

Abstract
Oscillations in the activation of multiple signalling pathways have never been shown before. Our results presented in the previous accompanying paper showed that TNF induces highly dynamic oscillations in mRNA production in ~13% of the mouse genome. Here, we further analyze the TNF time-series microarray data and find that multiple signaling components downstream of the TNF receptor undergo oscillations. Prior studies implicate IκBα and A-20 as the only oscillatory components in the TNF signaling cascade. We find however, that other components, such as TRAF1, displayed oscillations. This suggested the possibility that all signaling output from the TNF receptor may be oscillatory in nature. Indeed, we show that TNF triggers oscillations in the phosphorylation of three MAP kinases, as well as p65. Because Baltimore and colleagues had proposed that NF-κB drives the oscillatory nature of the IκBα/NF-κB feedback loop, we studied the effects of an NF-κB super-repressor on oscillations in MAPK phosphorylation; we find that the super-repressor altered the amplitude and frequency of MAP kinase phosphorylation, but failed to abolish oscillations. These results attest to a role for NF-κB as a modulator, but not the sole determinant of cyclical activation of signal transduction pathways. These results, together with those of the two accompanying papers, constitute a new paradigm through which cells orchestrate signaling molecules to produce highly dynamic physiological processes such as gene transcription and protein secretion. In view of the discovery that multiple phosphorylation pathways display dynamic oscillation, time-resolved, instead of static, measurements of kinase phosphorylation should become the experimental norm.
Keywords: TNF, MAPK, oscillations, NF-kappaB, JNK, ERK, p65, signaling, cyclical
INTRODUCTION
Oscillations in the phosphorylation of signaling molecules have been shown in only in three situations, Hes-1, p53 and IκB. In these three examples, the protein oscillations serve to govern feedback loops of the proteins involved. NF-κB regulates numerous genes involved in cellular stress responses, cell growth, cell survival and apoptosis. The oscillatory nature of NF-κB activation is governed by a negative feedback system where NF-κB proteins induce the expression of an inhibitor of their nuclear translocation, IκBα [1]. The NF-κB-IκBα complex is exported into the cytoplasm preventing further NF-κB action and thus creating a feedback loop [1].
In the proceeding accompanying paper, we showed that the inflammatory cytokine TNF triggered oscillations in mRNA production for >5,000 genes (~13% of the genome). Moreover, we showed that these continuous oscillations are not unique to TNF, but that TNF superfamily member cytokines such as RANK-L were also capable of inducing oscillations in gene transcription. We found that each cytokine had a distinctive mRNA induction profile over time, despite initial similarities in gene induction.
The results from the accompanying paper also showed that mRNA oscillation frequencies were as low as every 50 minutes. This suggests that mRNA production is highly dynamic. We hypothesize that this dynamic nature may be the result of rapidly changing events further upstream. Specifically, we hypothesize that multiple signaling components downstream of the TNF receptor may display dynamic oscillations in activation. In this paper, we first analyze the microarray data of the proceeding paper and find that several components of the TNF signaling pathway, such as TRAF1, undergo oscillations in their mRNA production. We then use phospho-flow cytometry to show that TNF triggers oscillations in the phosphorylation of MAP kinases. Through the use of an IκB super-repressor, we show that the oscillations in MAP kinase phosphorylation are independent of, but modulated by NF-κB. In the following paper, we show that the oscillations combine to recruit transcription factors to promoters in a cyclical fashion.
RESULTS
The preceeding paper showed that TNF induces dynamic changes in gene expression that have never been appreciated before. To investigate the mechanistic basis of these dynamic changes, we performed a pathway analysis on the two clusters containing early and late oscillator genes. KEGG pathway analysis, using a z-score cut-off of 2.0 to indicate significance, indentified several functional signaling pathways within the two clusters. Several key genes appeared in multiple KEGG pathways. For the early oscillators, these included IκBα, Ras-GRP1, TRAF-1, MALT1, SOCS3, PTK2 (FAK1) and TNF itself (Table 1). For the late oscillators, statistically significant association were noted with NF-κB p100, IAP, p15 and Fas (Table 1).
Table 1. Analysis of Gene Sub-Clusters Reveals Affected Pathways and Multiple Oscillatory Components in the TNF Signaling Cascade.
Primary macrophages were prepared as in Legend to Figure 1 and subject to microarray analysis. Early and late oscillator genes are defined in the proceeding paper. KEGG Pathway Analysis was used to derive z-scores, and statistically significant oscillators with a z-score >2 were identified.
| KEGG Pathway | Z-score | Oscillatory components |
|---|---|---|
| Small cell lung cancer | ||
| Early Oscillators | 4.04 | IκB α, TRAF1, Prostaglandin-endoperoxide synthase 2, FAK1 |
| Late Oscillators | 3.47 | NF-κB p100, p15, INK4b, IAP |
| T cell receptor signaling | ||
| Early Oscillators | 3.82 | IκBα, RasGRP1, TNF, and MALT1 |
| Late Oscillators | 1.14 | NF-κ p100 |
| Adipocytokine signaling | ||
| Early Oscillators | 3.27 | IκBα, IL-1, and TNF |
| Late Oscillators | 1.57 | NF-κ p100, and FACS (acetyl-CoA synthetase) |
| Apoptosis | ||
| Early Oscillators | 2.93 | IκBα, and TNF |
| Late Oscillators | 2.47 | NF-κ p100, IAP, and Fas |
| TGF-beta signaling | ||
| Early Oscillators | 2.93 | Smurf1, ID3, and TNF |
| Late Oscillators | 0.21 | p15 INK48 |
| Diabetes Pathways | ||
| Early Oscillators | 2.65–2.77 | SOCS3, IL-1, and TNF |
| Late Oscillators | 2.25–3/52 | Fas, MHCII, HNF6, and Nr5a2 |
Figure 1 shows a composite diagram of TNF receptor signalling containing the molecules annotated as early or late oscillators. It is obvious that while NF-κB components are most downstream, other oscillators, such as Ras-GRP1, TRAF1 and MALT1 are upstream. This oscillator profile prompted us to question whether the phosphorylation of NF-κB components, such as p65, and distal MAP kinases, such as JNK1/2, ERK and p38, was also oscillatory. Using a 96-well phospho-ELISA plate, we first measured phosphorylated p65 and JNK every 10 minutes for 0.5 hours and every 30 minutes thereafter for 8 hours following TNF application to primary murine macrophages. We found that the phosphorylation of both p65 and JNK oscillated over time, albeit with broad, somewhat poorly defined peaks (Figure 2A and 2B).
Figure 1. Occurrence of Early and Late Oscillator Genes Within the TNF Signaling Pathway.
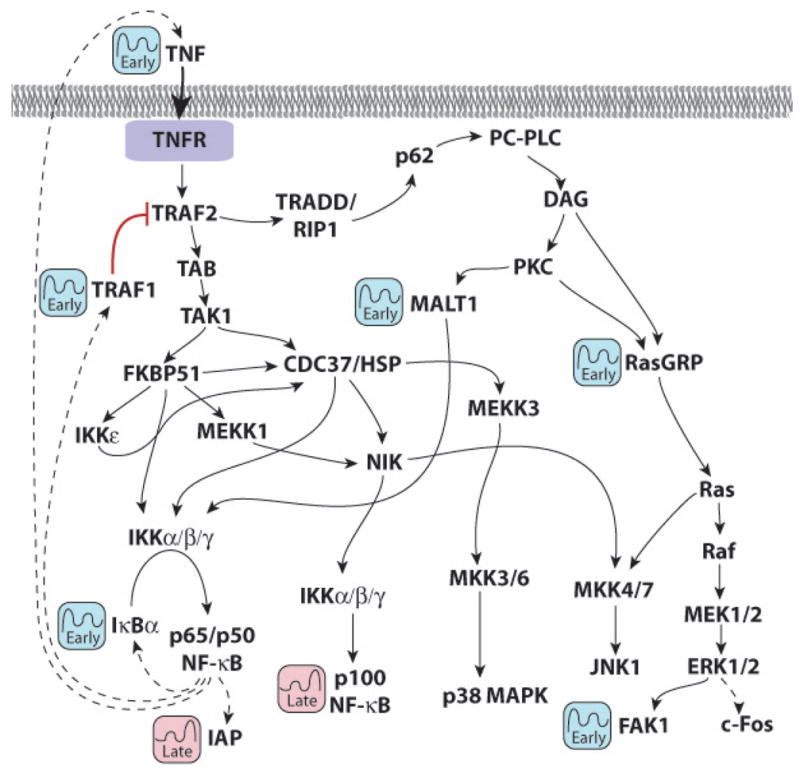
These oscillating components were discovered by performing a KEGG Pathway Analysis using Genesifter software of the two pre-selected clusters comprising early and late oscillators (see Table 1).
Figure 2. Phosphorylation of Signal Transduction Molecules is Oscillatory.

Temporal profiles at 30 minute intervals for 8 hours (with 10 and 20 minutes time points added) for p65 NF-κB and JNK1/2 phosphorylation in purified murine macrophages (A and B) using phospho-ELISAs following TNF addition (40 ng/mL). Data is plotted as absorbance (Abs) of the phosphorylated to non-phosphorylated protein (three replicate estimations per time point). In separate experiments, Jurkat cells were stimulated with TNF and fixed with formaldehyde at 10 minutes and at 30-minute intervals thereafter for 8 hours. After permeablization in methanol, the cells were stained for (p)-JNK1/2-Ax647 and (pS536)-p65-Ax488 or (p)-ERK1/2-Ax647 and (p)-p38 MAPK-Ax488 and analyzed by flow-cytometry to calculate the fold change in median fluorescence intensity (MFI). Increases in (pS536)-p65 (C) mirrored that of (p)-p38 MAPK (E), except that the trough for (pS536)-p65 remained significantly elevated in between the first two peaks. JNK1/2 phosphorylation (D) displayed a unique pattern for the first 3 hours, but thereafter became in unison with the phosphorylation pattern of p38 MAPK and p65. The phosphorylation of ERK1/2 (F) did not display oscillations, but rather underwent one wave of activation. The only time where all pathways were significantly activated was the initial signaling wave (G). Panel H shows similar data for JNK and ERK1/2 phosphorylation in Jurkat cells stably overexpressing the IκB super-repressor; these individual data are plotted together with that from wild type vector-transfected cells (I and J). Please refer to Supplementary Figure 2 for a representative plot, including a raw data set, from the flow cytometry experiments showing the calculation of a change in MFI at each time point.
To validate these results in another cell type, to obtain a better time resolution, and for further mechanistic experiments, intracellular phospho-flow cytometry was performed on a Jurkat cell line. The phosphorylation of p65, JNK and p38 was oscillatory for up to 8 hours (Figures 2C, 2D and 2E). The first phosphorylation peak for p65, JNK, p38 and ERK1/2 overlapped, after which ERK1/2 phosphorylation became rapidly attenuated (Figure 2F). Moreover, the phosphorylation profiles for p38 and p65 were in unison (Figure 2G). JNK phosphorylation also oscillated more rapidly than p65 or p38 phosphorylation in the first 3 hours. At around 4 hours, p65, p38 and JNK phosphorylation became simultaneously aligned (Figure 2G). This time point is significant in that it coincides with induction of late oscillator genes (see proceeding paper), but whether or not the coincidental phosphorylation of this trio mediates gene induction is evaluated in the following paper.
Because all three MAP kinases, p38, JNK and ERK1/2, displayed oscillations in phosphorylation, we theorized that a feedback mechanism within the early TNF signaling cascade likely mediated these oscillations. Using IκB null cells, Baltimore and co-workers first identified cyclical production of IκBα as a negative feedback loop to cyclically inhibit NF-κB activation [1]. We have verified the cyclical phosphorylation of p65, but find that this can also occur in wild type cells, not mutant cells as they have used. Using KEGG pathway analysis, we have further identified both TNF and TRAF1 as early oscillators identical to IκBα (Figure 1). TRAF1 is a known inhibitor of TNF receptor signalling [2], and is directly upstream of both p38 and JNK, but is more distantly related to ERK (Figure 1). Because TNF and TRAF1 are both NF-κB-dependent genes, we chose to examine whether abrogating NF-κB signaling could affect the phosphorylation of the two MAP kinases JNK and ERK1/2.
JNK and ERK phosphorylation were thus examined in Jurkat cells over-expressing an IκB super-repressor construct, which specifically suppresses p65 translocation. Figures 2I and 2J show comparisons of JNK and ERK phosphorylation in IκB-dominant negative (IκB-DN) overexpressing (solid lines) and wild type (broken lines) cells. Of note is that the oscillations in the phosphorylation of both MAP kinases persisted despite abrogation of NF-κB signaling. However, the amplitude of JNK phosphorylation was significantly increased and the oscillation frequency altered in IκB-DN cells (Figure 2I). Additionally, the frequency of ERK1/2 phosphorylation was converted from monophasic to oscillatory (Figures 2J).
Figure 2H shows that in IκB-DN cells, ERK1/2 and JNK oscillations were in unison for the first 3 hours following TNF. Furthermore, ERK1/2 did not show any appreciable phosphorylation after 4 hours (Figure 2K), whereas JNK continued to oscillate with a frequency and peak width distinct from that of empty vector transfectants (Figure 2I). Together, these data show that while NF-κB is not necessary for the genesis of MAP kinase oscillations, it regulates their amplitude and frequency.
DISCUSSION
The results show for the first time that TNF induces robust and rapid oscillations in the phosphorylation of the distal kinases JNK, ERK1/2 and p38, as well as p65. The pattern of these oscillations suggests that there are several key time points where all signaling pathways are simultaneously activated. Through the use of the NF-κB super-repressor, the amplitude and frequency of these oscillations is regulated, but is not generated by the previously demonstrated cyclic activation of NF-κB. Although decoding the mechanistic basis of these oscillations will represent a challenge, two key issues arise.
One issue is what is the mechanistic basis for oscillations in kinase activation? The other key issue is what are the purposes of oscillations in the regulation of cell function? As discussed in the proceeding paper, current thinking suggests that only a few critical molecular components are needed for generating oscillations. The cyclic activation of NF-κB has been attributed to the cyclic production of the inhibitor IκBα, which prevents the nuclear import of NF-κB subunits [1; 3]. In stark contrast, our results, presented here and in the proceeding paper, highlight a profound level of complexity in the generation and maintenance of TNF-induced oscillations in MAP kinase phosphorylation and gene expression, respectively. Our accompanying paper showed through affymetrix cluster analysis that >5000 genes had oscillatory behavior with different times of onset, response magnitudes and oscillation frequencies. We have shown here through pathway analysis that multiple components of the TNF signaling pathway were oscillatory. Rather than a two-component model where removing one component yields a clear genetic output, our results suggest that TNF signaling is much more complex than expected in terms of regulatory loops. Removing NF-κB for example does not abrogate the oscillations in MAP kinase phosphorylation. Instead, both magnitude and frequency of oscillations are affected, illustrating a further level of complexity in the molecular signals that precisely regulate these dynamic events.
There appears to be yet uncharacterized mechanisms, further hierarchical in complexity, which underlie the sustained oscillations in wild type cells. Dynamism in NF-κB has been previously demonstrated at various levels in different cell types. EMSA studies in mouse embryonic fibroblasts reveal dynamic oscillations in NF-κB activation at a frequency of one every 90 minutes [1]. This is consistent with our data presented in the accompanying paper, where we find that, in primary macrophages, the expression of some NF-κB regulated genes, including IκBα itself, occurs every 90 minutes. However, while we find that macrophages show continued oscillations, the oscillations in fibroblasts are dampened after 3 hours [1]. In a debate with the Baltimore group, Nelson et al. showed that the nuclear translocation of activated p65, which had been overexpressed in NIH3T3 fibroblasts, displayed continuous oscillations [3]. Their findings are concordant with our demonstration of continuous oscillations in p65 phosphorylation albeit we have used cells that do not overexpress NF-κB components.
Despite the demonstration of oscillations in NF-κB activation and IκB production, the purpose of these oscillations has remained unclear. Recent studies in the area of signal transduction involving the rapid oscillatory release of intracellular Ca2+ have highlighted distinct effects of different frequencies and amplitudes of an induced Ca2+ signal on the responsiveness of cells to stimuli (Figure 3A). For example, in naïve B cells, antigens evoke a Ca2+ signal of greater amplitude than self-tolerant B cells [4]. Due to differences in Ca2+ affinity of downstream signaling molecules, the output resulting from the two signal magnitudes is different with either NF-κB or NFATc becoming activated. Likewise, distinct frequencies in oscillatory Ca2+ signals trigger the cyclic release of hormones and cytokines in physiologically appropriate manner, for which enzymes such as CaM kinase II, decode the frequency of Ca2+ signalling [5].
Figure 3. Diagram Showing How Signaling Amplitudes or Synergism can Affect Gene Induction.

(A) Schematic of how the amplitude in a signal can give rise to distinct gene expression profiles. Specifically at high signal amplitude, signaling molecules with a low affinity for say Ca2+, can become activated and result in a distinct genetic output. (B) Schematic of how two signal transduction pathways can coordinate to lead to the expression of select genes, with one pathway mediating chromatin modification (for example, p38) and the other pathway initiating transcription (for example, p65).
Analogous to Ca2+ signaling, oscillatory changes in NF-κB and MAP kinase phosphorylation are likely beneficial. We propose that there may be added value of oscillatory behavior to the coordination of multiple pathways that regulate inflammatory gene expression. Many inflammatory genes require p38-mediated promoter processing for their induction, and despite NF-κB activation, disruption of p38 activation causes a failure of gene transcription [6]. Our demonstration that p38 is phosphorylated at the same time as p65 likely synergizes the resulting genetic output (Figure 3).
Moreover, in the preceeding paper, we showed that distinct genetic outputs can be triggered by closely related ligands that transduce almost identical early signaling cascades. We suggest that distinct outcomes in gene expression can arise from similar signal transduction molecules being orchestrated differently over time. Genes such as IL-6 appear to be a manifestation of total coincidence detection between signaling pathways. Our data showing that the single initial peak of IL-6 production coincides with the simultaneous activation of MAP kinases and NF-κB (Supplementary Figure 1) is consistent with the requirement of an enhancesome composed of transcription factors activated downstream of NF-κB, p38 MAPK, JNK and ERK [7]. One pathological setting for multiple-pathway coordination of IL-6 production is with H. pylori infection, where bacterial virulence factors simultaneously activate all three MAP kinase and NF-κB pathways to trigger IL-6 production in the gastric mucosa [8]. Similar to IL-6, we show in the subsequent paper that signaling pathways downstream of TNF and RANK-L differentially combine transcription factors on the CD38 promoter to lead to unique outcomes.
In summary, our results represent a paradigm shift in the understanding of how intracellular communication occurs. Previous work has focused identifying the players involved in the communication; here we show that there is a dynamic conversation amongst multiple players. Moreover, we show that certain players can leverage the conversation; for example, the dynamics of the signaling conversation changed when NF-κB was removed. This elaborate communication network and, in particular its dynamism, is thus in stark contrast with the scientific reductionism that has pared down biological processes to static biochemical and molecular outcomes. The 2.5 billion-year delay between the emergence of unicellular bacteria and evolution of the first multicellular organism reflects the elaborateness in cell communication that higher organisms must possess to survive [9]. Our study highlights the importance of measuring these fundamental processes, such as protein phosphorylation, as a function of time. This dynamic complexity lends cells the ability to precisely govern their own behavior and ensure survival of the organism.
METHODS
Cell Preparation
C57BL6J mice were sacrificed by cervical dislocation and both femurs and tibiae were surgically extracted and placed in media for bone marrow flushing. After pre-incubation and density purification, the cells were grown in Opti-MEM I (Invitrogen, CA) containing 5% FBS (Select USA stock, Invitrogen, CA), and 1% penicillin/streptomycin with 40 ng/mL recombinant murine M-CSF (R&D Systems, NJ).
Microarrays/Data Analysis
cDNA was prepared from cell extracts and then hybridized to 430.2 mouse genome arrays (Affymetrix, CA). The microarray data was quality-controlled and CEL files generated as previously described [10]. Subsequently, CEL files were uploaded into Genesifter (www.genesifter.net) for clustering and KEGG pathway analysis. The calculation and plotting of the derivative fold change was carried out in Microsoft Excel.
Phospho-Flow Cytometry
Jurkat cells, a generous gift from C. Friedman, were maintained in IMDM with 10% FBS (Invitrogen Select, CA) and 1% P/S. TNF was added and cells removed and fixed and then stained with Alexa 488/647 antibody combinations. More detailed information about the procedure of phospho-FACS can be obtained elsewhere [11].
Quantitative Real-Time PCR
Cells were stimulated, washed and shredded with a QaiShredder (Qaigen, MD). RNA was converted into cDNA using random primers and Superscript II cDNA synthesis kit (Invitrogen, CA). For quantitative PCR, Supermix with Rox (Bio-Rad, CA) was used with gene-specific primers on an ABI 7900HT.
Supplementary Material
Supplementary Figure 1. Time Series of TNF-Induced IL-6 mRNA Expression. IL-6 mRNA expression measured by quantitative PCR from macrophages treated with TNF (40 ng/mL). See Methods for details.
Supplemental Figure 2. Raw Data Analysis for Flow Cytometry Calculations. The excel sheet depicts the raw data and associated calculations for oscillations in p65 NF-κB phosphorylation triggered by TNF as measured by phospho-flow cytometry.
Acknowledgments
We sincerely thank A. Ting, C. Friedman and colleagues (MSSM) for supplying the Jurkat T-cells stably expressing the IκB super-repressor. We gratefully acknowledge support from the NIH: AG14907, DK70526, AG23176 (to MZ).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hoffmann A, Levchenko A, Scott ML, Baltimore D. The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science. 2002;298:1241–5. doi: 10.1126/science.1071914. [DOI] [PubMed] [Google Scholar]
- 2.Sasaki CY, Slemenda CF, Ghosh P, Barberi TJ, Longo DL. Traf1 induction and protection from tumor necrosis factor by nuclear factor-kappaB p65 is independent of serine 536 phosphorylation. Cancer Res. 2007;67:11218–25. doi: 10.1158/0008-5472.CAN-07-0922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nelson DE, Ihekwaba AE, Elliott M, Johnson JR, Gibney CA, Foreman BE, Nelson G, See V, Horton CA, Spiller DG, Edwards SW, McDowell HP, Unitt JF, Sullivan E, Grimley R, Benson N, Broomhead D, Kell DB, White MR. Oscillations in NF-kappaB signaling control the dynamics of gene expression. Science. 2004;306:704–8. doi: 10.1126/science.1099962. [DOI] [PubMed] [Google Scholar]
- 4.Berridge MJ. The AM and FM of calcium signalling. Nature. 1997;386:759–60. doi: 10.1038/386759a0. [DOI] [PubMed] [Google Scholar]
- 5.Dupont G, Houart G, De Koninck P. Sensitivity of CaM kinase II to the frequency of Ca2+ oscillations: a simple model. Cell Calcium. 2003;34:485–97. doi: 10.1016/s0143-4160(03)00152-0. [DOI] [PubMed] [Google Scholar]
- 6.Saccani S, Pantano S, Natoli G. p38-Dependent marking of inflammatory genes for increased NF-kappa B recruitment. Nat Immunol. 2002;3:69–75. doi: 10.1038/ni748. [DOI] [PubMed] [Google Scholar]
- 7.Tuyt LM, Dokter WH, Birkenkamp K, Koopmans SB, Lummen C, Kruijer W, Vellenga E. Extracellular-regulated kinase 1/2, Jun N-terminal kinase, and c-Jun are involved in NF-kappa B-dependent IL-6 expression in human monocytes. J Immunol. 1999;162:4893–902. [PubMed] [Google Scholar]
- 8.Lu H, Wu JY, Kudo T, Ohno T, Graham DY, Yamaoka Y. Regulation of interleukin-6 promoter activation in gastric epithelial cells infected with Helicobacter pylori. Mol Biol Cell. 2005;16:4954–66. doi: 10.1091/mbc.E05-05-0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alberts B. Molecular biology of the cell. Garland Science; New York: 2002. [Google Scholar]
- 10.Yang G, Zaidi M, Zhang W, Zhu LL, Li J, Iqbal J, Varbanov A, Gross G, Phipps R, Troen BR, Sun L. Functional grouping of osteoclast genes revealed through microarray analysis. Biochem Biophys Res Commun. 2007 doi: 10.1016/j.bbrc.2007.11.106. [DOI] [PubMed] [Google Scholar]
- 11.Irish JM, Hovland R, Krutzik PO, Perez OD, Bruserud O, Gjertsen BT, Nolan GP. Single cell profiling of potentiated phospho-protein networks in cancer cells. Cell. 2004;118:217–28. doi: 10.1016/j.cell.2004.06.028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Time Series of TNF-Induced IL-6 mRNA Expression. IL-6 mRNA expression measured by quantitative PCR from macrophages treated with TNF (40 ng/mL). See Methods for details.
Supplemental Figure 2. Raw Data Analysis for Flow Cytometry Calculations. The excel sheet depicts the raw data and associated calculations for oscillations in p65 NF-κB phosphorylation triggered by TNF as measured by phospho-flow cytometry.