

Abstract
Opposing theories of striatal hyper- and hypodopaminergic functioning have been suggested in the pathophysiology of externalizing behavior disorders. To test these competing theories, the authors used functional MRI to evaluate neural activity during a simple reward task in 12- to 16-year-old boys with attention-deficit/hyperactivity disorder and/or conduct disorder (n = 19) and in controls with no psychiatric condition (n = 11). The task proceeded in blocks during which participants received either (a) monetary incentives for correct responses or (b) no rewards for correct responses. Controls exhibited striatal activation only during reward, shifting to anterior cingulate activation during nonreward. In contrast, externalizing adolescents exhibited striatal activation during both reward and nonreward. Externalizing psychopathology appears to be characterized by deficits in processing the omission of predicted reward, which may render behaviors that are acquired through environmental contingencies difficult to extinguish when those contingencies change.
Keywords: conduct disorder, ADHD, striatum, anterior cingulate cortex, fMRI
Attention-deficit/hyperactivity disorder (ADHD) and conduct disorder (CD) co-occur up to 70% of the time in clinical samples (Klein et al., 1997). Although as many as 50% of children with ADHD do not develop CD, children who meet criteria for CD rarely present without a history of ADHD (Klein et al., 1997). High rates of comorbidity for these and related disorders suggest a common vulnerability for psychopathology that spans the externalizing spectrum. Indeed, research by Krueger and colleagues (Krueger et al., 2002; Krueger, Markon, Patrick, Benning, & Kramer, 2007) has indicated that externalizing behavior disorders, including conduct disorder, antisocial personality disorder, substance dependence, and disinhibited personality, share a common latent vulnerability that is almost entirely heritable. This latent vulnerability accounts for most of the covariance among externalizing disorders, whereas shared and nonshared environmental risk factors account for symptoms specific to each disorder. To date, the neural substrates of heritable vulnerability to externalizing psychopathology have not been fully elucidated.
Impulsivity may represent the core behavioral trait that is common across the externalizing spectrum. Although impulsivity has been defined in many ways, we use the term here to refer to a preference for immediate rewards over larger but delayed rewards (Sagvolden, Johansen, Aase, & Russell, 2005). Both ADHD and CD have been characterized in this way. CD in particular has been associated with perseveration for reward, even when reward contingencies become unfavorable (Newman & Wallace, 1993). Although researchers have speculated about shared neurobiological substrates for ADHD and CD that would predispose to this form of impulsivity (Beauchaine & Neuhaus, 2008; Gatzke-Kopp & Beauchaine, 2007), such theories have not been tested at the neurobiological level.
Consistent with shared liability models, it has been suggested that different environmental experiences shape common heritable risk for externalizing behaviors, resulting only in ADHD in protective environments, but escalating to CD/ADHD in high-risk environments (Beauchaine, Gatzke-Kopp, & Mead, 2007; Beauchaine & Neuhaus, 2008). This formulation, which is supported by data indicating that delinquency is potentiated by environmental risk among impulsive children (Lynam et al., 2000; Patterson, DeGarmo, & Knutson, 2000), might suggest a common neural deficiency for both disorders—at least before acquired antisocial behaviors become so well canalized that they alter long-term patterns of neural processing for those who progress to CD/ADHD.
Neurobiological Theories of Externalizing Behavior
Although a definitive neurobiological substrate of ADHD remains to be identified, considerable agreement exists that central dopamine (DA) dysfunction contributes to the disorder. This focus on DA arises in part from the effectiveness of methylphenidate, a central DA agonist (Patrick & Markowitz, 1997), as a treatment for ADHD. Additional support comes from molecular genetics studies (DiMaio, Frizenko, & Joober, 2003; Swanson & Castellanos, 2002), and from several structural and functional brain imaging studies identifying deficits in fronto-striatal circuitry that comprise the mesolimbic and mesocortical DA networks (Gatzke-Kopp & Beauchaine, 2007). Among the most frequently replicated findings for ADHD are structural deficits, abnormalities in asymmetry, metabolic deficiencies, and functional deficits in the caudate. The caudate and related structures of the basal ganglia are DA-rich regions implicated in the integration of reward cues toward facilitating goal-directed behavior (Marsden & Obeso, 1994; Tisch, Silberstein, Limousin-Dowsey, & Jahanshahi, 2004).
Independent literatures have also implicated—albeit somewhat indirectly—DA dysfunction as a primary deficit in the development of CD. Following from the work of Gray (1987) outlining neural networks involved in basic motivational systems governing approach and inhibition, several researchers have postulated involvement of the dopaminergically mediated behavioral approach system (BAS; also identified as behavioral activation system; Fowles, 1980) as the likely neural substrate for the excessive reward-seeking behaviors characteristic of CD (Beauchaine, 2001; Beauchaine, Katkin, Strassberg, & Snarr, 2001; Cloninger, Svrakic, & Svrakic, 1997; Quay, 1988).
DA and Reinforcement
Recent research has expanded our knowledge of the role of DA in reinforcement, beyond classic studies demonstrating its involvement in self-stimulation paradigms. This research indicates that phasic DA release into mesolimbic structures follows the receipt of both unanticipated rewards and the experience of novelty (Ljungberg, Apicella, & Schultz, 1992). Phasic DA release plays a critical role in learning, as behaviors that occur in close temporal proximity to rewards are reinforced, making those behaviors more likely to occur in the future. As the organism forms an association between a reward and a contingent behavior, DA release propagates backward to the stimulus signaling the reward (Schultz, Apicella, Scarnati, & Ljungberg, 1992). Should reward cease to follow a previously rewarded behavior, a significant decline in DA release signals a “prediction error,” motivating the animal to update its expectations and perhaps learn a new behavioral contingency (Ljungberg et al., 1992). Despite burgeoning research addressing the nature of DA in reinforcement learning, little research has incorporated these advances into theories of externalizing disorders. In fact, despite a confluence of hypotheses implicating DA dysfunction as a common neural substrate of ADHD and CD, considerable controversy exists regarding the precise nature of the deficit.
Translating Behavioral Observations Into Neurobiological Theories
Two antithetical theories of DA dysfunction have been proposed for the etiology of externalizing disorders. Following from descriptions of excessive approach or reward-seeking behaviors among those with CD (e.g., O’Brien & Frick, 1996), researchers have postulated overactive or hypersensitive central DA structures. This hypothesis follows from behavioral accounts of those with CD perseverating in their responding, despite changing contingencies in which previously rewarded behaviors become punished (Newman & Wallace, 1993; Quay, 1988).
Anatomical support for this theory comes from animal literature suggesting that infusions of DA into the nucleus accumbens lower the threshold for responding to rewarding stimuli and promote behavioral activity (Milner, 1999). This observation is consistent with the notion that overactivation of DA in the accumbens and other reward-related structures leads participants to respond for lower levels of reward, thus increasing overall approach behaviors, whereas DA antagonism often results in reduced behavioral activation. It is interesting to note that a similar hypothesis has been advanced for ADHD-like behaviors in a rodent model (Sullivan & Brake, 2003).
Accumulating evidence also supports theories of DA underresponding in the etiology of externalizing psychopathology. Research from a variety of disciplines suggests that underactivation of striatal and prefrontal DA projections leads to increased behavioral responding to raise activation levels within the mesolimbic system. Thus, what has been assumed to be reward hypersensitivity may in fact be reward insensitivity, which results in increased impulsive and perseverative responding, effectively upregulating a chronically underactive central reward system. Evidence from several sources has suggested that low levels of DA in the central reward system are experienced as aversive (De Witte, Pinto, Ansseau, & Verbanck, 2003; Laakso et al., 2003), and that increased DA activity is associated with pleasurable affective states (Ashby, Isen, & Turken, 1999). Moreover, research supports an association between low basal DA and a propensity to use DA agonist drugs of abuse (De Witte et al., 2003; Laine, Ahonen, Räsänen, & Tiihonen, 2001; Martin-Soelch et al., 2001). Furthermore, striatal DA dysfunction has been implicated in the personality trait of novelty seeking (Cloninger, 1987; Leyton et al., 2002). Thus, excessive approach behavior may be motivated by chronic under-activation of primary reward structures as the organism seeks to alleviate an aversive state of DA depletion.
The theory of DA deficiency as a neurobiological substrate of ADHD has been articulated elegantly by Sagvolden and colleagues (2005), who suggested that DA underresponding impedes the development of associations between rewards and contingent behaviors. For an individual with such a deficiency, phasic DA release following reinforcement is weak, resulting in a shortened temporal window for reinforcement associations to occur. As a result, only rewards that are delivered immediately after behaviors are effective reinforcers. This selects for behavioral patterns in which immediate gratification predominates, a characteristic frequently identified in children with both ADHD and CD (Sagvolden, Aase, Zeiner, & Berger, 1998).
Sagvolden et al. (2005) also commented on the role of DA in extinction. A central DA deficiency may create a floor effect whereby further decreases in neural activity that normally signify nonreinforcement (reward omission, extinction) cannot occur. This may lead to failure of extinction learning and perseveration of behaviors that are no longer reinforced. Anatomically, this phenomenon may be mediated by dopaminergic projections to the anterior cingulate cortex (ACC), a site shown to be associated with event-related potential (ERP) signals of reward prediction error (Holroyd & Coles, 2002). These DA-mediated signals appear to respond to omissions of reward, identifying situations in which error is more likely to occur, thereby eliciting cognitive control mechanisms over behavior in an effort to improve performance (Brown & Braver, 2005).
Taken together, the above discussion suggests that a chronic deficit in activity in the striatal-frontal pathway could result in an increased search for novel and exciting activities and a disregard for potential negative consequences. This pattern of approach behaviors reflects the symptoms of ADHD and CD, which may result from a dysfunctional central reward system. The striatum and cingulate may be the most likely anatomic regions underlying these deficiencies.
Although many imaging studies have implicated frontal lobe dysfunction in ADHD, frontal dysfunction is not likely to be the primary etiological source of externalizing symptoms common to ADHD and CD. Functional imaging research has identified the striatum as the site of action for the clinical effectiveness of methylphenidate (Brown & Braver, 2005), and both the dorsal and ventral striatum are critically involved in reinforcement learning (Wise, 2004). Furthermore, the caudate contains 100-fold more DA terminals than the medial prefrontal cortex and releases DA at a significantly higher rate (Garris & Rebec, 2002). Because these structures interact integrally with frontal structures, additional deficits may arise in prefrontal regions as a result of downstream deficiencies in striatal input. Furthermore, core symptoms of ADHD arise long before prefrontal DA networks mature (van Goozen, Fairchild, Snoek, & Harold, 2007). Accordingly, these frontal regions are not likely to be associated with the proposed deficiencies in reinforcement outlined here. In the current study, we therefore focus on striatal and anterior cingulate regions as sources of deficits in reward responding in adolescents with externalizing disorders.
Method
Participants
Following Institutional Review Board approval, we recruited participants through ads placed in newsletters throughout the Seattle Public School District, flyers distributed to local community centers and mental health service providers, postings on an Internet classified ad site, advertisements on buses, and a direct mailing to families in the local community. Parents who called in response to the ads (n = 216) were given a preliminary phone screen to determine their child’s potential eligibility. Parents answered questions from the CD, Oppositional Defiant Disorder (ODD), ADHD, Dysthymia, and Major Depressive Disorder subscales of the Adolescent Symptom Inventory (ASI; Gadow & Sprafkin, 1997), and the Aggression, Attention Problems, and Anxious/Depressed sub-scales of the Child Behavior Checklist (CBCL; Achenbach, 1991). The ASI provides scale scores and diagnostic cutoffs from the Diagnostic and Statistical Manual of Mental Disorders (4th ed., text rev.; American Psychiatric Association, 2000) for syndromes and was used to screen participants with a high likelihood of meeting criteria for an externalizing disorder. Those who met ASI criteria for CD and/or ADHD or who scored above the 98th percentile on the CBCL Aggression scale, suggesting that they might meet criteria for CD and/or ADHD in a more extensive interview, were invited to the laboratory for a diagnostic assessment. Potential participants who met criteria for major depression or who were currently taking an antidepressant or anxiolytic were excluded. Potential control participants were invited to the laboratory if they scored below the 60th percentile on all CBCL scales and exhibited no more than two symptoms of ADHD, ODD, or CD. Those with braces or implanted metal objects and those who expressed discomfort about confined spaces were excluded from enrollment. Because research suggests distinct psychophysiological correlates of CD for males and females (Beauchaine, Hong, & Marsh, 2008), only boys were enrolled because the small sample size precluded analyses by sex.
Assessments
Participants who met the above criteria (n = 66) were invited to the University of Washington for a 1–2 hour assessment, for which they were paid $50 and reimbursed for parking costs. Fourteen participants self-reported their racial/ethnic status as non-Caucasian, which is representative of the Seattle area. Participant parents and adolescents were met by two research assistants who explained the study, including the possibility that the family would be invited for a second visit involving a functional magnetic resonance imaging (fMRI) scan. Parents and adolescents signed consent and assent forms and were then separated. The adolescent then completed the Kaufman Brief Intelligence Test–Second Edition (KBIT-2; Kaufman & Kaufman, 2004), which was administered by a graduate research assistant trained in standardized testing. The KBIT-2 is a brief measure of verbal and nonverbal cognitive ability with excellent psychometric properties. The parent was asked to report on the adolescent’s ADHD, CD, and ODD symptoms using the Diagnostic Interview Schedule for Children (DISC; Shaffer, Fischer, Lucas, Mina, & Schwab-Stone, 2000). Finally, the adolescent was escorted to a mock scanner to confirm his willingness and ability to participate in the MRI scan.
Final sample
Participants who completed the initial session were evaluated for their eligibility to complete the fMRI protocol. Adolescents who met DISC criteria for ADHD and/or CD were admitted. Although a group-based comparison approach between ADHD and CD was originally planned, clean diagnostic separation between groups was not attainable. This is consistent with the majority of research on both ADHD and CD, in which diagnostic comorbidity is often so significant that the comorbidity is tolerated despite a primary focus on one or the other disorder. This clinical reality is consistent with the theories outlined in the introduction suggesting a common neurobiological vulnerability across the externalizing spectrum. Because the purpose of this article was to elucidate this general vulnerability, analyses were conducted with a broadly defined externalizing group.
Of the original 66 participants, 19 met full criteria for ADHD, and 2 met intermediate criteria for ADHD according to the DISC. Of these 2, both met criteria for CD, and 1 also had a past history of ADHD. Therefore, both were included in the externalizing group. In total, 12 of these 21 participants also met diagnostic criteria for CD according to the DISC, and 3 met intermediate criteria. Thus, the sample was characteristic of externalizing problems in general, with high levels of comorbidity. Two participants were subsequently eliminated from analyses due to excessive motion during the scan (see below). Of the remaining 19 participants with externalizing disorders, 16 reported having received a formal diagnosis of ADHD from a medical professional, and 10 were currently prescribed psychostimulant medication.
Among the 11 participants enrolled in the control group, none met diagnostic criteria for any externalizing disorder according the DISC interview, and none had ever received a diagnosis in a professional setting. Table 1 summarizes group differences in age, income, IQ, and diagnostic symptom scores.
Table 1.
Means (and Standard Deviations) of Diagnostic Criteria and Demographics by Group
| Characteristic or measure | Control (n = 11) | Externalizing (n = 19) | F(1, 29) | η |
|---|---|---|---|---|
| Age | 13.0 (1.0) | 13.6 (1.3) | 1.7 | .24 |
| Income (in thousands) | 70.5 (42.6) | 70.5 (63.1) | 0.0 | .00 |
| Full scale IQ | 115.0 (12.1) | 104.2 (14.8) | 4.2* | .36 |
| CBCL Attention | 51.0 (2.0) | 75.4 (8.8) | 81.4** | .86 |
| CBCL Aggression | 51.1 (2.4) | 74.1 (11.5) | 42.2** | .78 |
| ASI CD | 0.2 (0.4) | 8.5 (8.4) | 10.6** | .52 |
| ASI ODD | 2.8 (1.9) | 12.1 (6.4) | 21.5** | .66 |
| ASI ADHD | 6.2 (5.3) | 40.7 (10.7) | 100.3** | .88 |
| ASI MDD | 0.6 (1.0) | 5.4 (4.8) | 10.2** | .52 |
Note. CBCL = Child Behavior Checklist (Achenbach, 1991); ASI = Adolescent Symptom Inventory (Gadow & Sprafkin, 1997); CD = Conduct Disorder; ODD = Oppositional Defiant Disorder; ADHD = Attention-Deficit/Hyperactivity Disorder; MDD = Major Depressive Disorder. CBCL values represent T scores. ASI scores are cumulative symptom counts (scored as 0–4 per diagnostic criterion).
p ≤ .05.
p < .01.
fMRI
Behavioral task
Because of profound effects of psychostimulants on the brain regions of interest (Vles et al., 2003; Volkow, Fowler, Wang, Ding, & Gately, 2002), participants were asked to discontinue stimulant medications 36 hours prior to the scan. As expected, no participants in the control group were prescribed psychostimulant medication. However, 10 of the 19 participants in the externalizing group were taking psychostimulants. Previous research has suggested that medication washouts present minimal confounds. When participants who have experienced long-term stimulant treatment are compared in a medication washout state to participants who are medication naïve, no discernable differences in brain activation are found among ADHD groups who differ only on history of medication exposure (Pliszka et al., 2006).
In the scanner, participants engaged in a monetary incentive task presented using E-Prime software (Schneider, Eschman, & Zuccolotto, 2002). The task was similar to those used previously to evaluate reward- and extinction-related processes in children and adolescents with CD and ADHD (Beauchaine et al., 2001, 2007) and was designed to be very simple to minimize group differences in performance. This ensured that the task was equivalent for all participants, allowing for comparisons in neural correlates of responding, without a behavioral performance confound. Had there been significant group differences in task performance, any corresponding group differences in neural activation might be attributable to task difficulty rather than reward processing. In each trial, a green square was presented in pseudorandom order in either the left or right visual field. Participants were asked to indicate the side of the screen on which the square appeared by pressing a button with their corresponding thumb. The square was presented for 1,200 ms, and correct responses delivered within this interval were accompanied by a 500-ms tone and rewarded with a monetary increase presented at the fixation point of the screen. If the participant did not respond within this interval, or if he answered incorrectly, the task advanced to the next stimulus without feedback being presented.
Reward was administered in a block design. During reward blocks, participants were awarded $0.40 for each correct answer. During blocks of nonreward, the task was performed in the same manner, except (a) the monetary value was reset to $0.00 and did not change during the course of the block, and (b) correct responses were followed by a different 500-ms tone. During subsequent reward trials, the monetary value was reset, and rewards were added to the running total carried over from the previous reward block. Participants were told they would be rewarded during some but not all trials and that they should continue to respond during all trials. The task consisted of a total of 10 blocks. Each block consisted of 24 trials and lasted 30 s. Task blocks were separated by blocks of fixation (cross-hairs) that lasted 15 s. Blocks 1, 2, 4, 7, and 9 were reward blocks. The task was brief, lasting 7 min 45 s, to minimize demands on hyperactive children and reduce the incidence of data loss due to excessive motion.
Scan acquisition
Structural and functional MRI scans were performed on a 1.5 Tesla imaging system (General Electric, Waukesha, WI). The structural scan included 21-slice axial anatomic images in plane with functional data (TR/TE 200/2.2 ms, fast spoiled gradient recalled pulse sequence, 6 mm thick with 1-mm gap, 256 × 256 matrix). An fMRI series was collected following the structural scan by way of a two-dimensional gradient echo echoplanar pulse sequence (TR/TE 3000/50 ms, axial, 21 slices; 6 mm thick with 1-mm gap; 155 volumes). A high-resolution three-dimensional series was then acquired (TR/TE 11.1/2.0 ms, sagittal plane, fast spoiled gradient recalled pulse sequence, 124 slices; 1.4 mm, no gap, flip angle = 25 degrees, field of view = 24 cm).
Image processing
fMRI scans were analyzed using the Functional Magnetic Resonance Imaging of the Brain (FMRIB) Software Library (Smith et al., 2004).
Preprocessing
Preprocessing included motion correction (Jenkinson, Bannister, Brady, & Smith, 2002); nonbrain removal (Smith, 2002); spatial smoothing using a Gaussian kernel of full width half-maximum 5 mm; mean-based intensity normalization of all volumes by the same factor; and highpass temporal filtering (Gaussian-weighted least-squares straight line fitting, with σ = 50.0 s). Independent component analysis (ICA) was carried out with multivariate exploratory linear optimized decomposition into independent components (MELODIC; Beckmann & Smith, 2004) to investigate the possible presence of unexpected artifacts or activation. The individual ICA/MELODIC output components were analyzed by custom software to determine which components had large activation at the periphery of the brain or “rimness.” ICA components with large activation rimness were considered to be artifact that could arise in part from subject motion. The MELODIC filter option was used to filter out the artifact components that were identified in the previous step.
Statistical processing—individual-level analysis
The filtered 4D fMRI data for each participant were then processed using the FMRI Expert Analysis Tool, Version 5.63 (individual-level analysis), to find valid activation for the reward versus fixation and nonreward versus fixation conditions. Time-series statistical analysis was carried out using the FMRIB improved linear model with local autocorrelation correction (Woolrich, Ripley, Brady, & Smith, 2001). Z (Gaussianized T/F) statistic images were thresholded using clusters determined by z > 2.3 and a (corrected) cluster significance threshold of p = .05 (Worsley, Evans, Marrett, & Neelin, 1992). Results were coregistered with the Montreal Neurological Institute standard brain (MNI152; Evans et al., 1993) using the FMRIB Linear Image Registration Tool (Jenkinson et al., 2002; Jenkinson & Smith, 2001).
Statistical processing—group-level analysis
Results of regional activation were compared in a 2 (group: control vs. externalizing) × 2 (region: anterior cingulate vs. caudate) × 2 (condition: reward vs. nonreward) repeated measures analysis of variance (ANOVA).
A priori regions of interest were defined as the ACC, the caudate, and the putamen. Regions of interest were identified using the Automated Anatomical Labeling map (Tzourio-Mazoyer et al., 2002) applied to the standardized images. Only activations in the a priori identified regions were included in analyses. Results of whole-brain analysis are reported in Table 2 for the purposes of comparison. Group analyses were conducted using FMRIB’s local analysis of mixed effects (Beckmann, Jenkinson, & Smith, 2003). FMRIB’s local analysis of mixed effects models the intersession or intersubject random-effects component of the mixed-effects variance by aggregating the lower-level (e.g., subject) covariances to generate correct summary statistics for a group-level analysis. Linear modeling was conducted in a univariate manner, fitting each voxel’s time course independently.
Table 2.
Anatomical Locations of Significant Clusters of Activation for the Whole Brain for the Group × Condition Contrast of Noreward > Reward, Externalizing > Control
| Anatomical coordinatesb |
|||||
|---|---|---|---|---|---|
| Regiona | Cluster size (voxels) | Maximum z score | x | y | z |
| Cluster 1 | 1,641** | ||||
| L-sup. front. | 4.26 | −16 | 54 | 24 | |
| R-sup. front. | 4.02 | 16 | 34 | 32 | |
| L-mid. front. | 3.67 | −18 | 48 | 26 | |
| R-sup. med. front. | 3.48 | 14 | 46 | 28 | |
| Cluster 2 | 751* | ||||
| L-angular | 4.40 | −46 | −66 | 28 | |
| L-mid. Temporal | 4.13 | −52 | −58 | 20 | |
| L-inf. parietal | 3.20 | −56 | −58 | 36 | |
Note. The first entry indicates the cluster maxima followed by local maxima located in distinct anatomical regions within the contiguous activation of each cluster. Examinations of the group average maps suggest that the significant frontal activation documented above for the externalizing–control, reward–nonreward contrast was driven by greater frontal activation in the control group during the nonreward condition, essentially reflecting a larger anatomical area of activation than identified with the anterior cingulate region of interest. L = left; R = right; sup. = superior; front. = frontal; mid. = middle; med. = medial; inf. = inferior.
No significant activation was identified for the control–externalizing, reward–nonreward contrast.
Anatomical coordinates are given in Montreal Neurological Institute standard brain space (based on the MNI 152 brain).
p < .05.
p < .001. Whole-brain corrected.
Results
Behavioral Data
As expected, groups did not differ with respect to their behavioral responses. Externalizing participants did not differ from controls in the amount of money earned (M = $45.75, SD = 3.18 and M = $45.96, SD = 4.01, respectively), F(1, 28) = 0.02, p = .88, η2 × .001. A 2 (group) × 2 (condition: reward vs. nonreward) ANOVA indicated no main effect for group, and no Group × Condition interaction for either reaction time or accuracy, all Fs(1, 27) < 0.62, all ps ≥ 0.43, all partial η2s < .02. However, there was a main effect for condition in which accuracy declined in the nonreward conditions for all groups, F(1, 27) = 7.31, p = .01, partial η2 = .21. Means of the percentage of correct responses and standard deviations for both groups during the reward trials were as follows: control group (M = .96, SD = .08) and externalizing group (M = .95, SD = .07). For the nonreward trials, accuracy data were as follows: control group (M = .93, SD = .07) and externalizing group (M = .92, SD = .07). There was no main effect for condition in reaction time, F(1, 27) = .03, p = .86, partial η2 = .001, indicating that all participants continued to engage in the task during blocks of nonreward.
Imaging Data
Group contrast analyses
In the reward versus fixation conditions, activation maps for the control participants showed significant bilateral activation in the striatum. Similar activation was observed for the externalizing group, with no significant differences observed compared with controls. In the nonreward versus fixation condition, controls showed significantly increased bilateral activation in the anterior cingulate, whereas the externalizing group continued to exhibit significant activation in the striatum. Group average maps are presented by condition in Figure 1. Comparing conditions across groups, a significant effect was found for the nonreward–reward conditions in which participants in the control group showed greater activation in the anterior cingulate than participants in the externalizing group (see Figure 2). The locations of the significant clusters illustrated in each of the figures, and associated z scores, are presented in Tables 3 and 4, respectively. In addition to the masked region of interest analyses presented in Tables 3 and 4, Table 2 contains significant clusters of activation located outside of the anterior cingulate and basal ganglia regions of interest.
Figure 1.
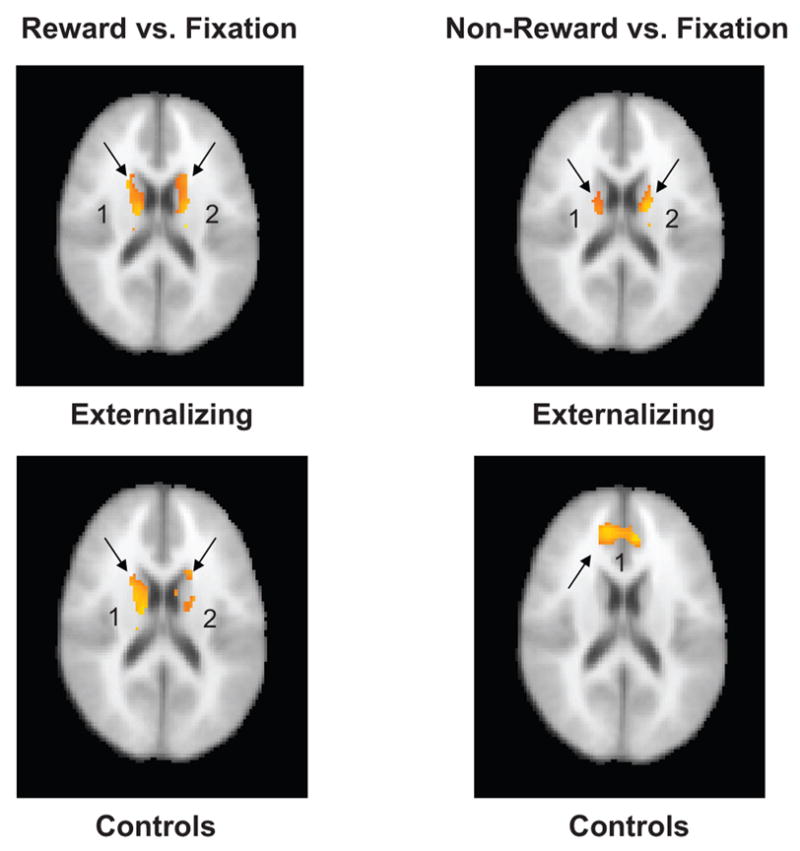
Group average maps of all participants during the reward versus fixation and nonreward versus fixation conditions. The identical axial slice (z voxel coordinate 45; 18 mm) was selected from each group map for comparison. Images are in radiologic convention (right = left, left = right). Numbers indicate, for each analysis, the cluster identified in Table 3.
Figure 2.
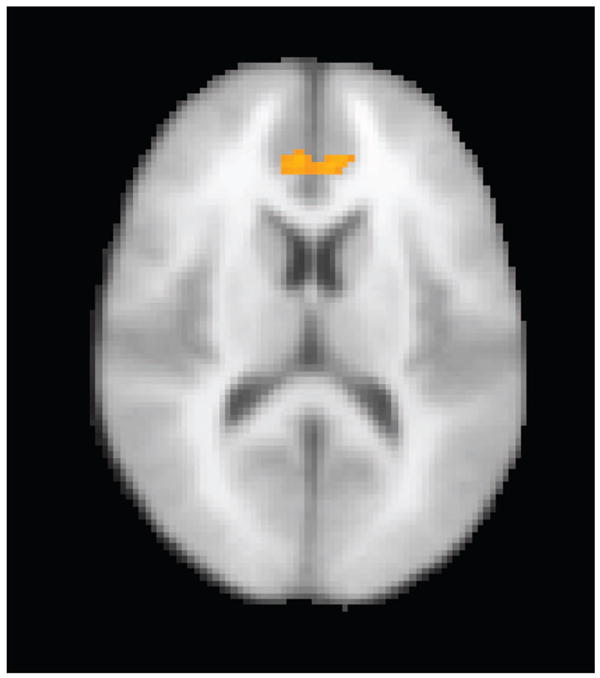
Nonreward–reward, controls–externalizing. Participants in the control group evidenced significantly greater anterior cingulate activation than participants in the externalizing group during nonreward blocks compared with reward blocks. Image is in radiologic convention (right = left, left = right).
Table 3.
Anatomical Locations of Significant Clusters of Activation Shown in Figure 1 (by Group) for Each Condition Within the Anterior Cingulate and Basal Ganglia Regions of Interest
| Anatomical coordinatesa |
|||||
|---|---|---|---|---|---|
| Region | Cluster size (voxels) | Maximum z score | X | y | z |
| Reward–fixation, externalizing | |||||
| Cluster 1 | 1,542† | ||||
| R-caudate | 5.33 | 12 | 24 | 4 | |
| R-putamen | 4.88 | 26 | 16 | 2 | |
| Cluster 2 | 1,596† | ||||
| L-caudate | 4.82 | −20 | −12 | 20 | |
| L-ant. cing. | 4.40 | −4 | 26 | −6 | |
| R-ant. cing. | 4.30 | 8 | 26 | −10 | |
|
| |||||
| Reward–fixation, control | |||||
| Cluster 1 | 315* | ||||
| R-caudate | 4.43 | 14 | 38 | 20 | |
| R-putamen | 2.95 | 24 | 4 | 14 | |
| Cluster 2 | 420*b | ||||
| R-caudate | 4.63 | 8 | 18 | 6 | |
| R-putamen | 3.60 | 26 | 16 | 2 | |
| Cluster 3 | 597** | ||||
| L-caudate | 4.01 | −8 | 20 | −4 | |
| L-putamen | 3.43 | −22 | 6 | 14 | |
|
| |||||
| Nonreward–fixation, externalizing | |||||
| Cluster 1 | 1,342† | ||||
| R-putamen | 5.51 | 30 | 16 | 2 | |
| R-caudate | 5.09 | 14 | 22 | 0 | |
| Cluster 2 | 1,383† | ||||
| L-caudate | 4.94 | −10 | 2 | 8 | |
| L-putamen | 4.37 | −20 | 20 | 2 | |
|
| |||||
| Nonreward–fixation, control | |||||
| Cluster 1 | 749* | ||||
| R-ant. cing. | 4.23 | 6 | 36 | 26 | |
| L-ant. cing. | 3.62 | −8 | 40 | 18 | |
Note. The first entry indicates the cluster maxima, followed by local maxima located in distinct anatomical regions within the contiguous activation of each cluster. R = right; L = left; ant. cing. = anterior cingulate.
Anatomical coordinates are given in Montreal Neurological Institute standard brain space (based on the MNI152 brain).
This cluster is not visible on the image in Figure 1.
p < .05.
p < .01.
p < .0001. Whole-brain corrected.
Table 4.
Anatomical Locations of Significant Cluster Activation (Shown in Figure 2) for the Group × Condition Contrast of Nonreward > Reward, Externalizing > Control Within the Anterior Cingulate Region of Interest
| Anatomical coordinatesa |
|||
|---|---|---|---|
| Maximum z score | x | y | z |
| 3.79 | 14 | 36 | 28 |
| 3.15 | 6 | 40 | 26 |
| 2.91 | 6 | 40 | 12 |
| 2.77 | −6 | 38 | 12 |
| 2.76 | −6 | 38 | 16 |
Note. Cluster size was 260 voxels. L = left; R = right. The first entry indicates the cluster maxima followed by local maxima in descending order.
Anatomical coordinates are given in Montreal Neurological Institute standard brain space (based on the MNI 152 brain).
Repeated measures analyses
A Group (controls vs. externalizing) × Region (caudate vs. anterior cingulate) × Condition (reward vs. nonreward) repeated measures ANOVA revealed a significant effect for region, F(1, 28) = 19.92, p < .001, partial η2 = .42, indicating that activation strength was different between the caudate and anterior cingulate. There was no main effect for condition or for group, both Fs < .25, both ps > .62, partial η2s < .01. However, a significant Region × Condition interaction was found, F(1, 28) = 35.04, p < .001, partial η2 = .56. As expected, the caudate and anterior cingulate showed differential patterns of activation dependent upon the delivery of reward. However, no additional second-order interactions (Region × Group or Condition × Group) were significant, all Fs < .73, all ps > .4, all partial η2s < .03. However, a significant Region × Condition × Group interaction was found, F(1, 28) = 9.16, p = .005, partial η2 = .25, suggesting that the relationship between the activation patterns in the anterior cingulate and caudate across the reward and nonreward conditions differed between groups. This pattern of results was unchanged when CD symptoms were entered as a covariate.
To elucidate the nature of this interaction, follow-up analyses were conducted separately for each condition to determine how the pattern of regional activation differed within the context of reward contingency. Analyses revealed no main effect for group and no Group × Region interaction in the reward condition, both Fs < .47, both ps > .4, both partial η2s < .02, indicating that the two groups showed comparable activation during reward. However, in the extinction condition there was a significant Group × Region interaction, F(1, 28) = 4.61, p = .04, partial η2 = .14, indicating greater activation in the anterior cingulate and lower activation in the caudate among children in the control group in comparison with children in the externalizing group. The interaction is illustrated in Figure 3.
Figure 3.

The 2 (group) × 2 (region) × 2 (condition) interaction. A: Relative activation in the anterior cingulate cortex (ACC) and caudate during the reward condition for each group. B: Relative activation in each region during the nonreward condition. Activation patterns do not differ between the groups in the reward condition but do differ significantly in the nonreward condition.
Discussion
Our primary objectives in conducting this study were to test competing theories of central nervous system reward processing in externalizing adolescents and to test the theory that externalizing psychopathology is associated with deficits in both reward and extinction processing. Results did not provide support for hypotheses regarding abnormalities in reward processing, as both groups showed comparable activation in striatal regions during receipt of incentives. However, significant differences were observed in how adolescents with externalizing disorders processed the omission of expected reward. Control participants shifted activation to the ACC when monetary incentives stopped following previously rewarded responses. In contrast, participants with externalizing disorders continued to show activation in the striatum and failed to recruit the ACC.
Previously advanced theories of reward sensitivity in children with externalizing disorders were based largely on observations of excessive stimulation-seeking behaviors (Quay, 1988). During simulated gambling tasks, children with externalizing disorders perseverate in previously rewarded responses after contingencies turn unfavorable, and they consequently earn less money than their typically developing peers (Fonseca & Yule, 1995). However, it has also been noted that externalizing children and adolescents demonstrate attenuated autonomic responses to extinction (Iaboni et al., 1997) and are less likely to respond to punishment cues once engaged in approach behaviors (e.g., Milich, Hartung, Martin, & Haigler, 1994; Newman & Wallace, 1993). Thus, it is unclear at the behavioral level whether or not these individuals are driven by an increase in appetitive motivation or a reduced sensitivity to cues of punishment.
Our study identifies some of the possible brain mechanisms responsible for perseverative responding for reward in children with externalizing disorders. In comparison with controls, adolescents with externalizing disorders failed to alter their neural activation patterns when monetary incentives were withdrawn. Thus, their continued responding to previously rewarded stimuli may be a function of aberrant processing of reward administration and a resulting failure of normal extinction processes. This finding is fully consistent with the theory of impaired extinction set forth by Sagvolden et al. (2005), resulting from a hypofunctioning mesolimbic DA system. The finding also supports a role of the anterior cingulate in processing feedback regarding the omission of expected rewards that could be used to modify behavior (Brown & Braver, 2005).
Although results from this study indicate dysfunction in processing changes in reward feedback that may reflect dopaminergic deficits, the results do not provide resolution to the hypo- versus hyperdopaminergic debate with regard to reward responsivity. Participants with externalizing disorders failed to evidence significant differences in activation during reward processing in comparison with controls, in either direction. Thus support was not provided for either theory. Rather, evidence suggests that behavioral manifestations of “reward sensitivity” may be due in part to a failure to process changes in reward contingencies.
However, it remains possible that dysfunction in reward process does characterize externalizing psychopathology in ways that were not detectable with the present design. Future research aimed at elucidating reward processing in externalizing pathology may benefit from event-related designs that allow for an examination of individual components of reward processing. Event-related designs are useful in isolating facets such as anticipation of reward and receipt of reward and in evaluating responses to relative reward amounts. For instance, the ACC is involved in coding associations between behavioral actions and reinforcement histories, whereas the orbitofrontal cortex appears to encode the emotional salience of reward and impact preferences among behavioral choices (Rushworth, Behrens, Rudebeck, & Walton, 2007), a distinction not made in the present design. With more extensive assessments, it is possible that hypo- versus hyperdopaminergic theories may prove overly simplistic with deficits in the dopaminergic response restricted to certain properties of reward processing and increased dopaminergic activation associated with other components of reward processing. Such designs will likely require imaging more extensive brain regions, assessing a broader network of structures involved in reward processing.
Several caveats should be considered in interpreting these and future research results examining theories of dysfunction in neurotransmitter systems. Although theories of CNS dysfunction advanced to explain ADHD and/or CD have focused largely on DA activity, actual neurochemical function can be difficult to assess in humans. Even though fMRI is safer and less invasive than alternatives, it assesses blood oxygenation rather than neurotransmitter activity.
Thus, care must be taken when using fMRI to address neurobiological theories developed at the cellular level of analysis, as this technique is not capable of specifying the type of neurotransmitter activity that is being altered. Data from animal models indicate that the arrival of a signal from a presynaptic cell into the synapse is responsible for the increased oxygenation identified through blood-oxygen-level–dependent fMRI (Logothetis, Pauls, Augath, Trinath, & Oeltermann, 2001). This suggests that this technique may not readily detect situations in which the neurotransmitter is released normally but fails to bind appropriately with the postsynaptic cell. Such failure could result from excessive downregulation of postsynaptic receptors or a deficiency in the receptor arising from a specific genetic anomaly. Thus, the lack of striatal differences between groups during the reward condition should not be taken as absolute evidence against such deficits proposed by the Sagvolden theory.
Indeed, psychophysiological measures of sympathetically mediated cardiac function that are thought to reflect dopaminergic striatal activity reveal deficiencies in children with CD during conditions of reward similar to the paradigm presented here (Beauchaine et al., 2007; Crone, Jennings, & van der Molen, 2003; Crowell et al., 2006; Iaboni et al., 1997). These cardiac data support the deficient DA response to reward hypothesis. However, event-related fMRI is useful in examining neural activity on a shorter time scale than cardiac psychophysiological techniques, which require averaging over longer epochs. Thus, multiple techniques may be required to fully elucidate the neural correlates of reward responding in humans and should be carefully matched to the specific question being addressed.
There are also developmental issues to consider in interpreting results. For instance, in at least some studies, longitudinal research examining the structural development of brain regions indicates that group differences in caudate structure that have been replicated across research studies in younger samples may normalize during adolescence (Castellanos et al., 2002). It is therefore possible that additional striatal deficits may be observed in younger samples, emphasizing the importance of considering age when comparing results across multiple studies.
The present study suggests that children with externalizing disorders have difficulty in processing changes in established reward contingencies. Although the pattern of results remained when CD symptoms were entered as a covariate, future research might compare subtypes of externalizing disorders to establish this as a common vulnerability across multiple externalizing pathologies. Future research will also be needed to determine whether or not this pattern is characteristic of females with externalizing disorders.
Acknowledgments
We thank Ly Nguyen, Ben Davis, Amelia Bachleda, and Jessica Chao for their contributions to the execution of this study, as well as Jeff Stevenson and Jenee O’Brien for their technical assistance and expertise in the imaging portion of the study. Work on this article was supported by Grant R01 MH63699 from the National Institute of Mental Health and Grant 3289 from the University of Washington Royalty Research Fund to Theodore P. Beauchaine.
Contributor Information
Lisa M. Gatzke-Kopp, Department of Human Development and Family Studies, Pennsylvania State University
Theodore P. Beauchaine, Department of Psychology, University of Washington
Katherine E. Shannon, Department of Psychology, University of Washington
Jane Chipman, Department of Psychology, University of Washington.
Andrew P. Fleming, Department of Psychology, University of Washington
Sheila E. Crowell, Department of Psychology, University of Washington
Olivia Liang, Department of Radiology, University of Washington.
Elizabeth Aylward, Department of Radiology, University of Washington.
L. Clark Johnson, Department of Psychosocial and Community Health, University of Washington.
References
- Achenbach T. Manual for the Child Behavior Checklist/4–18 and 1991 profile. Burlington: University of Vermont, Department of Psychiatry; 1991. [Google Scholar]
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 4. Washington DC: American Psychiatric Association; 2000. [Google Scholar]
- Ashby G, Isen A, Turken A. A neuropsychological theory of positive affect and its influence on cognition. Psychological Review. 1999;106:529–550. doi: 10.1037/0033-295x.106.3.529. [DOI] [PubMed] [Google Scholar]
- Beauchaine TP. Vagal tone, development, and Gray’s motivational theory: Toward an integrated model of autonomic nervous system functioning in psychopathology. Development and Psychopathology. 2001;13:183–214. doi: 10.1017/s0954579401002012. [DOI] [PubMed] [Google Scholar]
- Beauchaine TP, Gatzke-Kopp LM, Mead HK. Polyvagal theory and developmental psychopathology: Emotion dysregulation and conduct problems from preschool to adolescence. Biological Psychology. 2007;74:174–184. doi: 10.1016/j.biopsycho.2005.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauchaine TP, Hong J, Marsh P. Sex differences in autonomic correlates of conduct problems and aggression. Journal of the American Academy of Child and Adolescent Psychiatry. 2008;47:788–796. doi: 10.1097/CHI.0b013e318172ef4b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauchaine T, Katkin E, Strassberg Z, Snarr J. Disinhibitory psychopathology in male adolescents: Discriminating conduct disorder from attention-deficit/hyperactivity disorder through concurrent assessment of multiple autonomic states. Journal of Abnormal Psychology. 2001;110:610–624. doi: 10.1037//0021-843x.110.4.610. [DOI] [PubMed] [Google Scholar]
- Beauchaine TP, Neuhaus E. Impulsivity and vulnerability for psychopathology. In: Beauchaine TP, Hinshaw S, editors. Child psychopathology. Hoboken, NJ: Wiley; 2008. pp. 129–156. [Google Scholar]
- Beckmann C, Jenkinson M, Smith SM. General multi-level linear modeling for group analysis in fMRI. NeuroImage. 2003;20:1052–1063. doi: 10.1016/S1053-8119(03)00435-X. [DOI] [PubMed] [Google Scholar]
- Beckmann CF, Smith SM. Probabilistic independent component analysis for functional magnetic resonance imaging. IEEE Transactions in Medical Imaging. 2004;23:137–152. doi: 10.1109/TMI.2003.822821. [DOI] [PubMed] [Google Scholar]
- Brown JW, Braver TS. Learned predictions of error likelihood in the anterior cingulate cortex. Science. 2005;307:1059–1060. doi: 10.1126/science.1105783. [DOI] [PubMed] [Google Scholar]
- Castellanos FX, Lee PP, Sharp W, Jeffries NO, Greenstein DK, Clasen LS, et al. Developmental trajectories of brain volume abnormalities in children and adolescents with attention-deficit/hyperactivity disorder. Journal of the American Medical Association. 2002;288:1740–1748. doi: 10.1001/jama.288.14.1740. [DOI] [PubMed] [Google Scholar]
- Cloninger C. A systematic method for clinical description and classification of personality variants: A proposal. Archives of General Psychiatry. 1987;44:573–588. doi: 10.1001/archpsyc.1987.01800180093014. [DOI] [PubMed] [Google Scholar]
- Cloninger CR, Svrakic NM, Svrakic DM. Role of personality self-organization in development of mental order and disorder. Development and Psychopathology. 1997;9:881–906. doi: 10.1017/s095457949700148x. [DOI] [PubMed] [Google Scholar]
- Crone EA, Jennings JR, van der Molen MW. Sensitivity to interference and response contingencies in attention-deficit/hyperactivity disorder. Journal of Child Psychology and Psychiatry. 2003;44:214–226. doi: 10.1111/1469-7610.00115. [DOI] [PubMed] [Google Scholar]
- Crowell S, Beauchaine TP, Gatzke-Kopp LM, Sylvers P, Mead H, Chipman-Chacon J. Autonomic correlates of attention-deficit/hyperactivity disorder and oppositional defiant disorder in pre-school children. Journal of Abnormal Psychology. 2006;115:174–178. doi: 10.1037/0021-843X.115.1.174. [DOI] [PubMed] [Google Scholar]
- De Witte P, Pinto E, Ansseau M, Verbanck P. Alcohol and withdrawal: From animal research to clinical issues. Neuroscience and Biobehavioural Reviews. 2003;27:189–197. doi: 10.1016/s0149-7634(03)00030-7. [DOI] [PubMed] [Google Scholar]
- DiMaio S, Frizenko N, Joober R. Dopamine genes and attention deficit-hyperactivity disorder: A review. Journal of Psychiatry and Neuroscience. 2003;28:27–38. [PMC free article] [PubMed] [Google Scholar]
- Evans AC, Collins DL, Mills SR, Brown ED, Kelly RL, Peters TM. 3D statistical neuroanatomical models from 305 MRI volumes. Proceedings of the Institute of Electrical and Electronics Engineers—Nuclear Science Symposium and Medical Imaging Conference. 1993;3:1813–1817. [Google Scholar]
- Fonseca AC, Yule W. Personality and antisocial behavior in children and adolescents: An enquiry into Eysenck’s and Gray’s theories. Journal of Abnormal Child Psychology. 1995;23:767–781. doi: 10.1007/BF01447476. [DOI] [PubMed] [Google Scholar]
- Fowles DC. The three arousal model: Implications of Gray’s two-factor learning theory for heart rate, electrodermal activity, and psychopathy. Psychophysiology. 1980;17:87–104. doi: 10.1111/j.1469-8986.1980.tb00117.x. [DOI] [PubMed] [Google Scholar]
- Gadow K, Sprafkin J. Adolescent Symptom Inventory 4 screening manual. Stony Brook, NY: Checkmate Plus; 1997. [Google Scholar]
- Garris PA, Rebec GV. Modeling fast dopamine neurotransmission in the nucleus accumbens during behavior. Behavioural Brain Research. 2002;137:47–63. doi: 10.1016/s0166-4328(02)00284-x. [DOI] [PubMed] [Google Scholar]
- Gatzke-Kopp LM, Beauchaine TP. Central nervous system substrates of impulsivity. In: Coch D, Dawson G, Fischer K, editors. Human behavior, learning, and the developing brain: Atypical development. New York: Guilford Press; 2007. pp. 239–263. [Google Scholar]
- Gray J. The neuropsychology of emotion and personality. In: Stahl SM, Iversen SD, Goodman EC, editors. Cognitive neurochemistry. Oxford, England: Oxford University Press; 1987. pp. 171–190. [Google Scholar]
- Holroyd CB, Coles MGH. The neural basis of human error processing: Reinforcement learning, dopamine, and the error-related negativity. Psychological Review. 2002;109:679–709. doi: 10.1037/0033-295X.109.4.679. [DOI] [PubMed] [Google Scholar]
- Iaboni F, Douglas V, Ditto B. Psychophysiological response of ADHD children to reward and extinction. Psychophysiology. 1997;34:116–123. doi: 10.1111/j.1469-8986.1997.tb02422.x. [DOI] [PubMed] [Google Scholar]
- Jenkinson M, Bannister P, Brady M, Smith S. Improved optimisation for the robust and accurate linear registration and motion correction of brain images. Neuroimage. 2002;17:825–841. doi: 10.1016/s1053-8119(02)91132-8. [DOI] [PubMed] [Google Scholar]
- Jenkinson M, Smith SM. A global optimisation method for robust affine registration of brain images. Medical Image Analysis. 2001;5:143–156. doi: 10.1016/s1361-8415(01)00036-6. [DOI] [PubMed] [Google Scholar]
- Kaufman AS, Kaufman NL. Kaufman Brief Intelligence Test. 2. Circle Pines, MN: AGS Publishing; 2004. [Google Scholar]
- Klein RG, Abikoff H, Klass E, Ganeles D, Seese LM, Pollack S. Clinical efficacy of methylphenidate in conduct disorder with and without attention deficit hyperactivity disorder. Archives of General Psychiatry. 1997;54:1073–1080. doi: 10.1001/archpsyc.1997.01830240023003. [DOI] [PubMed] [Google Scholar]
- Krueger RF, Hicks BM, Patrick CJ, Carlson SR, Iacono WG, McGue M. Etiologic connections among substance dependence, antisocial behavior, and personality: Modeling the externalizing spectrum. Journal of Abnormal Psychology. 2002;111:411–424. [PubMed] [Google Scholar]
- Krueger RF, Markon KE, Patrick CJ, Benning SD, Kramer MD. Linking antisocial behavior, substance use, and personality: An integrative quantitative model of the adult externalizing spectrum. Journal of Abnormal Psychology. 2007;116:645–666. doi: 10.1037/0021-843X.116.4.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laakso A, Wallius E, Kajander J, Bergman J, Eskola O, Solin O, et al. Personality traits and striatal dopamine synthesis capacity in healthy subjects. American Journal of Psychiatry. 2003;160:904–910. doi: 10.1176/appi.ajp.160.5.904. [DOI] [PubMed] [Google Scholar]
- Laine TPJ, Ahonen A, Räsänen P, Tiihonen J. Dopamine transporter density and novelty seeking among alcoholics. Journal of Addictive Disease. 2001;20(4):95–100. doi: 10.1300/j069v20n04_08. [DOI] [PubMed] [Google Scholar]
- Leyton M, Boileau I, Benkelfat C, Diksic M, Baker G, Dagher A. Amphetamine-induced increases in extracellular dopamine, drug wanting and novelty seeking: A PET/[11C]Raclopride study in healthy men. Neuropsychopharmacology. 2002;27:1027–1035. doi: 10.1016/S0893-133X(02)00366-4. [DOI] [PubMed] [Google Scholar]
- Ljungberg T, Apicella P, Schultz W. Responses of monkey dopamine neurons during learning of behavioral reactions. Journal of Neurophysiology. 1992;67:145–163. doi: 10.1152/jn.1992.67.1.145. [DOI] [PubMed] [Google Scholar]
- Logothetis NK, Pauls J, Augath M, Trinath T, Oeltermann A. Neurophysiological investigation of the basis of the fMRI signal. Nature. 2001;412:128–130. doi: 10.1038/35084005. [DOI] [PubMed] [Google Scholar]
- Lynam D, Caspi A, Moffitt TE, Wikström POH, Loeber R, Novak S. The interaction between impulsivity and neighborhood context on offending: The effects of impulsivity are stronger in poorer neighborhoods. Journal of Abnormal Psychology. 2000;109:563–574. doi: 10.1037//0021-843x.109.4.563. [DOI] [PubMed] [Google Scholar]
- Marsden CD, Obeso JA. The functions of the basal ganglia and the paradox of stereotaxic surgery in Parkinson’s disease. Brain. 1994;117:877–897. doi: 10.1093/brain/117.4.877. [DOI] [PubMed] [Google Scholar]
- Martin-Soelch C, Leenders KL, Chevalley A-F, Missimer J, Kunig S, Magyar A, et al. Reward mechanisms in the brain and their role in dependence: Evidence from neurophysiological and neuroimaging studies. Brain Research Reviews. 2001;36:139–149. doi: 10.1016/s0165-0173(01)00089-3. [DOI] [PubMed] [Google Scholar]
- Milich R, Hartung CM, Martin CA, Haigler ED. Behavioral disinhibition and underlying processes in adolescents with disruptive behavior disorders. In: Routh DK, editor. Disruptive behavior disorders in childhood. New York: Plenum Press; 1994. pp. 109–138. [Google Scholar]
- Milner P. The autonomous brain. Mahwah, NJ: Erlbaum; 1999. [Google Scholar]
- Newman JP, Wallace JF. Diverse pathways to deficient self-regulation: Implications for disinhibitory psychopathology in children. Clinical Psychology Review. 1993;13:699–720. [Google Scholar]
- O’Brien B, Frick P. Reward dominance: Associations with anxiety, conduct problems, and psychopathy in children. Journal of Abnormal Child Psychology. 1996;24:223–241. doi: 10.1007/BF01441486. [DOI] [PubMed] [Google Scholar]
- Patrick KS, Markowitz JS. Pharmacology of methylphenidate, amphetamine enantiomers and pemoline in attention-deficit hyper-activity disorder. Human Psychopharmacology: Clinical and Experimental. 1997;12:527–546. [Google Scholar]
- Patterson GR, DeGarmo DS, Knutson N. Hyperactive and antisocial behaviors: Comorbid or two points in the same process? Development and Psychopathology. 2000;12:91–106. doi: 10.1017/s0954579400001061. [DOI] [PubMed] [Google Scholar]
- Pliszka SR, Glahn DC, Semrud-Clikeman M, Franklin C, Perez R, Xiong J, et al. Neuroimaging of inhibitory control areas in children with attention deficit hyperactivity disorder who were treatment naive or in long-term treatment. American Journal of Psychiatry. 2006;163:1052–1060. doi: 10.1176/ajp.2006.163.6.1052. [DOI] [PubMed] [Google Scholar]
- Quay H. The behavioral reward and inhibition system in childhood behavior disorder. In: Bloomingdale LM, editor. Attention deficit disorder: New research in attention, treatment and psychopharmacology. Vol. 3. Oxford, England: Pergamon Press; 1988. pp. 176–186. [Google Scholar]
- Rushworth MFS, Behrens TEJ, Rudebeck PH, Walton ME. Contrasting roles for cingulate and orbitofrontal cortex in decisions and social behavior. TRENDS in Cognitive Sciences. 2007;11:168–176. doi: 10.1016/j.tics.2007.01.004. [DOI] [PubMed] [Google Scholar]
- Sagvolden T, Aase H, Zeiner P, Berger D. Altered reinforcement mechanisms in attention-deficit/hyperactivity disorder. Behavioural Brain Research. 1998;94:61–71. [PubMed] [Google Scholar]
- Sagvolden T, Johansen E, Aase H, Russell V. A dynamic developmental theory of attention-deficit/hyperactivity disorder (ADHD) predominantly hyperactive/impulsive and combined subtypes. Behavioural and Brain Sciences. 2005;28:397–468. doi: 10.1017/S0140525X05000075. [DOI] [PubMed] [Google Scholar]
- Schneider W, Eschman A, Zuccolotto A. E-Prime (Version 1.1) Pittsburgh, PA: Psychology Software Tools, Inc; 2002. [Google Scholar]
- Schultz W, Apicella P, Scarnati E, Ljungberg T. Neuronal activity in monkey ventral striatum related to the expectation of reward. Journal of Neuroscience. 1992;12:4595–4610. doi: 10.1523/JNEUROSCI.12-12-04595.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer D, Fisher P, Lucas CP, Mina K, Schwab-Stone ME. NIMH Diagnostic Interview Schedule for Children Version IV (NIMH DISC-IV): Description, differences from previous versions, and reliability of some common diagnoses. Journal of the American Academy of Child and Adolescent Psychiatry. 2000;39:28–38. doi: 10.1097/00004583-200001000-00014. [DOI] [PubMed] [Google Scholar]
- Smith S. Fast robust automated brain extraction. Human Brain Mapping. 2002;17:143–155. doi: 10.1002/hbm.10062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SM, Jenkinson M, Woolrich MW, Beckmann CF, Behrens TEJ, Johansen-Berg H, et al. Advances in functional and structural MR image analysis and implementation as FSL. NeuroImage. 2004;23(Suppl 2):S208–S219. doi: 10.1016/j.neuroimage.2004.07.051. [DOI] [PubMed] [Google Scholar]
- Sullivan R, Brake W. What the rodent prefrontal cortex can teach us about attention-deficit/hyperactivity disorder: The critical role of early developmental events on prefrontal function. Behavioural Brain Research. 2003;146:43–55. doi: 10.1016/j.bbr.2003.09.015. [DOI] [PubMed] [Google Scholar]
- Swanson JM, Castellanos FX. Biological bases of ADHD—Neuroanatomy, genetics, and pathophysiology. In: Jenson PS, Cooper JR, editors. Attention deficit hyperactivity disorder. Kingston, NJ: Civic Research Institute; 2002. pp. 1–20. [Google Scholar]
- Tisch S, Silberstein P, Limousin-Dowsey P, Jahanshahi M. The basal ganglia: Anatomy, physiology, and pharmacology. Psychiatric Clinics of North America. 2004;27:757–799. doi: 10.1016/j.psc.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Tzourio-Mazoyer N, Landeau B, Papathanassiou D, Crivello F, Etard O, Delcroiz N, et al. Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. NeuroImage. 2002;15:273–289. doi: 10.1006/nimg.2001.0978. [DOI] [PubMed] [Google Scholar]
- van Goozen SHM, Fairchild G, Snoek H, Harold GT. The evidence for a neurobiological model of childhood antisocial behavior. Psychological Bulletin. 2007;133:149–182. doi: 10.1037/0033-2909.133.1.149. [DOI] [PubMed] [Google Scholar]
- Vles JS, Feron FJ, Hendriksen JG, Jolles J, van Kronnenburgh MJ, Weber WE. Methylphenidate down-regulates the dopamine receptor and transporter system in children with attention deficit hyperkinetic disorder (ADHD) Neuropediatrics. 2003;34:77–80. doi: 10.1055/s-2003-39602. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Fowler JS, Wang G, Ding Y, Gatley SJ. Mechanism of action of methylphenidate: Insights from PET imaging studies. Journal of Attention Disorders. 2002;6:S31–S43. doi: 10.1177/070674370200601s05. [DOI] [PubMed] [Google Scholar]
- Wise RA. Dopamine, learning and motivation. Nature Reviews Neuroscience. 2004;5:1–12. doi: 10.1038/nrn1406. [DOI] [PubMed] [Google Scholar]
- Woolrich MW, Ripley BD, Brady JM, Smith SM. Temporal autocorrelation in univariate linear modeling of fMRI data. Neuroimage. 2001;14:1370–1386. doi: 10.1006/nimg.2001.0931. [DOI] [PubMed] [Google Scholar]
- Worsley KJ, Evans AC, Marrett S, Neelin P. A three-dimensional statistical analysis for CBF activation studies in human brain. Journal of Cerebral Blood Flow Metabolism. 1992;12:900–918. doi: 10.1038/jcbfm.1992.127. [DOI] [PubMed] [Google Scholar]