

Abstract
Many cellular events are involved in ischemic neuronal death, and it has been difficult to identify those that play a critical role in the cascade triggered by lack of oxygen and glucose, although it has been widely recognized that overactivation of glutamate receptors represents one of the initiating factors. Different glutamate receptor antagonists, especially those for N-methyl-D-aspartate (NMDA) receptors, have achieved significant success in animal models of hypoxia/ischemia; however, these antagonists have failed in clinical trials. We previously reported that calpain-mediated truncation of metabotropic glutamate receptor 1α (mGluR1α) played a critical role in excitotoxicity, and that a TAT-mGluR1 peptide consisting of a peptide surrounding the calpain cleavage site of mGluR1α and the peptide transduction domain of the transactivating regulatory protein (TAT) of HIV was neuroprotective against excitotoxicity. In the present study we tested the effect of this peptide in in vitro and in vivo models of neonatal hypoxia/ischemia. TAT-mGluR1 peptide prevented oxygen/glucose deprivation- (OGD) and hypoxia/ischemia- (H/I) induced neuronal death in cultured hippocampal slices and neonatal rats, respectively. TAT-mGluR1 blocked H/I-induced mGluR1α degradation but had no effect on H/I-induced spectrin degradation, suggesting that neuroprotection was due to prevention of calpain-mediated mGluR1α truncation and not to calpain inhibition. Our results therefore suggest that mGluR1α truncation plays a critical role in neonatal hypoxia/ischemia and that blockade of this event may prevent the activation of many downstream cytotoxic cascades. Compared to glutamate receptor antagonists and general calpain inhibitors, TAT-mGluR1 may have limited side effects.
Keywords: oxygen/glucose deprivation, hypoxia/ischemia, metabotropic glutamate receptors, TAT-mGluR1, excitotoxicity, calpain
Introduction
In central nervous system (CNS), glutamatergic synapses represent the majority of excitatory synapses and play important roles in normal brain functions. However, overactivation of glutamatergic synapses leads to excitotoxicity, which contributes to neurodegeneration in several neurological diseases such as Huntington’s disease, amyotrophic lateral sclerosis, and Alzheimer’s disease (Estrada Sanchez et al., 2008; Corona et al., 2007; Cosman et al., 2007). Excitotoxicity is also a major factor in hypoxic/ischemic neuronal death, including that in neonatal brain (Alvarez-Diaz et al., 2007; McLean and Ferriero, 2004); during the early stages of cerebral hypoxia/ischemia, decreased supply of glucose and oxygen results in large decrease in ATP synthesis, elimination of membrane potential and failure of glutamate transport leading to accumulation of extracellular glutamate and excessive activation of glutamate receptors, especially of NMDA receptors (Gardoni and Di Luca, 2006; Camacho and Massieu, 2006). NMDA receptor antagonists have been shown to significantly reduce excitotoxicity in cellular and animal models of hypoxia/ischemia; however, complete blockade of NMDA receptors may cause various side effects, such as memory impairment, nausea, and psychosis, which has seriously limited the therapeutic potential of NMDA antagonists in excitotoxicity-related neurodegenerative diseases such as stroke (Gardoni and Di Luca, 2006; Villmann and Becker, 2007).
We recently reported that stimulation of NMDA receptors activated the calcium-dependent protease calpain, which in turn truncated the C-terminal domain of the metabotropic glutamate receptor 1α (mGluR1α) (Xu et al., 2007a). Calpain-mediated truncation of mGluR1α eliminated the link between mGluR1 and the neuroprotective PI3K-Akt signaling pathways while maintaining its neurodegenerative function through the release of calcium from endoplasmic reticulum (Xu et al., 2007b; Pellegrini-Giampietro, 2003; Rong et al., 2003). The importance of this interaction between NMDA and mGluR1α receptors in excitotoxicity was demonstrated by the finding that systemic injection of a peptide comprising the sequence surrounding the calpain truncation site of mGluR1α and the transduction peptide of the TAT protein of the HIV virus (TAT-mGluR1 peptide) could prevent mGluR1α truncation and excitotoxic neuronal death in the kainic acid model in mice. mGluR1α-induced enhancement in cytoplasmic calcium levels has also been associated with excitotoxic cell death in hypoxia/ischemia (H/I) (Hilton et al., 2006), and mGluR1 antagonists are neuroprotective in rat and gerbil stroke models (Kohara et al., 2008; Rao et al., 2000). On the other hand, mGluR1-elicited activation of PI3K-Akt pathway has been proposed as the molecular basis for the neuroprotective effects of mGluR1 agonists (Makarewicz, 2006). Therefore, like in the case of NMDA receptors, complete blockade of mGluR1 receptors might also interfere with mGluR1-mediated normal cellular functions and produce undesirable side effects in clinical applications. In the present study, we tested the effect of TAT-mGluR1 peptide on oxygen/glucose deprivation (OGD)- and H/I-induced cell death in cultured hippocampal slices and in 7-day old rats, respectively. Our results indicate that TAT-mGluR1 peptide significantly reduced OGD- and H/I-induced cell death by blocking calpain-mediated mGluR1α truncation and suggest a new therapeutic approach for H/I.
Materials and Methods
Materials
Monoclonal anti-mGluR1a antibody was obtained from BD Pharmingen (San Diego, CA, USA). Anti-spectrin antibody was obtained from Chemicon (Temecula, CA, USA). The TAT-mGluR1 peptide (YGRKKRRQRRRVIKPLTKSYQGSGK) was synthesized by USC/Norris Comprehensive Cancer Center Core Facilities. All other chemicals were purchased from Sigma Chemical Co. (St. Louis, MO, USA), unless indicated otherwise.
Organotypic hippocampal slice cultures
Organotypic hippocampal slice cultures (OHSC) were prepared from postnatal 7–9 day old Sprague-Dawley rats according to Stoppini et al (1991). Hippocampi were rapidly dissected and transverse slices (400 μm thick) were prepared with a McIlwain tissue chopper and placed on porous Millipore membrane inserts in a 6-well plate with each well containing 1 ml culture medium (50% basal medium eagle (BME), 25% horse serum (HS), 25% Earle’s Balanced Salts (EBSS), 1 mM glutamine, 25 mM HEPES, 15 mM glucose, 2 mM NaHCO3, 100 U/ml penicillin and 100 μg/ml streptomycin, pH 7.2). Culture medium was changed three times a week and slices were cultured for 2 weeks at 35 °C saturated with 5% CO2 before being used for OGD.
OGD treatment of cultured hippocampal slices
Cultured hippocampal slices were washed twice with sterile phosphate-buffered saline, and then transferred into new six-well plates with each well containing 1 ml of glucose- and serum-free DMEM pre-saturated with 95% N2–5% CO2 for 40 minutes. Hippocampal slices were incubated at 37 °C for 30 or 45 min in an anaerobic chamber saturated with 95% N2–5% CO2. At the end of the OGD period, hippocampal slices were washed twice in regular serum-free medium and returned to serum-free culture medium for 24 h before lactate dehydrogenase (LDH) or propidium iodide (PI) uptake assay.
Cell viability assays
LDH assay
Neuronal damage was assessed by measurement of LDH released into the medium (Bruce et al., 1995; Koh and Choi, 1987). At the end of the 24 h recovery, 0.3 ml of medium solution was mixed with 0.7 ml potassium phosphate buffer (100 mM K2HPO4, adjusted to pH 7.5 with KH2PO4). After 20 min, 0.5 ml freshly made sodium pyruvate/NADH solution was added to this solution immediately followed by measuring absorbance at 340 nm at 1 min interval. LDH release was normalized to protein levels and expressed as fold of control.
PI uptake assay
Neuronal damage was also assessed with PI uptake as previously described (Laake et al., 1999). PI (4.6 μg/ml) was added to culture medium at the beginning of recovery. PI uptake was visualized using a 5X objective with a microscope fitted with fluorescence detection, and images of PI-labeled slices were captured with a CCD camera; at this magnification, one image was sufficient to analyze an entire hippocampal slice. Fluorescence intensity was estimated by the following method: first, images were adjusted to gray levels and captured with Adobe Photoshop, with the background of images in white and PI-stained structures in black; second, modified images were analyzed quantitatively by densitometry with ImageJ software. Both assays are widely used and it has been clearly demonstrated that OGD treatment of cultured hippocampal slices results in relatively selective death of CA1 pyramidal neurons (see Bonde et al., 2005, for an extensive analysis of this model of cell death).
Hypoxia/ischemia (H/I)
Postnatal 7-day-old Sprague-Dawley neonatal rats were used for H/I treatment. Neonatal rats were anesthetized with 3% isoflurane, subjected to ligation of the right common carotid artery (ischemia), and after one h recovery were placed in a glass chamber filled with a humidified atmosphere of 8% O2/92% N2 (hypoxia) for 2.5 h. The chamber was placed in a water bath maintained at 37°C. For mGluR1-TAT treatment, mGluR1-TAT (150 mg/kg in dH2O) was injected intraperitoneally one h before hypoxia exposure. The same amount of distilled water was injected in H/I control animals. After hypoxia, rats were returned to their dams and sacrificed 24 h later for Nissl staining or western blots. In a separate experiment, animals were sacrificed six days after H/I and body and wet brain weights were measured.
Nissl staining and analysis of neuronal damage
Rats were killed under deep anesthesia by transcardiac perfusion with phosphate-buffered saline containing 4% paraformaldehyde. Brains were dissected and transferred to graded sucrose solutions (15% for 24 h and 30% for another 24 h) and sectioned using a frozen-microtome. Sections (50 μm) were then mounted onto slides and passed through different solutions in the following order: 100% EtOH (ethyl alcohol) 2 minutes, xylene 2 minutes, 100% EtOH 2 minutes, 70% EtOH 2 minutes, distilled water 5 minutes, cresyl violet 3 minutes, distilled water 2 dips, 70% EtOH 5 minutes, 80% EtOH 2 minutes, 90% EtOH 2 minutes, 95% EtOH 2 minutes, 100% EtOH 5 minutes, xylene 5 minutes, and then mounted with Permount and air-dried. Infarct area (stained pale) was measured using a digital image analysis system (ImageJ, National Institutes of Health) and total infarct volumes were calculated and expressed as mm3. Analysis of infarct volume was done blindly by investigators who had no knowledge of the experimental treatments.
Western blots
Hippocampal slices (from OGD experiments) or whole hippocampus (from H/I experiments) were homogenized in a lysis buffer containing: 150 mM NaCl, 5 mM EDTA, 1% SDS, 10 mM Tris-HCl (pH 7.4), 0.5 mM phenylmethylsulphonyl fluoride (PMSF), 2 μg/mL leupeptin, and 1:200 protease inhibitor cocktail. After sample preparation, proteins were separated by 8% or 12% SDS-PAGE gels and transferred onto PVDF membranes. The PVDF membranes were blocked with 5% non-fat milk at room temperature for 1 h and probed with different primary antibodies (actin, 1:10,000 dilution; mGluR1α, 1:1,000; spectrin 1:10000) at 4 °C overnight. Membranes were then incubated with corresponding secondary antibodies for 1 h and developed with ECL solutions. Western blots were scanned and analyzed quantitatively by densitometry with ImageJ software. Data were generally calculated as fold of control and expressed as means ± SEM. Student’s t-test was used for statistical analyses and only p values < 0.05 were considered as statistically significant.
Results
TAT-mGluR1 protected against OGD-induced neurodegeneration in cultured hippocampal slices
We first tested the effects of TAT-mGluR1 on OGD-induced cell death; cultured hippocampal slices were subjected to 30 or 45 min of OGD treatment followed by 24 h recovery in regular serum-free medium (with glucose and oxygen) with TAT-mGluR1 (10 μM) applied 2 h before and during OGD treatment. At the end of the recovery period, hippocampal slices and medium were collected for PI uptake or LDH release assays respectively. Compared to control, 30 min of OGD followed by 24 h recovery induced a 1.60 ± 0.14 fold increase in PI uptake, especially in CA1 and CA3 regions, an effect that was completely blocked by TAT-mGluR1 treatment (0.96 ± 0.05 fold of control, Fig. 1A&B, n=14). TAT-mGluR1 peptide not only reduced OGD-induced increases in LDH release (2.62 ± 0.20 fold of controls) but further decreased it to below control levels (p <0.01 as compared to OGD, Fig. 1C, n =5). TAT-mGluR1 peptide alone also decreased LDH release in control slices (not shown) and the levels of LDH release were similar to those observed with OGD +TAT-mGluR1. Compared to 30 min OGD, 45 min OGD induced severer cell death as measured by PI uptake (2.50 ± 0.14 compared to control, Fig. 2A&B, n =14) and LDH release (4.63 ± 0.33 compared to control, Fig. 2C, n =6); both effects were blocked by TAT-mGluR1 (p <0.01 as compared to OGD, Fig. 2).
Figure 1. Effects of TAT-mGluR1 on 30 min OGD-induced PI uptake and LDH release in organotypic hippocampal slice cultures.

A. PI uptake by cultured hippocampal slices subjected to OGD for 30 min followed by 24 h recovery in the absence or presence of TAT-mGluR1 (10 μM, applied 2 h before and during 30 min OGD).
B. Quantification of PI uptake. Images as shown in A were analyzed as described under Materials and Methods. Results are means ± S.E.M. of 14 slices obtained in 5 separate experiments. Results are expressed as fold increase over control values (* p < 0.01 as compared to control, † p < 0.01 as compared to OGD group, student’s t-test).
C. LDH release by cultured hippocampal slices subjected to OGD for 30 min followed by 24 h recovery in the absence or presence of TAT-mGluR1. LDH release was measured at the end of 24 h recovery. Results are means ± S.E.M. of 5 experiments and are expressed as fold increase over control values (* p < 0.01 as compared to control, † p < 0.01 as compared to OGD group, student’s t-test).
Figure 2. Effects of TAT-mGluR1 on 45 min OGD-induced PI uptake and LDH release in organotypic hippocampal slice cultures.

A. PI uptake by cultured hippocampal slices subjected to OGD for 45 min followed by 24 h recovery in the absence or presence of TAT-mGluR1 (10 μM, applied 2 h before and during 45 min OGD).
B. Quantification of PI uptake. Images as shown in A were analyzed as described under Materials and Methods. Results are means ± S.E.M. of 14 slices obtained in 6 separate experiments. Results are expressed as fold increase over control values (* p < 0.01 as compared to control, † p < 0.01 as compared to OGD group, student’s t-test).
C. LDH release by cultured hippocampal slices subjected to OGD for 45 min followed by 24 h recovery in the absence or presence of TAT-mGluR1. LDH release was measured at the end of 24 h recovery. Results are means ± S.E.M. of 6 experiments and are expressed as fold increase over control values (* p < 0.01 as compared to control, † p < 0.01 as compared to OGD group, student’s t-test).
TAT-mGluR1 protected against hypoxia/ischemia (H/I)-induced neurodegeneration in neonatal rats
To test whether TAT-mGluR1 also protected ischemic cell death in an in vivo system, 7-day-old neonatal rats were subjected to ligation of the right common carotid artery, followed by intraperitoneal (i.p.) injection of TAT-mGluR1 (150 mg/kg body weight) or vehicle (H2O) 1 h before exposure to an atmosphere of 8% O2 and 92% N2 for 2.5 h; animals were sacrificed 24 h later (see methods for details). Analysis of Nissl-stained brain sections indicated that H/I-induced infarct volume in the right hemisphere (ipsilateral) was 48.5 ± 8.6 mm3 in vehicle-treated rats but only 25.5 ± 5.9 mm3 in TAT-mGluR1-treated group (Fig. 3, p <0.05, n =7). Interestingly, the protective effect of the TAT-mGluR1 peptide was larger when only infarct volume in hippocampus was analyzed (Fig. 3C). Higher magnification images of Nissl-stained sections revealed widespread neuronal damage with condensed nuclei (arrows in Fig. 4) and evident swelling in hippocampal pyramidal layer in vehicle-treated H/I rats but not in TAT-mGluR1 treated, confirming the neuroprotective effect of the peptide (Fig. 4). In another set of experiments, postnatal-day 7 rats were pretreated with vehicle or 100 mg/kg of TAT-mGluR1 before being subjected to right carotid artery ligation followed by 90 min hypoxia (8% O2 plus 92% N2). Animals were sacrificed 6 days later and brain and body weights were measured and brain weight/body weight ratio was determined as an index of brain damage. The brain weight/body weight ratio was decreased from 3.65 ± 0.02% (control) to 3.10 ± 0.05% (H/I) in vehicle-treated H/I animals, and this decrease was also significantly reversed by TAT-mGluR1 treatment (3.33 ± 0.06%, means ± S.E.M; n =5, Fig. 5; One-way analysis of variance (ANOVA) followed by a Tukey’s post hoc test was used for pair-wise comparisons between experimental treatments; * p < 0.001 (compared to Control); † p < 0.05 compared to H/I alone).
Figure 3. Effects of TAT-mGluR1 on H/I-induced neurodegeneration in neonatal rats.

A. Nissl-staining. Seven day-old rat pups were injected i.p. with TAT-mGluR1 (150 mg/kg body weight) or H2O 1 h before hypoxia/ischemia as described under Materials and Methods. All pups were sacrificed 24 h after hypoxia/ischemia. Brains were processed for Nissl staining and the figure depicts representative sections at different anatomical levels from two animals in the H/I and H/I plus TAT-mGluR1 group.
B. Effect of TAT-mGluR1 on H/I-induced infarct volume in whole brain. Brain sections throughout the brain as shown in A were analyzed to obtain an estimate of the infarct volume. Results are means ± S.E.M. of 7 experiments (* p < 0.05 as compared to H/I, student’s t-test).
C. Effect of TAT-mGluR1 on H/I-induced infarct volume in hippocampus. Hippocampus in serial brain sections throughout the brain as shown in A were analyzed to obtain an estimate of the infarct volume. Results are means ± S.E.M. of 7 experiments (* p < 0.05 as compared to H/I, student’s t-test).
Figure 4. Effect of TAT-mGluR1 on H/I-induced neuronal loss in CA1 region of hippocampus.
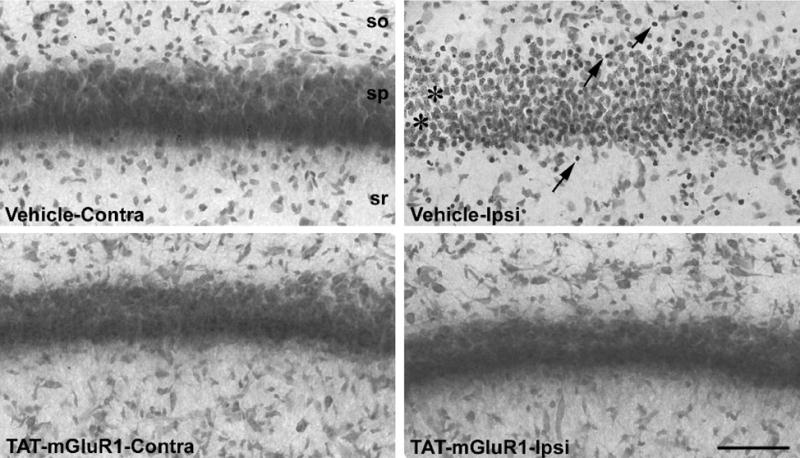
High magnification images of Nissl-stained CA1 pyramidal neurons from the contralateral (Contra) or ipsilateral (Ipsi) hippocampus of vehicle-treated or TAT-mGluR1-treated rats subjected to hypoxia/ischemia as described in Materials and Methods. Animals were sacrificed 24 h after H/I and brains processed for Nissl staining as described in Materials and Methods. Arrows in vehicle-Contra panel point to condensed nuclei while asterisks indicate gaps in the tissue possibly due to neuronal swelling. Note that the pyramidal layer is thicker and cells are less compact in vehicle-Contra panel than in other panels, which suggests possible edema. Note also that TAT-mGluR1 treatment markedly reduced neuronal damage. Scale bar=100 μm
Figure 5. Effects of TAT-mGluR1 on H/I-induced loss of brain weight in neonatal rats.
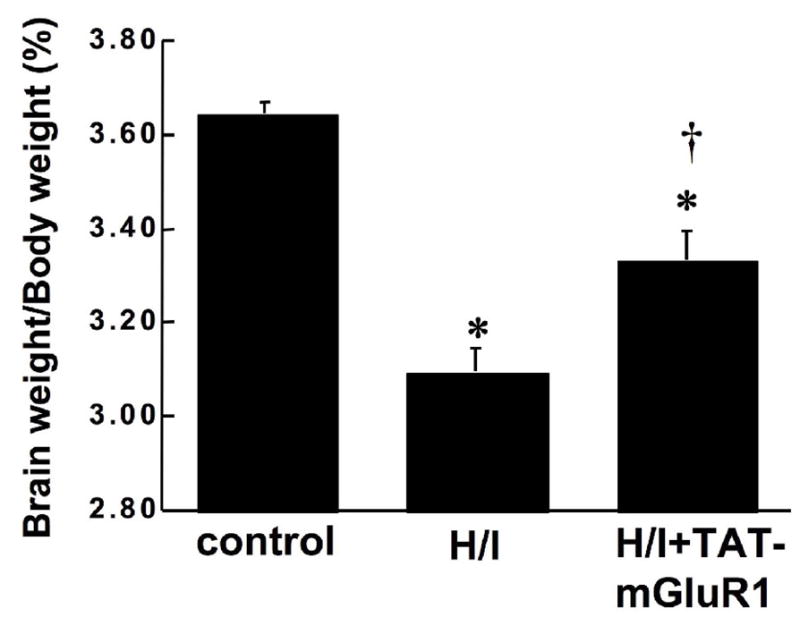
Seven day-old rat pups were injected i.p. with TAT-mGluR1 (H/I + TAT-mGluR1, 100 mg/kg body weight) or with H2O (H/I) 1 h before hypoxia as described under Materials and Methods. Rats in the control group were subjected to sham surgery w/o ligation of right common carotid artery. Six days after hypoxia/ischemia, rat pups were weighted and sacrificed; brains were dissected and weighted. Results are expressed as ratio of brain weight over body weight, and are means ± S.E.M. of 5 experiments (* p < 0.05 as compared to control, † p < 0.05 as compared to H/I, student’s t-test).
TAT-mGluR1 prevented hypoxia/ischemia (H/I)-induced mGluR1 truncation
Since TAT-mGluR1 peptide was previously shown to prevent calpain-mediated mGluR1α truncation (Xu et al. 2007a), we tested whether H/I-induced mGluR1α truncation could be prevented by TAT-mGluR1 peptide. Seven-day-old neonatal rats were subjected to H/I 1 h following i.p. injection of TAT-mGluR1 (150 mg/kg body weight) or vehicle (H2O), and sacrificed 24 h later for western blot analysis. Injection of the TAT-mGluR1 peptide did not have any effect on behavior before, during or after the H/I episode. Compared to control animals, mGluR1α levels in the right hemispheres of vehicle-treated H/I animals were reduced by 58 ± 7% (p <0.05, student’s t-test, n =6, means ± S.E.M.); in contrast, mGluR1α levels following TAT-mGluR1 injection and H/I were 95 ± 5% of control levels (Fig. 6, means ± S.E.M.). TAT-mGluR1 treatment also reversed OGD-induced mGluR1 truncation in cultured hippocampal slices (data not shown). On the other hand, TAT-mGluR1 injection did not alter H/I-induced calpain-mediated spectrin truncation (Fig. 7), suggesting that the protective effect of TAT-mGluR1 against ischemic cell death is not a consequence of a general inhibition of calpain activation. In addition, injection of the TAT-mGluR1 peptide did not have any effect on behavior before, during or after the H/I episode.
Figure 6. Effects of TAT-mGluR1 on H/I-induced mGluR1 truncation.
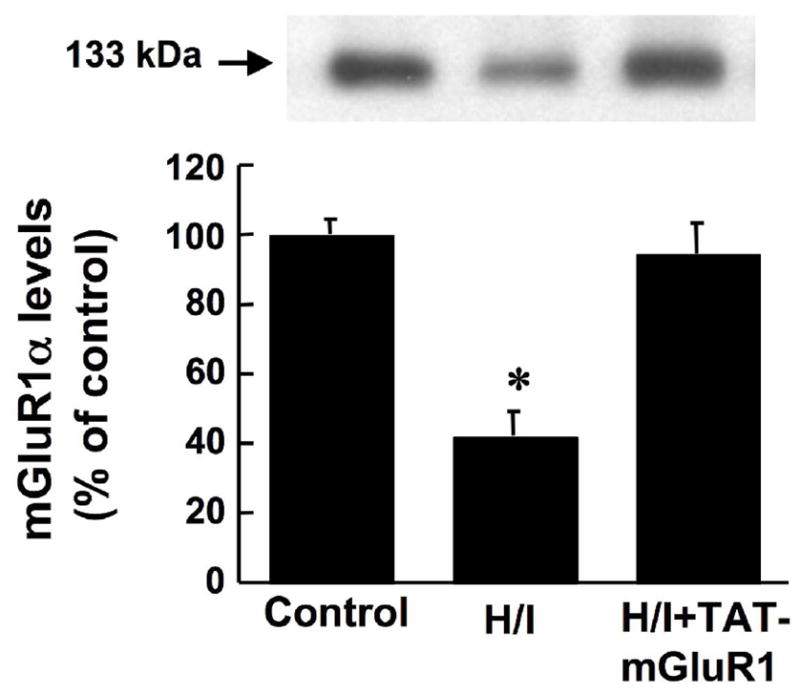
Seven day-old rat pups were injected i.p. with TAT-mGluR1 (H/I + TAT-mGluR1, 150 mg/kg body weight) or with H2O (H/I) 1 h before hypoxia, as described under Materials and Methods. Rats in the control group were subjected to sham surgery w/o ligation of right common carotid artery. Levels of mGluR1 were determined by western blots in the right hemisphere 24 h after hypoxia. Results are means ± S.E.M. of 6 animals. (* p < 0.05 as compared to control, student’s t-test).
Figure 7. Effects of TAT-mGluR1 on H/I-induced spectrin degradation.
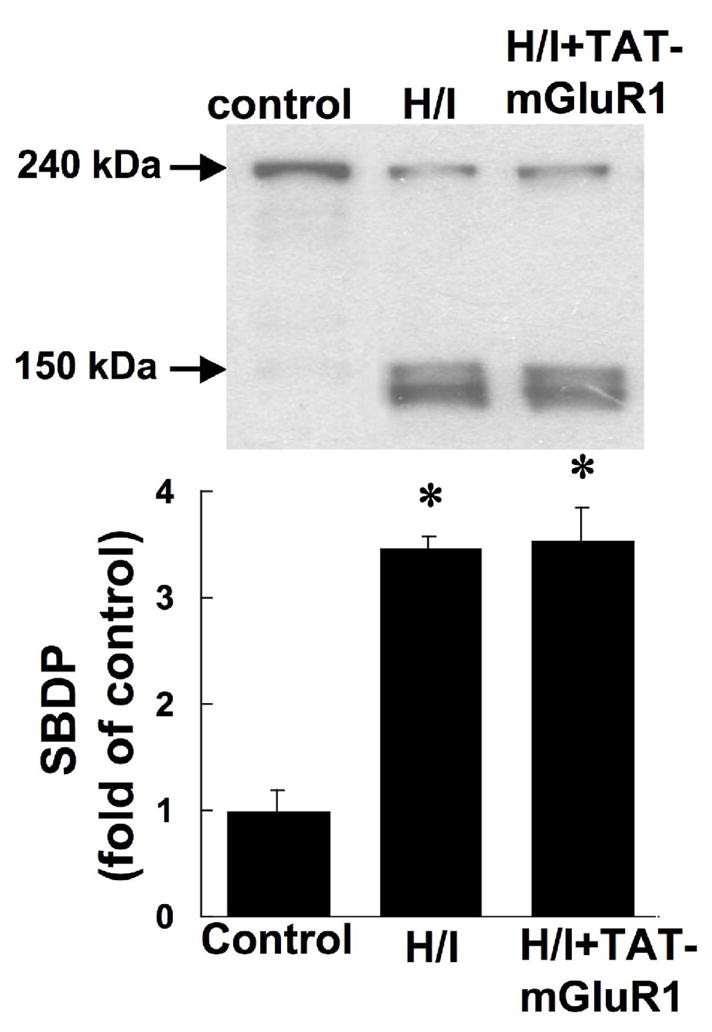
Seven day-old rat pups were injected i.p. with TAT-mGluR1 (H/I + TAT-mGluR1, 150 mg/kg body weight) or with ddH2O (H/I) 1 h before hypoxia, as described under Materials and Methods. Rats in the control group were subjected to sham surgery w/o ligation of right common carotid artery sham ligation surgery and hypoxia. Levels of spectrin breakdown products (SBDP, 145 and 150 kDa) in the right hemisphere 24 h after hypoxia were determined by western blots with antibody against spectrin. Results are means ± S.E.M. of 6 animals. (* p < 0.05 as compared to control, student’s t-test).
Discussion
Overactivation of glutamatergic receptors leads to excitotoxicity and is involved in several brain diseases including cerebral ischemia (Gardoni and Di Luca, 2006; Camacho and Massieu, 2006). Our previous study indicated that TAT-mGluR1 peptide was neuroprotective against NMDA-induced excitotoxicity in primary cultured neurons and kainic acid-induced neuronal death in vivo (Xu et al., 2007a). The present results indicate that TAT-mGluR1 peptide also provided significant neuroprotection against OGD- and H/I-induced neuronal death in cultured hippocampal slices and neonatal rats, respectively.
The massive release of glutamate coupled to decreased glutamate reuptake during hypoxia/ischemia is likely to activate both ionotropic glutamate receptors such as NMDA receptors and metabotropic glutamate receptors such as mGluR1α. The role of mGluR1α in excitotoxicity has been controversial, as mGluR1α agonists have been reported to protect neurons against oxidative stress (Chong et al., 2006; Sagara and Schubert, 1998) and to reduce OGD-induced neurotoxicity (Werner et al., 2007); on the other hand, mGluR1 antagonists also provided neuroprotection in animal models of transient focal cerebral ischemia (Kohara et al., 2008; Murotomi et al., 2008) or hypoxia/ischemia (Makarewicz et al., 2006). The complex effects of mGluR1α in ischemia result from the regulation of multiple cellular signaling pathways by mGluR1α, including phospholipase C (PLC)-β, protein kinase C (PKC), phosphoinositide 3-kinase (PI3K), and mitogen-activated protein kinase (MAPK) pathways (Rong et al., 2003; Wang et al., 2007; Skeberdis et al., 2001). Pharmacological studies indicated that activation of mGluR1α before ischemia resulted in neuroprotection, while mGluR1α activation after ischemia led to neurotoxicity (Pellegrini-Giampietro, 2003; Schroder et al., 1999). We proposed the hypothesis that calpain-mediated truncation of mGluR1α was a critical element to determine the role of mGluR1α in excitotoxicity and possibly hypoxia/ischemia; under normal conditions, intact mGluR1α activates both the neuroprotective PI3K-Akt pathway and the neurodegenerative pathway through PLC-β activation and increase in cytosolic calcium. However, calpain-mediated mGluR1α truncation eliminates the neuroprotective effects while leaving intact the calcium signaling/neurodegenerative component of mGluR1 activation (Xu et al., 2007a). We also provided evidence that the TAT-mGluR1 peptide, by specifically preventing calpain-mediated mGluR1α truncation produced neuroprotection. The present results further expend the neuroprotective effects of TAT-mGluR1 peptide to hypoxia/ischemia-induced neuronal damage.
TAT-mGluR1 completely blocked both 30 min and 45 min OGD-induced cell death in cultured hippocampal slices. Ischemic cell death involves multiple factors, including decreased ATP levels, excessive release of glutamate and prolonged activation of glutamate receptors, increased intracellular calcium and calpain activation, and excessive generation of reactive oxygen (ROS) (Koistinaho and Koistinaho, 2005; Zipp and Aktas, 2006; Won et al., 2002). We previously showed that mGluR1α was truncated within five minutes of 10 μM of NMDA treatment, which only induced mild cell toxicity in primary cultured neurons (Xu et al., 2007a), indicating that calpain-mediated mGluR1α truncation could occur rapidly following NMDA receptor and calpain activation during the early periods of ischemia. Therefore, inhibition of mGluR1α truncation provided by TAT-mGluR1 peptide might prevent the activation of multiple downstream processes that are implicated both in acute and delayed ischemic toxicity. Although we did not observe the accumulation of the 38 kD fragment of mGluR1a generated by calpain activation in the H/I model in neonatal rats, we did observe it in the same model in adult mice, suggesting that this fragment may be further processed in neonatal rat brain. While it is possible that other mechanisms could account for the decrease in mGluR1 levels, the most parsimonious explanation remains calpain-mediated truncation, as accumulation of SBDP clearly indicates that calpain is activated in this model. While the TAT-mGluR1 peptide prevents calpain-mediated truncation of mGluR1α, the exact mechanism underlying this effect is not completely understood. It is possible that the peptide provides a competitive interaction with calpain for mGluR1; however, it does not prevent calpain-mediated truncation of another calpain substrate, α-spectrin. The TAT peptide transduction domain has been widely used as a carrier for delivering peptides, proteins, or other cargos into neurons. It remains unclear how this short TAT sequence carries its cargo peptide or protein across cell membranes. In cell lines, the TAT-fusion protein enters cells through endocytosis and more specifically through lipid raft-dependent macropinocytosis (Wadia et al., 2004). It remains unknown though whether a similar mechanism applies to neurons and to short peptides. Previous studies with other TAT-coupled peptides have indicated that peripheral injection of these peptides lead to brain penetration, and it is therefore likely that the TAT-mGluR1 peptide we used did gain access to the brain (Bright et al., 2004; Bertaso et al., 2008).
Compared to its complete neuroprotection in the in vitro OGD model, TAT-mGluR1 peptide exhibited significant but incomplete protective effect in the in vivo hypoxia/ischemia model in neonate rats, as measured with Nissl staining. The effect was more complete when considering the hippocampus only, a result that might be related to a larger level of expression of the receptors in this brain structure as compared to the whole brain. Interestingly, TAT-mGluR1 peptide still completely prevented H/I-induced mGluR1α truncation, suggesting that other factors are also involved in H/I-induced neuronal death. In particular, it has recently been shown that oligodendrocytes are vulnerable to H/I due to the existence of NMDA receptors on their processes (Karadottir et al., 2005; Manning et al., 2008). Immature oligodendrocytes are also protected from H/I-mediated injury by agonists of mGluR1 and mGluR5 (Deng et al., 2004). In all these cases, activation of various cell death pathways has been evidenced and it is therefore likely that the calpain/mGluR1a pathway only represents one of the cell death pathways.
Calpain is a family of calcium-dependent neutral proteases that are ubiquitously distributed in the nervous system (Carafoli and Molinari, 1998). Although calpain has been implicated in synaptic modifications and plasticity under physiological conditions, it has also been shown to play critical roles in oxidative stress and neuronal death (Ray et al., 2000; Kelly and Ferreira, 2006; Lynch and Baudry, 1987). Calpain is also activated in cerebral ischemia (Yamashima et al., 2003), and calpain inhibitors have been shown to decrease both neuronal necrosis and apoptosis induced by hypoxia/ischemia in brains of newborn rats (Kawamura et al., 2005). However, calpain inhibitors are likely to exhibit severe side effects due to the broad functions of calpain. Unlike general calpain inhibitors, TAT-mGluR1 did not affect calpain activation as indicated by its lack of effects on H/I-induced accumulation of spectrin breakdown products, suggesting that the protective effect of TAT-mGluR1 was not through general calpain inhibition.
In conclusion, our results indicate that by blocking calpain-mediated mGluR1α truncation, TAT-mGluR1 protects hypoxic/ischemic cell death both in vitro and in vivo, suggesting that mGluR1α truncation might take place at early stages of hypoxia/ischemia and might play a key role in triggering further downstream cascades leading to neuronal death. By preventing the activation of these neurodegenerative cascades and preserving the neuroprotective effects of mGluR1α, TAT-mGluR1 might provide a new therapeutic approach for hypoxic/ischemic or other neurodegenerative diseases.
Acknowledgments
This research was supported by NINDS grants NS048521 to MB and NS048423 to XB. The authors wish to thank Anna Knize for her help in preparing cultured hippocampal slices.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alvarez-Diaz A, Hilario E, de Cerio FG, Valls-i-Soler A, Alvarez-Diaz FJ. Hypoxic-ischemic injury in the immature brain--key vascular and cellular players. Neonatology. 2007;92:227–235. doi: 10.1159/000103741. [DOI] [PubMed] [Google Scholar]
- Bertaso F, Zhang C, Scheschonka A, de Bock F, Fontanaud P, Marin P, Huganir RL, Betz H, Bockaert J, Fagni L, Lerner-Natoli M. PICK1 uncoupling from mGluR7a causes absence-like seizures. Nat Neurosci. 2008;11:940–948. doi: 10.1038/nn.2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonde C, Noraberg J, Noer H, Zimmer J. Ionotropic glutamate receptors and glutamate transporters are involved in necrotic neuronal cell death induced by oxygen-glucose deprivation of hippocampal slice cultures. Neuroscience. 2005;136:779–794. doi: 10.1016/j.neuroscience.2005.07.020. [DOI] [PubMed] [Google Scholar]
- Bright R, Raval AP, Dembner JM, Perez-Pinzon MA, Steinberg GK, Yenari MA, Mochly-Rosen D. Protein kinase C delta mediates cerebral reperfusion injury in vivo. J Neurosci. 2004;24:6880–6888. doi: 10.1523/JNEUROSCI.4474-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce AJ, Sakhi S, Schreiber SS, Baudry M. Development of kainic acid and N-methyl-D-aspartic acid toxicity in organotypic hippocampal cultures. Exp Neurol. 1995;132:209–219. doi: 10.1016/0014-4886(95)90026-8. [DOI] [PubMed] [Google Scholar]
- Camacho A, Massieu L. Role of glutamate transporters in the clearance and release of glutamate during ischemia and its relation to neuronal death. Arch Med Res. 2006;37:11–18. doi: 10.1016/j.arcmed.2005.05.014. [DOI] [PubMed] [Google Scholar]
- Carafoli E, Molinari M. Calpain: a protease in search of a function? Biochem Biophys Res Commun. 1998;247:193–203. doi: 10.1006/bbrc.1998.8378. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Li F, Maiese K. Group I metabotropic receptor neuroprotection requires Akt and its substrates that govern FOXO3a, Bim, and beta-catenin during oxidative stress. Curr Neurovasc Res. 2006;3:107–117. doi: 10.2174/156720206776875830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corona JC, Romo LB, Tapia R. Glutamate excitotoxicity and therapeutic targets for amyotrophic lateral sclerosis. Expert Opin Ther Targets. 2007;11:1415–1428. doi: 10.1517/14728222.11.11.1415. [DOI] [PubMed] [Google Scholar]
- Cosman KM, Boyle LL, Porsteinsson AP. Memantine in the treatment of mild-to-moderate Alzheimer’s disease. Expert Opin Pharmacother. 2007;8:203–214. doi: 10.1517/14656566.8.2.203. [DOI] [PubMed] [Google Scholar]
- Deng W, Wang H, Rosenberg PA, Volpe JJ, Jensen FE. Role of metabotropic glutamate receptors in oligodendrocyte excitotoxicity and oxidative stress. Proc Nat Acad Sci. 2004;101:7751–7756. doi: 10.1073/pnas.0307850101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrada Sanchez AM, Mejia-Toiber J, Massieu L. Excitotoxic neuronal death and the pathogenesis of Huntington’s disease. Arch Med Res. 2008;39:265–276. doi: 10.1016/j.arcmed.2007.11.011. [DOI] [PubMed] [Google Scholar]
- Gardoni F, Di Luca M. New targets for pharmacological intervention in the glutamatergic synapse. Eur J Pharmacol. 2006;545:2–10. doi: 10.1016/j.ejphar.2006.06.022. [DOI] [PubMed] [Google Scholar]
- Hilton GD, Nunez JL, Bambrick L, Thompson SM, McCarthy MM. Glutamate-mediated excitotoxicity in neonatal hippocampal neurons is mediated by mGluR-induced release of Ca++ from intracellular stores and is prevented by estradiol. Eur J Neurosci. 2006;24:3008–3016. doi: 10.1111/j.1460-9568.2006.05189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Káradóttir R, Cavelier P, Bergersen LH, Attwell D. NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature. 2005;2438:1162–1166. doi: 10.1038/nature04302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura M, Nakajima W, Ishida A, Ohmura A, Miura S, Takada G. Calpain inhibitor MDL 28170 protects hypoxic-ischemic brain injury in neonatal rats by inhibition of both apoptosis and necrosis. Brain Res. 2005;1037:59–69. doi: 10.1016/j.brainres.2004.12.050. [DOI] [PubMed] [Google Scholar]
- Kelly BL, Ferreira A. Beta-amyloid-induced dynamin 1 degradation is mediated by NMDA receptors in hippocampal neurons. J Biol Chem. 2006 doi: 10.1074/jbc.M605081200. [DOI] [PubMed] [Google Scholar]
- Koh JY, Choi DW. Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods. 1987;20:83–90. doi: 10.1016/0165-0270(87)90041-0. [DOI] [PubMed] [Google Scholar]
- Kohara A, Takahashi M, Yatsugi S, Tamura S, Shitaka Y, Hayashibe S, Kawabata S, Okada M. Neuroprotective effects of the selective type 1 metabotropic glutamate receptor antagonist YM-202074 in rat stroke models. Brain Res. 2008;1191:168–179. doi: 10.1016/j.brainres.2007.11.035. [DOI] [PubMed] [Google Scholar]
- Koistinaho M, Koistinaho J. Interactions between Alzheimer’s disease and cerebral ischemia--focus on inflammation. Brain Res Brain Res Rev. 2005;48:240–250. doi: 10.1016/j.brainresrev.2004.12.014. [DOI] [PubMed] [Google Scholar]
- Laake JH, Haug FM, Wieloch T, Ottersen OP. A simple in vitro model of ischemia based on hippocampal slice cultures and propidium iodide fluorescence. Brain Res Brain Res Protoc. 1999;4:173–184. doi: 10.1016/s1385-299x(99)00021-5. [DOI] [PubMed] [Google Scholar]
- Lynch G, Baudry M. Brain spectrin, calpain and long-term changes in synaptic efficacy. Brain Res Bull. 1987;18:809–815. doi: 10.1016/0361-9230(87)90220-6. [DOI] [PubMed] [Google Scholar]
- McLean C, Ferriero D. Mechanisms of hypoxic-ischemic injury in the term infant. Semin Perinatol. 2004;28:425–432. doi: 10.1053/j.semperi.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Makarewicz D, Duszczyk M, Gadamski R, Danysz W, Lazarewicz JW. Neuroprotective potential of group I metabotropic glutamate receptor antagonists in two ischemic models. Neurochem Int. 2006;48:485–490. doi: 10.1016/j.neuint.2005.12.022. [DOI] [PubMed] [Google Scholar]
- Manning SM, Talos DM, Zhou C, Selip DB, Park HK, Park CJ, Volpe JJ, Jensen FE. NMDA receptor blockade with memantine attenuates white matter injury in a rat model of periventricular leukomalacia. J Neurosci. 2008;28:6670–6678. doi: 10.1523/JNEUROSCI.1702-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murotomi K, Takagi N, Takayanagi G, Ono M, Takeo S, Tanonaka K. mGluR1 antagonist decreases tyrosine phosphorylation of NMDA receptor and attenuates infarct size after transient focal cerebral ischemia. J Neurochem. 2008;105:1625–1634. doi: 10.1111/j.1471-4159.2008.05260.x. [DOI] [PubMed] [Google Scholar]
- Pellegrini-Giampietro DE. The distinct role of mGlu1 receptors in post-ischemic neuronal death. Trends Pharmacol Sci. 2003;24:461–470. doi: 10.1016/S0165-6147(03)00231-1. [DOI] [PubMed] [Google Scholar]
- Rao AM, Hatcher JF, Dempsey RJ. Neuroprotection by group I metabotropic glutamate receptor antagonists in forebrain ischemia of gerbil. Neurosci Lett. 2000;293:1–4. doi: 10.1016/s0304-3940(00)01483-x. [DOI] [PubMed] [Google Scholar]
- Ray SK, Fidan M, Nowak MW, Wilford GG, Hogan EL, Banik NL. Oxidative stress and Ca2+ influx upregulate calpain and induce apoptosis in PC12 cells. Brain Res. 2000;852:326–334. doi: 10.1016/s0006-8993(99)02148-4. [DOI] [PubMed] [Google Scholar]
- Rong R, Ahn JY, Huang H, Nagata E, Kalman D, Kapp JA, Tu J, Worley PF, Snyder SH, Ye K. PI3 kinase enhancer-Homer complex couples mGluRI to PI3 kinase, preventing neuronal apoptosis. Nat Neurosci. 2003;6:1153–1161. doi: 10.1038/nn1134. [DOI] [PubMed] [Google Scholar]
- Sagara Y, Schubert D. The activation of metabotropic glutamate receptors protects nerve cells from oxidative stress. J Neurosci. 1998;18:6662–6671. doi: 10.1523/JNEUROSCI.18-17-06662.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder UH, Opitz T, Jager T, Sabelhaus CF, Breder J, Reymann KG. Protective effect of group I metabotropic glutamate receptor activation against hypoxic/hypoglycemic injury in rat hippocampal slices: timing and involvement of protein kinase C. Neuropharmacology. 1999;38:209–216. doi: 10.1016/s0028-3908(98)00180-4. [DOI] [PubMed] [Google Scholar]
- Skeberdis VA, Lan J, Opitz T, Zheng X, Bennett MV, Zukin RS. mGluR1-mediated potentiation of NMDA receptors involves a rise in intracellular calcium and activation of protein kinase C. Neuropharmacology. 2001;40:856–865. doi: 10.1016/s0028-3908(01)00005-3. [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Villmann C, Becker CM. On the hypes and falls in neuroprotection: targeting the NMDA receptor. Neuroscientist. 2007;13:594–615. doi: 10.1177/1073858406296259. [DOI] [PubMed] [Google Scholar]
- Wadia JS, Stan RV, Dowdy SF. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat Med. 2004;10:310–315. doi: 10.1038/nm996. [DOI] [PubMed] [Google Scholar]
- Wang JQ, Fibuch EE, Mao L. Regulation of mitogen-activated protein kinases by glutamate receptors. J Neurochem. 2007;100:1–11. doi: 10.1111/j.1471-4159.2006.04208.x. [DOI] [PubMed] [Google Scholar]
- Werner CG, Scartabelli T, Pancani T, Landucci E, Moroni F, Pellegrini-Giampietro DE. Differential role of mGlu1 and mGlu5 receptors in rat hippocampal slice models of ischemic tolerance. Eur J Neurosci. 2007;25:3597–3604. doi: 10.1111/j.1460-9568.2007.05614.x. [DOI] [PubMed] [Google Scholar]
- Won SJ, Kim DY, Gwag BJ. Cellular and molecular pathways of ischemic neuronal death. J Biochem Mol Biol. 2002;35:67–86. doi: 10.5483/bmbrep.2002.35.1.067. [DOI] [PubMed] [Google Scholar]
- Xu W, Wong TP, Chery N, Gaertner T, Wang YT, Baudry M. Calpain-mediated mGluR1alpha truncation: a key step in excitotoxicity. Neuron. 2007;53:399–412. doi: 10.1016/j.neuron.2006.12.020. [DOI] [PubMed] [Google Scholar]
- Xu W, Zhou M, Baudry M. Neuroprotection by Cell Permeable TAT-mGluR1 Peptide in Ischemia: Synergy Between Carrier and Cargo Sequences. Neuroscientist. 2008;14:409–414. doi: 10.1177/1073858407309762. [DOI] [PubMed] [Google Scholar]
- Yamashima T, Tonchev AB, Tsukada T, Saido TC, Imajoh-Ohmi S, Momoi T, Kominami E. Sustained calpain activation associated with lysosomal rupture executes necrosis of the postischemic CA1 neurons in primates. Hippocampus. 2003;13:791–800. doi: 10.1002/hipo.10127. [DOI] [PubMed] [Google Scholar]
- Zipp F, Aktas O. The brain as a target of inflammation: common pathways link inflammatory and neurodegenerative diseases. Trends Neurosci. 2006;29:518–527. doi: 10.1016/j.tins.2006.07.006. [DOI] [PubMed] [Google Scholar]