

Abstract
Non-tropane-based photoaffinity ligands for the dopamine transporter (DAT) are relatively unexplored in contrast to tropane-based compounds such as cocaine. In order to fill this knowledge gap, a ligand was synthesized in which the aromatic ring of pyrovalerone was substituted with a photoreactive azido group. The analog 1-(4-azido-3-iodophenyl)-2-pyrrolidin-1-yl-pentan-1-one demonstrated appreciable binding affinity for the DAT (Ki = 78 ± 18 nM), suggesting the potential utility of a radioiodinated version in structure-function studies of this protein.
Keywords: pyrovalerone, photoaffinity labeling, dopamine transporter, cocaine
1. Introduction
Despite decades of committed research, no FDA approved medications are clinically available to combat psychostimulant abuse and addiction.1 Pharmacological and behavioral studies indicate that the dopamine transporter (DAT) is the brain receptor chiefly responsible for the reward/reinforcing properties associated with amphetamines2 and cocaine.3 There are a plethora of structurally heterogeneous ligands that are known to bind to the DAT and inhibit the uptake of dopamine;4 however, details regarding the transport inhibition mechanism and the discrete ligand binding pockets remain poorly understood. As a result, the synthesis of compounds towards elucidating conformational states and structural elements associated with the DAT, namely via probing the interactions of substrates and inhibitors with this protein, remains an important objective in the search for psychostimulant abuse therapeutics.
Results from structure-activity relationship (SAR) studies and site-directed mutagenesis experiments imply that structurally disparate inhibitors bind to different domains or binding sites within the DAT.5–7 Additionally, it has been suggested that the binding of inhibitors to distinct DAT domains could affect their behavioral profile in cocaine abuse animal models.6 As a result, radiolabeled (3H, 125I) affinity (-NCS) and photoaffinity (-N3) irreversible ligands continue to remain pertinent towards mapping substrate and inhibitor DAT binding sites at the amino acid level. The chemical development of DAT irreversible ligands to date has predominantly focused on tropane-based ligands8–16 and their conformationally flexible piperidine17 and piperazine18–22 analogs. [125I]-MFZ-2-24 (1, Figure 1), an irreversible tropane-based cocaine analog, demonstrated covalent ligation via photoaffinity labeling to the more intracellular-proximal half of transmembrane domain (TM) 1 within a thirteen-amino acid sequence.23 However, [125I]-RTI-82 (2), which possesses the same tropane pharmacophore found in MFZ-2-24 but features the photoreactive azide moiety anchored off the ester rather than the tropane nitrogen, demonstrated incorporation into TM6.24 Collectively these probes provide evidence of the close 3-D proximity of DAT TMs 1 and 6. Additionally, piperazine analog [125I]-DEEP (3), a conformationally flexible analog of the tropane-based benztropine class of DAT inhibitors,6 also demonstrated incorporation into TM 1–2.25
Figure 1.

Representative examples of tropane- and piperazine-based DAT photoaffinity ligands.
Structurally heterologous non-tropane-based compounds have received significantly less attention versus tropane-based ligands in terms of their development into DAT irreversible probes. In an effort to expand the arsenal of complementary chemical probes useful for characterizing DAT 3-D structure, we report herein the design, synthesis, and preliminary labeling studies of a photoaffinity probe based on pyrovalerone (5, Figure 2), a modestly selective inhibitor of the dopamine transporter over the norepinephrine transporter with little effect upon serotonin trafficking.
Figure 2.

Structural relationship between bupropion (4), pyrovalerone (5), and target photoaffinity compound 6.
Our interest in pyrovalerone stems from its structural resemblance to bupropion (4), a well-known drug marketed as a smoking-cessation agent (Zyban) and antidepressant (Wellbutrin). More recently, bupropion as a dual norepinephrine and dopamine reuptake inhibitor has attracted significant attention as a pharmacotherapeutic for methamphetamine dependence.1, 26 However, determination of the DAT conformational states and binding sites for bupropion and structurally related compounds is in its infancy.27 With respect to development of this structural class of inhibitors into potential DAT photoaffinity probes, it is demonstrated herein that pyrovalerone displays markedly higher binding affinity than bupropion at the DAT. Additionally, structure-activity relationship studies performed by Meltzer and colleagues indicate that pyrovalerone’s aromatic ring is able to tolerate a wide range of substitutions in terms of retaining appreciable DAT affinity.28 Thus, the present study seeks to develop pyrovalerone into a compact DAT photoaffinity probe for providing high resolution structural information regarding its binding site. This work also further explores the therapeutic potential of its analogs.
2. Results and Discussion
2.1. Chemistry
Target compound 6 was prepared in six overall steps via synthetic methodology common to the construction of DAT photoaffinity ligands (Scheme 1). First, N-[4-(2-pyrrolidin-1-yl-pentanoyl)phenyl acetamide (7) was synthesized via Friedel-Crafts acylation, α-bromination, and pyrrolidine displacement as previously described.28 Amide hydrolysis of 7 under acidic conditions provided the aniline pyrovalerone derivative 8 in moderate yield (43%). Regioselective electrophilic aromatic iodination was then achieved using ICl in acetic acid as previously described.10 Finally, conversion of 9 to target azido-iodo analog 6 was accomplished in good yield (93%) via diazotization with nitrous acid and displacement with sodium azide.
Scheme 1.

Synthesis of target compound 6.
2.2. Pharmacology
With pyrovalerone derivatives 6 – 9 in hand, ligand affinities (Ki values) were determined for inhibition of [3H]-WIN-35,428 (a cocaine analog) binding to hDAT in N2A neuroblastoma cells. [3H]-Dopamine uptake inhibition potencies in the same cells under the same conditions were also determined (Table 1). Racemic bupropion (4) and pyrovalerone (5) were also synthesized28, 29 and pharmacologically evaluated for comparison to the novel compounds. Replacement of the 4-Me group in pyrovalerone with 4-NHAc slightly reduced DAT binding affinity.28 However, hydrolysis of the amide to the corresponding aniline results in a compound (8) with high DAT affinity comparable to pyrovalerone. Addition of the 3-I group resulted in ~6-fold decrease in binding affinity for the DAT while replacing the aniline NH2 with the N3 group further decreased affinity by 2.5-fold. The 78 nM DAT affinity for target compound 6 was 6-fold higher than bupropion yet 10-fold less than pyrovalerone, retaining substantial DAT affinity that justified its further development into a potential DAT photoaffinity probe. Uptake inhibition IC50 values (Table 1) were typically 3 to 4-fold higher than the binding Ki values for each compound (using the Cheng-Prusoff equation, conversion of uptake inhibition constants from IC50 to Ki did not significantly change the value, allowing for direct comparison of binding and uptake results). This 3 to 4-fold shift was previously observed with rDAT/CHO cells in this laboratory for WIN 35,428, cocaine, mazindol, and methylphenidate.7 Interestingly, compound 8 displayed essentially the same value for binding and uptake inhibition (Table 1), a pattern previously seen for benztropine and the related compounds GBR-12,909 and rimcazole.7 Cocaine and benztropine have been suggested to occupy nonidentical DAT sites or conformations;5–7 the present result may imply that compound 8 also interacts with the DAT in a fashion different from the other compounds in Table 1.
Table 1.
Inhibition of [3H]-WIN 35,428 binding and [3H]-dopamine uptake of compounds at hDAT N2A neuroblastoma cells.
| Compound # | Aromatic Substituent | [3H]-WIN Binding Inhibition Ki (nM)a | [3H]-DA Uptake Inhibition IC50 (nM)a |
|---|---|---|---|
| 4 (bupropion) | 3-Cl | 441 ± 174 | 1567 ± 716 |
| 5 (pyrovalerone) | 4-Me | 8 ± 2 | 32 ± 8 |
| 6 | 4-N3-3-I | 78 ± 18 | 264 ± 78 |
| 7 | 4-NHAc | 30.2 ± 2.0b | 67.9 ± 8.4b |
| 8 | 4-NH2 | 5 ± 1 | 7 ± 2 |
| 9 | 4-NH2-3-I | 28 ± 8 | 175 ± 59 |
Each Ki or IC50 value represents data from at least three independent experiments with each data point on the curve performed in duplicate.
From Reference 28.
2.3. Radiosynthesis
Given that 6 demonstrated reasonably high DAT affinity, and that wash-resistant binding experiments on nonradioactive azido compounds frequently give false positives in the assessment of covalent attachment,14 [125I]-6 was directly synthesized. The radioiodo compound could then be used to determine if photoactivation produced covalent ligation to the DAT. As shown in Scheme 2, a one-flask synthesis of [125I]-6 was performed using methodology previously described in detail for the preparation of radioiodinated cocaine analogs as DAT photoaffinity labels.11 Briefly, electrophilic radioiodination of 8 with [125I]-NaI (1.67 mCi) under no-carrier-added conditions using Chloramine-T as the oxidant was followed by diazotization and subsequent treatment with sodium azide. Although this sequence ending with reversed-phase HPLC isolation provided [125I]-6 in only 20% isolated yield, high purity (>99%) and high specific activity (1946 mCi/μmol) were achieved. The radioligand exhibited a chromatographic profile identical to that of non-radioactive 6 (Figure 3) and showed good stability upon prolonged storage at −20 °C in the dark (92% radiochemical purity after 25 days). Figure 3A shows the preparative HPLC trace where [125I]-6 (tR = 20.0 min) was well resolved from radioactive and nonradioactive side-products. The major non-radioactive materials are assigned as the azide (tR = 6.2 min) and chloroazide (tR = 13.0 min) congeners based upon model studies conducted in the presence and in the absence of Chloramine-T. Figure 3B shows co-elution of purified [125I]-6 with a fully characterized sample of non-radioactive 6 under the same reversed-phase HPLC conditions.
Scheme 2.

Synthesis of [125I]-6.
Figure 3.

Panel A. Reversed-phase HPLC chromatogram for isolation of [125I]-6. The radioligand exhibited an appropriate retention time based on the non-radioactive standard and was well resolved from radioactive and non-radioactive side-products. Panel B. Co-elution of purified [ 125I]-6 and a standard sample of 6 under the same HPLC conditions used for preparative work.
2.4. DAT Photoaffinity Labeling
We next turned our attention to preliminary photoaffinity labeling of DAT with [125I]-6. LLC-PK1 cells expressing rDAT and HEK 293 cells expressing 6Xhis-hDAT were photoaffinity labeled with [125I]-6 in the absence or presence of 100 μM (−)-cocaine. The cells were then solubilized and the lysates were subjected to immunoprecipitation followed by SDS-PAGE and autoradiography according to previously described procedures.30 Labeled proteins of ~80 kDa were obtained from each lysate, demonstrating the incorporation of [125I]-6 into the DAT (Figure 4). For both samples, photolabeling was blocked by cocaine, suggesting that [125I]-6 labels the DAT as opposed to a nonspecific cell surface site.30
Figure 4.
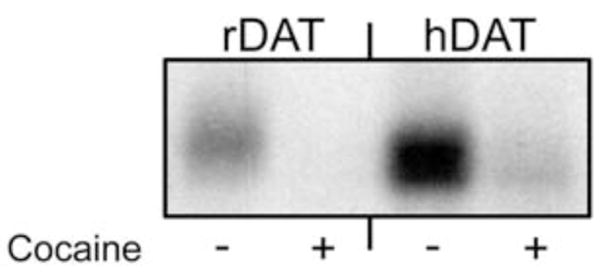
Photoaffinity labeling of DAT with [125I]-6. Cells expressing rDAT or 6X-his- hDAT were photoaffinity-labeled with [125I]-6 in the absence or presence of 100 μM (−)-cocaine. Cells were solubilized and DATs were immunoprecipitated with DAT antibody 16 (rDAT) or anti-his antibody (hDAT) followed by SDS-PAGE and autoradiography.
It should be noted that in addition to a photoreactive azide functional group, compound 6 also contains a phenyl ketone structural motif. The carbonyl of such a group could also result in electron disproportionation upon photolysis, as such chemistry is traditionally displayed by benzophenone functional groups in photoaffinity labeling.31 Thus, the involvement of this carbonyl carbon (versus the presumed nitrene resulting from azide photolysis) remains a possibility with respect to covalent ligation to the DAT.
Additionally, successful adduction of [125I]-6 to the DAT suggests that this non-tropane ligand tolerates direct substitution of a photoreactive azido group on the aromatic ring of the inhibitor scaffold. This contrasts with designs of tropane-based irreversible compounds in which the azide is placed at a distance (usually via a linker) from the inhibitor pharmacophore in order to achieve successful protein labeling.10 Thus, compact irreversible ligands based on a pyrovalerone scaffold may offer the advantage of a shorter tether between compound functional groups and DAT residues in or near the inhibitor binding site.
3. Conclusion
We have synthesized and pharmacologically evaluated a novel photoaffinity ligand (6) based on the non-tropane DAT inhibitor pyrovalerone. This is the first example of a DAT irreversible ligand based on the 2-substituted aminopentanophenone class of monoamine uptake inhibitors, thus representing an important contribution to the growing battery of probes for characterizing DAT function and 3-D structure. There is evidence that DAT inhibitors bind to nonidentical DAT sites or conformations,5–7 suggesting that novel crosslinking agents based on this structural class may yield new monoamine transporter structure-function information. Ligand 6 bound with relatively high affinity to the DAT and its 125I analog was shown to bind covalently to rDAT and hDAT expressed in cultured cells. It should be noted that the S-isomer of pyrovalerone has been found to be the more biologically active compound;28 thus, attempts to resolve racemic 6 into its enantiomerically pure components are underway toward obtaining a specific and improved DAT photoaffinity probe. Future directions include development of additional DAT irreversible ligands based on the 2-substituted aminopentanophenone structural class, their pharmacological characterization, and detailed elucidation of DAT binding domains for comparison to established tropane-based probes.
4. Experimental
All solvents and chemicals were purchased from Aldrich Chemical Co. or Fisher Scientific and used without further purification. All column chromatography was performed using Fisher S826-25 silica gel sorbent (70–230 mesh) and eluting solvent mixtures as specified. Thin-layer chromatography (TLC) was performed using TLC silica gel 60 F254 plates obtained from EMD Chemicals, Inc. and compounds were visualized under UV light. Proportions of solvents used for TLC are by volume. The 1H and 13C NMR spectra were recorded on a Bruker 400 MHz spectrometer. Chemical shifts for 1H and 13C NMR spectra are reported as parts per million (δ ppm) relative to tetramethylsilane (0.00 ppm) as an internal standard. Coupling constants are measured in Hz. HRMS samples were analyzed by positive ion electrospray on a Bruker 12 Tesla APEX -Qe FTICR-MS with an Apollo II ion source. Chemical names follow IUPAC nomenclature. A radioisotope dose calibrator (Capintec CRC-15W) was used for radioactivity measurements and similar counting geometries were employed for each determination.
4.1. 1-(4-Aminophenyl)-2-pyrrolidin-1-yl-pentan-1-one (8)
A solution of N-[4-(2-pyrrolidin-1-yl-pentanoyl)phenyl]acetamide28 (7, 183 mg, 0.63 mmol) in 1M aq HCl (7 ml) was refluxed for 15 h. The mixture was cooled to room temperature, diluted with water, carefully alkalinized with K2CO3, and extracted with EtOAc. The organic layer was washed with brine, dried (MgSO4), filtered, concentrated, then chromatographed (hexanes:EtOAc:Et3N, 60:38:2) to provide 67 mg of aniline 8 as a yellow oil (43%). Rf = 0.24 (hexanes:EtOAc:Et3N, 60:38:2). 1H NMR (CDCl3, 400 MHz) δ 7.89 (d, 2H, J= 8.7 Hz), 6.56 (d, 2H, J= 8.8 Hz), 4.53 (bs, 2H), 3.78 (m, 1H), 2.60 (m, 2H), 2.48 (m, 2H), 1.82 (m, 1H), 1.67 (m, 5H), 1.17 (m, 2H), 0.77 (t, 3H, J= 7.3 Hz). 13C NMR (CDCl3, 100 MHz) δ 199.3, 151.9, 131.1, 126.9, 113.5, 68.1, 51.3, 33.9, 23.2, 19.3, 14.3. HRMS calcd for C15H23N2O+ 247.1805, found 247.1804.
4.2. 1-(4-Amino-3-iodophenyl)-2-pyrrolidin-1-yl-pentan-1-one (9)
ICl (426 mg, 2.62 mmol) was added to a solution of 8 (577 mg, 2.34 mmol) in glacial AcOH (41.5 mL). The mixture was stirred for 16 h at room temperature then concentrated to dryness. The residue was partitioned between H2O and CHCl3 and the aqueous layer was basified with sat. aq. NaHCO3 to pH 9. The aqueous layer was then extracted with CHCl3 (X3) and the combined organic layers were dried (MgSO4), filtered, and concentrated in vacuo to provide 620 mg of iodo aniline 9 (70%). Rf = 0.2 (hexanes:EtOAc:Et3N, 80:18:2). 1H NMR (CDCl3, 400 MHz) δ 8.47 (d, 1H, J= 1.9 Hz), 7.98 (dd, 1H, J= 1.9, 8.5 Hz), 6.71 (d, 1H, J= 8.5 Hz), 4.65 (bs, 2H), 3.75 (m, 1H), 2.66 (m, 2H), 2.55 (m, 2H), 1.91-1.84 (m, 1H), 1.78-1.70 (m, 5H), 1.29-1.20 (m, 2H), 0.87 (t, 3H, J= 7.3 Hz). 13C NMR (CDCl3, 100 MHz) δ 198.0, 150.9, 140.7, 130.7, 128.8, 113.0, 82.8, 68.8, 51.3, 33.5, 23.4, 19.3, 14.3. HRMS calcd for C15H21IN2ONa+ 395.0591, found 395.0582.
4.3. 1-(4-Azido-3-iodophenyl)-2-pyrrolidin-1-yl-pentan-1-one (6)
A 0°C solution of 9 (248 mg, 0.67 mmol) in trifluoroacetic acid (3.3 mL) was treated with NaNO2 (113 mg, 1.64 mmol). The mixture was stirred in the dark for 45 min at 0 °C, then carefully treated with NaN3 (532 mg, 8.18 mmol) and Et2O (3.3 mL). The reaction was stirred in the dark at 0 °C for 2 h, then diluted with H2O and Et2O. The organic layer was separated, washed with brine, dried (MgSO4), filtered, and concentrated. Chromatography (hexanes:EtOAc:Et3N, 80:18:2) provided 247 mg of azido iodo 6 (93%). Rf = 0.37 (hexanes:EtOAc:Et3N, 80:18:2). 1H NMR (CDCl3, 400 MHz) δ 8.59 (d, 1H, J= 1.9 Hz), 8.27 (dd, 1H, J= 1.9, 8.4Hz), 7.18 (d, 1H, J= 8.4 Hz), 3.73 (m, 1H), 2.67 (m, 2H), 2.54 (m, 2H), 1.92-1.82 (m, 1H), 1.80-1.70 (m, 5H), 1.28-1.17 (m, 2H), 0.88 (t, 3H, J= 7.3 Hz). 13C NMR (CDCl3, 100 MHz) δ 198.2, 146.0, 140.8, 134.4, 130.2, 117.9, 87.6, 69.9, 60.4, 51.1, 32.5, 23.4, 21.0, 19.4, 14.2. HRMS calcd for C15H20IN4O+ 399.0676, found 399.0669.
4.4. Pharmacology
[3H]-WIN-35,428 binding inhibition assays were performed with N2A neuroblastoma cells stably transfected with human wildtype DAT cDNA. Cell monolayers were grown to confluence at 37 °C, 5% CO2 in 24 well plates with Opti-MEM media supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 U/ml streptomycin, and 100 μg/ml G-418 (all from Fisher Scientific). Confluent monolayers were first washed 2 × 1 ml with 22°C “KRH buffer” (25 mM HEPES, pH 7.3, 125 mM NaCl, 4.8 mM KCl, 1.3 mM CaCl2, 1.2 mM MgSO4, 1.2 mM KH2PO4, and 5.6 mM glucose) supplemented with 50 mM ascorbic acid (KRH/AA). The washed and aspirated monolayers were incubated with 500 ml [3H]-WIN-35,428 (1 nM) and nonradioactive competitor (0.1 nM – 10 μM) for 15 minutes at 22°C, followed by removal by aspiration and 2 × 1 ml KRH/AA washes. Non-specific binding was determined by using 10 μM mazindol as the competitor. Monolayers were solubilized by incubation with 1 ml 1% SDS at 22°C for 1 hour with gentle shaking. Cell lysates were transferred into vials containing 5 ml ScintiSafe fluid (Fisher), mixed thoroughly by inversion and vortexing, and analyzed via scintillation counting for determination of remaining tritium radioactivity.
[3H]-Dopamine uptake inhibition assays were conducted identical to the [3H]-WIN 35,428 binding inhibition assays described above with the exceptions that 10 nM [3H]-dopamine replaced the WIN radioligand and the nonradioactive inhibitor drug was added 10 min before the 5 min [3H]-dopamine uptake interval commenced.
Ki values for nonlinear regression of [3H]-WIN-35,428 displacement curves were determined with GraphPad Prism 5.0 (GraphPad, La Jolla, CA). The algorithm converts Ki values from IC50 values using the Cheng-Prusoff equation: Ki = IC50/1 + [Ligand]/Kd.32 The regression best fit the data points using one site competitive binding curves. Irreversible binding was not otherwise addressed in these assays (it is unknown whether the time course employed is adequate for covalent linkage, nor was UV irradiation employed).
4.5. [125I]-1-(4-Azido-3-iodophenyl)-2-pyrrolidin-1-yl-pentan-1-one ([125I]-6)
Aniline 8 (40 μL, 5.0 mM) in NaOAc buffer (pH 4.0; 0.2 M) was treated with no-carrier-added [125I]-NaI (20 μL, 1.67 mCi) followed by N-chloro-4-toluenesulfonamide (Chloramine-T) trihydrate (10 μL, 7.0 mM). After 30 min at ambient temperature the mixture was cooled to −5 °C, and treated sequentially with cold HOAc (100 μL, 3.0 M) and NaNO2 (30 μL, 0.5 M). After 15 min, sodium azide (30 μL, 0.5 M) was added and the mixture allowed to warm to ambient temperature over 20 min. The reaction mixture was then quenched with Na2S2O5 (10 μL, 50 mM) and taken up in a syringe along with a rinse (200 μL) of the vessel with HPLC mobile phase: MeOH (16.5%), CH3CN (16.5%) and an aqueous solution (67%) containing Et3N (2.1% v/v) and HOAc (2.8% v/v). The HPLC system was equipped with a UV absorbance detector (280 nm), a flow-through radioactivity detector, and a Waters C-18 Nova-Pak column (radial compression module, 8 × 100 mm, 4 μm). Using a flow rate of 3 mL/min, radioactive material (tR = 20.0 min) corresponding to 6 was resolved from non-radioactive and radioactive side products. [125I]-6 was collected (6.5 mL), diluted to 30 mL with distilled water, and passed through an activated (EtOH/water) solid phase extraction cartridge (Waters Sep-Pak Light t-C-18) that was flushed with water (2.0 mL) to remove residual salts, and then with air. The cartridge retained >98% of the radioactivity and then was fully eluted with EtOH (1.5 mL) to give [125I]-6 (0.33 mCi; 20%) as a concentrated solution. The material co-eluted with 6 under the HPLC conditions described above and displayed 99% radiochemical purity. After 25 days of storage at −20 °C in the dark, 92% radiochemical purity was observed by HPLC. The radioligand was accompanied by a single, less lipophilic, decomposition product (8%). A specific radioactivity of 1946 mCi/μmol was determined for [125I]-6 using HPLC methods to determine the mass associated with the absorbance of carrier in a sample of known radioactivity. The UV response for non-radioactive 6 was linear (r2 = 0.99) for a five-point standard curve ranging from 38 – 450 pmol. The major non-radioactive products observed during HPLC purification were tentatively assigned as the azide (tR = 6.2 min) and the chloroazide (tR = 13.0 min) based upon HPLC analyses of model reactions. These were performed as described above without radioiodine. In brief, aniline 8 was treated with only NaNO2 and NaN3 to produce the azide, and then again with NaNO2, NaN3 and varying amounts of Chloramine-T to allow identification of chloroazide.
4.6. DAT Photoaffinity Labeling
LLC-PK1 cells expressing rDAT and HEK-293 cells expressing 6Xhis hDAT were grown to 90% confluency in 6-well plates. Medium was removed and 1 ml of [125I]-6 prepared in KRH buffer was added to a final concentration of 30 nM for 1 h at 4°C. For pharmacological displacement, 100 μM (−)-cocaine was included in the binding mixture. Cells were irradiated with shortwave ultraviolet light (254 nm, Fotodyne UV Lamp model 3–6000) for 45 s at a distance of 15–20 mm to photoactivate the radioligand. Medium was removed and the cells were washed twice with 1 ml of ice-cold KRH buffer. The photolabeled cells were lysed with 0.6 ml/well RIPA buffer (50 mM NaF, 2 mM EDTA, 125 mM Na3PO4, 1.25% Triton X-100, and 1.25% sodium deoxycholate) for 15 min at 0 °C, followed by centrifugation at 20,000 × g for 15 min at 4 °C. The supernatant fraction was transferred to clean tubes for immunoprecipitation. Lysates were subjected to immunoprecipitation as described previously23,24 using antiserum 16 generated against amino acids 42–59 of rDAT or anti-his monoclonal antibody (Sigma) for his-tagged hDAT. Immunoprecipitated samples were separated on 4–20% SDS-polyacrylamide gels followed by autoradiography using HyperfilmTM MP film for 1–4 days at −80 °C.
Acknowledgments
This work was funded by a Hunkele Dreaded Disease Award (D.J.L.), the Mylan School of Pharmacy at Duquesne University (D.J.L.), and NIH grants DA16604 (C.K.S.) and DA15175 (R.A.V.). We thank NIDA Drug Supply for contribution of certain nonradioactive DAT ligand compounds.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kampman KM. Addiction Science & Clinical Practice. 2008;4:28. doi: 10.1151/ascp084228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fleckenstein AE, Volz TJ, Riddle EL, Gibb JW, Hanson GR. Annu Rev Pharmacol Toxicol. 2007;47:681. doi: 10.1146/annurev.pharmtox.47.120505.105140. [DOI] [PubMed] [Google Scholar]
- 3.Woolverton WL, Johnson KM. Trends Pharmacol Sci. 1992;13:193. doi: 10.1016/0165-6147(92)90063-c. [DOI] [PubMed] [Google Scholar]
- 4.Runyon SP, Carroll FI. Curr Top Med Chem. 2006;6:1825. doi: 10.2174/156802606778249775. [DOI] [PubMed] [Google Scholar]
- 5.Reith MEA, Berfield JL, Wang L, Ferrer JV, Javitch JA. J Biol Chem. 2001;276:29012. doi: 10.1074/jbc.M011785200. [DOI] [PubMed] [Google Scholar]
- 6.Newman AH, Kulkarni SS. Med Res Rev. 2002;22:1. doi: 10.1002/med.10014. [DOI] [PubMed] [Google Scholar]
- 7.Ukairo OT, Bondi CD, Newman AH, Kulkarni SS, Kozikowski AP, Pan S, Surratt CK. J Pharmacol Exp Ther. 2005;314:575. doi: 10.1124/jpet.105.085829. [DOI] [PubMed] [Google Scholar]
- 8.Murthy V, Davies HML, Hedley SJ, Childers SR. Biochem Pharmacol. 2007;74:336. doi: 10.1016/j.bcp.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Y, Hajipour AR, Sievert MK, Arbabian M, Ruoho AE. Biochemistry. 2007;46:3532. doi: 10.1021/bi061727o. [DOI] [PubMed] [Google Scholar]
- 10.Newman AH, Cha JH, Cao J, Kopajtic T, Katz JL, Parnas ML, Vaughan R, Lever JR. J Med Chem. 2006;49:6621. doi: 10.1021/jm0603973. [DOI] [PubMed] [Google Scholar]
- 11.Lever JR, Zou M-F, Parnas ML, Duval RA, Wirtz SE, Justice JB, Vaughan RA, Newman AH. Bioconjugate Chem. 2005;16:644. doi: 10.1021/bc0497214. [DOI] [PubMed] [Google Scholar]
- 12.Zou MF, Kopajtic T, Katz JL, Newman AH. J Med Chem. 2003;46:2908. doi: 10.1021/jm0300375. [DOI] [PubMed] [Google Scholar]
- 13.Zou MF, Kopajtic T, Katz JL, Wirtz S, Justice JB, Newman AH. J Med Chem. 2001;44:4453. doi: 10.1021/jm0101904. [DOI] [PubMed] [Google Scholar]
- 14.Agoston GE, Vaughan R, Lever JR, Izenwasser S, Terry PD, Newman AH. Bioorg Med Chem Lett. 1997;7:3027. [Google Scholar]
- 15.Kline RH, Eshleman AJ, Wright J, Eldefrawi ME. J Med Chem. 1994;37:2249. doi: 10.1021/jm00040a019. [DOI] [PubMed] [Google Scholar]
- 16.Carroll FI, Gao Y, Abraham P, Lewin AH, Lew R, Patel A, Boja JW, Kuhar MJ. J Med Chem. 1992;35:1813. doi: 10.1021/jm00088a017. [DOI] [PubMed] [Google Scholar]
- 17.Dutta AK, Fei XS, Vaughan RA, Gaffaney JD, Wang NN, Lever JR, Reith MEA. Life Sci. 2001;68:1839. doi: 10.1016/s0024-3205(01)00981-x. [DOI] [PubMed] [Google Scholar]
- 18.Cao J, Lever JR, Kopajtic T, Katz JL, Pham AT, Holmes ML, Justice JB, Newman AH. J Med Chem. 2004;47:6128. doi: 10.1021/jm049670w. [DOI] [PubMed] [Google Scholar]
- 19.Husbands SM, Izenwasser S, Loeloff RJ, Katz JL, Bowen WD, Vilner BJ, Newman AH. J Med Chem. 1997;40:4340. doi: 10.1021/jm9705519. [DOI] [PubMed] [Google Scholar]
- 20.Berger SP, Martenson RE, Laing P, Thurkauf A, Decosta B, Rice KC, Paul SM. Mol Pharmacol. 1991;39:429. [PubMed] [Google Scholar]
- 21.Sallee FR, Fogel EL, Schwartz E, Choi SM, Curran DP, Niznik HB. FEBS Lett. 1989;256:219. doi: 10.1016/0014-5793(89)81752-1. [DOI] [PubMed] [Google Scholar]
- 22.Grigoriadis DE, Wilson AA, Lew R, Sharkey JS, Kuhar MJ. J Neurosci. 1989;9:2664. doi: 10.1523/JNEUROSCI.09-08-02664.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parnas ML, Gaffaney JD, Zou MF, Lever JR, Newman AH, Vaughan RA. Mol Pharmacol. 2008;73:1141. doi: 10.1124/mol.107.043679. [DOI] [PubMed] [Google Scholar]
- 24.Vaughan RA, Sakrikar DS, Parnas ML, Adkins S, Foster JD, Duval RA, Lever JR, Kulkarni SS, Newman AH. J Biol Chem. 2007;282:8915. doi: 10.1074/jbc.M610633200. [DOI] [PubMed] [Google Scholar]
- 25.Vaughan RA, Agoston GE, Lever JR, Newman AH. J Neurosci. 1999;19:630. doi: 10.1523/JNEUROSCI.19-02-00630.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rose ME, Grant JE. Annals of Clinical Psychiatry. 2008;20:145. doi: 10.1080/10401230802177656. [DOI] [PubMed] [Google Scholar]
- 27.Schmitt KC, Zhen J, Kharkar P, Mishra M, Chen N, Dutta AK, Reith MEA. J Neurochem. 2008;107:928. doi: 10.1111/j.1471-4159.2008.05667.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meltzer PC, Butler D, Deschamps JR, Madras BK. J Med Chem. 2006;49:1420. doi: 10.1021/jm050797a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perrine DM, Ross JT, Nervi SJ, Zimmerman RH. J Chem Ed. 2000;77:1479. [Google Scholar]
- 30.Vaughan RA, Parnas ML, Gaffaney JD, Lowe MJ, Wirtz S, Pham A, Reed B, Dutta SM, Murray KK, Justice JB. J Neurosci. 2005;143:33. doi: 10.1016/j.jneumeth.2004.09.022. [DOI] [PubMed] [Google Scholar]
- 31.Vodovozova EL. Biochemistry (Moscow) 2007;72:1. doi: 10.1134/s0006297907010014. [DOI] [PubMed] [Google Scholar]
- 32.Cheng Y, Prusoff WH. Biochem Pharmacol. 1973;22:3099. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]