we performed quantitative ChIP assays following either TNF or RANK-L stimulation. Figures 1A and B show that following TNF stimulation at around 3 hours both the AP-1 transcription factor c-fos and the NF-κB transcription factors p65 and p50 were recruited. Additionally, the same transcription factors appear to be re-recruited at around 6 hours. While both transcription factor classes appeared synchronous, AP-1 appeared to last longer on the promoter than NF-κB. Interestingly, we found that waves of JNK activation, while occurring roughly in unison with p65 activation, also appear to last slightly longer (see accompanying paper on MAPK oscillations).
Figure 1. TNF but not RANK-L Induces Oscillatory Transcription Factor Recruitment to the CD38 Promoter in Macrophages.

Primary hematopoetic stem cells were purified from murine long bones and induced to form macrophages by growth in the presence of M-CSF (40 ng/mL). TNF (40 ng/mL) or RANK-L (80 ng/mL) was added and DNA extracted at 30-minute intervals for 6 hours following stimulation. Chromatin immunoprecipitation (ChIP) was performed with anti-p50, anti-p65 or anti-fos antibodies. The resulting precipitates were analyzed by quantitative PCR and expressed as percent maximal fold change.
While stimulation with TNF led to two distinct waves of simultaneous AP-1 and NF-κB recruitment to the CD38 promoter, RANK-L, in contrast, did not lead to any synchronous recruitment and failed to recruit NF-κB p65 to the promoter (Figure 1B). RANK-L did, however, cause one wave of AP-1 recruitment early at 1-hour post stimulation, and produced mild increases in NF-κB p50 recruitment, which were not synchronous with AP-1 recruitment (Figure 1B). Thus, in agreement with previous work showing that both JNK and NF-κB were necessary to produce the resulting increase in CD38 expression [4], we find both AP-1 and NF-κB are recruited to the CD38 promoter, starting at 3 hours following TNF stimulation.
Because of the seemingly simultaneous recruitment of both AP-1 and NF-κB transcription factors, we next assessed whether these factors aggregated into a single transcriptional complex. We performed re-ChIP assays by first immunoprecipitating for either NF-κB or AP-1 and then re-immunoprecipitating for the other transcription factor. As a marker for transcriptional induction we included the co-factor CBP in the analysis. In the basal, unstimulated state, we found that either NF-κB proteins or AP-1 (c-jun) were bound to the CD38 promoter, but neither transcription factor class was bound at the same time as the other (Figure 2A). Interestingly, basal CD38 expression appears to be primarily driven by NF-κB as these proteins were bound to CBP (Figure 2A). This finding fits with previous data showing that treating cells in the basal state with an NF-κB inhibitor appears to slightly decrease basal CD38 expression [4].
Figure 2. Re-ChIP Analysis of the CD38 Promoter in the Basal State and After TNF Stimulation.
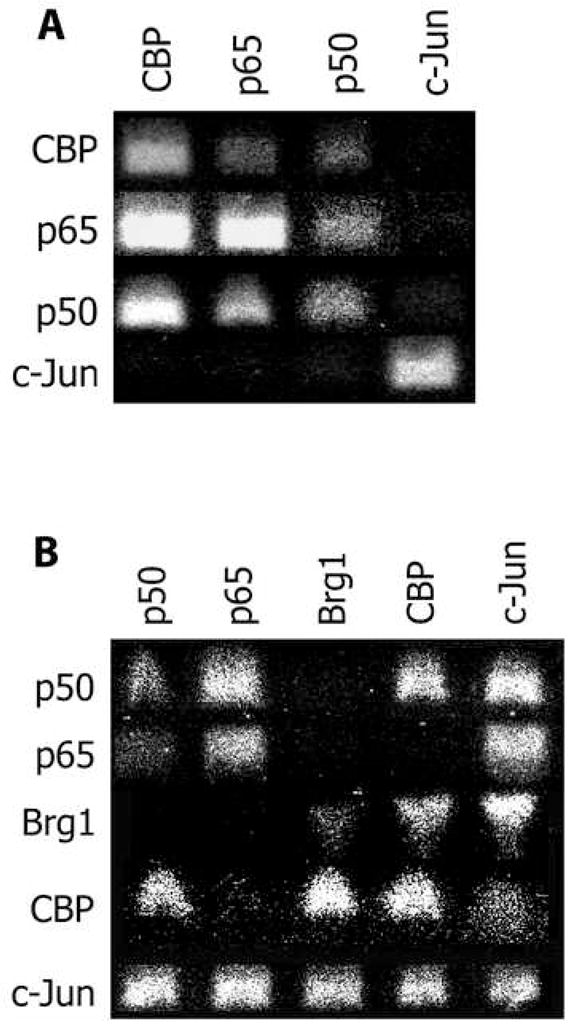
Primary macrophages were prepared as in Legend to Figure 1 and subject to ChIP analysis/After the first round of ChIP, the samples were split and subject to a second round of ChIP. The resulting matrix shows the antibodies used in the first round at the top, and those in the second round along the side. The resulting DNA was run on an aragose gel and a picture taken with a Polaroid camera and subsequently scanned. (A) The protein complexes found in the basal state on the CD38 promoter. Note that either AP-1 proteins or NF-κB proteins are bound at any one time, but they are not bound together. (B) The protein complexes present on the CD38 promoter after 3 hours of TNF stimulation. Note that in contrast to the basal state, after TNF stimulation both AP-1 proteins and NF-κB proteins are bound together.
For the TNF-stimulated state, we carried out the same re-ChIP analysis, but also added a marker for chromatin remodeling, Brg-1. As expected from the hypothesis generated from Figure 1, following TNF stimulation both NF-κB protein and AP-1 protein were found in a complex together (Figure 2B). However, there appeared to be several distinct complexes present on the promoter. One complex consisted of simultaneous p65/p50/c-jun binding; however, this complex surprisingly was not associated with either CBP or Brg-1 (Figure 2B). There were only two apparent complexes containing CBP. One consisted of p50/c-jun/CBP, but excluded both Brg-1 and p65 (Figure 2B). The other complex consisted of c-jun/CBP/Brg-1, but excluded both NF-κB proteins (Figure 2B). Thus, following TNF stimulation, both NF-κB and AP-1 proteins are recruited to form numerous complexes on the CD38 promoter, some of which are involved in chromatin remodeling and transcriptional induction.
Discussion
CD38, an ADPribosyl cyclase, has previously been shown to be strongly upregulated by TNF, but not by the related cytokine RANK-L [4]. This differential upregulation is significant in that it primes macrophages to respond to immune stimuli, while inhibiting signaling that induces osteoclastogenesis [1]. The specific signaling pathways downstream of TNF needed to produce CD38 upregulation involve JNK, NF-κB and PKC [4]. Our central premise as to why TNF, but not RANK-L, is able to induce differential gene induction is related to time-specific differences in the combinatorial recruitment of transcription factors to promoters. In this scenario, even though RANK-L can induce similar pathways, it does so in a temporally different manner, thus failing to simultaneously accumulate all requisite factors needed for induction. Data in Figure 1 support this hypothesis. We find that RANK-L causes AP-1 recruitment and mild changes in p50 NF-κB recruitment, but importantly that these recruitment waves are asynchronous. In contrast, TNF produces synchronous recruitment of AP-1 and NF-κB proteins. The data presented here substantiate the previous findings that TNF-induced JNK and NF-κB signaling pathways are needed to produce CD38 upregulation [4]. While it is clear that these pathways temporally combine on the CD38 promoter, we sought to discern next what specific complex mediated transcription.
Recent experiments using the BiFC-based FRET (BiFC-FRET) assay for visualization of complexes in living cells has shown that NF-κB and AP-1 (jun/fos) interact in the nucleus to form tertiary complexes [9]. In the TNF-stimulated state, we show using re-ChIP analysis that AP-1 factors and NF-κB combine to form a complex not seen in the basal state. Somewhat surprisingly, we found that p65 was not found in complex with CBP following stimulation with TNF. Rather, it appeared that p50, either bound as homodimers or to other NF-κB proteins or that c-jun was preferentially utilized for CBP-containing complexes. This finding suggests that CD38 upregulation is non-typical and indeed previous reports have shown that its mRNA induction time is significantly later than “traditional” NF-κB-regulated genes such as IκBα. Our findings are in agreement with a recent report showing that p50 is able to directly bind CBP, and that a p50/CBP complex, exclusive of p65, is responsible for IL-10 induction by LPS in murine macrophages [10]. Our results further suggest that a p50/c-jun/CBP complex is the one that mediates transcriptional induction; this finding is in accord with the fact that CBP is required for jun-mediated induction [11].
To further examine transcriptional induction, we studied Brg-1 recruitment. While CBP and RNA polymerase II are required for jun-mediated induction, previous studies have implicated SWI/SNF protein recruitment as the key initiator of transcription. We find that the SWI/SNF protein Brg-1 was recruited only in complexes containing c-jun and excluding NF-κB. This finding was surprising because it suggests that the initiation of transcription occurred after NF-κB p65 and p50 had left the promoter. There are two possible explanations for this. One possibility is that the general transcriptional complex is recruited by a combination of NF-κB and AP-1 factors, but that the actual start of transcription occurs only when AP-1 is present and NF-κB has departed. This hypothesis is supported by our time course studies showing that both NF-κB and AP-1 factors are first recruited to the promoter at the same time, but that AP-1 factors remain bound for a short period after NF-κB proteins have been removed (Figure 1A).
Alternatively, the situation may be similar to what has been observed previously in lymphocytes with Bfl-1 expression, where Brg-1 was temporally correlated with jun and CBP recruitment and not with p65 or p50 recruitment [12]. However, in those ChIP experiments, c-Rel was temporally correlated to both Brg-1 and CBP [12]; we have not studied c-Rel recruitment here, thus it remains a possibility that c-Rel and not p65 or p50 is critical to the initiation of CD38 transcription. Thus, for TNF-induced expression of CD38, a multi-protein complex of NF-κB and AP-1 proteins appears to be initially formed and then subsequently replaced by an AP-1 SWI/SNF complex that excludes NF-κB (Figure 3).
Figure 3. Diagram of the transcription complexes present on the CD38 promoter in the basal and TNF-stimulated states.

Panel A is a pictorial diagram representing the complexes from Figure 2 that are present on the CD38 promoter in the basal state. The pictures in Panel B provide a possible interpretation of the sequence of events that are occurring at the CD38 promoter following TNF stimulation.
In summary, we have shown that the previously observed differential regulation of CD38 expression by TNF and RANK-L does indeed involve differences in the utilization of effectors downstream of NF-κB and JNK signaling pathways. Specifically, we have shown that TNF and RANK-L have temporal differences in their induction of oscillations of NF-κB and AP-1 transcription factor activation to the CD38 promoter. We also find that these two transcription factor classes, while not associated in the basal state, come together in a complex following TNF stimulation.
Methods
Cell Preparation
C57BL6J mice, inbreed from founders obtained from Jackson Laboratory (Maine), were housed in the Annenberg building animal care facility (Mount Sinai School of Medicine, NY). Mice were sacrificed by cervical dislocation and both femurs and tibea were surgically extracted and placed in media for bone marrow flushing. After cutting the ends of each bone, the marrow was flushed with media into a clean dish using a 23-guage needle and 3 mL syringe. The total marrow from all animals was pooled and spun down at 2,500 rpm for 5 minutes. The pellet was subsequently resuspended in Opti-MEM I (Invitrogen, CA) containing 5% FBS (Select USA stock, Invitrogen, CA), and 1% penicillin/streptomycin. 5 ng/mL recombinant murine M-CSF (R&D Systems, NJ) was added and the cells incubated for one day in a 37°C, 5% CO2, water-jacketed tissue culture incubator. As a result of this incubation, most of the CD11b+ macrophages from the bone marrow became adherent to the dish.
The non-adherent cells were collected and spun down in plain media at 2,500 rpm for 5 minutes. This cell pellet was resuspended in 9 mL of plain media, filtered through a 70μ cell strainer (BD Biosciences, CA), and gently layered over 6 mL of Ficoll-Hypaque (Amersham/GE Health Sciences, NJ). This dual-layered tube was then centrifuged at 1,500 rpm for 15 minutes at 4°C. This density centrifugation resulted in the separation of cells, most were pelleted at the bottom of the tube, but some cells formed a cloudy-white interface layer. The cells from this interface layer were gently collected and spun down in plain media at 3,000 rpm for 5 minutes. The resulting pellet was resuspended in the Opti-MEM media (described above).
For all experiments, an appropriate amount of M-CSF was added such that the final plated concentration was 40 ng/mL. For ChIP analysis, the cells were plated in 20cm dishes. To enhance the specificity of the response to TNF and to remove contaminating cells, after 2 days of culture, the media was removed and fresh Opti-MEM containing M-CSF was added. Experiments were then performed after 1–2 days after this media change; at this time >99% of the cells were non-proliferating, as assessed by BrdU incorporation (APC BrDU flow kit, BD Biosciences, CA), and ~99% of the total cells expressed markers specific for monocyte-macrophages.
Chromatin Immunoprecipitation (ChIP)
The procedure for ChIP analysis was kindly provided by Dr. G Natoli (Switzerland). The general procedure is as follows: hematopoetic stem cell-derived macrophages were grown in 15 cm dishes as described above. After stimulation, the cells are fixed directly in their culture medium by adding in an appropriate amount of 37% paraformaldehyde to achieve a 1.7% final concentration. The cells were fixed for 15 minutes at room temperature and then washed twice with ice-cold 1x PBS containing Complete EDTA-free protease inhibitors (Roche, NJ) and scraped using a plastic cell lifter. The cells in suspension were spun down in a 15 mL tube and resuspended for counting.
Cells were diluted with SDS lysis buffer (Upstate Biotech, VA) and then sonicated using a Misonex 3000 on power setting 1.5 for 4 rounds of 15 seconds; all samples were kept on ice at all times. Following sonication, a portion of the sonicated solution was uncrosslinked for analysis of proper shearing of genomic DNA. The remainder was pre-cleared by incubation with non-immune rabbit IgG, which was pre-conjugated to an anti-rabbit IgG magnetic bead conjugated antibody (New England Biotech, MA) by overnight incubation. The “contaminants” were cleared by 3 rounds of incubation on a BD Magnetic Cell Separator (BD Bioscience, CA), where the non-bound solution was transferred each time to a clean tube for further incubation against the magnet.
After pre-clearing, the resulting extract solution was diluted using ChIP dilution buffer (Upstate, VA) and aliquotted for the immunprecipitation of various proteins. One aliquot was set aside to serve as an input control. For each protein, the primary antibody (either rabbit or mouse; all from Santa Cruz Biotech, CA) was attached to the secondary antibody (either anti-rabbit or anti-mouse; New England Biotech, MA) by overnight incubation at 4°C; the primary-secondary antibody conjugate was then added into the sample and incubated for 2 hours at 4°C, following which, the positive (magnet-bound) fraction was separated using the procedure described above for pre-clearing, except that the non-bound fraction was discarded each time. The same procedure was followed for Re-ChIP analysis, except that after the first IP, a separation solution was used (Upstate Biotech, VA) to obtain the samples for IP with a second antibody.
To reverse the paraformaldehyde crosslinks, samples were incubated with 5 mM NaCl at 60°C overnight. The DNA from the resulting solution was subsequently purified using Qaigen PCR purification kits following manufacturer’s directions. The obtained DNA was eluted with Buffer EB (Qaigen, CA) and stored at −80°C until analysis. Quantization of the immunoprecipitated DNA samples was carried out in an analogous fashion as described above for “Quantitative PCR” in the preceding paper.
Acknowledgments
We gratefully acknowledge support from the NIH: AG14907, DK70526, AG23176 (to MZ).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Iqbal J, Zaidi M. CD38 is required for priming by TNF-alpha: a mechanism for extracellular coordination of cell fate. Am J Physiol Renal Physiol. 2007;292:F1283–90. doi: 10.1152/ajprenal.00381.2006. [DOI] [PubMed] [Google Scholar]
- 2.Tliba O, Panettieri RA, Jr, Tliba S, Walseth TF, Amrani Y. Tumor necrosis factor-alpha differentially regulates the expression of proinflammatory genes in human airway smooth muscle cells by activation of interferon-beta-dependent CD38 pathway. Mol Pharmacol. 2004;66:322–9. doi: 10.1124/mol.104.001040. [DOI] [PubMed] [Google Scholar]
- 3.Yang G, Zaidi M, Zhang W, Zhu LL, Li J, Iqbal J, Varbanov A, Gross G, Phipps R, Troen BR, Sun L. Functional grouping of osteoclast genes revealed through microarray analysis. Biochem Biophys Res Commun. 2008;366:352–9. doi: 10.1016/j.bbrc.2007.11.106. [DOI] [PubMed] [Google Scholar]
- 4.Iqbal J, Kumar K, Sun L, Zaidi M. Selective upregulation of the ADP-ribosyl cyclases CD38 and CD157 by TNF but not by RANK-L reveals differences in downstream signaling. Am J Physiol Renal Physiol. 2006;291:F557–66. doi: 10.1152/ajprenal.00066.2006. [DOI] [PubMed] [Google Scholar]
- 5.Djavaheri-Mergny M, Amelotti M, Mathieu J, Besancon F, Bauvy C, Souquere S, Pierron G, Codogno P. NF-kappaB activation represses tumor necrosis factor-alpha-induced autophagy. J Biol Chem. 2006;281:30373–82. doi: 10.1074/jbc.M602097200. [DOI] [PubMed] [Google Scholar]
- 6.Hoberg JE, Popko AE, Ramsey CS, Mayo MW. IkappaB kinase alpha-mediated derepression of SMRT potentiates acetylation of RelA/p65 by p300. Mol Cell Biol. 2006;26:457–71. doi: 10.1128/MCB.26.2.457-471.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosenfeld MG, Lunyak VV, Glass CK. Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev. 2006;20:1405–28. doi: 10.1101/gad.1424806. [DOI] [PubMed] [Google Scholar]
- 8.Natoli G. Tuning up inflammation: how DNA sequence and chromatin organization control the induction of inflammatory genes by NF-kappaB. FEBS Lett. 2006;580:2843–9. doi: 10.1016/j.febslet.2006.02.072. [DOI] [PubMed] [Google Scholar]
- 9.Shyu YJ, Suarez CD, Hu CD. Visualization of AP-1 NF-kappaB ternary complexes in living cells by using a BiFC-based FRET. Proc Natl Acad Sci U S A. 2008;105:151–6. doi: 10.1073/pnas.0705181105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cao S, Zhang X, Edwards JP, Mosser DM. NF-kappaB1 (p50) homodimers differentially regulate pro- and anti-inflammatory cytokines in macrophages. J Biol Chem. 2006;281:26041–50. doi: 10.1074/jbc.M602222200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bannister AJ, Oehler T, Wilhelm D, Angel P, Kouzarides T. Stimulation of c-Jun activity by CBP: c-Jun residues Ser63/73 are required for CBP induced stimulation in vivo and CBP binding in vitro. Oncogene. 1995;11:2509–14. [PubMed] [Google Scholar]
- 12.Edelstein LC, Lagos L, Simmons M, Tirumalai H, Gelinas C. NF-kappa B-dependent assembly of an enhanceosome-like complex on the promoter region of apoptosis inhibitor Bfl-1/A1. Mol Cell Biol. 2003;23:2749–61. doi: 10.1128/MCB.23.8.2749-2761.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]