

Abstract
1,2-Dibromoethane and 1,3-butadiene are cancer suspects present in the environment and have been used widely in industry. The mutagenic properties of 1,2-dibromoethane and the 1,3-butadiene oxidation product diepoxybutane are thought to be related to the bis-electrophilic character of these chemicals. The discovery that overexpression of O6-alkylguanine alkyltransferase (AGT) enhances bis-electrophile-induced mutagenesis prompted a search for other proteins that may act by a similar mechanism. A human liver screen for nuclear proteins that cross-link with DNA in the presence of 1,2-dibromoethane identified histones H2b and H3 as candidate proteins. Treatment of isolated histones H2b and H3 with diepoxybutane resulted in DNA-protein cross-links and produced protein adducts, and DNA-histone H2b cross-links were identified (immunochemically) in Escherichia coli cells expressing histone H2b. However, heterologous expression of histone H2b in E. coli failed to enhance bis-electrophile-induced mutagenesis. These results are similar to those found with the cross-link candidate glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Loecken, E. M. and Guengerich, F. P. (2008) Chem. Res. Toxicol. 21, 453−458) but, in contrast to GAPDH, histone H2b bound DNA with even higher affinity than AGT. The extent of DNA cross-linking of isolated histone H2b was similar to that of AGT, suggesting that differences in post-cross-linking events explain the difference in mutagenesis.
Introduction
Modification of DNA is a major pathway by which carcinogens can exert their harmful effects (1). The resulting DNA adducts can prevent accurate replication of the genome, leading to the accumulation of mutations, which can give rise to cancer (2). Understanding the mechanisms by which DNA adducts contribute to mutagenesis is critical for developing potential chemoprevention strategies and discovering biomarkers.
1,2-Dibromoethane and 1,3-butadiene are both mutagens and potential human hazards due to environmental contamination and occupational exposure (3) (www.atsdr.cdc.gov/toxprofiles/tp37.html), (4) (www.atsdr.cdc.gov/toxprofiles/tp28.html). 1,2-Dibromoethane was widely used as an anti-knock additive in leaded gasoline and as a pesticide before its mutagenic and carcinogenic properties were discovered (5-7). 1,3-Butadiene is an important industrial chemical widely used in the production of rubber and plastic and is also present in automobile exhaust and cigarette smoke (8). Diepoxybutane is a minor but highly mutagenic product resulting from epoxidation of 1,3-butadiene by P450 enzymes (9). The oxidation products resulting from 1,3-butadiene are thought to be responsible for the mutagenic and carcinogenic properties observed in experimental animals treated with 1,3-butadiene (10-12).
In addition to mono-alkylating biological molecules, 1,2-dibromoethane and diepoxybutane also cross-link biological molecules through reactions at the two electrophilic centers. This phenomenon was initially observed in systems overexpressing glutathione S-transferase (13) or O6-alkylguanine-DNA alkyltransferase (AGT) (14-16), where an increase in mutagenesis was observed with treatment by bis-electrophiles. Isolated DNA-protein cross-links provided evidence for the tethering of reactive cysteine residues to nucleophilic sites on DNA by bis-electrophiles (17, 18). These lesions are known to be mutagenic, which may be a consequence of their bulk and DNA-distorting behavior that can block replicative polymerases, disrupting normal DNA processing (19, 20). DNA-protein cross-links have also been shown to be relatively stable in comparison with other adducts, but the comprehensive characterization of these DNA lesions has not been achieved in part due to difficulties isolating these complexes (21-23).
In order to determine if the formation of DNA-protein cross-links is a general mechanism of mutagenesis by bis-electrophiles, we employed a mass spectrometry-based search of DNA-binding proteins. From the candidate proteins identified, human histones H2b and H3 were examined in detail because of their DNA-binding ability and basic nature (24). Although reactions were observed between isolated histones and diepoxybutane, heterologous expression of histone H2b in treated Escherichia coli cells did not lead to mutagenic enhancement in contrast to AGT, although in vivo DNA-histone H2b cross-links were observed. These findings are similar to those observed with glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (25) and provide some insight into the molecular properties required for proteins to enhance bis-electrophile-induced mutagenesis.
Experimental Procedures
Materials
1,2-Dibromoethane, (1,2,3,4)-diepoxybutane (racemic), and CH2Br2 were purchased from Aldrich Chemical Co. (Milwaukee, WI). The oligonucleotide 5'-GGAGGAGGAGGAGGAG-3' was synthesized by Midland Certified Reagent Co. (Midland, TX) and purified by denaturing gel electrophoresis. Purified human histone H2b was purchased from New England Biolabs (Ipswich, MA). Bovine DNase I and single-stranded calf thymus DNA-cellulose was purchased from Sigma-Aldrich (St. Louis, MO).
Isolation of Nuclear Proteins from Human Liver
Liver samples were from six individuals (Tennessee Donor Service, Nashville, TN). All donors were males between the age of 8 and 56, with various causes of death (motor vehicle accident [4], respiratory arrest [1], and gun shot wound to the head [1]). The frozen samples (10 g each sample) were thawed on ice, pooled, and homogenized in four volumes of buffer (0.10 M Tris-acetate (pH 7.4) containing 0.10 M KCl, 1.0 mM EDTA, and 25 μM butylated hydroxytoluene) using a Potter-Elvehjem homogenizer, filtered through cheesecloth, and centrifuged at 103 × g for 10 min. The nuclei pellet was resuspended in 300 mL of 2.2 M sucrose containing 5 mM MgCl2 and centrifuged at 7.5 × 104 × g for 1 h. The nuclei pellet was resuspended in 300 mL of 0.32 M sucrose containing 5 mM MgCl2 and centrifuged again at 103 × g for 10 min. The resulting pellet was resuspended in 60 mL of 20 mM Tris-HCl buffer (pH 7.4) containing 10 mM MgCl2, 1 mM EDTA, 2 mM CaCl2, and 1 mM β-mercaptoethanol and sonicated for 90 s. DNase I was added to a final concentration of 100 μg/mL and the mixture was incubated at 15 °C for 45 min. EDTA was added to a final concentration of 20 mM before freezing the samples overnight at −20 °C. The nuclear homogenate was thawed and dialyzed overnight in 20 mM Tris-HCl buffer (pH 7.5) containing 50 mM NaCl, 1 mM β-mercaptoethanol, and 2 mM EDTA at 4 °C. DNA-binding nuclear proteins were enriched using a column (2 × 20 cm) containing 2 g DNA-cellulose, which was prewashed with 200 mL of Buffer A (20 mM Tris-HCl (pH 7.4) containing 50 mM NaCl, 1 mM EDTA, 1 mM β-mercaptoethanol, and 10% glycerol (v/v)). The dialyzed extract was centrifuged for 20 min at 103 × g to remove cellular debris, and glycerol was added to 10% (v/v). Fifteen-mL aliquots were added to the column, which was then eluted sequentially with 20 mL solutions of Buffer A containing 0.1 M, 0.5 M, 1.0 M, and 2.2 M NaCl. Fractions from the 2.2 M NaCl wash were collected (∼ 23 mL) and dialyzed overnight in 50 volumes of 50 mM Tris-HCl buffer (pH 7.4) containing 0.1 mM EDTA at 4 °C. The samples were concentrated using Amicon 5,000 MWCO spin filters (Millipore, Billerica, MA) at 4 × 103 × g for 45 min at 4 °C. Protein concentrations were determined using a bicinchromonic acid (BCA) assay according to the manufacturers’ protocol (Thermo Fisher, Rockford, IL).
Screening for Dibromoethane-Induced Cross-links
Reactions containing 100 μL of concentrated nuclear protein solution (77 μg/mL), 5 mg DNA-cellulose, and 20 mM 1,2-dibromoethane or dimethyl sulfoxide (DMSO) were incubated at 37 °C for 1 h, with gentle vortex mixing every 5 to 10 min. Samples were pelleted by centrifugation and the beads were washed four times with 50 mM Tris-HCl buffer (pH 7.4) containing 1 mM EDTA, followed by two washes with 0.10 M NH4HCO3, three washes with 2.2 M NH4HCO3, and finally three washes with 0.10 M NH4HCO3 (26, 27). Reduction, alkylation, and digestion of the mixtures with trypsin were performed as previously described (28), with the exception of frequent gentle mixing. After pelleting the beads by centrifugation, the peptide solution was concentrated and desalted with P10 C18 ZipTips (Millipore) according to the manufacturer's instructions (www.millipore.com/userguides.nsf/docs/pr02358).
Analysis of Human Liver Screens by LC-MS/MS
LC-MS/MS analysis of the peptides was performed using a Thermo LTQ ion trap mass spectrometer equipped with a Thermo MicroAS autosampler and Thermo Surveyor HPLC pump, Nanospray source, and Xcalibur 2.0 SR2 instrument control. Peptides were resolved on a fused silica capillary column, 100 μm × 11 cm, packed with C18 resin (Jupiter C18, 5 μm, 300 Å, Phenomonex, Torrance, CA) using an inline solid phase extraction column (100 μm × 4 cm) packed with the same C18 resin (using a frit generated with liquid silicate Kasil 1 (29)) and utilizing a “vented column” setup similar to that previously described (30) except that the flow from the HPLC pump was split prior to the injection valve. The flow rate during the loading and desalting phase of the gradient was 1 μL/min and was 700 nL/min during the separation phase. Mobile phase A was 0.1% HCO2H in H2O and mobile phase B was 0.1% HCO2H in CH3CN (v/v). A 95 min LC separation was performed with a 15 min washing period diverted to waste after the pre-column (100% A for the first 10 min, followed by a gradient to 98% A at 15 min) to allow for the removal of any residual salts. After the initial washing period, a 60 min gradient was applied in which the first 35 min was a slow, linear gradient from 98% A to 75% A (v/v), followed by a faster gradient to 10% A (v/v) (at 65 min) and an isocratic phase (at 10% A, v/v) to 75 min. MS/MS scans were acquired using an isolation width of 2 m/z, an activation time of 30 ms, and 30% normalized collision energy using 1 microscan. The MS/MS spectra of the peptides were acquired using data-dependent scanning in which one full MS spectra, using a full mass range of 400−2000 a.m.u., was followed by three MS/MS spectra. Dynamic exclusion was enabled for 60 s with an exclusion list size of 50 and a repeat count of one.
Database Searching, Filtering, and False-Discovery Rate Determination
The “ScanDenser” algorithm was used to extract tandem mass spectra from Thermo RAW files and transcode them to DTA files. Spectra containing fewer than 25 peaks or had less than 2e1 measured total ion current were not extracted. Singly-charged DTA files were created if 90% of the total ion current occurred below the precursor ion, and all other spectra were processed as both doubly and triply charged DTA files. Proteins were identified using the TurboSEQUEST v.27 (rev. 12) algorithm (Thermo Electron, San Jose, CA) searching against the unihuman2−1205_rev database (96858) sequences with an appended reversed version of the database for a total of 193,716 sequences. Searches were performed allowing the following differential modifications: +57 on cysteine (for carboxyimidomethylation from iodoacetamide), +16 on methionine (oxidation). Peptide and fragment ion tolerances were set to 2.5 and 1.0 Da, respectively. Protein matches were preliminarily filtered using the following criteria, based on the parent charge state of the peptide: minimum cross correlation scores (Xcorr) of 1.0, 1.8, and 2.5 were required for charge states 1, 2, and 3, respectively. Additionally, a minimum preliminary score (Sp) of 350 was required, as well as placement within the top 5 (Sp) scores for that search. After application of filtering criteria, false-discovery rates were estimated from peptide matches to the reverse database, in which the total number of reverse peptides was multiplied by two and divided by the total number of peptide hits. These filtering criteria achieved a false positive rate of ≤1% in all datasets.
Gel Shift Assays
Gel mobility shift assays were performed as previously described in order to detect cross-links, with the exception of using larger 20 × 20 cm 15% sodium dodecyl sulfate (w/v) polyacrylamide gels (17). Reactions (10 μL) containing 1 μg recombinant purified histone H2b or H3 and 5’-end 32P-labeled oligonucleotide 5’-GGAGGAGGAGGAGGAG-3’ were incubated overnight at 37 °C with bis-electrophiles prior to analysis. In vitro cross-linking assays using double-stranded DNA utilized the same 16-mer oligonucleotide annealed to a complimentary 16-mer oligonucleotide.
Sample Preparation for Mass Spectral Analysis of Histone H2b and H3 Adducts
Samples (10 μL) consisting of 1 μg purified recombinant histone H2b protein or 0.8 μg histone H3 in 50 mM Tris-HCl buffer (pH 7.4) containing 0.1 mM EDTA were treated in order to identify protein adducts resulting from reactions with bis-electrophiles. Samples were incubated with 10 nM, 100 μM, or 20 mM of each bis-electrophile and incubated for 2 h at 37 °C. The reduction, alkylation, and tryptic digestion of the samples were carried out as previously described (28). Likewise, the sites directly involved in DNA cross-linking were analyzed by adding 16-mer oligonucleotides (5’-GGAGGAGGAGGAGGAG-3’) to the reactions. After incubation these samples were dried in vacuo and reconstituted in a solution containing 100 mM NaCl and 15 mM sodium citrate (pH 7.0). The labile adducts were then subject to neutral thermal hydrolysis by heating the samples to 100 °C for 30 min and peptides were digested as above.
Analysis of Histone Peptides by LC-MS/MS
LC-MS/MS analysis of the peptides was performed using an LTQ-Orbitrap mass spectrometer equipped with an Eksigent NanoLC-AS1 Autosampler 2.08 and the Eksigent NanoLC-1D plus HPLC pump nanospray source. The peptides were resolved on a packed fused silica capillary column, 100 μm × 15 cm, packed with C18 resin (Jupiter C18, 5 μm, 300 Å, Phenomonex, Torrance, CA) and coupled with an inline trapping column that was 100 μm × 4 cm packed with the same C18 resin (using a frit generated with liquid silicate Kasil 1 (30)). Liquid chromatography was carried out at ambient temperature with a flow rate of 0.5 μL/min using a gradient mixture of 0.1% (v/v) HCO2H in H2O (solvent A) and 0.1% (v/v) HCO2H in CH3CN (solvent B). The flow rate during the loading and desalting phase of the gradient was 1.5 μL/min and during separation phase was 500 nL/min. A 95 min gradient was performed with a 10 min washing period diverted to waste after the precolumn (98% A for the first 10 min followed by a gradient to 98% A at 15 min, all v/v) to allow for removal of any residual salts. After the initial washing period, a 60 min gradient was performed where the first 35 min was a slow, linear gradient from 98% A to 75% A (v/v), followed by a faster gradient to 10% A at 65 min and an isocratic phase at 10% A to 75 min. Centroided MS/MS scans were acquired on the LTQ-Orbitrap using an isolation width of 2 m/z, an activation time of 30 ms, an activation q of 0.25, and 30% normalized collision energy using 1 microscan with a maximum injection time of 0.1 s for each MS/MS scan and 1 microscan with a maximum injection time of 1 s for each full orbitrap MS scan. The tune parameters were as follows: spray voltage of 1.9 kV, a capillary temperature of 160 °C, a capillary voltage of 48 V, and tube lens of 125 V. The AGC target values were set at 106 for full MS and 104 for MS/MS spectra. A full scan obtained for eluting peptides in the range of 400−2000 a.m.u. was collected on the Orbitrap at a resolution of 6 × 104, followed by five data-dependent MS/MS scans on the LTQ portion of the instrument with a minimum threshold of 103 set to trigger MS/MS spectra. A dynamic exclusion list of the 50 previously analyzed precursors was maintained for 60 s in which time MS/MS was not performed on those masses.
Database Search Pipeline for Protein Adducts
Tandem mass spectra were converted from Thermo RAW format to the mzML format by the msconvert tool of ProteoWizard (31). Peptides were identified against a database containing the two histone sequences along with 19 other proteins identified in the mixture (mostly keratins and E. coli proteins). The full database was then doubled in size by adding the reversed version of each sequence. The MyriMatch database search algorithm version 1.5.2 (32) identified tandem mass spectra to peptide sequences. Semi-tryptic peptide candidates were included as possible matches. Potential modifications included formation of N-terminal pyroglutamine, and the following additions to peptide N-termini, lysine, arginine, or cysteine: 44.026215, 86.036779, 104.041151, 178.07, 238.09, or 220.08 a.m.u. (Figure 2). Precursors were required to be within 0.1 m/z of the peptide monoisotopic mass, or of the monoisotopic mass plus or minus one neutron. Fragment ions were uniformly required to fall within 0.5 m/z of the monoisotope. IDPicker v2.2.4 (33) filtered peptide matches to a 0 % FDR (including only those matches that outscored the best-scoring reversed match) and applied parsimony to the protein lists, requiring all proteins to match at least two distinct peptides. The summation of two independent searches of the data differing only in the presence of + 238 a.m.u. of the search criterial were used to report the findings.
Figure 2.

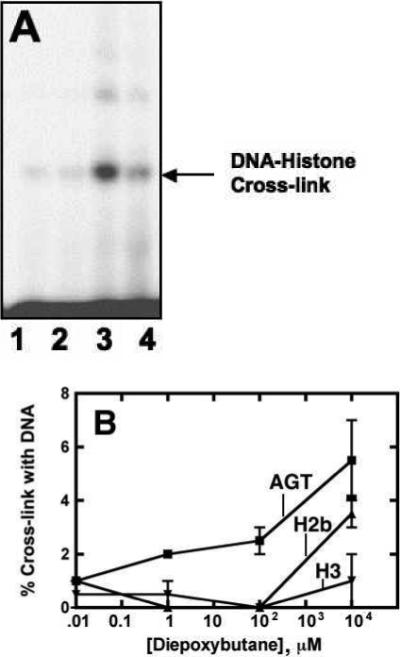
Cross-linking of purified histone H2b to oligonucleotides by diepoxybutane. (A) Gel shift assays were performed by incubating histone H2b (1 μg) and 32P-5’-endlabeled 16-mer oligonucleotide in reactions containing DMSO (<1%, v/v) (lane 1), 20 mM CH2Br2 (lane 2), 20 mM diepoxybutane (lane 3), or 20 mM 1,2-dibromoethane (lane 4) for 1 h at 37 °C. Samples were separated by SDS-polyacrylamide gel (15% w/v) electrophoresis. (B) DNA-protein cross-links were detected via autoradiography and quantified using Quantity One software (BioRad).
Human Histone H2b cDNA Synthesis and Expression
The amino acid sequences and native cDNA sequences for human histones H2b and H3 were obtained from GenBank (NM_003528 and NM_003532, respectively). Sixteen overlapping oligonucleotides (30- to 40-mers) were synthesized (Operon, Huntsville, AL) according to DNAWorks results (http://helixweb.nih.gov/dnaworks/) (34), with codons optimized automatically for E. coli type B. Melting temperatures of the designed oligonucleotides were selected to be 62 ± 3 °C. The 5′- and 3′-flanking sequences were 5′-CCGAATT-3′ (sense) and 5′-GACCCCTGGATCCCGC-3′ (sense) respectively. Flanking sequences were engineered to contain an EcoRI restriction site on the 5’-end and a BamHI restriction site on the 3’-end of the cDNA. Polymerase cycling assembly and polymerase chain reaction (PCR) amplification of the gene was performed as described elsewhere (34). The PCR product was digested with BamHI and EcoRI restriction enzymes (New England Biolabs) and purified by agarose gel electrophoresis. The resulting fragment was ligated into a pINIII-A3(lppp-5) vector, which was also digested with EcoRI and BamHI restriction enzymes. One Shot Top10 E. coli cells (Invitrogen, Carlsbad, CA) were transformed with the ligation mixture and selected for with Luria-Bertani medium plates containing 50 μg/mL ampicillin. Purification of vectors from surviving colonies was performed with a QIAprep Miniprep kit (Qiagen, Valencia, CA), and digested with EcoRI and BamHI restriction enzymes in order to screen for ligated vectors. Digested plasmids (from selected colonies that yielded 0.5 and 7 kb DNA fragments upon restriction digestion) were analyzed in the Vanderbilt DNA Sequencing facility using an Applied Biosystems Model 3700 fluorescence sequencing unit with a Taq dye terminator kit (PE Applied Biosystems, Foster City, CA). The sequenced plasmid was amplified in One Shot Top10 cells (Invitrogen) and isolated before transformation of TRG8 E. coli cells. Luria-Bertani plates containing both ampicillin (50 μg/mL) and kanamycin (50 μg/mL) were used to select colonies for both cell type and pINIII vector. Histone H2b expression was verified in E. coli TRG8 cells induced with 0.2 mM isopropyl β-d-thiogalactopyranoside by immunoblot analysis using polyclonal human anti-histone H2b antibodies (New England Biolabs) according to the manufacturer's instructions. Secondary antibodies with infrared fluorescence tags were used for detection on a LI-COR Odyssey Infrared Imaging System (LI-COR, Lincoln, NE).
Mutagenesis and Survival Assays
Overnight cultures of TRG8 cells containing AGT, histone H2b, or empty pINIII vector were used to inoculate 5 mL of Luria-Bertani media containing ampicillin (50 μg/mL) and kanamycin (50 μg/mL). Cultures were grown at 37 °C while shaking at 250 rpm until an OD600 of ∼ 0.4 was reached. Expression was induced with 0.2 mM isopropyl β-D-thiogalactopyranoside for 90 min prior to treatment. Mutagenesis and survival assays were carried out as previously described (16), but with 60 min reaction time and his+ plate dilutions of 1:106.
DNA Binding Assays
Electrophoretic mobility shift assays were carried out as previously described (25, 35), but using a 15% (w/v) native polyacrylamide gel. Briefly, various concentrations of recombinant AGT or histone H2b were added to 32P-labeled 16-mer oligonucleotides (5’-GGAGGAGGAGGAGGAG-3’) in 10 mM Tris-acetate buffer (pH 7.4) containing 100 mM NaCl. Mixtures were incubated at 23 °C for 30 min and glycerol was added to 10% (v/v) glycerol before loading on a 20 cm gel, which was run at 8 V/cm.
In Vivo Cross-link Detection
Identification of in vivo cross-links was performed as previously described with some modifications (36). Cultures of E. coli TRG8 cells containing pINIII-histone H2b or the empty pINIII vector were grown in Luria-Bertani medium containing ampicillin (50 μg/mL) and kanamycin (50 μg/mL) to an OD600 of ∼ 0.5 and induced with 0.2 mM isopropyl β-D-thiogalactopyranoside for 1 h at 37 °C while shaking at 250 rpm. Cultures were then treated for 90 min with 0.032 or 0.2 mM diepoxybutane dissolved in DMSO (< 1% v/v) or a DMSO vehicle control. One-mL aliquots were centrifuged and washed with 1 mL of 1X M9 salt solution before isolating genomic DNA with Promega Wizard Genomic DNA purification kit (Madison, WI) according to manufacturer's protocol for Gram-negative bacteria. The isolated DNA was washed with 2 M NaCl for 30 min at 37 °C and precipitated with 2.5 volumes of isopropanol. Pellets containing the DNA were suspended in 0.5 mL of 50 mM Tris-HCl (pH 7.4) buffer containing 10 mM MgCl2 and 20 μg/mL DNase I and digested for 30 min at 37 °C. The solutions were then dried in vacuo prior to reconstitution with 25 μL H2O and 5 μL 5X SDS-PAGE loading buffer. Samples (20 μL) were heated at 95 °C for 10 min and loaded onto a 15% SDS-PAGE gel and immunoblotting as described above for expression analysis. Quantification was performed using secondary antibodies with infrared fluorescence tags that were detected with a LI-COR Odyssey Infrared Imaging System.
Results and Discussion
Screening for Crosslink Candidates
The ability of DNA-protein cross-links to contribute to bis-electrophile-mediated mutagenesis prompted our examination of other proteins that might form mutagenic cross-links (17, 36). In order to identify other proteins cross-linked to DNA by 1,2-dibromoethane, human liver nuclei extract was enriched for DNA binding proteins by collecting the high-salt eluting fractions from a DNA-cellulose column. The protein mixture was then incubated with DNA-cellulose and 20 mM 1,2-dibromoethane. The proteins bound to the beads were then digested with trypsin and the resulting peptides were analyzed by multidimensional LC-MS/MS, using a Sequest software search. In two independent screens, variants of human histones were identified from multiple peptides and from these we selected human histone H2b as a candidate (Table S1, Supporting Information). The analyzed peptides should represent covalently bound proteins, but it is impossible to rule out false positives because the cross-linked peptides remain bound to the beads. This type of analysis is also biased toward high abundance proteins and those that can efficiently ionize in MS (37). AGT was only identified in one of the screens, presumably due to its low abundance (38).
Cross-linking of Histones H2b and H3 to DNA by bis-Electrophiles
In vitro cross-linking of AGT to oligonucleotides was observed in the presence of bis-electrophiles and shown be involved in mutagenesis (17, 36, 39). Accordingly, histones H2b and H3 were incubated with varying concentrations of bis-electrophiles and radiolabeled oligonucleotide. Gel mobility shift assays revealed that both histones H2b and H3 cross-link to DNA with diepoxybutane in a concentration-dependent manner. In comparison with AGT (39), histone H2b and H3 cross-links appeared to require moderately higher concentrations of bis-electrophile (Figure 1). Comparisons between DNA-protein cross-link formation involving double-stranded or single-stranded DNA oligonucleotides revealed that histone H2b is only slightly more reactive toward single-stranded DNA (Figure S3, Supporting Information). The reactivity bias towards diepoxybutane was unexpected because 1,2-dibromoethane was used in the initial screen. One possible explanation is carryover of non-covalently bound histones on the DNA-cellulose beads. However, the only cross-link identified between DNA and peptide was the result of a reaction with 1,2-dibromoethane (vide infra), possibly as a result of different reaction conditions that lead to biased reactivity towards bis-electrophiles. These results are similar to previous findings with GAPDH (25).
Figure 1.
Proposed products of reactions between lysine, diepoxybutane, and guanine. A targeted MS search for protein adducts was performed on the tryptic peptides from samples of purified histone H2b incubated with diepoxybutane. Reactions containing histone H2b, diepoxybutane, and 16-mer oligonucleotides from the in vitro cross-linking assays were subjected to neutral thermal hydrolysis prior to digestion. In addition to lysine residues, changes in a.m.u. corresponding to the diepoxybutane adducts and guanine cross-links were monitored on arginines, cysteines, and N-termini.
MS Analysis of bis-Electrophile-Treated Histones H2b and H3
Recombinant protein was incubated with bis-electrophiles and subjected to tryptic digestion before analysis by high resolution LC-MS/MS. Data collected were analyzed with IDPicker software in targeted searches for hydrolyzed histone H2b and H3 protein adducts formed by 1,2-dibromoethane (+44 a.m.u.) or diepoxybutane (+86 and +104 a.m.u.). DNA-protein cross-links induced by bis-electrophiles were also analyzed in samples containing oligonucleotides that were digested with trypsin after neutral thermal hydrolysis (Figure 2). This procedure yielded labile N7-guanyl cross-links between histone amino acids and DNA corresponding to +220 and +238 a.m.u. (diepoxybutane) as well as +178 a.m.u. (1,2-dibromoethane) adducts (36). Although precedence exists for these particular adducts being formed (40), it is possible that other adducts are produced that the targeted search would not identify, such as those reacting at sites other than the guanine N7 and adenine N3 atoms (36). Analysis of digested purified protein yielded ∼ 90% protein coverage of histone H2b and ∼ 80% histone H3 protein coverage. Of the 14 residues not observed in the MS data from histone H2b, six were lysines and two were arginines. There were two lysines, six arginines, and one cysteine in the 29 residues of histone H3 not identified in the samples.
Treatment of histone H2b with diepoxybutane resulted in nine adducted lysine residues, with the relative amount of +86 a.m.u. adducts approximately equaling that of the +104 a.m.u. protein adducts. Nearly all ions containing these modifications were found in samples treated with the highest concentration of diepoxybutane (20 mM). The most commonly modified residue on histone H2b was Lys6 with a total of 24 ions observed in the 20 mM diepoxybutane-treated samples, which represented a total of ∼ 7 % of all Lys6 ions identified. Treatment of histone H2b with diepoxybutane yielded numerous ions corresponding to N-terminus modifications, as well as adduction on Arg73. Diepoxybutane-induced histone H2b-guanine cross-links where not observed on lysine, arginine, or the N-terminus proline residues.
Treatment of histone H3 with diepoxybutane yielded protein adducts on five lysine residues, with approximately twice as many epoxide (+86 a.m.u.) as triol (+104 a.m.u.) adducts observed. Two-thirds of all diepoxybutane adducts corresponded to samples treated with 20 mM bis-electrophile. Lys28 was found to be the major target for modification by diepoxybutane with ∼16% of all observed ions being adducted. Modification of 3 arginine residues (Arg27, Arg43, Arg127) was observed, as well as 11 N-terminal adducts in samples treated with diepoxybutane. While arginine residues were not anticipated to be major targets for alkylation, there is precedence for post-translational modification of these sites in histone (41), and evidence exists that arginine undergoes carcinogen-induced alkylation in treated proteins (42). Although no modifications corresponding to diepoxybutane protein adducts (+86, +104 a.m.u.) were observed on Cys111, two ions indicate that this residue cross-links to DNA (+220 a.m.u.) in 20 mM diepoxybutane.
Treatment of H2b with 1,2-dibromoethane resulted in identification of only one +44 a.m.u.-modified ion at Lys109, which was also the only lysine found to be cross-linked to DNA by 1,2-dibromoethane (+178 a.m.u.). The only histone H3 residue found to be adducted by 1,2-dibromoethane (+44 a.m.u.) was Cys111, which yielded one ion. Two lysines (Lys57, Lys80) were found to cross-link to DNA with 1,2-dibromoethane although protein adducts were not observed with 1,2-dibromoethane treatment alone (+44 a.m.u.). The Lys57 and Lys80 residues, however, did form protein adducts with diepoxybutane.
Expression of Human Histone H2b in E. coli and Mutagenesis Assays with bis-Electrophiles
Overexpression of the DNA repair protein AGT has been shown to paradoxically enhance mutagenesis by bis-electrophiles (17, 43). The identification of human histones in both liver screens (Table S1, Supporting Information) suggested that DNA-protein cross-links may form in part because of their basic charge and DNA binding ability. Following demonstration that histones H2b and H3 were reactive with diepoxybutane in vitro, the ability of H2b to enhance in vivo mutagenesis via heterologous expression in E. coli cells was assessed (Figure 4A).
Figure 4.
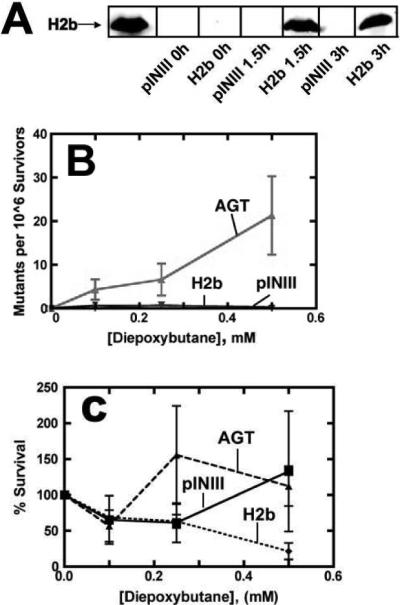
Histone H2b expression in E. coli does not enhance mutagenesis by bis-electrophiles. (A) Recombinant expression of histone H2b in TRG8 cells was quantified at various time points after induction by immunoblotting. Expression of histone H2b (100 nM) was compared to the extract of TRG8 cells containing pINIII empty vector, which contains no detectable histone protein. (B) TRG8 cells expressing AGT or histone H2b or containing the control pINIII vector were treated with varying concentrations of diepoxybutane for 30 min at 37 °C. Mutant colonies that grew on his− plates were quantified visually, and mutagenesis was assessed after determining the quantity of viable cells grown on his+ plates. (C) AGT, H2b, and pINIII TRG8 cells were treated with diepoxybutane on his+ plates. Colonies were quantified and compared with untreated plates to determine survivorship.
Heterologous expression of histone H2b in E. coli TRG8 cells was quantified by immunoblotting and showed ∼ 5-fold higher protein expression for histone H2b (100 nM) compared to AGT (17 nM) (data not shown). Histones are not found in prokaryotes, so the observation that no histone H2b protein was detected in blots of TRG8 extract containing empty pINIII vector was expected (44). Attempts to express histone H3 cDNA were unsuccessful for reasons that are unclear.
Cells induced to express histone H2b were assessed for survival and mutagenesis upon treatment with varying concentrations of bis-electrophiles. TRG8 cells containing an empty pINIII vector or expressing human AGT served as the negative and positive controls, respectively. Treated cells were plated on agar lacking histidine (his−) to select for cells with reversion mutations in the hisG gene. As expected (17), AGT expression resulted in more mutant colonies forming than in untreated controls. Neither the pINIII-containing cells nor those expressing histone H2b displayed enhanced mutagenesis (Figure 4B). Survival of diepoxybutane-treated cells was not affected in either pINIII- or histone-H2b containing cells, while cells expressing AGT displayed increased sensitivity to bis-electrophiles (Figure 4C). Similar results were observed following treatment with 1,2-dibromoethane (Figure S2, Supporting Information).
DNA Binding Ability of Histone H2b and AGT
Previous work with the cross-link candidate GAPDH showed that reactivity toward diepoxybutane in vitro is not necessarily an indication of the ability of a protein to enhance mutagenesis in vivo (25). We proposed that the difference in DNA-binding ability between AGT and GAPDH could contribute to the lack of mutagenic enhancement by diepoxybutane. In contrast, histone H2b is a critical component of chromatin and is known to bind DNA with high affinity (24). We demonstrated that, unlike GAPDH, histone H2b binds DNA with much higher affinity than AGT (Figure 5). While histones are primarily basic proteins containing a high proportion of lysine residues, they lack a highly nucleophilic active site such as the reactive cysteine residue necessary to enhance mutagenesis by AGT (18).
Figure 5.
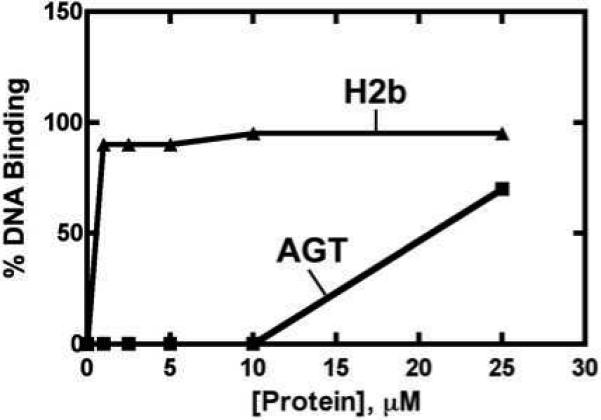
DNA binding assays. Samples containing purified histone H2b or AGT were incubated with 32P-5’-end-labeled 16-mer at 23 °C for 30 min. A 15% (w/v) native polyacrylamide gel electrophoresis system was used to separate protein-DNA complexes from unmodified oligonucleotides. A voltage of 8 V/cm was used to separate DNA-protein cross-links, which were then quantified with autoradiography.
Detection of In Vivo DNA-Histone H2B Cross-links
The ability of AGT to enhance mutagenesis of bis-electrophiles by cross-linking to DNA has been demonstrated in vivo (36). GAPDH was also found to react with diepoxybutane in a manner similar to that of AGT; however, in vivo mutagenic enhancement was not observed (25). This finding was partly attributed to the reduced DNA-binding ability of GAPDH in comparison with AGT, which could potentially prevent the formation of DNA-GAPDH cross-links in vivo. Like GAPDH, purified human histone H2b cross-linked to DNA and yielded protein adducts with diepoxybutane treatment (Figures 1, 3), but expression of histone H2b in treated E. coli did not enhance mutagenesis by bis-electrophiles (Figure 4). However, treatment of TRG8 cells expressing histone H2b with diepoxybutane yielded detectable DNA-protein cross-links upon analysis of genomic DNA (Figure 6). These findings suggest the cellular processing of these lesions differs from that of AGT-DNA cross-links in E. coli cells. Identifying the exact mechanism responsible for this disparity is complicated by the fact that information is limited about the processing, repair, and bypass of specific DNA-protein cross-links (20).
Figure 3.
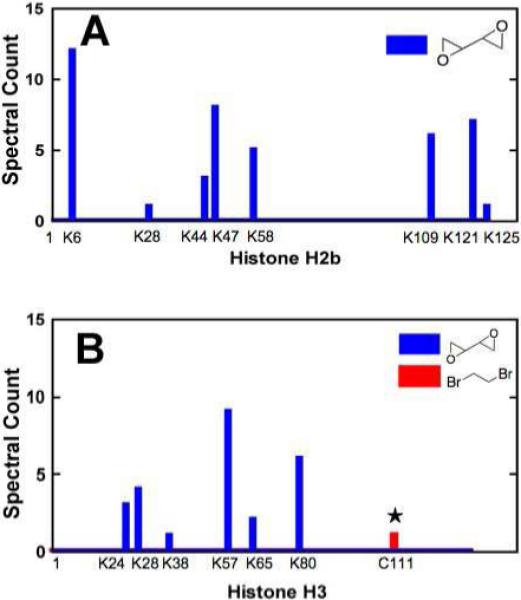
MS analysis of reactions between bis-electrophiles and histones H2b and H3. (A) Purified histone H2b (1 μg) or (B) histone H3 (1 μg) was incubated with 10 nM to 10 mM 1,2-dibromoethane or diepoxybutane for 1 h at 37 °C and digested with trypsin. Peptides were analyzed with a high-resolution mass spectrometer (Thermo Orbitrap) and the data were mined for adducts using Myrimatch and IDPicker software. The single peptide-DNA cross-link to be identified (☆) formed between DNA and a histone H3 cysteine with 1,2-dibromoethane treatment.
Figure 6.

Detection of in vivo DNA-histone H2b cross-links. E. coli TRG8 cells expressing histone H2b or containing an empty pINIII vector were treated with 0, 0.032, or 0.2 mM diepoxybutane for 90 min at 37 °C. The genomic DNA from 1 mL cultures was isolated, washed, and digested with DNase I prior to separation on a 15% (w/v) SDS-polyacrylamide gel and immunoblot analysis. Samples were quantified using a LI-COR Odyssey Infrared Imaging System.
Conclusion
Characterization of DNA-protein cross-links formed by bis-electrophiles is required to understand the mechanism of mutagenesis and is especially critical because of the potential for human exposure to these chemicals. The ability of other proteins to behave in a similar manner toward bis-electrophiles was addressed in our human liver screen. We established that purified histones H2b and H3 were able to cross-link with DNA using in vitro gel shift assays, using AGT as a positive control (Figure 1). In accord with these findings, DNA adducts resulting from protein alkylation and protein-DNA cross-links were identified (Figure 2, 3). However, treatment of histone H2b-overexpressing E. coli cells with bis-electrophiles did not elevate mutation levels (Figure 4). The higher DNA binding ability of histone H2b in comparison with AGT (Figure 5) suggests that the absence of enhanced mutagenesis was not the result of reduced protein-DNA interactions. However, it is reasonable to at least partially attribute differences in mutagenic enhancement to the innate differences in reactivity of histone H2b and AGT with bis-electrophiles (Figure 2B), including the ability of AGT to flip out damaged bases from the DNA duplex (45). The identification of in vivo histone H2b-DNA cross-links supports the hypothesis that lack of mutagenesis in E. coli cells is not due to the absence of DNA-protein cross-links (Figure 6). This may be the result of differential processing of DNA-protein cross-links. The mechanisms by which cells deal with these large lesions are poorly understood but are thought to involve proteolysis, nucleotide excision repair, and homologous recombination (20). The processing of cross-linked proteins may begin with the cleavage of cross-linked proteins to peptides, which could go on to elicit DNA polymerase bypass, possibly resulting in mutations. The biochemical differences between histone and AGT cross-links may lead to differential processing and repair. Indeed, DNA-peptide cross-links that vary in their amino acid sequences have distinctly different mutagenic potentials (46, 47). To date AGT remains unique in its ability to enhance bis-electrophile-induced mutagenesis, most likely due to the reactivity of its active site residue and distinct DNA-binding capabilities (9) and possibly to post-cross-linking phenomena.
Supplementary Material
Acknowledgments
This work was supported in part by NIH grants R01 ES010546, P30 ES000267, and T32 ES007028. We thank A. J. Ham for analyzing mass spectra screens, D. C. Liebler for his input in this project, and R. L. Eoff for his help with mass spectrometry. We are also grateful to W. H. McDonald and D. Overstreet for their invaluable assistance with our mass spectrometry analysis. A. E. Pegg and N. A. Loktionova generously provided AGT protein.
Abbreviations
- AGT
O6-alkylguanine-DNA alkyltransferase
- DMSO
dimethyl sulfoxide
- DTA
Desktop Auditor
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- PCR
polymerase chain reaction
References
- 1.Hemminki K. DNA adducts and mutations in occupational and environmental biomonitoring. Environ. Health Perspect. 1997;105:823–827. doi: 10.1289/ehp.97105s4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Poirier MC, Beland FA. DNA adduct measurements and tumor incidence during chronic carcinogen exposure in animal models: implications for DNA adduct-based human cancer risk assessment. Chem. Res. Toxicol. 1992;5:749–755. doi: 10.1021/tx00030a003. [DOI] [PubMed] [Google Scholar]
- 3.National Toxicology Program . Report on Carcinogens. Eleventh Edition 2002. pp. 81–82. U.S. Department of Health and Human Services, Public Health Service, National Toxicology Program. 1,2-Dibromoethane (Ethylene Dibromide).
- 4.National Toxicology Program . Report on Carcinogens. Eleventh Edition 2002. pp. 36–39. U.S. Department of Health and Human Services, Public Health Service, National Toxicology Program. 1,3-Butadiene.
- 5.Olson WA, Habermann RT, Weisburger EK, Ward JM, Weisburger JH. Induction of stomach cancer in rats and mice by halogenated aliphatic fumigants. J. Natl. Cancer Inst. 1973;51:1993–1995. doi: 10.1093/jnci/51.6.1993. [DOI] [PubMed] [Google Scholar]
- 6.Letz GA, Pond SM, Osterloh JD, Wade RL, Becker CE. Two fatalities after acute occupational exposure to ethylene dibromide. J. Am. Med. Assoc. 1984;252:2428–2431. [PubMed] [Google Scholar]
- 7.Hill DL, Shih TW, Johnston TP, Struck RF. Macromolecular binding and metabolism of the carcinogen 1,2-dibromoethane. Cancer Res. 1978;38:2438–2442. [PubMed] [Google Scholar]
- 8.Himmelstein MW, Acquavella JF, Recio L, Medinsky MA, Bond JA. Toxicology and epidemiology of 1,3-butadiene. Crit. Rev. Toxicol. 1997;27:1–108. doi: 10.3109/10408449709037482. [DOI] [PubMed] [Google Scholar]
- 9.Guengerich FP. Principles of covalent binding of reactive metabolites and examples of activation of bis-electrophiles by conjugation. Arch. Biochem. Biophys. 2005;433:369–378. doi: 10.1016/j.abb.2004.07.035. [DOI] [PubMed] [Google Scholar]
- 10.Melnick RL, Sills RC. carcinogenicity of 1,3-butadiene, isoprene, and chloroprene in rats and mice. Chem.-Biol. Interact. 1990;135−136:27–42. doi: 10.1016/s0009-2797(01)00213-7. [DOI] [PubMed] [Google Scholar]
- 11.Henderson RF, Thornton-Manning JR, Bechtold WE, Dahl AR. Metabolism of 1,3-butadiene: species differences. Toxicology. 1996;113:17–22. doi: 10.1016/0300-483x(96)03422-1. [DOI] [PubMed] [Google Scholar]
- 12.Rice JM, Boffetta P. 1,3-Butadiene, isoprene and chloroprene: reviews by the IARC monographs programme, outstanding issues, and research priorities in epidemiology. Chem.-Biol. Interact. 2001;135−136:11–26. doi: 10.1016/s0009-2797(01)00175-2. [DOI] [PubMed] [Google Scholar]
- 13.Rannug U, Sundvall A, Ramel C. The mutagenic effect of 1,2-dichloroethane on Salmonella typhimurium. I. Activation through conjugation with glutathione in vitro. Chem.-Biol. Interact. 1978;20:1–16. doi: 10.1016/0009-2797(78)90076-5. [DOI] [PubMed] [Google Scholar]
- 14.Abril N, Luqueromero FL, Prieto-Alamo MJ, Margison GP, Pueyo C. ogt alkyltransferase enhances dibromoalkane mutagenicity in excision repair-deficient Escherichia coli K-12. Mol. Carcinogen. 1995;12:110–117. doi: 10.1002/mc.2940120208. [DOI] [PubMed] [Google Scholar]
- 15.Abril N, Luque-Romero FL, Prieto-Alamo M-J, Rafferty JA, Margison GP, Pueyo C. Bacterial and mammalian DNA alkyltransferases sensitize Escherichia coli to the lethal and mutagenic effects of dibromoalkanes. Carcinogenesis. 1997;18:1883–1888. doi: 10.1093/carcin/18.10.1883. [DOI] [PubMed] [Google Scholar]
- 16.Liu H, Xu-Welliver M, Pegg AE. The role of human O6-alkylguanine-DNA alkyltransferase in promoting 1,2-dibromoethane-induced genotoxicity in Escherichia coli. Mutat. Res. 2000;452:1–10. doi: 10.1016/s0027-5107(00)00062-2. [DOI] [PubMed] [Google Scholar]
- 17.Liu L, Pegg AE, Williams KM, Guengerich FP. Paradoxical enhancement of the toxicity of 1,2-dibromoethane by O6-alkylguanine-DNA alkyltransferase. J. Biol. Chem. 2002;277:37920–37928. doi: 10.1074/jbc.M205548200. [DOI] [PubMed] [Google Scholar]
- 18.Guengerich FP, Fang Q, Liu L, Hachey DL, Pegg AE. O6-Alkylguanine-DNA alkyltransferase: low pKa and high reactivity of cysteine 145. Biochemistry. 2003;42:10965–10970. doi: 10.1021/bi034937z. [DOI] [PubMed] [Google Scholar]
- 19.Costa M, Zhitkovich A, Toniolo P. DNA-protein cross-links in welders: molecular implications. Cancer Res. 1993;53:460–463. [PubMed] [Google Scholar]
- 20.Barker S, Weinfeld M, Murray D. DNA-protein crosslinks: their induction, repair, and biological consequences. Mutat. Res. 2005;589:111–135. doi: 10.1016/j.mrrev.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 21.Bjorklund CC, Davis WB. Stable DNA-protein cross-links are products of DNA charge transport in a nucleosome core particle. Biochemistry. 2007;46:10745–10755. doi: 10.1021/bi700475b. [DOI] [PubMed] [Google Scholar]
- 22.Voitkun V, Zhitkovich A. Analysis of DNA-protein crosslinking activity of malondialdehyde in vitro. Mutat. Res. 1999;424:97–106. doi: 10.1016/s0027-5107(99)00011-1. [DOI] [PubMed] [Google Scholar]
- 23.Minko IG, Kurtz AJ, Croteau DL, Van Houten B, Harris TM, Lloyd RS. Initiation of repair of DNA-polypeptide cross-links by the UvrABC nuclease. Biochemistry. 2005;44:3000–3009. doi: 10.1021/bi0478805. [DOI] [PubMed] [Google Scholar]
- 24.Peterson CL, Laniel MA. Histones and histone modifications. Curr. Biol. 2004;14:R546–551. doi: 10.1016/j.cub.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 25.Loecken EM, Guengerich FP. Reactions of glyceraldehyde 3-phosphate dehydrogenase sulfhydryl groups with bis-electrophiles produce DNA-protein cross-links but not mutations. Chem. Res. Toxicol. 2008;21:453–458. doi: 10.1021/tx7003618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alberts B, Herrick G. DNA-cellulose chromatography. Methods Enzymol. 1971;21D:198–217. [Google Scholar]
- 27.Sjåstad K, Haarr L, Kleppe K. Characterization of the DNA-cellulose-binding proteins from Escherichia coli K 12. Biochim. Biophys. Acta. 1983;739:8–16. doi: 10.1016/0167-4781(83)90038-6. [DOI] [PubMed] [Google Scholar]
- 28.Dennehy MK, Richards KA, Wernke GR, Shyr Y, Liebler DC. Cytosolic and nuclear protein targets of thiol-reactive electrophiles. Chem. Res. Toxicol. 2006;19:20–29. doi: 10.1021/tx050312l. [DOI] [PubMed] [Google Scholar]
- 29.Cortes HJ, Pfeiffer C, Richter B, Stevens T. Porous ceramic bed supports for fused-silica packed capillary columns used in liquid-chromatography. High Resol. Chromatogr. Chromatogr. Commun. 1987;10:446–448. [Google Scholar]
- 30.Licklider LJ, Thoreen CC, Peng J, Gygi SP. Automation of nanoscale microcapillary liquid chromatography-tandem mass spectrometry with a vented column. Anal. Chem. 2002;74:3076–3083. doi: 10.1021/ac025529o. [DOI] [PubMed] [Google Scholar]
- 31.Kessner D, Chambers M, Burke R, Agus D, P. M. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics. 2008;24:2534–2536. doi: 10.1093/bioinformatics/btn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tabb DL, Fernando CG, Chambers MC. MyriMatch: highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. J. Proteome Res. 2007;6:654–661. doi: 10.1021/pr0604054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang B, Chambers MC, Tabb DL. Proteomic parsimony through bipartite graph analysis improves accuracy and transparency. J. Proteome Res. 2007;6:3549–3557. doi: 10.1021/pr070230d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu Z-L, Bartleson CJ, Ham A-JL, Guengerich FP. Heterologous expression, purification, and properties of human cytochrome P450 27C1. Arch. Biochem. Biophys. 2006;445:138–146. doi: 10.1016/j.abb.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 35.Rasimas JJ, Kar SR, Pegg AE, Fried MG. Interactions of human O6-alkylguanine-DNA alkyltransferase (AGT) with short single-stranded DNAs. J. Biol. Chem. 2007;282:3357–3366. doi: 10.1074/jbc.M608876200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu L, Hachey DL, Valadez JG, Williams KM, Guengerich FP, Loktionova NA, Kanugula S, Pegg AE. Characterization of a mutagenic DNA adduct formed from 1,2-dibromoethane by O6-alkylguanine-DNA alkyltransferase. J. Biol. Chem. 2004;279:4250–4259. doi: 10.1074/jbc.M311105200. [DOI] [PubMed] [Google Scholar]
- 37.Smith BC, Denu JM. Chemical mechanisms of histone lysine and arginine modifications. Biochim. Biophys. Acta. 2009;1789:45–57. doi: 10.1016/j.bbagrm.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O'Connor TR, Laval J. Physical association of the 2,6-diamino-4-hydroxy-5N-formamidopyrimidine-DNA glycosylase of Escherichia coli and an activity nicking DNA at apurinic/apyrimidinic sites. Proc. Natl. Acad. Sci. U. S. A. 1989;86:5222–5226. doi: 10.1073/pnas.86.14.5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu S, Guttman A. Electrophoresis microchips for DNA analysis. Trends Anal. Chem. 2004;23:422–431. [Google Scholar]
- 40.Kaina B, Christmann M, Naumann S, Roos WP. MGMT: key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair. 2007;6:1079–1099. doi: 10.1016/j.dnarep.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 41.Valadez JG, Liu L, Loktionova NA, Pegg AE, Guengerich FP. Human O6-alkylguanine-DNA alkyltransferase activation of a series of bis-electrophiles to produce mutagens. Chem. Res. Toxicol. 2004;17:972–982. doi: 10.1021/tx049897u. [DOI] [PubMed] [Google Scholar]
- 42.Goggin M, Anderson C, Park S, Swenberg J, Walker V, Tretyakova N. Quantitative high-performance liquid chromatography-electrospray ionization-tandem mass spectrometry analysis of the adenine-guanine cross-links of 1,2,3,4-diepoxybutane in tissues of butadiene-exposed B6C3F1 mice. Chem. Res. Toxicol. 2008;21:1163–1170. doi: 10.1021/tx800051y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lambert C, Li J, Jonscher K, Yang T-C, Reigan P, Quintana M, Harvey J, Freed BM. Acrolein inhibits cytokine gene expression by alkylating cysteine and arginine residues in the NF-kB1 DNA binding domain. J. Biol. Chem. 2007;282:19666–19675. doi: 10.1074/jbc.M611527200. [DOI] [PubMed] [Google Scholar]
- 44.Peterson LA, Harris TM, Guengerich FP. Evidence for an episulfonium ion intermediate in the formation of S-[2-(N7-guanyl)ethyl]glutathione in DNA. J. Am. Chem. Soc. 1988;110:3284–3291. [Google Scholar]
- 45.Sandman K, Pereira SL, Reeve JN. Diversity of prokaryotic chromosomal proteins and the origin of the nucleosome. Cell Mol. Life Sci. 1998;54:1350–1364. doi: 10.1007/s000180050259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tubbs JL, Pegg AE, Tainer JA. DNA binding, nucleotide flipping, and the helix-turn-helix motif in base repair by O6-alkylguanine-DNA alkyltransferase and its implications for cancer chemotherapy. DNA Repair (Amsterdam) 2007;6:1100–1115. doi: 10.1016/j.dnarep.2007.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Minko IG, Yamanaka K, Kozekov ID, Kozekova A, Indiani C, O'Donnell ME, Jiang Q, Goodman MF, Rizzo CJ, Lloyd RS. Replication bypass of the acrolein-mediated deoxyguanine DNA-peptide cross-links by DNA polymerases of the DinB family. Chem. Res. Toxicol. 2008;21:1983–1990. doi: 10.1021/tx800174a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Minko IG, Kozekov ID, Kozekova A, Harris TM, Rizzo CJ, Lloyd RS. Mutagenic potential of DNA-peptide crosslinks mediated by acrolein-derived DNA adducts. Mutat. Res. 2008;637:161–172. doi: 10.1016/j.mrfmmm.2007.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.