

Abstract
CD4+ T cells orchestrate innate and adaptive immunity. In the central nervous system they modulate immune responses including cell trafficking and glial neuroregulatory functions through an array of soluble molecules cell-cell interactions affecting tissue homeostasis. During disease their roles evolve to an auto-aggressive or, alternatively, protective phenotype. How such a balance is struck in the setting of neurodegenerative disorders may reflect a dichotomy between regulatory T cell, anti-inflammatory and neuroprotective activities versus effector T cell inflammation and neurodegeneration. Interestingly, such roles may show commonalities amongst neurodegenerative diseases. Herein we focus on strategies to modulate such CD4+ T cell responses for therapeutic gain.
Keywords: MGC, Microglia; PD, Parkinson’s disease; AD, Alzheimer’s disease; HAND, HIV-1 associated neurocognitive disorders; MP, mononuclear phagocytes; Treg, regulatory T cells; Teff, effector T cells
1. Introduction
CD4+ helper T lymphocytes (Th), CD4+ regulatory T lymphocytes (Treg) and CD8+ cytotoxic T lymphocytes (CTL) are subset classifications that have undergone significant phenotypic and functional changes in recent years (Germain, 2002; Shevach, 2002). In regards to the former and despite its profound functional roles in immunoregulation, CD4+ T cells, have no purported phagocytic activity and do not kill pathogens directly, but serve to activate and direct the functions of other cells to do its bidding (Zhu and Paul, 2008). These activities include antibody class switching for B cells, CTL activation and mobilization, as well as mononuclear phagocytes (MP; blood borne monocytes, tissue macrophages and microglia) phagocytic and intracellular killing activities (Hume et al., 2002; Parker, 1993; Van Ginderachter et al., 2006; Yamamoto et al., 2008; Zhu and Paul, 2008). As described, CD4+ T cells possess limited mechanisms for cell killing such as granulysin and Fas-mediated apoptosis. Such immune functions are central tenets held for conventional immunology (Bevan, 2004; Castellino and Germain, 2006; Zhu and Paul, 2008). Starting in the mid to later 1980s, CD4+ helper T cells were divided into Th1 and Th2 subsets based on their cytokine profiles and effector functions (Mosmann et al., 1986; Mosmann and Coffman, 1989). Now, more than twenty years later, the phenotype and function of T cell subsets have evolved once again with the description of regulatory (Tr1, Th3, Treg) and Th17 cell phenotypes (Fontenot et al., 2003; Hori et al., 2003; Sakaguchi et al., 1995; Weaver et al., 2006). Moreover, CD4+ T cell function in health and disease has evolved significantly following the discovery of its central role as a cellular target for human immunodeficiency virus (HIV) infections (De Boer, 2007; Grossman et al., 2002; Ho et al., 1995).
In regards to the brain, the role played by CD4+ T cells have only recently been appreciated beginning less than two decades ago (Hickey et al., 1991). Indeed, the brain was previously regarded as an immune privileged organ, but only recently has it been shown that such privilege is limited (Bechmann et al., 2007; Chen and Palmer, 2008). The idea of such ‘immune privilege’ was based previously on its limited abilities to withstand damage during inflammation and its relatively poor regenerative capacity. However, both innate and adaptive immunity are vibrant despite any limitations conferred by the blood-brain barrier (Bechmann et al., 2007; Chen and Palmer, 2008). The known active role of both afferent and efferent arms of adaptive immunity results in antigen drainage to the cervical lymph nodes and active immune responses that affect neurodegenerative activities during disease and is now well appreciated (Galea et al., 2007; Kierdorf et al., 2009; Perry et al., 2007; Theodore et al., 2008). There are several reasons for this. First, in an activated state, CD4+ T cells can readily cross the blood-brain-barrier (BBB) (Engelhardt and Ransohoff, 2005; Gonzalez-Scarano and Martin-Garcia, 2005). Second, in regards to function, recruitment of peripheral leukocytes into the central nervous system (CNS) during neuroinflammatory, infectious and neurodegenerative diseases (for example, Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), and HIV-1 associated neurocognitive disorders (HAND, and stroke) serves both in protective and disease-accelerating processes (Appel, 2009; Banerjee et al., 2008; Benner et al., 2008; Brochard et al., 2009; Carrithers et al., 2000; Liu et al., 2009; Planas and Chamorro, 2009; Theodore et al., 2008). Third, although much is known in regards to the role played by CD4+ T cells as immune modulators and surveillance of microbial pathogens and neoplasia, it is only within the past five years that a dialogue between peripheral immunity and degenerative diseases of the CNS has been appreciated (Appel, 2009; Benner et al., 2008; Benner et al., 2004; Brochard et al., 2009; Byram et al., 2004; Clausen et al., 2007; Garg et al., 2008). Thus, this review focuses on the latter and serves to outline and describe a newly advancing field of adaptive immunity for neurodegenerative diseases. Particularly, we will outline how and under what conditions CD4+ T cells could elicit neuroprotection or neurotoxicity and how might these responses be utilized for diagnostic and therapeutic gain.
2. Neurodegenerative disorders and immunity
Neurodegenerative disorders are defined neuropathologically within specific brain regions by significant neuronal loss accompanied by neuroinflammatory astroglio- and microgliosis (Selkoe, 1994). Commonly, dysfunction of selected brain circuits initiates synaptic damage prior to neuronal loss. Indeed, decreased neuronal numbers follows neuronal dysfunction (Mosley et al., 2006). The clinical manifestations of disease that include impairments of memory, behavior, personality, judgment, attention, language (amongst other executive functions) as well as movement and gait characteristic of AD, PD, ALS, and HAND (Appel, 2009; Appel et al., 2008; Beers et al., 2008; Gonzalez-Scarano and Martin-Garcia, 2005; Vesce et al., 2007). Although the causes of each are divergent, cell-mediated processes in disease pathogenesis are similar. Moreover, the brain is limited in the ability to respond to injuries including regulation of inflammation, first and foremost (Appel, 2009; Peterson and Fujinami, 2007; Tansey et al., 2007). In recent years, relationships between neuroinflammation, adaptive immunity, and neurodegenerative have been sought (Appel, 2009; Mosley et al., 2006; Tansey et al., 2007). Nonetheless, the major focus of research and therapeutic development remain on innate immunity and the primary roles of astrocytes and microglia in disease (Farina et al., 2007; Hanisch and Kettenmann, 2007). Indeed, relationships between adaptive immunity and neurodegeneration or neuroprotection were until very recently, less well pursued. Thus, the role of effector and regulatory CD4+ T cells should be and is herein a primary focus. We begin with a brief review of common neurodegenerative disorders and previous knowledge regarding the role the immune system plays in neurodegenerative disease.
AD is the first most common neurodegenerative disease and the leading cause of senile dementia worldwide (Kawas and Brookmeyer, 2001). It is a progressive dementia first described by German psychiatrist Alois Alzheimer in 1906 (Graeber et al., 1997). Clinically, AD symptoms of cognitive and behavioral deficits are concordant with neuronal loss and atrophy in brain regions linked to learning and memory (Caselli et al., 2006; Goedert and Spillantini, 2006; Graeber et al., 1997). Such impairment ultimately results in severe disability for social and occupational functions (Harrington et al., 2007; Markesbery and Lovell, 1998; Tabert et al., 2005). The multifaceted causes of AD include genetic, environmental, and dietary factors but the precise etiology awaits discovery (Goedert and Spillantini, 2006; Graeber et al., 1997). Currently, there are three competing hypotheses for disease neuroopathogenesis. The first is cholinergic, which proposes that AD is caused by reduced synthesis of the neurotransmitter acetylcholine (Francis et al., 1999); the second is amyloidogenic, which postulates that accumulation of amyloid beta (Aβ) deposits resulting from protein aggregation and/or over-expression are the fundamental cause of neuronal impairments (Bishop and Robinson, 2002; Hardy and Selkoe, 2002; Lee et al., 2006); the third is tauogenic, which suggests that the tau protein abnormalities initiate the cascade of neurofibrillary tangles and plaques with concomitant alterations in dendritic arbor and neural physiology (Mudher and Lovestone, 2002). Based on these, two distinct, but not mutually exclusive theories on AD, other neuropathogenesis theories were introduced and included the “positive feed-forward” and “insult” theories. The former proposes that aberrant Aβ activates MPs, stimulating the release of various inflammatory cytokines, chemokines and excitotoxic factors that stimulate neurons to produce more Aβ. The latter theory suggests that some “insults”, including genetic predisposition, cause an initial neuronal damage with deposition of debris that could not be digested inefficiently by microglia, which result in microglia over-activation and chronic inflammation. In addition, complement deposition and lymphocyte infiltration might amplify the inflammatory response and cause more neuronal death (Goldgaber et al., 1989; Kadiu et al., 2005; McGeer and McGeer, 1995; Schmechel et al., 1988). Both theories highlight the importance of neuroinflammation (Hanisch and Kettenmann, 2007; McGeer and McGeer, 1995; Rogers et al., 2007; Walker and Lue, 2005) and genetic factors including mutation of presenilin-1 (PS-1) (Chen et al., 2008b; Das, 2008; Wang et al., 2007; Yukioka et al., 2008), presenilin-2 (PS-2) (Lleo et al., 2001; Schellenberg, 2003; Steiner et al., 1999; Wolozin et al., 1996), and apolipoprotein E (ApoE) polymorphisms (Pericak-Vance et al., 1991). Neuroinflammation is initiated by resident glial cells (microglia, astrocyte and perivascular macrophage) and potentially amplified by infiltration of peripheral immunocytes (T lymphocyte, B lymphocyte and dendritic cells) (Benner et al., 2008; Blasko and Grubeck-Loebenstein, 2003). The interplay between immunity, genetics, and environment do affect AD pathobiology; however, the relative influences of each over the other are not yet well appreciated.
PD is second to AD in disease prevalence (Nussbaum and Ellis, 2003). Like AD, PD is progressive and does show, in some forms, clear genetic linkages (Tansey et al., 2007). In 1817, the clinical features of PD were first described by James Parkinson in his classic monograph “An Essay on the Shaking Palsy” (Lees, 2007; Parkinson, 2002). However, after almost two centuries, the available therapeutic strategies remain symptomatic due, amongst other factors, to a limited understanding of the molecular neurobiology and pathobiology of the disease. In regards to the neuropathology, dopaminergic neurons in the substantia nigra pars compacta (SNpc) are lost along with their connections to the striatum (comprised by the caudate nucleus and putamen) and the presence of intraneuronal proteinacious cytoplasmic inclusions termed “Lewy bodies” (Barzilai and Melamed, 2003; Dauer and Przedborski, 2003; Fiskum et al., 2003; Michel et al., 2002; Paleologou et al., 2009). Recently, studies of PD pathogenesis have led to the prevailing hypothesis that misfolded proteins and dysfunction of the ubiquitin-proteasome pathway is pivotal to PD pathogenesis (Bennett et al., 1999; Dauer and Przedborski, 2003; Fishman and Oyler, 2002; Hattori et al., 2003; Hattori et al., 2000; Tanaka et al., 2004). In addition, mitochondrial dysfunction and oxidative stress is linked to PD development (Mosley et al., 2006; Tansey et al., 2007), which also have been shown to contribute dopaminergic neuronal loss (Albers and Beal, 2000; Benner et al., 2004; Ogawa et al., 2004; Reynolds et al., 2007a; Zhu et al., 2004). Like in AD, neuroinflammation is a strict feature associated with disease pathogenesis and progression (Bartels and Leenders, 2007; Benner et al., 2008; Mosley et al., 2006; Rogers et al., 2007; Tansey et al., 2008; Tansey et al., 2007; Wahner et al., 2007; Whitton, 2007). Neuroinflammation can accelerate dopaminergic neuronal death (necrosis and/or apoptosis), which can itself amplify neuroinflammation in a paracrine manner (Tansey et al., 2007).
Genetic factors and environmental cues are thought to play roles in familial and sporadic PD. The latter is dominant in post-encephalitic and chemical-induced PD, such as rotenone and 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine (MPTP). Currently, six genes have been identified to be associated with high penetrant autosomal dominant or recessive PD, including mutations in the gene encoding α-synuclein, parkin, DJ-1, ubiquitin c-terminal hydrolase-1, PTEN-induced kinase 1, and leucine rich repeat kinase 2 (Lee and Liu, 2008; Shimura et al., 2000; Volles and Lansbury, 2002; Volles et al., 2001). Such mutations have provided insight into the pathogenesis of PD. For example loss of function of parkin results in dysfunction of the ubiquitin-proteasome system; whereas, mutations in PTEN-induced kinase 1 and DJ-1 result in mitochondrial dysfuction and oxidative stress (Rogaeva et al., 2004; Takahashi-Niki et al., 2004). These observations support common pathogenic mechanisms associated with protein aggregation, mitochondrial dysfunction, and oxidative stress in AD and PD, linked in severity to neuroinflammatory responses (Rogers et al., 2007).
The neuropathology of ALS, also known as Lou Gehrig’s disease, is characterized by weakness and atrophy of muscles, which are denervated from both upper and lower motor neurons in the brainstem and spinal cord (Charles and Swash, 2001; Martin et al., 2000; Pradat and Bruneteau, 2006). Studies using transgenic mouse models and patient tissue delineated that the causes of ALS appear to be multifactorial, including environmental and genetic factors (Shaw and Wilson, 2003). The former implicates factors associated with viral infection, heavy metals, trauma, and experimental neurotoxins, while the latter suggests that mutations in the genes encoding Cu/Zn superoxide dismutase (SOD1), neurofilament H gene, the astrocytic-specific glutamate transporter, excitatory amino acid transporter (EAAT2, GLT1), and most recently, fused in sarcoma (FUS) (Bruijn et al., 2004; Cluskey and Ramsden, 2001; Kwiatkowski et al., 2009; Lee et al., 2002; Tu et al., 1997; Vance et al., 2009). Other risk factors, such as oxidative stress, glutaminergic excitotoxicity, abnormal enzyme metabolites, aberrant protein aggregation, and peripheral immune abnormalities may directly participate in the pathogenesis of ALS (Appel, 2009; Banerjee et al., 2008; Bruijn et al., 2004). Finally, evidence of neuroinflammation in ALS patients and experimental models demonstrates a pivotal role in disease and its progression (McGeer and McGeer, 2002).
HAND commonly follows progressive HIV-1 infection. A central feature of HAND is “severe neuronal dysfunction without loss”, which ranges in clinical severity from subtle deficits to incapacitating dementia: asymptomatic neurocognitive impairment (ANI), mild neurocognitive disorder (MND) and HIV-associated dementia (HAD) (Ellis et al., 2007; Gonzalez-Scarano and Martin-Garcia, 2005). Early in the AIDS epidemic, about 20–30% of individuals infected with HIV-1 developed a range of cognitive and motor symptoms, including impaired short-term memory, reduced concentration and muscle weakness (Gendelman et al., 1997; Gonzalez-Scarano and Martin-Garcia, 2005; Williams et al., 2001). These symptoms often occur concomitant with behavioral symptoms, such as personality changes, apathy and social withdrawal, and even led to a near vegetative state referred to as HAD. However, the advent and widespread use of combinatorial antiretroviral therapy (ART) markedly reduced the incidence of overt disease. Simultaneously, the prevalence of ANI and MND continue to increase, which suggests that onset and progression of HAND is linked, in part, to viral load and diminished adaptive immunity (Ellis et al., 2007; Gendelman et al., 1997; Gendelman et al., 1998; Gonzalez-Scarano and Martin-Garcia, 2005; Peterson and Fox, 2001; Reynolds et al., 2007a; Reynolds, 2007). Clinical manifestation of HAND evolves into the triad of cognitive, behavior and motor dysfunction induced by axonal disruption and “pruning” or aberrant sprouting of synaptodendritic connections (Ellis et al., 2007; Ghorpade et al., 1998; Gonzalez-Scarano and Martin-Garcia, 2005; Zheng and Gendelman, 1997).
HIV-1 enters the brain and is found in cerebrospinal fluid after primary infection, however, the mechanism by which HIV-1 invades the CNS is still limited. The CNS is separated by the BBB and the cerebrospinal fluid is separated by choroid-plexus epithelium. Both barriers are selectively permeable to regulate the traffic of cells and substances from bloodstream into the CNS. Even the “invasion” of peripheral immune cells across the BBB as normal immune surveillance is carefully regulated. Entry of HIV-1 to the CNS has been studied using animal models and cell culture systems for more than 20 years. Several hypotheses of CNS infection have been postulated from those studies. The “Trojan horse” hypothesis proposes that HIV-1 passes the BBB as sequestered passengers in infected cells that are trafficking to the brain (Meltzer et al., 1990). Other studies have suggested that HIV-1 might enter the CNS as free virus, as well as through transcytosis of endothelial cells (Buescher, 2007; Ellis et al., 2007; Gendelman et al., 1997; Gonzalez-Scarano and Martin-Garcia, 2005). After entry into the brain, HIV-1 disrupts disrupts neuronal function either directly through interaction with HIV-derived proteins/nucleic acid (direct effect) (Brenneman et al., 1994; D’Aversa et al., 2005; Eugenin et al., 2005; Spector and Zhou, 2008) or indirectly by HIV-induced cytokines and chemokines form MPs (Buckner et al., 2006; Chadwick et al., 2008; Mollace et al., 2001; Roberts et al., 2003; Wesselingh and Thompson, 2001; Yeh et al., 2000). Which factors are most deleterious and the extent by that each affects neuronal function remain central questions for the pathobiology of neurodegenerative disease.
3. Neural stem cells and innate immunity
Neurons, astrocytes, oligodendrocytes and microglia are the principal cell populations in the brain. Recent studies on neuronal plasticity have challenged paradigms established by Santiago Ramón y Cajal and others (Lopez-Munoz et al., 2006). One study indicated that microglia progenitors can transdifferentiate into neurons, astrocytes and oligodendrocytes, suggesting a novel role of microglia as multipotential stem cells (Yokoyama et al., 2004); whereas, astrocyte and oligodendrocyte progenitors can transdifferentiate into neurons (Doetsch et al., 2002), suggesting a novel role for these cell populations as multipotential stem cells. Neurogenesis requires two important growth factors, epidermal growth factor (EGF) (Doetsch et al., 2002) and fibroblast growth factor (FGF). Recently, evidence demonstrates roles for vascular endothelial growth factor (VEGF) in neurogenesis and neurovascular signaling deficits in neurodegeneration (Zacchigna et al., 2008).
Innate immunity is the first defense against pathogenic insults (Takeuchi and Akira, 2007). Several advantages of the innate immune system over the adaptive immune system include mechanisms that are germline-encoded, constitutively present, and immediately reactive, as well as mechanisms that have broad molecular-pattern recognition, non-specific effector function, and do not require immune memory formation (Akira et al., 2006; Farina et al., 2007). Innate molecules, such as pattern recognition receptors (PRRs) and the complement system are also included (Akira et al., 2006; Klotman and Chang, 2006; Rivest, 2003). In other words, the innate immune system is hard-wired for immediate and broad reactivity against foreign insult. The innate immune system consists of mast cells, neutrophils, natural killer cells (NK), gamma-delta T cells, natural killer T cells (NKT) and mononuclear phagocytes (MPs; monocyte, macrophages and dendritic cells) (Calandra and Roger, 2003; Ghorpade, 2008; Hammad and Lambrecht, 2008; Hume et al., 2002; Sternberg, 2006; Yamamoto et al., 2008). In the following part of this review, we will emphasize the function of MPs and their PRRs in health and disease (Kadiu et al., 2005; Okun et al., 2008; Seitz, 2003).
The function of innate immunity is to coordinate among all innate cells and molecules the proper cooperation for systemic scanning necessary for immune surveillance and homeostasis maintenance (Hanisch and Kettenmann, 2007; Mosser and Edwards, 2008). After being activated by injury, tumor or pathogen, the innate immune system functions focus on clearance of pathogens and debris, tissue repair, and in orchestrating adaptive immune responses (Hanisch and Kettenmann, 2007; Mosser and Edwards, 2008; Pichlmair and Reis e Sousa, 2007). Within the CNS main executors of innate immunity are perivascular macrophages and microglia (Kadiu et al., 2005). The primary function of ‘resting’ microglia is to engulf cellular debris and metabolic waste; whereas, the adjunct role of ‘resting’ microglia is to synthesize and secret neurotrophins; usually attributed to the primary role of astrocytes (Hanisch and Kettenmann, 2007; Streit, 2006). In response to pathological conditions, microglia transform into reactive state (Glanzer et al., 2007; Hanisch and Kettenmann, 2007) and the role of microglia switches from a phagocytic and neurotrophic phenotype to a reactive phenotype expressing toxic and reparative functions (Streit, 2006). The intent of inflammation is to eliminate pathogen and repair injury (Schwartz et al., 2006; Streit, 2006). However, the consequences of uncontrolled inflammation cause energy exhaustion and injury exacerbation have been reported (Tansey et al., 2008; Tansey et al., 2007). To date, the role of neuroinflammation for CNS disease is still argued.
Astrocytes are the most abundant cells in the CNS and together with microglia actively participate in local innate immunity triggered by injury and disease (Farina et al., 2007). Usually, in contrary to microglia, the job of the astrocyte is to support neuronal structure and function by neurotrophin production, ion maintenance and neurotransmitter modulation (Dong and Benveniste, 2001; Farina et al., 2007). In addition, astrocytes possess a series of receptors involved in innate immunity such as toll-like receptors and scavenger receptors, and soluble mediators including cytokines and chemokines (Dong and Benveniste, 2001; Farina et al., 2007). Using those sensors, astrocytes sense injurious insults. Therefore, following CNS insult, astrocytes work together with microglia to remove pathogens and restore homeostasis, actively and passively (Falsig et al., 2006; Farina et al., 2007). Astrocytes also promote leukocyte recruitment and amplify CNS inflammatory responses (Farina et al., 2007). The appearance of astrogliosis and microgliosis in specific brain regions where neurodegenerative scars form are thought to be primary or secondary culprits of neurodegeneration, regardless of their contribution to protective acute neuroinflammation as a primary function (Tansey et al., 2007). To some extent, such prejudiced idea was born out of misconceptions about the nature and function of reactive gliosis was rooted in cell-culture studies, in which endotoxin-treated microglia produce potential secretory neurotoxins. These include, but are not limited to, proinflammatory cytokines, quinolinic acid, arachidonic acid and its metabolites, and reactive oxygenic species (ROS). These have branded microglia as inducers of neurodegeneration rather than simple sensory and versatile effector cells (Schwartz et al., 2006; Streit, 2006). A neurodegenerative microglial reaction may occur as a consequence of the microglia’s primary role to eliminate misfolded protein or to attempt repair infectious injury as in HAND or an attempt to heal injury. What appears to worsen disease, however, is neurodestructive chronic neuroinflammation. A singular strategy in designing protective immune therapies rests in coping with the aberrantly altered homeostatic environment as the immune responses, under disease conditions, become both ineffective and disease progressive. These processes thus warrant therapeutic intervention with anti-inflammatory drugs or through induction of regulatory adaptive immune responses (Tansey et al., 2007).
As the most important molecules in innate immunity, pattern recognition receptors (PRRs) are constitutively expressed in the host and are highly conserved among species. Members of PRR families, such as toll-like receptors (TLRs) and NOD-like receptors (NLRs), recognize the evolutionarily conserved motifs of pathogen-associated molecular patterns (PAMPs), which include bacterial carbohydrates, nucleic acids, lipotechoic acids and fungal glycans (Akira et al., 2001; Farina et al., 2007; Imler and Hoffmann, 2001; Medzhitov, 2001). TLRs are expressed by both immune and non-immune cells (Farina et al., 2007; Hou et al., 2008; Medzhitov, 2001). Within the CNS, microglia, astrocytes, oligodendrocytes and neurons express different TLR profiles (Hanisch et al., 2008). Those TLRs function cooperatively to trigger and tailor innate immune response, as well as adaptive immune response at various points during a response (Akira et al., 2001; Pasare and Medzhitov, 2004). As sensors of danger signals, the nature of TLRs is supposed to be neuroprotective in an evolutionary context (Hanisch et al., 2008; Pasare and Medzhitov, 2004). For example, TLRs are regarded as means to prevent pathogen infection and promote plaque clearance (Hanisch et al., 2008). The observations that activation of immune cells through TLR ligation can induce the production of copious amounts of proinflammatory cytokines, especially when over-stimulated, promoted the notion of a more neurotoxic phenotype (Farina et al., 2007; Okun et al., 2008). Those cytokines, though essential for pathogen elimination and immune modulation, are detrimental for neuronal vitality when bystander damage outweighs the benefits of clearance mechanisms (Farina et al., 2007; Okun et al., 2008).
4. Adaptive immunity
Adaptive immunity, which is also called acquired immunity, is involved in later phases of infection. In contrast to innate immunity, several features particular to adaptive immunity which include mechanisms of somatic-rearrangement, induciblity, delayed reactivity, atomic-pattern recognition, high specificity, and memory (Vivier and Malissen, 2005). The adaptive immune system consists of two main cell populations, T lymphocytes and B lymphocytes, and two huge molecular repertoires, T cell receptors (TCRs) and B cell receptors (BCRs) provided by a capacity for somatic recombination of their antigen receptor genes (Vivier and Malissen, 2005). The generation of diversity among TCRs and BCRs is provided from segment rearrangement for combinatorial diversity and nucleotide recombination for junctional diversity (Nadel and Feeney, 1995). With diversity plus degeneracy, the adaptive immune system has the capacity to generate receptors capable of recognizing highly specific epitopes (Margulies, 2003; Martinez-Hackert et al., 2006; Reiser et al., 2003).
4.1. Adaptive immunity, homeostasis and surveillance
There are two forms of adaptive immunity. Humoral immunity is mediated by antibodies secreted by B lymphocytes, while cell-mediated immunity is executed by T lymphocytes (Vivier and Malissen, 2005). In health, both humoral immunity and cell-mediated immunity play important roles in immune surveillance and homeostasis maintenance (Vivier and Malissen, 2005). In disease, they work cooperatively to clear pathogens and form immunological memory (Vivier and Malissen, 2005). Neutralizing effects and opsonizing effects are typical effector mechanisms of antibodies secreted by B cells, while helper and cytotoxicity are typical effector mechanisms of T cells. The two types work collaboratively to exert adaptive immunity (Vivier and Malissen, 2005). Normally, the brain was considered as “immune-privileged” organ whereby T and B cells were thought to be excluded. However, as “immune-privileged” in the brain is not absolute, this notion has been rejected (Chen and Palmer, 2008). Both T cells and B cells are found to be present in both healthy and diseased brain (Chen and Palmer, 2008).
4.2. B cells
B cells carry out humoral immunity. Albeit seldom reported to be present in the brain during homeostatic conditions, during disease evidence of humoral immune responses in the initiation and modulation of neurodegeneration came from findings of experimental models wherein dopaminergic neurodegeneration was triggered by passive transfer of immunoglobulin from PD patients to mice (He et al., 2002). It was estimated that this effect was mediated by interaction of IgG and FcgammaR1, which are expressed by activated B cells and microglia, respectively.
4.3. T cells
T cells play a central role in adaptive immunity, not only in cell-mediated immunity, but also in humoral immunity (Vivier and Malissen, 2005). They are widely distributed in all tissues. Typically, T cells circulate in the body fluid until recognition by their TCR of its cognate antigen presented in the context of the MHC molecules by antigen presenting cells (APCs) located within secondary lymphoid organs. After activation, T cells undergo massive proliferation and expansion, and elicit effector functions either by cell-cell contact or cytokine release, including mediating activation of B cells, modulation of innate immune cell function, or inducing target cell death (Vivier and Malissen, 2005). According to different phenotypes and functions, T cells are divided into two populations, Th and CTL (Vivier and Malissen, 2005). Moreover, CD4+ T cells can be subdivided into conventional T cells (effector T cells and memory T cells) and regulatory T cells (Tregs, including natural occurring Tregs and adaptively induced Tregs) (Grossman et al., 2004a). Because HIV-1 infects these cells, intense study on CD4+ T cell behavior and function has evolved (De Boer, 2007; Grossman et al., 2002; Ho et al., 1995).
Few T lymphocytes patrol for immune surveillance of the normal brain (Chen and Palmer, 2008). Under injurious or disease conditions, more T lymphocytes, but not B lymphocytes, infiltrate into the site of brain injury (Appel, 2008; Brochard et al., 2009; Engelhardt and Ransohoff, 2005). Leukocyte recruitment is cell- and site-specific, thus infiltration is not likely due to massive disruption of the BBB (Appel, 2008; Brochard et al., 2009; Engelhardt and Ransohoff, 2005). Under neurodegenerative conditions, a high ratio of activated T lymphocytes is found circulating within the peripheral blood, thus raising the question as to whether those activated T lymphocytes came from the brain, or are they destined to infiltrate the brain. Currently, no related study has been posed to address those possibilities. Additionally, the presence of activated peripheral T cells, suggests a role in neurodegeneration. As for the contribution of infiltrating T lymphocytes to neurodegeneration, different groups have opposite opinions. Brochard et al reported that the presence of CD4+ T cells in postmortem substantia nigra of PD patients as well as in the MPTP-induced PD model, is neurodestructive (Brochard et al., 2009). Previous reports however show that while infiltration of effector CD4+ T cells is neurodestructive, infiltration of CD4+ Treg is neuroprotective (Reynolds et al., 2007b). Moreover, extravasation of CD4+ T cells in the mutant SOD1 transgenic ALS mouse model is neuroprotective (Beers et al., 2008).
5. Effector T cells and neurodegenerative diseases
After activation, naïve CD4+ T cells (Th0) begin clonal expansion. Depending on different stimuli and APC signals, they differentiate into different subtypes and are classified by different cytokine profiles and effector functions, such as Th1 (expressing IL-2, IFN-γ, and TNF-α), Th2 (expressing IL-4, IL-5, and IL-13) and Th17 (expressing IL-17 and IL-22) (Bodles and Barger, 2004; Kaiko et al., 2008; Mosmann et al., 1986; Mosmann et al., 2005; Wilson et al., 2009). According to the Th1/Th2 paradigm, cytokines secreted by Th1 and Th2 antagonize each other to the extent that Th1 and Th2 development are considered mutually exclusive (Mosmann and Coffman, 1989). Recently, following the discovery of a new lineage of IL-17 producing effector T cells (Th17) which exhibit unique effector functions distinct from Th1 and Th2 cells has redefined the Th1/Th2 paradigm (Bettelli et al., 2006). Through expression of IL-17A, Th17 cells are able to amplify clearance of pathogenic cell types via increased cell-mediated immunity by T cells, antibody production by B cells as well as increase production of antimicrobial peptides such as defensins and S100 proteins, proinflammatory cytokines and chemokines, as well as matrix metalloproteinases in a variety of cells including fibroblasts, endothelial cells, and epithelial cells (Iwakura et al., 2008). In addition to involvement in mutual immuneregulation, Th1, Th2 and Th17 regulate mononuclear phagocytes, B lymphocytes and polymorphonuclear cells, respectively. Thus such regulation functions exhibit several common characteristics including redundancy, pleiotropy, synergy, and antagonism (Korn et al., 2009). This level of activity must be tightly regulated, lest these T cells become over-stimulated and hyper-activated leading to dysregulation, pathological sequelae, or overt autoimmune disease. In order to avoid detrimental effects, new homeostatic balances must be attained with shifts in Th1/Th2/Th17 predominance within disease progression (Kaiko et al., 2008).
Since interactions along the neuroendocrine-immune axis are analogous to a scale-free network, rather than a single-direction axis, communication is controlled via using the soluble molecules of each system (neurotransmitters, hormones and cytokines) as common signals (Fabris et al., 1995; Miller, 1998). Here, we address only how immune system affects the CNS in both in both maintenance of homeostasis and in disease.
In the healthy brain, CD4+ T cells are indispensable for homeostasis; whereas, during disease Th1, Th2 and Th17 cells play different roles in neuroprotection and neurodestruction (Chen et al., 2008a). Signaling by innate immune cells differentially activated by recognition of various pathogenic types or different traumas to the brain, different effector T cells can dictate the functional potential for orchestrating pathogen clearance and wound repair. During this process, Th1 and Th17, as producers of pro-inflammatory cytokines, directly contribute to neuroinflammation by secreting IL-1, IL-6, IL-17, TNF-α and IFN-γ, as well as indirectly enhance microglia-mediated neurotoxicity by up-regulating the release of reactive oxygen species (ROS) and nitric oxide (NO) from microglia (Weiner, 2008). Th2, as producers of anti-inflammatory cytokines, up-regulate the release of insulin-like growth factor-1 (IGF-1) from microglia and enhance microglia-mediated neuroprotection (Appel et al., 2008). Moreover, recent studies show that effector T cells could enhance neuroinflammation and accelerate neurodegeneration in the MPTP-induced PD model and the HIV-1/VSV-infected HIVE model (Benner et al., 2008; Liu et al., 2009; Reynolds et al., 2007b). These observations have led to the theoretical basis to skew the neuroimmune response away from a pro-inflammatory cytokine profile toward an anti-inflammatory cytokine pattern as a therapeutic strategy for neurodegenerative diseases.
6. Regulatory T cells and neurodegenerative diseases
Natural occurring regulatory T cells (Tregs) are characterizied by several constitutively expressed markers, including CD4, CD25, CD62L, CD103, CD152, glucocorticoid-induced tumor necrosis factor receptor (GITR), latency-associated peptide (LAP), the forkhead homeobox domain P3 (FoxP3) transcription factor (Battaglia et al., 2003; Chen et al., 2008a; Igarashi et al., 2008; Nishizuka and Sakakura, 1969). FoxP3 transcription factor is essential for the phenotype and function of Tregs (Huehn et al., 2009; Sakaguchi, 2004). A primary role of Tregs is to provide immune tolerance to self (Onishi et al., 2008; Sakaguchi, 2004). Self-tolerance is achieved and maintained by “recessive” and “dominant” mechanisms (Sakaguchi, 2004; von Boehmer, 2003). In recessive mechanisms, self-reactive lymphocytes are programmed to die by apoptosis when exposed to self-antigen at an immature developmental stage in the thymus (T cells) and bone marrow (B cells); a phenomenon referred to as clonal deletion. Other recessive mechanisms may render the cell functionally inactive (anergic) after exposure to self-antigen. In dominant mechanisms, T cells actively inhibit activation and expansion of aberrant or over-reactive lymphocytes (active suppression), in particular other types of T cells. Evidence now indicates that each adaptive immune response involving T or B effector cells yields an appropriate regulatory response to maintain balance between the two populations. This is seemingly critical for proper control of adaptive immune responses, both in quality and magnitude, and for the establishment or breaking of tolerance to self and non-self antigens. Indeed the absence of Tregs early in postnatal development of mice results in systemic multi-organ autoimmunity, in “recessive” self-tolerance,
Studies on the immunological synapse indicate that while Teffs are induced through cell-cell contact at the synapse in the context of soluble mediators or signals (Huppa and Davis, 2003), Tregs inhibit Teff activation at the immunological synapse through via cell-cell contact, secretion of immunosuppressive cytokines such as IL-10 and TGF-β, and direct effector cell killing. Increasing evidence indicate since Tregs operate primarily at sites of inflammation, all their diverse mechanisms are available to modulate immune reactions under variable inflammatory conditions. Their mechanistic complexity and diversity evolves from diverse tasks performed by various Treg cell subsets in different stages of the immune reaction. (Huehn and Hamann, 2005). However, in different disease states, Treg function results in vastly different outcomes, wherein autoimmunity and neurodegeneration Tregs may protectively attenuate inflammation; whereas, in infectious and neoplastic disease Tregs may suppress the immune response necessary to eliminate foreign entities (Alyanakian et al., 2003; Belkaid et al., 2002; Ghiringhelli et al., 2004; Sakaguchi et al., 2001; Smyth et al., 2001; Viglietta et al., 2004; von Herrath and Harrison, 2003; Wang et al., 2004). Thus, Treg function for neuroregulation may be considered a double-edged sword whereby as a regulator of innate immunity or microglial-mediated neurotoxicity, which inhibits neuroinflammation, may also prevent the necessary clearance of misfolded proteins, neuronal debris, or viral infected reservoirs.
Nonetheless, whether Tregs are beneficial or detrimental in HIV-1 infection has not been entirely resolved. Some have suggested a detrimental role for Tregs since they suppress the immune response to HIV and allow HIV-1 progression (Seddiki and Kelleher, 2008; Tenorio et al., 2009), however others suggest a more beneficial role for Tregs due to their ability to suppress chronic immune activation and neuroinflammation, both main initiators of neuronal pathology in HIV (Seddiki and Kelleher, 2008). For example, SIV-infected natural hosts, sooty mangabeys (SM) do not develop AIDS, because they maintain substantive Treg levels and show limited immune activation while simian immunodeficiency virus (SIV)-infected Asian rhesus macaques (RM) readily progress to AIDS and poorly suppress chronic immune activation (Klatt et al., 2008; O’Connell and Siliciano, 2008; Pereira et al., 2007; Seddiki and Kelleher, 2008; Silvestri et al., 2007). Thus, differences in clinical outcome in disease-resistant SM and disease-susceptible RM may be linked, in some manner, to Treg responses, however this is not absolute. Indeed, the numbers of peripheral Tregs in chronically SIV-infected SM are maintained while a significant loss occurs in chronically SIV-infected RM. Further works, however, showed correlations between plasma viral load and the level of Tregs in SIV-infected RM, but not SM. Treg-depleted PBMC from RM led to a significant enhancement of CD4+ and CD8+ T-cell responses to SIV peptides, there was no detectable T-cell response to the same pool in Treg-depleted cells from SIV-infected SM (Klatt et al., 2008; O’Connell and Siliciano, 2008; Pereira et al., 2007; Seddiki and Kelleher, 2008; Silvestri et al., 2007).
Using dialectic views, it is possible that Tregs play divergent roles early in infection than late in disease. Early in infection, potent HIV-specific immune responses are suppressive, whereas later in infection, Tregs suppress the chronic immune activation (Klatt et al., 2008; O’Connell and Siliciano, 2008; Pereira et al., 2007; Seddiki and Kelleher, 2008; Silvestri et al., 2007). Taken together these results suggest new theories on pathogenesis of HIV diseases and new strategies on HIV/AIDS therapies such as a generalized immune over-activation hypothesis versus a tolerance induction strategy (Bourinbaiar and Abulafia-Lapid, 2005; Brenchley, 2006; Brenchley et al., 2006; Grossman et al., 2006; McMichael, 2006; Mehandru et al., 2004; Neutra and Kozlowski, 2006; Pearson, 2004; Rizzardi et al., 2002). Most interestingly, the conventional immunotherapeutic approaches aimed at enhancing the immune response against HIV have repeatedly failed when applied in clinical practice, while clinical benefit has been invariably associated with usage of vaccines that act in accord with the principles of alloimmunization. Thus immunotherapeutic strategies incorporating alloimmunization, which primarily induce tolerance rather than immune activation, might better yield clinical outcomes in HIV/AIDS (Bourinbaiar and Abulafia-Lapid, 2005; Bourinbaiar et al., 2006).
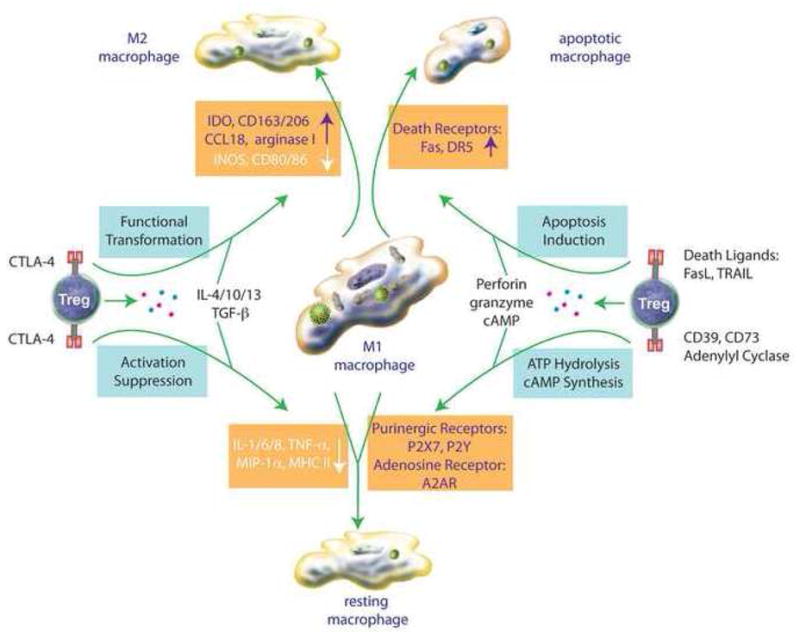
Since chronic neuroinflammation seemingly plays a demonstrable role in progression of neurodegeneration, Tregs, having the capacity to attenuate inflammation, represent an attractive therapeutic target in neurodegenerative disorders (Kipnis et al., 2004; Liesz et al., 2009; O’Connor and Anderton, 2008; Planas and Chamorro, 2009; Reynolds et al., 2007b). Mechanisms by which Tregs attenuate neuroinflammation include suppression of Teff activation and induction of Teff apoptosis (Grossman et al., 2004a; Grossman et al., 2004b; Pandiyan et al., 2007; Scheffold et al., 2007). Recent data also demonstrate that Tregs provide neuroprotective activity by upregulation of brain-derived neurotrophic factor (BDNF) and glial cell-derived neurotrophic factor (GDNF), downregulation of pro-inflammatory cytokines and ROS production, as well as direct induction of apoptosis in α-synuclein activated microglia and HIV-1/VSV infected macrophages (Liu et al., 2009; Reynolds, 2009). These findings present Tregs and Treg function in a new light relative to neurodegeneration, whereby multiple and new regulatory mechanisms have been determined (Figures 1 and 2). Tregs use not only Fas/FasL pathways, but also perforin/granzyme pathway to destroy target cells (Grossman et al., 2004a). Additionally, Tregs utilize other immunosuppressive mechanisms including production of IL-10 and TGF-β (O’Connor and Anderton, 2008), downregulation or aggregation of microglial MHC and CD80/CD86 molecules (Onishi et al., 2008; Reynolds, 2009), hydrolysis of ATP released by necrotic cells and synthesis of cAMP by highly active adenylyl cyclase (Bopp et al., 2007). Moreover, Tregs also induce the transformation of brain MP from M1 phenotype to M2 phenotypes as an effective mechanism to attenuate neuroinflammation (Tiemessen et al., 2007) (Figure 2).
Figure 1.
The potential for multifaceted roles of Treg in the immunoregulation of the HIV-1 infected macrophage. Such roles may include activation-suppression, phenotypic modulation, functional transformation or induction of apoptosis and may substantively affect the onset and tempo of neurodegeneration in the setting of HAND.
Figure 2.

The multifaceted roles of Treg and Teff in neurodegenerative disorders. For neurodegenerative disease (HAND, PD, AD, ALS), Teff and Treg exert opposing influences on neuronal function and speeding neurodestruction or neuroprotection, respectively. However, in early stage disease immunoregulatory control responses are operative and initiated by Treg to modulate MP homeostatic function and promote neuronal integrity. However, as disease progresses Teff up-regulate neurotoxic molecules and exacerbate MP activation and dysfuction, while Treg attenuate these neurotoxic responses. Treg modulate neuronal survival and function and could occur through changes the phenotype of the M1, pro-inflammatory brain MP to an M2 phenotype. The latter is an alternatively activated macrophage that possesses anti-inflammatory and potential neuroprotective functions.
In models that recapitulate the effects of Tregs on α-synuclein stimulated microglia in early asymptomatic and later overt disease stages in PD, Tregs have been shown to exhibit distinct functions (Banerjee et al., 2008; Reynolds et al., 2007b; Reynolds, 2009). Co-culture of microglia with Tregs prior to stimulation (early asymptomatic stage) converts a neurotoxic microglial phenotype to neuroprotective by inhibition of oxidative stress and diminution of cathepsin activity. On the other hand, co-culture of Tregs after microglial stimulation (late, overt disease) induces microglial apoptosis by upregulation of CD95 ligand and increased caspase-3 activity. Proteomic studies revealed that Tregs alter the α-synuclein-stimulated microglial proteome whereby modulated cellular proteins were associated with metabolism, migration, phagocytosis, protein transport and degradation, redox biology, and cytoskeletal and bioenergetic cell functions (Reynolds, 2009). These data support the importance of adaptive immunity in the regulation of PD-associated microglial inflammation and suggest a novel hypothesis for Treg regulation of microglial functions during the asymptomatic and overt disease stages in PD. During early, asymptomatic PD, regulatory adaptive immune responses attenuate microglial activation and neuroinflammatory responses, as well as control ROS and retard degenerative activities and disease progression.. Tregs at this stage, modulate microglia to be actively phagocytic and produce a broad spectrum of regulatory factors that maintain CNS homeostasis and limit α-synuclein accumulation. In the overt stage, regulatory immune responses break down, leading to hyperactive Teff function, loss of neuroimmune surveillance. Moreover, as aging is a major risk factor for PD, microglial senescence may lead to functional deficiency and chronic microgliosis. Treg mechanisms at this stage would then preferentially favor induction of microglial programmed cell death over anti-inflammatory activity (Reynolds, 2009).
In recent studies in models of HIVE, Tregs suppress ROS production and pro-inflammatory cytokine secretion by HIV-1 infected macrophages (Liu et al., 2009). In addition, Tregs reduce virus replication and induce macrophage apoptosis. Beneficially, this would inhibit macrophage activation and migration, decrease viral load, and destroy viral reservoirs in HIV-1 infected individuals. Furthermore, extended preservation of Tregs in the peripheral blood of HIV-1 infected elite suppressors (those that maintain CD4+ counts and controlled viremia) has been suggested as one mechanism to control progression of HIV-1 infection (Chase et al., 2008) (Figure 1). Based on those findings, upregulation of Treg numbers or Treg function represent an attractive strategy for therapeutic vaccine study in HIV-1 infection. In HIVE, adoptive transfer of Tregs attenuates astrogliosis and microgliosis with concomitant neuroprotection, upregulation of neurotrophic factors, downregulation proinflammatory cytokines, oxidative stress, and viral replication (Liu et al., 2009). Additionally, co-culture of human peripheral blood monocytes derived macrophage (MDM) with Tregs induces accelerated MDM cytotoxicity upon homotypic cell aggregation and fusion, while co-cultivation with Teffs enhances MDM activation with increased activated stellate and elongated morphologies (data from our laboratory).
Using the G93A-SOD1 transgenic mouse model of ALS, adoptive transfer of Tregs delays onset of neurological symptoms, while Teffs increases the latency between onset and entry into late stage, although both Tregs and Teffs delay the loss of motor function and extend survival (Banerjee et al., 2008). Although the precise mechanisms for this dual protective role of both effector and regulatory T cell populations has not been fully elucidated, subsequent studies support the observation that CD4+T cells have a protective role in the etiology of ALS (Appel, 2009) and supports an adaptive immune abnormality in CD4 T cell function in the G93A SOD1 transgenic mouse model (Appel, 2009; Banerjee et al., 2008).
In addition, one study indicated that Tregs only in the CNS, not in the other secondary lymphoid organs, retain a remarkable proliferative capacity which could drive accumulation of resident Tregs, but not peripheral Tregs, in inflamed CNS (O’Connor and Anderton, 2008). In light of those findings, passive transfer or active induction of Tregs is a potential attractive strategy for therapeutic intervention of neurodegeneration, whereby reactive neurotoxic T cell subpopulations are converted to anti-inflammatory and neurotrophic..
7. CTL and neurodegenerative diseases
CD8+ CTLs are well known for killing of target cells, which include viral-infected cells, tumor cells and hyperactivated cells. CTLs kill targets cells via three non-mutually exclusive pathways, which utilize perforin/granzyme, Fas/FasL, and TNF/TNFR. Few CTLs are found in normal CNS during immune surveillance, however under pathological conditions such as stroke, tumor, infection, neurodegeneration, or autoimmunity increased numbers of peripheral CTL proliferate and accumulate in and around brain lesions. Whle seemingly necessary for insult clearance, excessive CTL numbers or CTL activity, as found in multiple sclerosis, could be deleterious (Neumann et al., 2002).
8. Immune-based pathways for neurodegeneration
Accumulating evidence indicates that the kynurenine pathway (KP) and quinolinic acid (QA) play a role in neurodegeneration (Moresco et al., 2008) possibly via mechanisms of excitotoxicity and astrocytic apoptosis (Dantzer et al., 2008). Indolamine 2, 3-dioxygenase (IDO), which converts tryptophan into kynurenine (Ghiringhelli et al., 2004), was first recognized as a vital immunomodulatory enzyme in immunosuppression (Dantzer et al., 2008; Kwidzinski and Bechmann, 2007). Its role IDO in the brain has more recently been highlighted through characterization of the QA pathway, for which QA is a product of the KP. In addition, IDO is found to be highly inducible by pro-inflammatory cytokines, including IFN-γ and TNF-α (Dantzer et al., 2008; Kwidzinski and Bechmann, 2007), which augments IDO with exacerbatory effects. As a result, IDO has evolved as a novel target for therapeutic intervention of neurodegeneration as well as depression.
Arachidonic acid (AA), as the other key metabolite involved in neurodegeneration, is the product of phospholipases A2 (PLA2) hydrolyzation of membrane phospholipids and is the precursor for various proinflammatory mediators, including prostaglandins, leukotrienes, and thromboxanes. Under homeostatic conditions, PLA2 regulation provides balanced AA conversion into proinflammatory mediators and AA reincorporation into the membrane. Loss of strict control of PLA2 activity under disease conditions, yields the disproportionate production of proinflammatory mediators which exacerbates neuroinflammation and neurodegeneration in AD, ALS and MS (Farooqui et al., 2006; Pinto et al., 2003; Sun et al., 2004). Treatment with PLA2 inhibitors has proven effective in animal models suggesting another attractive therapeutic candidate in neurodegenerative diseases (Farooqui et al., 2006).
The ubiquitin-proteasome system (UPS) and autophagy-lysosome pathway (ALP) (Tansey et al., 2007) are two important mechanisms involved in elimination of intracellular pathogens and misfolded proteins, and antigen presentation by MHC II molecules (Pan et al., 2008). Alteration of UPS or autophagy diminishes protein degradation and leads to abnormal protein aggregation; a principal histopathological finding in neurodegenerative diseases. Thus, dysfunctional UPS and ALP, which play pivotal roles in protein clearance mechanisms under homeostatic conditions and misfolded protein aggregation in the pathogenesis of neurodegeneration, will provided another promising target for therapeutic intervention (Mayer et al., 1996; Mayer et al., 1994; Pan et al., 2008).
9. Conclusion
We hypothesize that regardless of the initiating etiological insult or affected brain area particular for each neurodegenerative disorder, a “two-stage” neuroinflammatory pathway is a common element in neuropathogenesis and disease progression. Early, during asymptomatic stages of disease, regulatory T cells maintain, in large part, control over neuroinflammation, and although unable to totally inhibit disease progression, secondary neurodegeneration due to over-active neuroinflammation is maintained at minimal levels. Phagocytic microglia engulf and digest neuronal debris and misfolded proteins with occasional inflammatory events, but inflammation is relatively contained. With disease progression, regulatory control is gradually lost, resulting in increased microglial hyperactivity and neurotoxicity leading to greater insult and secondary neurodegeneration that is more characteristic of overt disease during the symptomatic stage. Thus therapeutic strategies that enhance regulatory control over hyperactive immune responses either through suppression of microglial or Teff functions would target the neuroinflammatory component commonly associated with neurodegenerative disorders.
Acknowledgments
We thank Ms. Robin Taylor for outstanding administrative and computer support. National Institute of Health grants P01MH645702, R37 NS36126, P01 NS31492, 2R01 NS034239, P20RR 15635, P20RR 21937, P20 DA026146 and P01 NS43985 supported this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Albers DS, Beal MF. Mitochondrial dysfunction and oxidative stress in aging and neurodegenerative disease. J Neural Transm Suppl. 2000;59:133–154. doi: 10.1007/978-3-7091-6781-6_16. [DOI] [PubMed] [Google Scholar]
- Alyanakian MA, You S, Damotte D, Gouarin C, Esling A, Garcia C, Havouis S, Chatenoud L, Bach JF. Diversity of regulatory CD4+T cells controlling distinct organ-specific autoimmune diseases. Proc Natl Acad Sci U S A. 2003;100:15806–15811. doi: 10.1073/pnas.2636971100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appel SH. CD4+ T cells mediate cytotoxicity in neurodegenerative diseases. J Clin Invest. 2008 doi: 10.1172/JCI38096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appel SH. CD4+ T cells mediate cytotoxicity in neurodegenerative diseases. J Clin Invest. 2009;119:13–15. doi: 10.1172/JCI38096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appel SH, Beers DR, Henkel JS, Zhao W. Novel therapeutic targets in neurodegenerative diseases: lessons from amyotrophic lateral sclerosis. Curr Neurol Neurosci Rep. 2008;8:353–355. doi: 10.1007/s11910-008-0054-6. [DOI] [PubMed] [Google Scholar]
- Banerjee R, Mosley RL, Reynolds AD, Dhar A, Jackson-Lewis V, Gordon PH, Przedborski S, Gendelman HE. Adaptive immune neuroprotection in G93A-SOD1 amyotrophic lateral sclerosis mice. PLoS ONE. 2008;3:e2740. doi: 10.1371/journal.pone.0002740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels AL, Leenders KL. Neuroinflammation in the pathophysiology of Parkinson’s disease: evidence from animal models to human in vivo studies with [11C]-PK11195 PET. Mov Disord. 2007;22:1852–1856. doi: 10.1002/mds.21552. [DOI] [PubMed] [Google Scholar]
- Barzilai A, Melamed E. Molecular mechanisms of selective dopaminergic neuronal death in Parkinson’s disease. Trends Mol Med. 2003;9:126–132. doi: 10.1016/s1471-4914(03)00020-0. [DOI] [PubMed] [Google Scholar]
- Battaglia A, Ferrandina G, Buzzonetti A, Malinconico P, Legge F, Salutari V, Scambia G, Fattorossi A. Lymphocyte populations in human lymph nodes. Alterations in CD4+ CD25+ T regulatory cell phenotype and T-cell receptor Vbeta repertoire. Immunology. 2003;110:304–312. doi: 10.1046/j.1365-2567.2003.01742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechmann I, Galea I, Perry VH. What is the blood-brain barrier (not)? Trends Immunol. 2007;28:5–11. doi: 10.1016/j.it.2006.11.007. [DOI] [PubMed] [Google Scholar]
- Beers DR, Henkel JS, Zhao W, Wang J, Appel SH. CD4+ T cells support glial neuroprotection, slow disease progression, and modify glial morphology in an animal model of inherited ALS. Proc Natl Acad Sci U S A. 2008;105:15558–15563. doi: 10.1073/pnas.0807419105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkaid Y, Piccirillo CA, Mendez S, Shevach EM, Sacks DL. CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature. 2002;420:502–507. doi: 10.1038/nature01152. [DOI] [PubMed] [Google Scholar]
- Benner EJ, Banerjee R, Reynolds AD, Sherman S, Pisarev VM, Tsiperson V, Nemachek C, Ciborowski P, Przedborski S, Mosley RL, Gendelman HE. Nitrated alpha-synuclein immunity accelerates degeneration of nigral dopaminergic neurons. PLoS ONE. 2008;3:e1376. doi: 10.1371/journal.pone.0001376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benner EJ, Mosley RL, Destache CJ, Lewis TB, Jackson-Lewis V, Gorantla S, Nemachek C, Green SR, Przedborski S, Gendelman HE. Therapeutic immunization protects dopaminergic neurons in a mouse model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2004;101:9435–9440. doi: 10.1073/pnas.0400569101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MC, Bishop JF, Leng Y, Chock PB, Chase TN, Mouradian MM. Degradation of alpha-synuclein by proteasome. J Biol Chem. 1999;274:33855–33858. doi: 10.1074/jbc.274.48.33855. [DOI] [PubMed] [Google Scholar]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- Bevan MJ. Helping the CD8(+) T-cell response. Nat Rev Immunol. 2004;4:595–602. doi: 10.1038/nri1413. [DOI] [PubMed] [Google Scholar]
- Bishop GM, Robinson SR. The amyloid hypothesis: let sleeping dogmas lie? Neurobiol Aging. 2002;23:1101–1105. doi: 10.1016/s0197-4580(02)00050-7. [DOI] [PubMed] [Google Scholar]
- Blasko I, Grubeck-Loebenstein B. Role of the immune system in the pathogenesis, prevention and treatment of Alzheimer’s disease. Drugs Aging. 2003;20:101–113. doi: 10.2165/00002512-200320020-00002. [DOI] [PubMed] [Google Scholar]
- Bodles AM, Barger SW. Cytokines and the aging brain - what we don’t know might help us. Trends Neurosci. 2004;27:621–626. doi: 10.1016/j.tins.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Bopp T, Becker C, Klein M, Klein-Hessling S, Palmetshofer A, Serfling E, Heib V, Becker M, Kubach J, Schmitt S, Stoll S, Schild H, Staege MS, Stassen M, Jonuleit H, Schmitt E. Cyclic adenosine monophosphate is a key component of regulatory T cell-mediated suppression. J Exp Med. 2007;204:1303–1310. doi: 10.1084/jem.20062129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourinbaiar AS, Abulafia-Lapid R. Autoimmunity, alloimmunization and immunotherapy of AIDS. Autoimmun Rev. 2005;4:403–409. doi: 10.1016/j.autrev.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Bourinbaiar AS, Root-Bernstein RS, Abulafia-Lapid R, Rytik PG, Kanev AN, Jirathitikal V, Orlovsky VG. Therapeutic AIDS vaccines. Curr Pharm Des. 2006;12:2017–2030. doi: 10.2174/138161206777442119. [DOI] [PubMed] [Google Scholar]
- Brenchley JM. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Retrovirology. 2006;3:-. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- Brenchley JM, Price DA, Douek DC. HIV disease: fallout from a mucosal catastrophe? Nat Immunol. 2006;7:235–239. doi: 10.1038/ni1316. [DOI] [PubMed] [Google Scholar]
- Brenneman DE, McCune SK, Mervis RF, Hill JM. gp120 as an etiologic agent for NeuroAIDS: neurotoxicity and model systems. Adv Neuroimmunol. 1994;4:157–165. doi: 10.1016/s0960-5428(06)80252-4. [DOI] [PubMed] [Google Scholar]
- Brochard V, Combadiere B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, Bonduelle O, Alvarez-Fischer D, Callebert J, Launay JM, Duyckaerts C, Flavell RA, Hirsch EC, Hunot S. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest. 2009;119:182–192. doi: 10.1172/JCI36470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruijn LI, Miller TM, Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci. 2004;27:723–749. doi: 10.1146/annurev.neuro.27.070203.144244. [DOI] [PubMed] [Google Scholar]
- Buckner CM, Luers AJ, Calderon TM, Eugenin EA, Berman JW. Neuroimmunity and the blood-brain barrier: molecular regulation of leukocyte transmigration and viral entry into the nervous system with a focus on neuroAIDS. J Neuroimmune Pharmacol. 2006;1:160–181. doi: 10.1007/s11481-006-9017-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buescher JL, Gross S, Gendelman HE, Ikezu T. Handbook of clinical neurology. North-Holland Pub. Co; Netherlands: 2007. pp. 45–67. [DOI] [PubMed] [Google Scholar]
- Byram SC, Carson MJ, DeBoy CA, Serpe CJ, Sanders VM, Jones KJ. CD4-positive T cell-mediated neuroprotection requires dual compartment antigen presentation. J Neurosci. 2004;24:4333–4339. doi: 10.1523/JNEUROSCI.5276-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calandra T, Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol. 2003;3:791–800. doi: 10.1038/nri1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrithers MD, Visintin I, Kang SJ, Janeway CA., Jr Differential adhesion molecule requirements for immune surveillance and inflammatory recruitment. Brain. 2000;123 (Pt 6):1092–1101. doi: 10.1093/brain/123.6.1092. [DOI] [PubMed] [Google Scholar]
- Caselli RJ, Beach TG, Yaari R, Reiman EM. Alzheimer’s disease a century later. J Clin Psychiatry. 2006;67:1784–1800. doi: 10.4088/jcp.v67n1118. [DOI] [PubMed] [Google Scholar]
- Castellino F, Germain RN. Cooperation between CD4(+) and CD8(+) T cells: When, where, and how. Annual Review of Immunology. 2006;24:519–540. doi: 10.1146/annurev.immunol.23.021704.115825. [DOI] [PubMed] [Google Scholar]
- Chadwick W, Magnus T, Martin B, Keselman A, Mattson MP, Maudsley S. Targeting TNF-alpha receptors for neurotherapeutics. Trends Neurosci. 2008;31:504–511. doi: 10.1016/j.tins.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles T, Swash M. Amyotrophic lateral sclerosis: current understanding. J Neurosci Nurs. 2001;33:245–253. doi: 10.1097/01376517-200110000-00005. [DOI] [PubMed] [Google Scholar]
- Chase AJ, Yang HC, Zhang H, Blankson JN, Siliciano RF. Preservation of FoxP3+ regulatory T cells in the peripheral blood of human immunodeficiency virus type 1-infected elite suppressors correlates with low CD4+ T-cell activation. J Virol. 2008;82:8307–8315. doi: 10.1128/JVI.00520-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ML, Yan BS, Bando Y, Kuchroo VK, Weiner HL. Latency-associated peptide identifies a novel CD4+CD25+ regulatory T cell subset with TGFbeta-mediated function and enhanced suppression of experimental autoimmune encephalomyelitis. J Immunol. 2008a;180:7327–7337. doi: 10.4049/jimmunol.180.11.7327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Nakajima A, Choi SH, Xiong X, Tang YP. Loss of presenilin function causes Alzheimer’s disease-like neurodegeneration in the mouse. J Neurosci Res. 2008b;86:1615–1625. doi: 10.1002/jnr.21601. [DOI] [PubMed] [Google Scholar]
- Chen Z, Palmer TD. Cellular repair of CNS disorders: an immunological perspective. Hum Mol Genet. 2008;17:R84–92. doi: 10.1093/hmg/ddn104. [DOI] [PubMed] [Google Scholar]
- Clausen F, Lorant T, Lewen A, Hillered L. T lymphocyte trafficking: a novel target for neuroprotection in traumatic brain injury. J Neurotrauma. 2007;24:1295–1307. doi: 10.1089/neu.2006.0258. [DOI] [PubMed] [Google Scholar]
- Cluskey S, Ramsden DB. Mechanisms of neurodegeneration in amyotrophic lateral sclerosis. Mol Pathol. 2001;54:386–392. [PMC free article] [PubMed] [Google Scholar]
- D’Aversa TG, Eugenin EA, Berman JW. NeuroAIDS: contributions of the human immunodeficiency virus-1 proteins Tat and gp120 as well as CD40 to microglial activation. J Neurosci Res. 2005;81:436–446. doi: 10.1002/jnr.20486. [DOI] [PubMed] [Google Scholar]
- Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das HK. Transcriptional regulation of the presenilin-1 gene: implication in Alzheimer’s disease. Front Biosci. 2008;13:822–832. doi: 10.2741/2723. [DOI] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- De Boer RJ. Time scales of CD4+ T cell depletion in HIV infection. PLoS Med. 2007;4:e193. doi: 10.1371/journal.pmed.0040193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doetsch F, Petreanu L, Caille I, Garcia-Verdugo JM, Alvarez-Buylla A. EGF converts transit-amplifying neurogenic precursors in the adult brain into multipotent stem cells. Neuron. 2002;36:1021–1034. doi: 10.1016/s0896-6273(02)01133-9. [DOI] [PubMed] [Google Scholar]
- Dong Y, Benveniste EN. Immune function of astrocytes. Glia. 2001;36:180–190. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- Ellis R, Langford D, Masliah E. HIV and antiretroviral therapy in the brain: neuronal injury and repair. Nat Rev Neurosci. 2007;8:33–44. doi: 10.1038/nrn2040. [DOI] [PubMed] [Google Scholar]
- Engelhardt B, Ransohoff RM. The ins and outs of T-lymphocyte trafficking to the CNS: anatomical sites and molecular mechanisms. Trends Immunol. 2005;26:485–495. doi: 10.1016/j.it.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Eugenin EA, Dyer G, Calderon TM, Berman JW. HIV-1 tat protein induces a migratory phenotype in human fetal microglia by a CCL2 (MCP-1)-dependent mechanism: possible role in NeuroAIDS. Glia. 2005;49:501–510. doi: 10.1002/glia.20137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabris N, Mocchegiani E, Provinciali M. Pituitary-thyroid axis and immune system: a reciprocal neuroendocrine-immune interaction. Horm Res. 1995;43:29–38. doi: 10.1159/000184234. [DOI] [PubMed] [Google Scholar]
- Falsig J, Porzgen P, Lund S, Schrattenholz A, Leist M. The inflammatory transcriptome of reactive murine astrocytes and implications for their innate immune function. J Neurochem. 2006;96:893–907. doi: 10.1111/j.1471-4159.2005.03622.x. [DOI] [PubMed] [Google Scholar]
- Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007;28:138–145. doi: 10.1016/j.it.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Farooqui AA, Ong WY, Horrocks LA. Inhibitors of brain phospholipase A2 activity: their neuropharmacological effects and therapeutic importance for the treatment of neurologic disorders. Pharmacol Rev. 2006;58:591–620. doi: 10.1124/pr.58.3.7. [DOI] [PubMed] [Google Scholar]
- Fishman PS, Oyler GA. Significance of the parkin gene and protein in understanding Parkinson’s disease. Curr Neurol Neurosci Rep. 2002;2:296–302. doi: 10.1007/s11910-002-0004-7. [DOI] [PubMed] [Google Scholar]
- Fiskum G, Starkov A, Polster BM, Chinopoulos C. Mitochondrial mechanisms of neural cell death and neuroprotective interventions in Parkinson’s disease. Ann N Y Acad Sci. 2003;991:111–119. doi: 10.1111/j.1749-6632.2003.tb07469.x. [DOI] [PubMed] [Google Scholar]
- Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- Francis PT, Palmer AM, Snape M, Wilcock GK. The cholinergic hypothesis of Alzheimer’s disease: a review of progress. J Neurol Neurosurg Psychiatry. 1999;66:137–147. doi: 10.1136/jnnp.66.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galea I, Bernardes-Silva M, Forse PA, van Rooijen N, Liblau RS, Perry VH. An antigen-specific pathway for CD8 T cells across the blood-brain barrier. J Exp Med. 2007;204:2023–2030. doi: 10.1084/jem.20070064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg SK, Banerjee R, Kipnis J. Neuroprotective immunity: T cell-derived glutamate endows astrocytes with a neuroprotective phenotype. J Immunol. 2008;180:3866–3873. doi: 10.4049/jimmunol.180.6.3866. [DOI] [PubMed] [Google Scholar]
- Gendelman HE, Persidsky Y, Ghorpade A, Limoges J, Stins M, Fiala M, Morrisett R. The neuropathogenesis of the AIDS dementia complex. AIDS. 1997;11(Suppl A):S35–45. [PubMed] [Google Scholar]
- Gendelman HE, Zheng J, Coulter CL, Ghorpade A, Che M, Thylin M, Rubocki R, Persidsky Y, Hahn F, Reinhard J, Jr, Swindells S. Suppression of inflammatory neurotoxins by highly active antiretroviral therapy in human immunodeficiency virus-associated dementia. J Infect Dis. 1998;178:1000–1007. doi: 10.1086/515693. [DOI] [PubMed] [Google Scholar]
- Germain RN. T-cell development and the CD4-CD8 lineage decision. Nat Rev Immunol. 2002;2:309–322. doi: 10.1038/nri798. [DOI] [PubMed] [Google Scholar]
- Ghiringhelli F, Larmonier N, Schmitt E, Parcellier A, Cathelin D, Garrido C, Chauffert B, Solary E, Bonnotte B, Martin F. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur J Immunol. 2004;34:336–344. doi: 10.1002/eji.200324181. [DOI] [PubMed] [Google Scholar]
- Ghorpade A, Gendelman HE, Kipnis J. Neuroimmune Pharmacology. Springer; New York: 2008. pp. 89–104. [Google Scholar]
- Ghorpade A, Nukuna A, Che M, Haggerty S, Persidsky Y, Carter E, Carhart L, Shafer L, Gendelman HE. Human immunodeficiency virus neurotropism: an analysis of viral replication and cytopathicity for divergent strains in monocytes and microglia. J Virol. 1998;72:3340–3350. doi: 10.1128/jvi.72.4.3340-3350.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glanzer JG, Enose Y, Wang T, Kadiu I, Gong N, Rozek W, Liu J, Schlautman JD, Ciborowski PS, Thomas MP, Gendelman HE. Genomic and proteomic microglial profiling: pathways for neuroprotective inflammatory responses following nerve fragment clearance and activation. J Neurochem. 2007;102:627–645. doi: 10.1111/j.1471-4159.2007.04568.x. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG. A century of Alzheimer’s disease. Science. 2006;314:777–781. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- Goldgaber D, Harris HW, Hla T, Maciag T, Donnelly RJ, Jacobsen JS, Vitek MP, Gajdusek DC. Interleukin 1 regulates synthesis of amyloid beta-protein precursor mRNA in human endothelial cells. Proc Natl Acad Sci U S A. 1989;86:7606–7610. doi: 10.1073/pnas.86.19.7606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Scarano F, Martin-Garcia J. The neuropathogenesis of AIDS. Nat Rev Immunol. 2005;5:69–81. doi: 10.1038/nri1527. [DOI] [PubMed] [Google Scholar]
- Graeber MB, Kosel S, Egensperger R, Banati RB, Muller U, Bise K, Hoff P, Moller HJ, Fujisawa K, Mehraein P. Rediscovery of the case described by Alois Alzheimer in 1911: historical, histological and molecular genetic analysis. Neurogenetics. 1997;1:73–80. doi: 10.1007/s100480050011. [DOI] [PubMed] [Google Scholar]
- Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity. 2004a;21:589–601. doi: 10.1016/j.immuni.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Grossman WJ, Verbsky JW, Tollefsen BL, Kemper C, Atkinson JP, Ley TJ. Differential expression of granzymes A and B in human cytotoxic lymphocyte subsets and T regulatory cells. Blood. 2004b;104:2840–2848. doi: 10.1182/blood-2004-03-0859. [DOI] [PubMed] [Google Scholar]
- Grossman Z, Meier-Schellersheim M, Paul WE, Picker LJ. Pathogenesis of HIV infection: what the virus spares is as important as what it destroys. Nature Medicine. 2006;12:289–295. doi: 10.1038/nm1380. [DOI] [PubMed] [Google Scholar]
- Grossman Z, Meier-Schellersheim M, Sousa AE, Victorino RM, Paul WE. CD4+ T-cell depletion in HIV infection: are we closer to understanding the cause? Nat Med. 2002;8:319–323. doi: 10.1038/nm0402-319. [DOI] [PubMed] [Google Scholar]
- Hammad H, Lambrecht BN. Dendritic cells and epithelial cells: linking innate and adaptive immunity in asthma. Nat Rev Immunol. 2008;8:193–204. doi: 10.1038/nri2275. [DOI] [PubMed] [Google Scholar]
- Hanisch UK, Johnson TV, Kipnis J. Toll-like receptors: roles in neuroprotection? Trends Neurosci. 2008;31:176–182. doi: 10.1016/j.tins.2008.01.005. [DOI] [PubMed] [Google Scholar]
- Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Harrington WW, C SB, J GW, N OM, J GB, D CL, W RO, M CL, D MI. The Effect of PPARalpha, PPARdelta, PPARgamma, and PPARpan Agonists on Body Weight, Body Mass, and Serum Lipid Profiles in Diet-Induced Obese AKR/J Mice. PPAR Res. 2007;2007:97125. doi: 10.1155/2007/97125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori N, Kobayashi H, Sasaki-Hatano Y, Sato K, Mizuno Y. Familial Parkinson’s disease: a hint to elucidate the mechanisms of nigral degeneration. J Neurol. 2003;250(Suppl 3):III2–10. doi: 10.1007/s00415-003-1302-y. [DOI] [PubMed] [Google Scholar]
- Hattori N, Shimura H, Kubo S, Suzuki T, Tanaka K, Mizuno Y. [Autosomal recessive juvenile parkinsonism: its pathogenesis is involved in the ubiquitin-proteasome pathway] Rinsho Shinkeigaku. 2000;40:1293–1296. [PubMed] [Google Scholar]
- He Y, Le WD, Appel SH. Role of Fcgamma receptors in nigral cell injury induced by Parkinson disease immunoglobulin injection into mouse substantia nigra. Exp Neurol. 2002;176:322–327. doi: 10.1006/exnr.2002.7946. [DOI] [PubMed] [Google Scholar]
- Hickey WF, Hsu BL, Kimura H. Lymphocyte-T Entry into the Central-Nervous-System. Journal of Neuroscience Research. 1991;28:254–260. doi: 10.1002/jnr.490280213. [DOI] [PubMed] [Google Scholar]
- Ho DD, Neumann AU, Perelson AS, Chen W, Leonard JM, Markowitz M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature. 1995;373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- Hou B, Reizis B, DeFranco AL. Toll-like receptors activate innate and adaptive immunity by using dendritic cell-intrinsic and -extrinsic mechanisms. Immunity. 2008;29:272–282. doi: 10.1016/j.immuni.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huehn J, Hamann A. Homing to suppress: address codes for Treg migration. Trends Immunol. 2005;26:632–636. doi: 10.1016/j.it.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Huehn J, Polansky JK, Hamann A. Epigenetic control of FOXP3 expression: the key to a stable regulatory T-cell lineage? Nat Rev Immunol. 2009;9:83–89. doi: 10.1038/nri2474. [DOI] [PubMed] [Google Scholar]
- Hume DA, Ross IL, Himes SR, Sasmono RT, Wells CA, Ravasi T. The mononuclear phagocyte system revisited. J Leukoc Biol. 2002;72:621–627. [PubMed] [Google Scholar]
- Huppa JB, Davis MM. T-cell-antigen recognition and the immunological synapse. Nat Rev Immunol. 2003;3:973–983. doi: 10.1038/nri1245. [DOI] [PubMed] [Google Scholar]
- Igarashi H, Cao Y, Iwai H, Piao J, Kamimura Y, Hashiguchi M, Amagasa T, Azuma M. GITR ligand-costimulation activates effector and regulatory functions of CD4+ T cells. Biochem Biophys Res Commun. 2008;369:1134–1138. doi: 10.1016/j.bbrc.2008.03.024. [DOI] [PubMed] [Google Scholar]
- Imler JL, Hoffmann JA. Toll receptors in innate immunity. Trends Cell Biol. 2001;11:304–311. doi: 10.1016/s0962-8924(01)02004-9. [DOI] [PubMed] [Google Scholar]
- Iwakura Y, Nakae S, Saijo S, Ishigame H. The roles of IL-17A in inflammatory immune responses and host defense against pathogens. Immunol Rev. 2008;226:57–79. doi: 10.1111/j.1600-065X.2008.00699.x. [DOI] [PubMed] [Google Scholar]
- Kadiu I, Glanzer JG, Kipnis J, Gendelman HE, Thomas MP. Mononuclear phagocytes in the pathogenesis of neurodegenerative diseases. Neurotox Res. 2005;8:25–50. doi: 10.1007/BF03033818. [DOI] [PubMed] [Google Scholar]
- Kaiko GE, Horvat JC, Beagley KW, Hansbro PM. Immunological decision-making: how does the immune system decide to mount a helper T-cell response? Immunology. 2008;123:326–338. doi: 10.1111/j.1365-2567.2007.02719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawas CH, Brookmeyer R. Aging and the public health effects of dementia. N Engl J Med. 2001;344:1160–1161. doi: 10.1056/NEJM200104123441509. [DOI] [PubMed] [Google Scholar]
- Kierdorf K, Wang Y, Neumann H. Immune-Mediated CNS Damage. Results Probl Cell Differ. 2009 doi: 10.1007/400_2008_15. [DOI] [PubMed] [Google Scholar]
- Kipnis J, Avidan H, Caspi RR, Schwartz M. Dual effect of CD4+CD25+ regulatory T cells in neurodegeneration: a dialogue with microglia. Proc Natl Acad Sci U S A. 2004;101(Suppl 2):14663–14669. doi: 10.1073/pnas.0404842101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klatt NR, Villinger F, Bostik P, Gordon SN, Pereira L, Engram JC, Mayne A, Dunham RM, Lawson B, Ratcliffe SJ, Sodora DL, Else J, Reimann K, Staprans SI, Haase AT, Estes JD, Silvestri G, Ansari AA. Availability of activated CD4+ T cells dictates the level of viremia in naturally SIV-infected sooty mangabeys. J Clin Invest. 2008;118:2039–2049. doi: 10.1172/JCI33814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klotman ME, Chang TL. Defensins in innate antiviral immunity. Nat Rev Immunol. 2006;6:447–456. doi: 10.1038/nri1860. [DOI] [PubMed] [Google Scholar]
- Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009 doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski TJ, Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, Siddique T, McKenna-Yasek D, Sapp PC, Horvitz HR, Landers JE, Brown RH., Jr Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- Kwidzinski E, Bechmann I. IDO expression in the brain: a double-edged sword. J Mol Med. 2007;85:1351–1359. doi: 10.1007/s00109-007-0229-7. [DOI] [PubMed] [Google Scholar]
- Lee FJ, Liu F. Genetic factors involved in the pathogenesis of Parkinson’s disease. Brain Res Rev. 2008;58:354–364. doi: 10.1016/j.brainresrev.2008.02.001. [DOI] [PubMed] [Google Scholar]
- Lee HG, Zhu X, Nunomura A, Perry G, Smith MA. Amyloid-beta vaccination: testing the amyloid hypothesis? : heads we win, tails you lose! Am J Pathol. 2006;169:738–739. doi: 10.2353/ajpath.2006.060633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KW, Kim HJ, Sung JJ, Park KS, Kim M. Defective neurite outgrowth in aphidicolin/cAMP-induced motor neurons expressing mutant Cu/Zn superoxide dismutase. Int J Dev Neurosci. 2002;20:521–526. doi: 10.1016/s0736-5748(02)00052-7. [DOI] [PubMed] [Google Scholar]
- Lees AJ. Unresolved issues relating to the shaking palsy on the celebration of James Parkinson’s 250th birthday. Mov Disord. 2007;22(Suppl 17):S327–334. doi: 10.1002/mds.21684. [DOI] [PubMed] [Google Scholar]
- Liesz A, Suri-Payer E, Veltkamp C, Doerr H, Sommer C, Rivest S, Giese T, Veltkamp R. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med. 2009;15:192–199. doi: 10.1038/nm.1927. [DOI] [PubMed] [Google Scholar]
- Liu J, Gong N, Huang X, Reynolds AD, Mosley RL, Gendelman HE. Neuromodulatory activities of CD4+CD25+ regulatory T cells in a murine model of HIV-1-associated neurodegeneration. J Immunol. 2009;182:3855–3865. doi: 10.4049/jimmunol.0803330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lleo A, Blesa R, Gendre J, Castellvi M, Pastor P, Queralt R, Oliva R. A novel presenilin 2 gene mutation (D439A) in a patient with early-onset Alzheimer’s disease. Neurology. 2001;57:1926–1928. doi: 10.1212/wnl.57.10.1926. [DOI] [PubMed] [Google Scholar]
- Lopez-Munoz F, Boya J, Alamo C. Neuron theory, the cornerstone of neuroscience, on the centenary of the Nobel Prize award to Santiago Ramon y Cajal. Brain Res Bull. 2006;70:391–405. doi: 10.1016/j.brainresbull.2006.07.010. [DOI] [PubMed] [Google Scholar]
- Margulies DH. Molecular interactions: stiff or floppy (or somewhere in between?) Immunity. 2003;19:772–774. doi: 10.1016/s1074-7613(03)00331-5. [DOI] [PubMed] [Google Scholar]
- Markesbery WR, Lovell MA. Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer’s disease. Neurobiol Aging. 1998;19:33–36. doi: 10.1016/s0197-4580(98)00009-8. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Price AC, Kaiser A, Shaikh AY, Liu Z. Mechanisms for neuronal degeneration in amyotrophic lateral sclerosis and in models of motor neuron death (Review) Int J Mol Med. 2000;5:3–13. doi: 10.3892/ijmm.5.1.3. [DOI] [PubMed] [Google Scholar]
- Martinez-Hackert E, Anikeeva N, Kalams SA, Walker BD, Hendrickson WA, Sykulev Y. Structural basis for degenerate recognition of natural HIV peptide variants by cytotoxic lymphocytes. J Biol Chem. 2006;281:20205–20212. doi: 10.1074/jbc.M601934200. [DOI] [PubMed] [Google Scholar]
- Mayer RJ, Tipler C, Arnold J, Laszlo L, Al-Khedhairy A, Lowe J, Landon M. Endosome-lysosomes, ubiquitin and neurodegeneration. Adv Exp Med Biol. 1996;389:261–269. doi: 10.1007/978-1-4613-0335-0_33. [DOI] [PubMed] [Google Scholar]
- Mayer RJ, Tipler C, Laszlo L, Arnold J, Lowe J, Landon M. Endosome-lysosomes and neurodegeneration. Biomed Pharmacother. 1994;48:282–286. doi: 10.1016/0753-3322(94)90173-2. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res Brain Res Rev. 1995;21:195–218. doi: 10.1016/0165-0173(95)00011-9. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. Inflammatory processes in amyotrophic lateral sclerosis. Muscle Nerve. 2002;26:459–470. doi: 10.1002/mus.10191. [DOI] [PubMed] [Google Scholar]
- McMichael AJ. Hiv Vaccines. Annual Review of Immunology. 2006;24:227–255. doi: 10.1146/annurev.immunol.24.021605.090605. [DOI] [PubMed] [Google Scholar]
- Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1:135–145. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- Mehandru S, Poles MA, Tenner-Racz K, Horowitz A, Hurley A, Hogan C, Boden D, Racz P, Markowitz M. Primary HIV-1 infection is associated with preferential depletion of CD4+ T lymphocytes from effector sites in the gastrointestinal tract. J Exp Med. 2004;200:761–770. doi: 10.1084/jem.20041196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meltzer MS, Skillman DR, Gomatos PJ, Kalter DC, Gendelman HE. Role of mononuclear phagocytes in the pathogenesis of human immunodeficiency virus infection. Annu Rev Immunol. 1990;8:169–194. doi: 10.1146/annurev.iy.08.040190.001125. [DOI] [PubMed] [Google Scholar]
- Michel PP, Hirsch EC, Agid Y. [Parkinson’s disease: cell death mechanisms] Rev Neurol (Paris) 2002;158:24–32. [PubMed] [Google Scholar]
- Miller AH. Neuroendocrine and immune system interactions in stress and depression. Psychiatr Clin North Am. 1998;21:443–463. doi: 10.1016/s0193-953x(05)70015-0. [DOI] [PubMed] [Google Scholar]
- Mollace V, Nottet HS, Clayette P, Turco MC, Muscoli C, Salvemini D, Perno CF. Oxidative stress and neuroAIDS: triggers, modulators and novel antioxidants. Trends Neurosci. 2001;24:411–416. doi: 10.1016/s0166-2236(00)01819-1. [DOI] [PubMed] [Google Scholar]
- Moresco RM, Lavazza T, Belloli S, Lecchi M, Pezzola A, Todde S, Matarrese M, Carpinelli A, Turolla E, Zimarino V, Popoli P, Malgaroli A, Fazio F. Quinolinic acid induced neurodegeneration in the striatum: a combined in vivo and in vitro analysis of receptor changes and microglia activation. Eur J Nucl Med Mol Imaging. 2008;35:704–715. doi: 10.1007/s00259-007-0651-7. [DOI] [PubMed] [Google Scholar]
- Mosley RL, Benner EJ, Kadiu I, Thomas M, Boska MD, Hasan K, Laurie C, Gendelman HE. Neuroinflammation, Oxidative Stress and the Pathogenesis of Parkinson’s Disease. Clin Neurosci Res. 2006;6:261–281. doi: 10.1016/j.cnr.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136:2348–2357. [PubMed] [Google Scholar]
- Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. 1986. J Immunol. 2005;175:5–14. [PubMed] [Google Scholar]
- Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. 1989;7:145–173. doi: 10.1146/annurev.iy.07.040189.001045. [DOI] [PubMed] [Google Scholar]
- Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudher A, Lovestone S. Alzheimer’s disease-do tauists and baptists finally shake hands? Trends Neurosci. 2002;25:22–26. doi: 10.1016/s0166-2236(00)02031-2. [DOI] [PubMed] [Google Scholar]
- Nadel B, Feeney AJ. Influence of coding-end sequence on coding-end processing in V(D)J recombination. J Immunol. 1995;155:4322–4329. [PubMed] [Google Scholar]
- Neumann H, Medana IM, Bauer J, Lassmann H. Cytotoxic T lymphocytes in autoimmune and degenerative CNS diseases. Trends Neurosci. 2002;25:313–319. doi: 10.1016/s0166-2236(02)02154-9. [DOI] [PubMed] [Google Scholar]
- Neutra MR, Kozlowski PA. Mucosal vaccines: the promise and the challenge. Nat Rev Immunol. 2006;6:148–158. doi: 10.1038/nri1777. [DOI] [PubMed] [Google Scholar]
- Nishizuka Y, Sakakura T. Thymus and reproduction: sex-linked dysgenesia of the gonad after neonatal thymectomy in mice. Science. 1969;166:753–755. doi: 10.1126/science.166.3906.753. [DOI] [PubMed] [Google Scholar]
- Nussbaum RL, Ellis CE. Alzheimer’s disease and Parkinson’s disease. N Engl J Med. 2003;348:1356–1364. doi: 10.1056/NEJM2003ra020003. [DOI] [PubMed] [Google Scholar]
- O’Connell K, Siliciano RF. Immune alteration fends off AIDS. Nat Med. 2008;14:1016–1018. doi: 10.1038/nm1008-1016. [DOI] [PubMed] [Google Scholar]
- O’Connor RA, Anderton SM. Foxp3+ regulatory T cells in the control of experimental CNS autoimmune disease. J Neuroimmunol. 2008;193:1–11. doi: 10.1016/j.jneuroim.2007.11.016. [DOI] [PubMed] [Google Scholar]
- Ogawa N, Asanuma M, Miyoshi K. [Mechanism of specific dopaminergic neuronal death in Parkinson’s disease] Nippon Rinsho. 2004;62:1629–1634. [PubMed] [Google Scholar]
- Okun E, Griffioen KJ, Lathia JD, Tang SC, Mattson MP, Arumugam TV. Toll-like receptors in neurodegeneration. Brain Res Rev. 2008 doi: 10.1016/j.brainresrev.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onishi Y, Fehervari Z, Yamaguchi T, Sakaguchi S. Foxp3+ natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc Natl Acad Sci U S A. 2008;105:10113–10118. doi: 10.1073/pnas.0711106105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paleologou KE, Kragh CL, Mann DM, Salem SA, Al-Shami R, Allsop D, Hassan AH, Jensen PH, El-Agnaf OM. Detection of elevated levels of soluble {alpha}-synuclein oligomers in post-mortem brain extracts from patients with dementia with Lewy bodies. Brain. 2009 doi: 10.1093/brain/awn349. [DOI] [PubMed] [Google Scholar]
- Pan T, Kondo S, Le W, Jankovic J. The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson’s disease. Brain. 2008;131:1969–1978. doi: 10.1093/brain/awm318. [DOI] [PubMed] [Google Scholar]
- Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol. 2007;8:1353–1362. doi: 10.1038/ni1536. [DOI] [PubMed] [Google Scholar]
- Parker DC. T cell-dependent B cell activation. Annu Rev Immunol. 1993;11:331–360. doi: 10.1146/annurev.iy.11.040193.001555. [DOI] [PubMed] [Google Scholar]
- Parkinson J. An essay on the shaking palsy. 1817. J Neuropsychiatry Clin Neurosci. 2002;14:223–236. doi: 10.1176/jnp.14.2.223. discussion 222. [DOI] [PubMed] [Google Scholar]
- Pasare C, Medzhitov R. Toll-like receptors: linking innate and adaptive immunity. Microbes Infect. 2004;6:1382–1387. doi: 10.1016/j.micinf.2004.08.018. [DOI] [PubMed] [Google Scholar]
- Pearson H. Scientists seeking HIV in all the wrong places. Nature Medicine. 2004;10:1145–1145. [Google Scholar]
- Pereira LE, Villinger F, Onlamoon N, Bryan P, Cardona A, Pattanapanysat K, Mori K, Hagen S, Picker L, Ansari AA. Simian immunodeficiency virus (SIV) infection influences the level and function of regulatory T cells in SIV-infected rhesus macaques but not SIV-infected sooty mangabeys. J Virol. 2007;81:4445–4456. doi: 10.1128/JVI.00026-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pericak-Vance MA, Bebout JL, Gaskell PC, Jr, Yamaoka LH, Hung WY, Alberts MJ, Walker AP, Bartlett RJ, Haynes CA, Welsh KA, et al. Linkage studies in familial Alzheimer disease: evidence for chromosome 19 linkage. Am J Hum Genet. 1991;48:1034–1050. [PMC free article] [PubMed] [Google Scholar]
- Perry VH, Cunningham C, Holmes C. Systemic infections and inflammation affect chronic neurodegeneration. Nat Rev Immunol. 2007;7:161–167. doi: 10.1038/nri2015. [DOI] [PubMed] [Google Scholar]
- Peterson LK, Fujinami RS. Inflammation, demyelination, neurodegeneration and neuroprotection in the pathogenesis of multiple sclerosis. J Neuroimmunol. 2007;184:37–44. doi: 10.1016/j.jneuroim.2006.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson PK, Fox HS. NeuroAIDS: retroviral pathology and drugs of abuse. Adv Exp Med Biol. 2001;493:259–261. doi: 10.1007/0-306-47611-8_32. [DOI] [PubMed] [Google Scholar]
- Pichlmair A, Reis e Sousa C. Innate recognition of viruses. Immunity. 2007;27:370–383. doi: 10.1016/j.immuni.2007.08.012. [DOI] [PubMed] [Google Scholar]
- Pinto F, Brenner T, Dan P, Krimsky M, Yedgar S. Extracellular phospholipase A2 inhibitors suppress central nervous system inflammation. Glia. 2003;44:275–282. doi: 10.1002/glia.10296. [DOI] [PubMed] [Google Scholar]
- Planas AM, Chamorro A. Regulatory T cells protect the brain after stroke. Nat Med. 2009;15:138–139. doi: 10.1038/nm0209-138. [DOI] [PubMed] [Google Scholar]
- Pradat PF, Bruneteau G. Classical and atypical clinical features in amyotrophic lateral sclerosis. Rev Neurol (Paris) . 2006;162(Spec No 2):4S17–14S24. [PubMed] [Google Scholar]
- Reiser JB, Darnault C, Gregoire C, Mosser T, Mazza G, Kearney A, van der Merwe PA, Fontecilla-Camps JC, Housset D, Malissen B. CDR3 loop flexibility contributes to the degeneracy of TCR recognition. Nat Immunol. 2003;4:241–247. doi: 10.1038/ni891. [DOI] [PubMed] [Google Scholar]
- Reynolds A, Laurie C, Mosley RL, Gendelman HE. Oxidative stress and the pathogenesis of neurodegenerative disorders. Int Rev Neurobiol. 2007a;82:297–325. doi: 10.1016/S0074-7742(07)82016-2. [DOI] [PubMed] [Google Scholar]
- Reynolds AD, Banerjee R, Liu J, Gendelman HE, Mosley RL. Neuroprotective activities of CD4+CD25+ regulatory T cells in an animal model of Parkinson’s disease. J Leukoc Biol. 2007b;82:1083–1094. doi: 10.1189/jlb.0507296. [DOI] [PubMed] [Google Scholar]
- Reynolds AD, Kanmogne G, Kadiu I, Gendelman HE. Comprehensive Textbook of AIDS Psychiatry. Oxford Press; United Kingdom: 2007. pp. 207–253. [Google Scholar]
- Reynolds AD, Stone DK, Mosley RL, Gendelman HE. Nitrated alphasynuclein induced alterations in microglial immunity are regulated by CD4+ T cell subsets. J immunol. 2009 doi: 10.4049/jimmunol.0803982. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivest S. Molecular insights on the cerebral innate immune system. Brain Behav Immun. 2003;17:13–19. doi: 10.1016/s0889-1591(02)00055-7. [DOI] [PubMed] [Google Scholar]
- Rizzardi GP, Harari A, Capiluppi B, Tambussi G, Ellefsen K, Ciuffreda D, Champagne P, Bart PA, Chave JP, Lazzarin A, Pantaleo G. Treatment of primary HIV-1 infection with cyclosporin A coupled with highly active antiretroviral therapy. Journal of Clinical Investigation. 2002;109:681–688. doi: 10.1172/JCI14522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts ES, Zandonatti MA, Watry DD, Madden LJ, Henriksen SJ, Taffe MA, Fox HS. Induction of pathogenic sets of genes in macrophages and neurons in NeuroAIDS. Am J Pathol. 2003;162:2041–2057. doi: 10.1016/S0002-9440(10)64336-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogaeva E, Johnson J, Lang AE, Gulick C, Gwinn-Hardy K, Kawarai T, Sato C, Morgan A, Werner J, Nussbaum R, Petit A, Okun MS, McInerney A, Mandel R, Groen JL, Fernandez HH, Postuma R, Foote KD, Salehi-Rad S, Liang Y, Reimsnider S, Tandon A, Hardy J, St George-Hyslop P, Singleton AB. Analysis of the PINK1 gene in a large cohort of cases with Parkinson disease. Arch Neurol. 2004;61:1898–1904. doi: 10.1001/archneur.61.12.1898. [DOI] [PubMed] [Google Scholar]
- Rogers J, Mastroeni D, Leonard B, Joyce J, Grover A. Neuroinflammation in Alzheimer’s disease and Parkinson’s disease: are microglia pathogenic in either disorder? Int Rev Neurobiol. 2007;82:235–246. doi: 10.1016/S0074-7742(07)82012-5. [DOI] [PubMed] [Google Scholar]
- Sakaguchi S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol. 2004;22:531–562. doi: 10.1146/annurev.immunol.21.120601.141122. [DOI] [PubMed] [Google Scholar]
- Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–1164. [PubMed] [Google Scholar]
- Sakaguchi S, Sakaguchi N, Shimizu J, Yamazaki S, Sakihama T, Itoh M, Kuniyasu Y, Nomura T, Toda M, Takahashi T. Immunologic tolerance maintained by CD25+ CD4+ regulatory T cells: their common role in controlling autoimmunity, tumor immunity, and transplantation tolerance. Immunol Rev. 2001;182:18–32. doi: 10.1034/j.1600-065x.2001.1820102.x. [DOI] [PubMed] [Google Scholar]
- Scheffold A, Murphy KM, Hofer T. Competition for cytokines: T(reg) cells take all. Nat Immunol. 2007;8:1285–1287. doi: 10.1038/ni1207-1285. [DOI] [PubMed] [Google Scholar]
- Schellenberg GD. Alzheimer disease genes: presenilin 2 mutation number 9 and still counting. Arch Neurol. 2003;60:1521–1522. doi: 10.1001/archneur.60.11.1521. [DOI] [PubMed] [Google Scholar]
- Schmechel DE, Goldgaber D, Burkhart DS, Gilbert JR, Gajdusek DC, Roses AD. Cellular localization of messenger RNA encoding amyloid-beta-protein in normal tissue and in Alzheimer disease. Alzheimer Dis Assoc Disord. 1988;2:96–111. doi: 10.1097/00002093-198802020-00002. [DOI] [PubMed] [Google Scholar]
- Schwartz M, Butovsky O, Bruck W, Hanisch UK. Microglial phenotype: is the commitment reversible? Trends Neurosci. 2006;29:68–74. doi: 10.1016/j.tins.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Seddiki N, Kelleher AD. Regulatory T cells in HIV Infection: Who’s Suppressing What? Curr Infect Dis Rep. 2008;10:252–258. doi: 10.1007/s11908-008-0041-8. [DOI] [PubMed] [Google Scholar]
- Seitz M. Toll-like receptors: sensors of the innate immune system. Allergy. 2003;58:1247–1249. doi: 10.1046/j.1398-9995.2003.00225.x. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Cell biology of the amyloid beta-protein precursor and the mechanism of Alzheimer’s disease. Annu Rev Cell Biol. 1994;10:373–403. doi: 10.1146/annurev.cb.10.110194.002105. [DOI] [PubMed] [Google Scholar]
- Shaw CA, Wilson JM. Analysis of neurological disease in four dimensions: insight from ALS-PDC epidemiology and animal models. Neurosci Biobehav Rev. 2003;27:493–505. doi: 10.1016/j.neubiorev.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Shevach EM. CD4+ CD25+ suppressor T cells: more questions than answers. Nat Rev Immunol. 2002;2:389–400. doi: 10.1038/nri821. [DOI] [PubMed] [Google Scholar]
- Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25:302–305. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- Silvestri G, Paiardini M, Pandrea I, Lederman MM, Sodora DL. Understanding the benign nature of SIV infection in natural hosts. J Clin Invest. 2007;117:3148–3154. doi: 10.1172/JCI33034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth MJ, Godfrey DI, Trapani JA. A fresh look at tumor immunosurveillance and immunotherapy. Nat Immunol. 2001;2:293–299. doi: 10.1038/86297. [DOI] [PubMed] [Google Scholar]
- Spector SA, Zhou D. Autophagy: an overlooked mechanism of HIV-1 pathogenesis and neuroAIDS? Autophagy. 2008;4:704–706. doi: 10.4161/auto.6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner H, Duff K, Capell A, Romig H, Grim MG, Lincoln S, Hardy J, Yu X, Picciano M, Fechteler K, Citron M, Kopan R, Pesold B, Keck S, Baader M, Tomita T, Iwatsubo T, Baumeister R, Haass C. A loss of function mutation of presenilin-2 interferes with amyloid beta-peptide production and notch signaling. J Biol Chem. 1999;274:28669–28673. doi: 10.1074/jbc.274.40.28669. [DOI] [PubMed] [Google Scholar]
- Sternberg EM. Neural regulation of innate immunity: a coordinated nonspecific host response to pathogens. Nat Rev Immunol. 2006;6:318–328. doi: 10.1038/nri1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ. Microglial senescence: does the brain’s immune system have an expiration date? Trends Neurosci. 2006;29:506–510. doi: 10.1016/j.tins.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Sun GY, Xu J, Jensen MD, Simonyi A. Phospholipase A2 in the central nervous system: implications for neurodegenerative diseases. J Lipid Res. 2004;45:205–213. doi: 10.1194/jlr.R300016-JLR200. [DOI] [PubMed] [Google Scholar]
- Tabert MH, Liu X, Doty RL, Serby M, Zamora D, Pelton GH, Marder K, Albers MW, Stern Y, Devanand DP. A 10-item smell identification scale related to risk for Alzheimer’s disease. Ann Neurol. 2005;58:155–160. doi: 10.1002/ana.20533. [DOI] [PubMed] [Google Scholar]
- Takahashi-Niki K, Niki T, Taira T, Iguchi-Ariga SM, Ariga H. Reduced anti-oxidative stress activities of DJ-1 mutants found in Parkinson’s disease patients. Biochem Biophys Res Commun. 2004;320:389–397. doi: 10.1016/j.bbrc.2004.05.187. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Akira S. [Pathogen recognition by innate immunity] Arerugi. 2007;56:558–562. [PubMed] [Google Scholar]
- Tanaka K, Suzuki T, Hattori N, Mizuno Y. Ubiquitin, proteasome and parkin. Biochim Biophys Acta. 2004;1695:235–247. doi: 10.1016/j.bbamcr.2004.09.026. [DOI] [PubMed] [Google Scholar]
- Tansey MG, Frank-Cannon TC, McCoy MK, Lee JK, Martinez TN, McAlpine FE, Ruhn KA, Tran TA. Neuroinflammation in Parkinson’s disease: is there sufficient evidence for mechanism-based interventional therapy? Front Biosci. 2008;13:709–717. doi: 10.2741/2713. [DOI] [PubMed] [Google Scholar]
- Tansey MG, McCoy MK, Frank-Cannon TC. Neuroinflammatory mechanisms in Parkinson’s disease: potential environmental triggers, pathways, and targets for early therapeutic intervention. Exp Neurol. 2007;208:1–25. doi: 10.1016/j.expneurol.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenorio AR, Spritzler J, Martinson J, Gichinga CN, Pollard RB, Lederman MM, Kalayjian RC, Landay AL. The effect of aging on T-regulatory cell frequency in HIV infection. Clin Immunol. 2009;130:298–303. doi: 10.1016/j.clim.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodore S, Cao S, McLean PJ, Standaert DG. Targeted overexpression of human alpha-synuclein triggers microglial activation and an adaptive immune response in a mouse model of Parkinson disease. J Neuropathol Exp Neurol. 2008;67:1149–1158. doi: 10.1097/NEN.0b013e31818e5e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiemessen MM, Jagger AL, Evans HG, van Herwijnen MJ, John S, Taams LS. CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc Natl Acad Sci U S A. 2007;104:19446–19451. doi: 10.1073/pnas.0706832104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu PH, Gurney ME, Julien JP, Lee VM, Trojanowski JQ. Oxidative stress, mutant SOD1, and neurofilament pathology in transgenic mouse models of human motor neuron disease. Lab Invest. 1997;76:441–456. [PubMed] [Google Scholar]
- Van Ginderachter JA, Movahedi K, Hassanzadeh Ghassabeh G, Meerschaut S, Beschin A, Raes G, De Baetselier P. Classical and alternative activation of mononuclear phagocytes: picking the best of both worlds for tumor promotion. Immunobiology. 2006;211:487–501. doi: 10.1016/j.imbio.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al-Saraj S, Al-Chalabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo JM, Miller CC, Shaw CE. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vesce S, Rossi D, Brambilla L, Volterra A. Glutamate release from astrocytes in physiological conditions and in neurodegenerative disorders characterized by neuroinflammation. Int Rev Neurobiol. 2007;82:57–71. doi: 10.1016/S0074-7742(07)82003-4. [DOI] [PubMed] [Google Scholar]
- Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med. 2004;199:971–979. doi: 10.1084/jem.20031579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivier E, Malissen B. Innate and adaptive immunity: specificities and signaling hierarchies revisited. Nat Immunol. 2005;6:17–21. doi: 10.1038/ni1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volles MJ, Lansbury PT., Jr Vesicle permeabilization by protofibrillar alpha-synuclein is sensitive to Parkinson’s disease-linked mutations and occurs by a pore-like mechanism. Biochemistry. 2002;41:4595–4602. doi: 10.1021/bi0121353. [DOI] [PubMed] [Google Scholar]
- Volles MJ, Lee SJ, Rochet JC, Shtilerman MD, Ding TT, Kessler JC, Lansbury PT. Vesicle permeabilization by protofibrillar alpha-synuclein: Implications for the pathogenesis and treatment of Parkinson’s disease. Biochemistry. 2001;40:7812–7819. doi: 10.1021/bi0102398. [DOI] [PubMed] [Google Scholar]
- von Boehmer H. Dynamics of suppressor T cells: in vivo veritas. J Exp Med. 2003;198:845–849. doi: 10.1084/jem.20031358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Herrath MG, Harrison LC. Antigen-induced regulatory T cells in autoimmunity. Nat Rev Immunol. 2003;3:223–232. doi: 10.1038/nri1029. [DOI] [PubMed] [Google Scholar]
- Wahner AD, Sinsheimer JS, Bronstein JM, Ritz B. Inflammatory cytokine gene polymorphisms and increased risk of Parkinson disease. Arch Neurol. 2007;64:836–840. doi: 10.1001/archneur.64.6.836. [DOI] [PubMed] [Google Scholar]
- Walker DG, Lue LF. Investigations with cultured human microglia on pathogenic mechanisms of Alzheimer’s disease and other neurodegenerative diseases. J Neurosci Res. 2005;81:412–425. doi: 10.1002/jnr.20484. [DOI] [PubMed] [Google Scholar]
- Wang HY, Lee DA, Peng G, Guo Z, Li Y, Kiniwa Y, Shevach EM, Wang RF. Tumor-specific human CD4+ regulatory T cells and their ligands: implications for immunotherapy. Immunity. 2004;20:107–118. doi: 10.1016/s1074-7613(03)00359-5. [DOI] [PubMed] [Google Scholar]
- Wang Y, Greig NH, Yu QS, Mattson MP. Presenilin-1 mutation impairs cholinergic modulation of synaptic plasticity and suppresses NMDA currents in hippocampus slices. Neurobiol Aging. 2007 doi: 10.1016/j.neurobiolaging.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity. 2006;24:677–688. doi: 10.1016/j.immuni.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Weiner HL. A shift from adaptive to innate immunity: a potential mechanism of disease progression in multiple sclerosis. J Neurol. 2008;255(Suppl 1):3–11. doi: 10.1007/s00415-008-1002-8. [DOI] [PubMed] [Google Scholar]
- Wesselingh SL, Thompson KA. Immunopathogenesis of HIV-associated dementia. Curr Opin Neurol. 2001;14:375–379. doi: 10.1097/00019052-200106000-00018. [DOI] [PubMed] [Google Scholar]
- Whitton PS. Inflammation as a causative factor in the aetiology of Parkinson’s disease. Br J Pharmacol. 2007;150:963–976. doi: 10.1038/sj.bjp.0707167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KC, Corey S, Westmoreland SV, Pauley D, Knight H, deBakker C, Alvarez X, Lackner AA. Perivascular macrophages are the primary cell type productively infected by simian immunodeficiency virus in the brains of macaques: implications for the neuropathogenesis of AIDS. J Exp Med. 2001;193:905–915. doi: 10.1084/jem.193.8.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CB, Rowell E, Sekimata M. Epigenetic control of T-helper-cell differentiation. Nat Rev Immunol. 2009;9:91–105. doi: 10.1038/nri2487. [DOI] [PubMed] [Google Scholar]
- Wolozin B, Iwasaki K, Vito P, Ganjei JK, Lacana E, Sunderland T, Zhao B, Kusiak JW, Wasco W, D’Adamio L. Participation of presenilin 2 in apoptosis: enhanced basal activity conferred by an Alzheimer mutation. Science. 1996;274:1710–1713. doi: 10.1126/science.274.5293.1710. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Kiyota T, Walsh SM, Liu J, Kipnis J, Ikezu T. Cytokine-mediated inhibition of fibrillar amyloid-beta peptide degradation by human mononuclear phagocytes. J Immunol. 2008;181:3877–3886. doi: 10.4049/jimmunol.181.6.3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh MW, Kaul M, Zheng J, Nottet HS, Thylin M, Gendelman HE, Lipton SA. Cytokine-stimulated, but not HIV-infected, human monocyte-derived macrophages produce neurotoxic levels of l -cysteine. J Immunol. 2000;164:4265–4270. doi: 10.4049/jimmunol.164.8.4265. [DOI] [PubMed] [Google Scholar]
- Yokoyama A, Yang L, Itoh S, Mori K, Tanaka J. Microglia, a potential source of neurons, astrocytes, and oligodendrocytes. Glia. 2004;45:96–104. doi: 10.1002/glia.10306. [DOI] [PubMed] [Google Scholar]
- Yukioka F, Matsuzaki S, Kawamoto K, Koyama Y, Hitomi J, Katayama T, Tohyama M. Presenilin-1 mutation activates the signaling pathway of caspase-4 in endoplasmic reticulum stress-induced apoptosis. Neurochem Int. 2008;52:683–687. doi: 10.1016/j.neuint.2007.08.017. [DOI] [PubMed] [Google Scholar]
- Zacchigna S, Lambrechts D, Carmeliet P. Neurovascular signalling defects in neurodegeneration. Nat Rev Neurosci. 2008;9:169–181. doi: 10.1038/nrn2336. [DOI] [PubMed] [Google Scholar]
- Zheng J, Gendelman HE. The HIV-1 associated dementia complex: a metabolic encephalopathy fueled by viral replication in mononuclear phagocytes. Curr Opin Neurol. 1997;10:319–325. [PubMed] [Google Scholar]
- Zhu J, Paul WE. CD4 T cells: fates, functions, and faults. Blood. 2008;112:1557–1569. doi: 10.1182/blood-2008-05-078154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Smith MA, Perry G, Aliev G. Mitochondrial failures in Alzheimer’s disease. Am J Alzheimers Dis Other Demen. 2004;19:345–352. doi: 10.1177/153331750401900611. [DOI] [PMC free article] [PubMed] [Google Scholar]