

Abstract
Autophagy is a lysosomal degradation pathway that is essential for survival, differentiation, development, and homeostasis. Autophagy principally serves an adaptive role to protect organisms against diverse pathologies, including infections, cancer, neurodegeneration, aging, and heart disease. However, in certain experimental disease settings, the self-cannibalistic or, paradoxically, even the prosurvival functions of autophagy may be deleterious. This Review summarizes recent advances in understanding the physiological functions of autophagy and its possible roles in the causation and prevention of human diseases.
Introduction
Fasting has been an integral part of health and healing practices throughout the recorded history of mankind. This ancient tradition may be partially rooted in a cellular process we are now beginning to understand in modern scientific terms. One of the most evolutionarily conserved cellular responses to organismal fasting is the activation of the lysosomal degradation pathway of autophagy, a process in which the cell self-digests its own components. This self-digestion not only provides nutrients to maintain vital cellular functions during fasting but also can rid the cell of superfluous or damaged organelles, misfolded proteins, and invading micro-organisms. Interestingly, self-digestion by autophagy—a process that is potently triggered by fasting—is now emerging as a central biological pathway that functions to promote health and longevity.
The Autophagic Pathway
Autophagy (from the Greek, “auto” oneself, “phagy” to eat) refers to any cellular degradative pathway that involves the delivery of cytoplasmic cargo to the lysosome. At least three forms have been identified—chaperone-mediated autophagy, microautophagy, and macroautophagy—that differ with respect to their physiological functions and the mode of cargo delivery to the lysosome. This Review will focus on macroautophagy (herein referred to as autophagy), the major regulated catabolic mechanism that eukaryotic cells use to degrade long-lived proteins and organelles. This form of autophagy involves the delivery of cytoplasmic cargo sequestered inside double-membrane vesicles to the lysosome (Figure 1). Initial steps include the formation (vesicle nucleation) and expansion (vesicle elongation) of an isolation membrane, which is also called a phagophore. The edges of the phagophore then fuse (vesicle completion) to form the autophagosome, a double-membraned vesicle that sequesters the cytoplasmic material. This is followed by fusion of the autophagosome with a lysosome to form an autolysosome where the captured material, together with the inner membrane, is degraded (Figure 1).
Figure 1. The Cellular, Molecular, and Physiological Aspects of Autophagy.

The cellular events during autophagy follow distinct stages: vesicle nucleation (formation of the isolation membrane/phagophore), vesicle elongation and completion (growth and closure), fusion of the double-membraned autophagosome with the lysosome to form an autolysosome, and lysis of the autophagosome inner membrane and breakdown of its contents inside the autolysosome. This process occurs at a basal level and is regulated by numerous different signaling pathways (see text for references). Shown here are only the regulatory pathways that have been targeted pharmacologically for experimental or clinical purposes. Inhibitors and activators of autophagy are shown in red and green, respectively. At the molecular level, Atg proteins form different complexes that function in distinct stages of autophagy. Shown here are the complexes that have been identified in mammalian cells, with the exception of Atg13 and Atg17 that have only been identified in yeast. The autophagy pathway has numerous proposed physiological functions; shown here are functions revealed by in vivo studies of mice that cannot undergo autophagy (see Table 1).
Autophagy occurs at low basal levels in virtually all cells to perform homeostatic functions such as protein and organelle turnover. It is rapidly upregulated when cells need to generate intracellular nutrients and energy, for example, during starvation, growth factor withdrawal, or high bioenergetic demands. Autophagy is also upregulated when cells are preparing to undergo structural remodeling such as during developmental transitions or to rid themselves of damaging cytoplasmic components, for example, during oxidative stress, infection, or protein aggregate accumulation. Nutritional status, hormonal factors, and other cues like temperature, oxygen concentrations, and cell density are important in the control of autophagy. The molecular cascade that regulates and executes autophagy has been the subject of recent, comprehensive reviews (Klionsky, 2007; Maiuri et al., 2007a; Mizushima and Klionsky, 2007; Rubinsztein et al., 2007).
One of the key regulators of autophagy is the target of rapamycin, TOR kinase, which is the major inhibitory signal that shuts off autophagy in the presence of growth factors and abundant nutrients. The class I PI3K/Akt signaling molecules link receptor tyrosine kinases to TOR activation and thereby repress autophagy in response to insulin-like and other growth factor signals (Lum et al., 2005). Some of the other regulatory molecules that control autophagy include 5′-AMP-activated protein kinase (AMPK), which responds to low energy; the eukaryotic initiation factor 2α (eIF2α), which responds to nutrient starvation, double-stranded RNA, and endoplasmic reticulum (ER) stress; BH3-only proteins that contain a Bcl-2 homology-3 (BH3) domain inhibition of the Beclin 1/class III PI3K and disrupt Bcl-2/Bcl-XL complex; the tumor suppressor protein, p53; death-associated protein kinases (DAPk); the ER-membrane-associated protein, Ire-1; the stress-activated kinase, c-Jun-N-terminal kinase; the inositoltrisphosphate (IP3) receptor (IP3R); GTPases; Erk1/2; ceramide; and calcium (Criollo et al., 2007; Maiuri et al., 2007a; Meijer and Codogno, 2006; Rubinsztein et al., 2007).
Downstream of TOR kinase, there are more than 20 genes in yeast (known as the ATG genes) that encode proteins (many of which are evolutionarily conserved) that are essential for the execution of autophagy (Mizushima and Klionsky, 2007) (Figure 1). These include a protein serine/threonine kinase complex that responds to upstream signals such as TOR kinase (Atg1, Atg13, Atg17), a lipid kinase signaling complex that mediates vesicle nucleation (Atg6, Atg14, Vps34, and Vps15), two ubiquitin-like conjugation pathways that mediate vesicle expansion (the Atg8 and Atg12 systems), a recycling pathway that mediates the disassembly of Atg proteins from mature autophagosomes (Atg2, Atg9, Atg18), and vacuolar permeases that permit the efflux of amino acids from the degradative compartment (Atg22). In mammals, proteins that act more generally in lysosomal function are required for proper fusion with autophagosomes—such as the lysosomal transmembrane proteins, LAMP-2 and CLN3—and for the degradation of autophagosomal contents, such as the lysosomal cysteine proteases, cathepsins B, D, and L (Table 1).
Table 1.
Phenotypes of Mice with Mutations in Autophagy Genes
| Genotype (Organ) | Phenotype | Reference |
|---|---|---|
| Mutations Affecting Formation of Autophagosomes | ||
| atg4C−/− (all tissues) | Diaphragm-specific autophagy defect during starvation. Increased chemically induced fibrosarcomas. | Marino et al., 2007 |
| atg5−/− (all tissues) | Death within 24 hr after birth presumably due to nutrient and energy depletion. Suckling defect. Increased numbers of apoptotic cells in embryos. | Kuma et al., 2004; Qu et |
| atg5F/F:nestin-Cre (neurons) | Progressive neurodegeneration associated with ubiquitinated protein aggregates and inclusion bodies. | Hara et al., 2006 |
| atg5F/F:MLC2v-Cre (cardiomyocytes) | Normal hearts under basal conditions but increased pressure load-induced ventricular dilatation and heart failure. | Nakai et al., 2007 |
| atg5F/F:MerCreMer (cardiomyocytes, tamoxifeninducible) | Ventricular dilatation, contractile dysfunction, disorganized sarcomeres, and misaligned/aggregated mitochondria after tamoxifen injection. | Nakai et al., 2007 |
| atg5−/− (dendritic cells) | Normal dendritic cell development but impaired autophagic delivery to endosomal Toll-like receptors and interferon production during virus infection. | Lee et al., 2007 |
| atg5−/− (T cells) | Increased spontaneous apoptosis in vivo (CD8+ T cells) and defective activation-induced proliferation in vitro (CD4+ and CD8+ T cells). | Pua et al., 2007 |
| atg6/beclin 1−/− (all tissues) | Abnormal ectodermal layer with reduced cavitation and early embryonic lethality. | Yue et al., 2003; Qu et al., 2007 |
| atg6/beclin 1+/− (all tissues) | Increased frequency of spontaneous malignancies (especially lymphomas) and mammary neoplasia. Decreased pressure overload-induced heart failure. Decreased cardiac injury during ischemia/reperfusion. | Qu et al., 2003; Yue et al., 2003; Zhu et al., 2007; Matsui et al., 2007 |
| atg7−/− (all tissues) | Death within 24 hr after birth presumably due to nutrient and energy depletion. | Komatsu et al., 2005 |
| atg7F/F:nestin-Cre (neurons) | Progressive neurodegeneration associated with ubiquitinated protein aggregates and inclusion bodies. Increased frequency of TUNEL+ neurons. | Komatsu et al., 2006 |
| atg7F/F:Mx1-Cre (liver) | Ubiquitinated protein aggregates, deformed mitochondria, and aberrant membranous structures in hepatocytes. Reduced removal of peroxisomes after chemical treatment. | Komatsu et al., 2005; Iwata et al., 2006 |
| ambra1−/− (all tissues) | Decreased autophagy, increased apoptosis, and increased cell proliferation in fetal brain. Neural tube defects and embryonic death. | Fimia et al., 2007 |
| bif-1−/− | Increased frequency of spontaneous lymphomas and solid tumors. | Takahashi et al., 2007 |
|
| ||
| Mutations Affecting Lysosomal Clearance of Autophagosomes | ||
| lamp-2−/− (all tissues) | Autophagosome accumulation in multiple tissues. Impaired hepatocyte long-lived protein degradation. Vacuolar cardiomyopathy and skeletal myopathy. | Tanaka et al., 2000 |
| cln3 (all tissues) | Juvenile neuronal ceroid lipofuscinosis, with autophagic vacuolization and LC3-I to LC3-II conversion. | Cao et al., 2006 |
| cathepsin D−/− (all tissues) | Neuronal ceroid lipofuscinosis with autophagic vacuolization and LC3-I to LC3-II conversion. Bax knockout reduces enhanced apoptosis but not autophagic degeneration and neuronal loss. | Koike et al., 2005; Shacka et al., 2007 |
| cathepsin B−/−L−/− (all tissues) | Severe brain atrophy with enhanced apoptosis, autophagic vacuolization, and LC3-I to LC3-II conversion. | Felbor et al., 2002; Koike et al., 2005 |
The identification of signals that regulate autophagy and genes that execute autophagy has facilitated detection and manipulation of the autophagy pathway. Phosphatidylethanolamine (PE) conjugation of yeast Atg8 or mammalian LC3 during autophagy results in a nonsoluble form of Atg8 (Atg8-PE) or LC3 (LC3-II) that stably associates with the autophagosomal membrane (Figure 1). Consequently, autophagy can be detected biochemically (by assessing the generation of Atg8-PE or LC3-II) or microscopically (by observing the localization pattern of fluorescently tagged Atg8 or LC3) (Mizushima and Klionsky, 2007). These approaches must be coupled with ancillary measures to discriminate between two physiologically distinct scenarios—increased autophagic flux without impairment in autophagic turnover (i.e., an increased “on-rate”) versus impaired clearance of autophagosomes (i.e., a “decreased off-rate”), which results in a functional defect in autophagic catabolism (Figure 2).
Figure 2. Alterations in Different Stages of Autophagy Have Different Consequences.

An increased on-rate of autophagy occurs in response to stress signals, resulting in increased autophagosomal and autolysosomal accumulation and successful execution of the adaptive physiological functions of autophagy. In certain disease states or upon treatment with lysosomal inhibitors, there is a reduced off-rate resulting in impaired lysosomal degradation of autophagosomes. This results in increased autophagosomal accumulation and adverse pathophysiological consequences related to unsuccessful completion of the autophagy pathway. A decreased on-rate is observed if signaling activation of autophagy is defective or mutations are present in ATG genes. This results in decreased autophagosomal accumulation, the accumulation of protein aggregates and damaged organelles, and pathophysiological consequences related to deficient protein and organelle turnover. The physiological and pathophysiological consequences listed for “increased on-rate,” “reduced off-rate,” and “decreased on-rate” are based on knockout studies of the ATG genes in model organisms.
Autophagy can be pharmacologically induced by inhibiting negative regulators such as TOR with rapamycin (Rubinsztein that et al., 2007); the antiapoptotic proteins Bcl-2 and Bcl-XL bind to the mammalian ortholog of yeast Atg6, Beclin 1, with ABT-737 (Maiuri et al., 2007b); IP3R with xestospongin B, an levels IP3R antagonist; or lithium, a molecule that lowers IP3 (Criollo et al., 2007). Autophagy can be pharmacologically inhibited by targeting the class III PI3K involved in autophagosome formation with 3-methyladenine or by targeting the fusion of autophagosomes with lysosomes, using inhibitors of the lysosomal proton pump such as bafilomycin A1 or lysosomotropic alkalines such as chloroquine and 3-hydroxychloroquine (Rubinsztein et al., 2007) (Figure 1). It should be noted that all of these pharmacological agents lack specificity for the autophagy pathway. Therefore, although some of these agents such as rapamycin, lithium, and chloroquine are clinically available and may be helpful for treating diseases associated with autophagy deregulation, genetic approaches to inhibiting autophagy—for example, knockout of ATG genes by homologous recombination or knockdown by small-interfering RNA (siRNA)—have yielded more conclusive information about the biologic roles of autophagy in health and disease.
Physiological Functions of Autophagy
Autophagy Defends against Metabolic Stress
Autophagy is activated as an adaptive catabolic process in response to different forms of metabolic stress, including nutrient deprivation, growth factor depletion, and hypoxia. This bulk form of degradation generates free amino and fatty acids that can be recycled in a cell-autonomous fashion or delivered systemically to distant sites within the organism. Presumably, the amino acids generated are used for the de novo synthesis of proteins that are essential for stress adaptation. The molecular basis for the recycling function of autophagy has only recently begun to be defined with the identification of yeast Atg22 as a vacuolar permease required for the efflux of amino acids resulting from autophagic degradation (Mizushima and Klionsky, 2007). It is presumed that the recycling function of autophagy is conserved in mammals and other higher organisms, although direct data proving this concept are lacking.
The amino acids liberated from autophagic degradation can be further processed and, together with the fatty acids, used by the tricarboxylic acid cycle (TCA) to maintain cellular ATP production. The importance of autophagy in fueling the TCA cycle is supported by studies showing that certain phenotypes of autophagy-deficient cells can be reversed by supplying them with a TCA substrate such as pyruvate (or its membrane-permeable derivative methylpyruvate). For example, methylpyruvate can maintain ATP production and survival in growth factor-deprived autophagy-deficient cells that would otherwise quickly die (Lum et al., 2005). It can also restore ATP production, the generation of engulfment signals, and effective corpse removal in autophagy-deficient cells during embryonic development (Qu et al., 2007).
This role of autophagy in maintaining macromolecular synthesis and ATP production is likely a critical mechanism underlying its evolutionarily conserved prosurvival function. Gene knockout or knockdown studies in diverse phyla provide strong evidence that autophagy plays an essential function in organismal survival during nutrient stress (Maiuri et al., 2007a). Yeast cells lacking ATG genes display reduced tolerance to nitrogen or carbon deprivation and are defective in starvation-induced sporulation. Similarly, null mutations in ATG genes in slime molds limit viability and differentiation during nutrient deprivation. Loss-of-function mutations in ATG genes in plants reduce tolerance to nitrogen or carbon depletion, resulting in enhanced chlorosis, reduced seed set, and accelerated leaf senescence (Bassham et al., 2006). Furthermore, siRNA-mediated knockdown of atg genes in nematodes decreases survival during starvation (Kang et al., 2007). Autophagy also enables mammals to withstand nutrient depletion (Table 1). Mice lacking either atg5−/− or atg7−/− are born at normal Mendelian ratios yet die within hours after birth, presumably due to their inability to adapt to the neonatal starvation period.
Thus, a critical physiological role of autophagy appears to be the mobilization of intracellular energy resources to meet cellular and organismal demands for metabolic substrates. The requirement for this function of autophagy is not limited to settings of nutrient starvation. Because growth factors are often required for nutrient uptake, loss of growth factor signaling can result in reduced intracellular metabolite concentrations and activation of autophagy-dependent survival mechanisms (Lum et al., 2005). It is also possible that in certain settings, especially when cells suddenly have high metabolic needs, autophagy may be needed in a cell-autonomous fashion to generate sufficient intracellular metabolic substrates to maintain cellular energy homeostasis. This hypothesis may explain why there are high levels of autophagy in the mouse heart and diaphragm immediately following birth (Kuma et al., 2004).
Autophagy Works as a Cellular Housekeeper
The repertoire of routine housekeeping functions performed by autophagy includes the elimination of defective proteins and organelles, the prevention of abnormal protein aggregate accumulation, and the removal of intracellular pathogens. Such functions are likely critical for autophagy-mediated protection against aging, cancer, neurodegenerative diseases, and infection. Although some of these functions overlap with those of the ubiquitin-proteosome system—the other major cellular proteolytic system—the autophagy pathway is uniquely capable of degrading entire organelles such as mitochondria, peroxisomes, and ER as well as intact intracellular microorganisms. Further, the relative role of the autophagy-lysosome system in protein quality control—i.e., in preventing the intra-cellular accumulation of altered and misfolded proteins—may be greater than previously anticipated.
Tissue-specific disruption of ATG genes has revealed a critical role for basal autophagy in protein quality control in murine postmitotic cells (Table 1). Atg7 deletion in hepatocytes, atg5 and atg7 deletion in neurons, and atg5 deletion in cardiomyocytes result in the accumulation of ubiquitin-positive protein aggregates in inclusion bodies that are associated with cellular degeneration. Such abnormalities have not been reported for atg5-deficient dendritic cells or T lymphocytes, perhaps because autophagy is less important for the waste management of rapidly proliferating cells. Moreover, the underlying mechanism for the accumulation of ubiquitin-positive aggregates in certain autophagy knockout mouse tissues remains unknown. Cytoplasmic accumulation of diffuse ubiquitinated proteins precedes the accumulation of aggregates in atg5-deficient neurons (Hara et al., 2006). Thus, aggregate formation may be a secondary result of a general defect in protein turnover rather than a failure of basal autophagy to clear aggregates that are formed constitutively in normal conditions (Mizushima and Klionsky, 2007). According to such a model, in the absence of autophagy, the turnover of cytosolic proteins is impaired, increasing their propensity to become damaged and misfolded and subsequently ubiquitinated and aggregated (Figure 3). It is not yet clear whether the ubiquitinated proteins are autophagically sequestered in a random, nonselective fashion, or whether they are selectively targeted to the autophagosome by a mechanism involving p62/SQSTM1, an adaptor protein that binds both ubiquitin and LC3 (Pankiv et al., 2007).
Figure 3. Autophagy, Protein Quality Control, and Neurodegeneration.
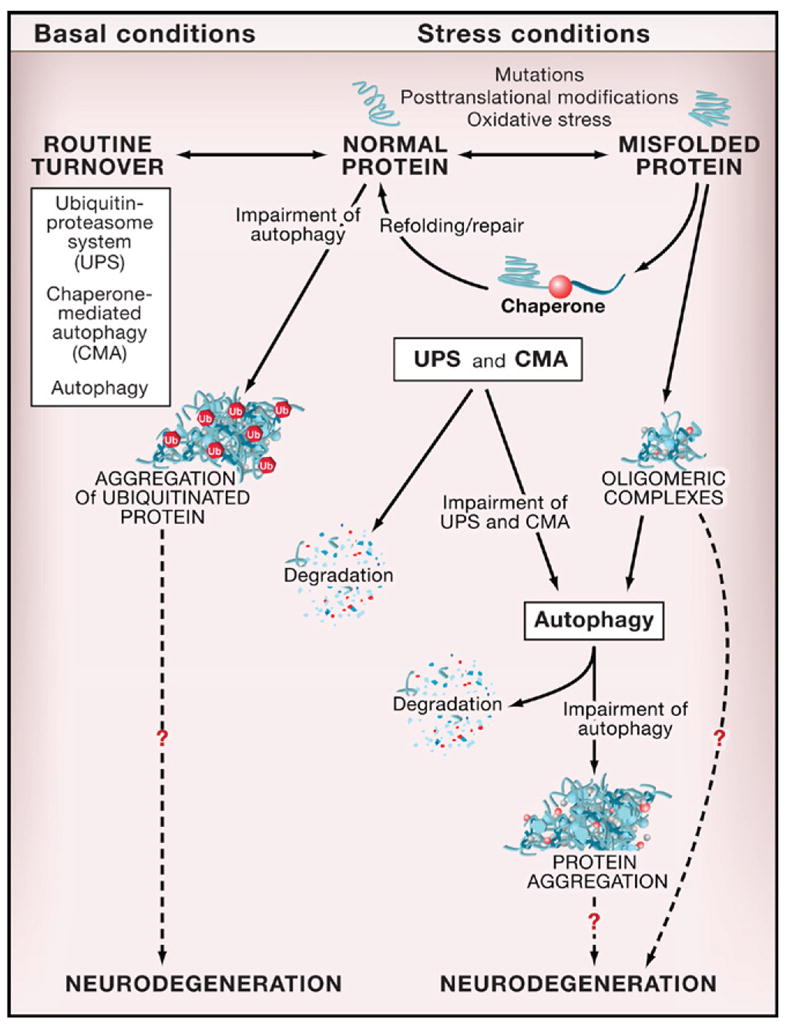
Normal proteins are routinely turned over by different protein degradation systems, including the ubiquitin-proteasome system (UPS), chaperone-mediated autophagy (CMA), and macroautophagy (referred to herein as “autophagy”). In autophagy-deficient neurons, there is an accumulation of ubiquitinated protein aggregates that is associated with neurodegeneration. Similar effects of autophagy deficiency are observed in other postmitotic cells (hepatocytes, cardiomyocytes) under basal conditions. Proteins altered by mutations (such as polyglutamine expansion tracts), posttranslational modifications, or stress (such as oxidative stress, UV irradiation, toxins) undergo a conformational change, are recognized by molecular chaperones, and are either refolded and repaired or delivered to protein degradation systems (usually UPS or CMA). If these protein degradation systems are impaired or if the altered proteins form oligomeric complexes that cannot be recognized by the UPS or CMA, autophagy may be the primary route for the removal of these abnormal and potentially toxic proteins. Impaired autophagy is associated with the formation of protein aggregates and increased neurodegeneration. The mechanisms by which abnormal proteins and impaired autophagy result in neurodegeneration are not known.
Unlike proteasomal degradation, the autophagic breakdown of substrates is not limited by steric considerations and therefore autophagy can sequester and degrade entire organelles. In yeast, autophagy participates in the selective removal of superfluous peroxisomes (pexophagy) generated when cells adapt to glucose metabolism (Nair and Klionsky, 2005) and perhaps in the elimination of damaged mitochondria (mitophagy), as atg mutant yeast accumulate dysfunctional mitochondria (Zhang et al., 2007). Selective degradation of peroxisomes or mitochondria was reported in hepatocytes isolated from clofibrate-treated or starved rats, respectively (Kim et al., 2007), but until recently the importance of these forms of selective autophagy in mammalian physiology was unclear. However, under steady-state conditions, atg7-deficient mouse hepatocytes accumulate peroxisomes, deformed mitochondria, and aberrant concentric membranous structures that are contiguous with the ER, and during chemical treatment, atg7-deficient mouse livers display a defect in the removal of excess peroxisomes (Table 1). Furthermore, agents that promote ER stress induce the selective autophagy of ER membranes (reticulophagy) both in yeast and in mammalian cells (Klionsky, 2007). Taken together, these observations indicate that basal and induced autophagy are likely important for the physiological control of number and quality of organelles across diverse phyla and function to eliminate superfluous and damaged organelles. Defined or candidate signals for selective organelle recognition by autophagy include the peroxisome membrane tag, Pex14, an outer mitochondrial membrane protein, Uth1p, in yeast (Mizushima and Klionsky, 2007), and the mitochondrial permeability transition in mammalian cells (Kim et al., 2007).
Autophagy May Be a Guardian of the Genome
Recent studies in ATG gene-deficient immortalized epithelial cells indicate that the autophagic machinery can limit DNA damage and chromosomal instability (Mathew et al., 2007a). Because these studies used cells with simultaneous defects in DNA checkpoints and apoptosis pathways, it is not yet known whether autophagy plays a primary function in preventing genomic instability in normal cells. However, in view of known functions of autophagy in energy homeostasis and in protein and organelle quality control, this seems likely. Such a role of autophagy would mechanistically link effects on the prevention of tumor initiation, tumor progression, aging, and neurodegeneration. The precise mechanisms by which deficient autophagy compromises genomic stability are unclear. Failure to control the damage of checkpoint or repair proteins, deregulated turnover of centrosomes, insufficient energy for proper DNA replication and repair, and excessive generation of reactive oxygen species due to inefficient removal of damaged mitochondria are possible alterations that may contribute to genomic instability in autophagy-defective cells (Jin and White, 2007; Mathew et al., 2007a).
Autophagy in Life and Death Decisions of the Cell
Under most circumstances, autophagy constitutes a stress adaptation pathway that promotes cell survival. An apparent paradox is that autophagy is also considered a form of nonapoptotic programmed cell death called “type II” or “autophagic” cell death. This type of cell death has been historically defined by morphological criteria, but it is now clear that the mere presence of autophagosomes in dying cells is insufficient to distinguish “cell death with autophagy” from “cell death by autophagy.” The knockdown of ATG genes has recently defined whether autophagy functions in the execution of cell death in different settings (see Maiuri et al., 2007a for detailed review).
It is not yet understood what factors determine whether autophagy is cytoprotective or cytotoxic and whether cytotoxicity occurs as the result of self-cannibalism, the specific degradation of cytoprotective factors, or other as of yet undefined mechanisms (for an extensive discussion, see Maiuri et al., 2007a). The most intuitive mechanism is self-cannibalism. However, cells subjected to prolonged growth factor deprivation or shortage of glucose and oxygen can lose the majority of their mass via autophagy and fully recover when placed in optimal culture conditions (Degenhardt et al., 2006; Lum et al., 2005), suggesting that cell death via autophagy may not be simply a matter of crossing a quantitative threshold of self-digestion. Although autophagy can independently influence life and death decisions of the cell (by being cytoprotective or self-destructive), it is also intricately linked to apoptotic death pathways. Factors that may control the cellular “decision” between the two responses include potentially variable thresholds for each process, molecular links that coordinately regulate apoptosis and autophagy, and mutual inhibition or activation of each pathway by the other (see Maiuri et al., 2007a for details).
There is no evidence currently that the ATG genes promote programmed cell death that occurs physiologically in vivo, for instance during development. In fact, nematodes lacking bec-1, an ortholog of atg6/beclin 1, and mice lacking beclin 1 or atg5 display increased, rather than decreased, numbers of apoptotic cells in embryonic tissues (Qu et al., 2007; Takacs-Vellai et al., 2005; Yue et al., 2003). Given the recently identified role of ATG genes in facilitating the heterophagic removal of apoptotic corpses (Qu et al., 2007) (Figure 1C), it is not yet certain whether the increased numbers of apoptotic cells in autophagy-deficient embryos represent increased cell death events, delayed clearance of dead cells, or a combination of the two. Perhaps clearer evidence for a prosurvival function of autophagy in vivo is provided by tissue-specific ATG gene knockout studies—for example the neuron-specific knockout of atg5 or atg7 and T cell-specific knockout of atg5—where increased apoptosis is observed in mature animals (Table 1).
The intricate interplay between autophagy and life and death decisions of the cell mirrors some of the complexities in deciphering the roles of autophagy in human diseases and their treatments. For decades, pathologists have noted ultrastructural features of autophagy in a cornucopia of human diseases, including infections, neurodegenerative and myodegenerative diseases, cardiomyopathies, and cancer (de Duve and Wattiaux, 1966; Martinez-Vicente and Cuervo, 2007). These findings were largely either ignored or presumed to reflect a causative role of autophagy in cellular degeneration and disease. The inability to distinguish between defective autophagy (with decreased removal of autophagosomes) and increased autophagic activity (with increased formation of autophagosomes) further confounded the pathophysiological interpretation of autophagosome accumulation in tissue samples. Now, with the identification of signaling pathways that regulate autophagy, evolutionarily conserved gene products that mediate autophagy, and methods to distinguish between increased on-rates versus decreased off-rates of autophagy, pharmacological, genetic, and biochemical approaches are being used to redefine the role of autophagy in the pathogenesis of human diseases.
Autophagy in Disease
Autophagy and Neurodegenerative Diseases
Early reports demonstrating that autophagosomes accumulate in the brains of patients with diverse neurodegenerative diseases, including Alzheimer’s disease, transmissible spongiform encephalopathies, Parkinson’s disease, and Huntington’s disease (reviewed in Rubinsztein et al., 2007; Williams et al., 2006), led to the initial hypothesis that autophagy contributed to the pathogenesis of these disorders. In mice with cerebellar degeneration due to mutations in glutamate receptor, autophagy was also postulated to be a mechanism of nonapoptotic cell death (Yue et al., 2002). In contrast, more recent studies provide compelling evidence that at least in model organisms autophagy protects against diverse neurodegenerative diseases and that the accumulation of autophagosomes primarily represents the activation of autophagy as a beneficial physiological response or, in the case of Alzheimer’s disease, the consequence of a defect in autophagosomal maturation (Martinez-Vicente and Cuervo, 2007; Rubinsztein et al., 2007; Williams et al., 2006).
Beyond its role in the clearance of misfolded proteins spontaneously generated during routine protein turnover (discussed above), autophagy likely plays an important role in the clearance of aggregate-prone mutant proteins associated with several different neurodegenerative diseases (Figure 3). These include proteins with polyglutamine (polyQ) expansion tracts such as those seen in Huntington’s disease and spinocerebellar ataxia, mutant α-synucleins that cause familial Parkinson’s disease, and different forms of tau including mutations causing frontotemporal dementia (Williams et al., 2006). Because substrates need to be unfolded to pass through the narrow pore of the proteasomal barrel, oligomeric and aggregated proteins are poor substrates for proteasomal degradation and better targets for autophagic degradation. The mechanism by which these proteins exert their cellular toxicity is still controversial, but it is generally believed that they are particularly toxic in oligomeric complexes and that higher-order protein aggregates may be formed as a last attempt to prevent toxicity in the absence of a properly functioning quality-control system (Martinez-Vicente and Cuervo, 2007). This view is consistent with the model that autophagy functions as a quality-control system that targets oligomeric proteins and with the evidence that autophagy activation reduces, whereas autophagy inhibition increases, the formation of protein aggregates and the neurotoxicity of aggregate-prone proteins.
Pharmacological activation of autophagy reduces the levels of soluble and aggregated forms of mutant huntingtin protein, proteins mutated in spinocerebellar ataxia, mutant forms of α-synuclein, and mutant tau; it also reduces their cellular toxicity in vitro and their neurotoxicity in either mouse or Drosophila models (Rubinsztein et al., 2007). ATG gene knockdown or knockout increases aggregate formation and toxicity of polyQ expansion proteins in C. elegans (Jia et al., 2007). Autophagy induced by overexpression of histone deacetylase 6 also compensates for impairment in the ubiquitin-proteasome system in a fly model of spinobulbar muscle dystrophy (Pandey et al., 2007). In these models, autophagy-mediated neuroprotection may be due to a quantitative reduction in the amounts of the toxic protein species as well as antiapoptotic effects (Rubinsztein et al., 2007).
The development of neurodegenerative disease in patients with proteinopathies implies that the autophagy may reach a saturation point in which its capacity to degrade the mutant aggregate-prone proteins is exceeded, or that concurrent defects may occur in the autophagy pathway. Acquired defects in autophagosome formation may result from the sequestration of autophagy proteins in aggregates formed by mutant proteins, the age-related decline that occurs in Beclin 1 and potentially other autophagy protein expression in human brain, and other as-of-yet unidentified factors (Shibata et al., 2006). In addition to defects that result in decreased autophagic activity, genetic or functional alterations may occur that impair delivery of autophagosomes to the lysosome. For example, mutations that affect the dynein motor machinery impair autophagosome-lysosome fusion, leading to decreased autophagic clearance of aggregate proteins and enhanced toxicity of the huntingtin mutant protein in Drosophila and mouse models (Ravikumar et al., 2005). It is thus possible that impaired autophagic clearance may contribute to the pathogenetic mechanism by which dynein mutations cause motor neuron diseases in humans (Table 2). Other mutations involved in lysosomal function also reduce autophagic turnover (with an observed increase in autophagosomal accumulation) and are associated with neuronal ceroid lipofuscinosis (Batten disease) in mice, such as CLN3 partial deletion, cathepsin D knockout, and double cathepsin B/L knockout (Table 1). The interrelationship between autophagy and Alzheimer’s disease may also involve an impairment in autophagolysosomal maturation. Autophagosome-like structures accumulate in dystrophic neurons of Alzheimer’s disease patients, presumably as a result of impairment in autophagolysosomal maturation and, intriguingly, may constitute a unique site of production/accumulation of the pathogenic amyloid β protein (Yu et al., 2005).
Table 2.
Germline or Somatic Mutations that Affect Autophagy in Human Disease
| Affected Protein | Disease and Putative Link to Autophagy | Reference |
|---|---|---|
| Gene Products Required for Autophagy | ||
| Atg16L1 (germline) | Crohn’s disease (a type of inflammatory bowel disease), possibly due to altered autophagy-dependent immune regulation or bacterial clearance. | Massey and Parkes, 2007 |
| Beclin 1a (somatic; monoallelic deletions) | Breast, ovarian, and prostate cancer, possibly due to genomic instability or altered cell growth control associated with reduced autophagy. | Liang et al., 1999 |
| UVRAG (somatic; monoallelic deletions) | Colon cancer, possibly due to mechanisms similar to those observed with beclin 1 deletion. | Liang et al., 2006 |
|
| ||
| Genes Products Involved in Autophagosomal Sequestration, Movement, or Maturation | ||
| P62/SQSTM1 (mutations in ubiquitin-binding site) | Paget disease (abnormal bone loss associated with increased osteoclast activity), possibly due to impaired autophagy of ubiquitinated proteins. | Lucas et al., 2006 |
| Dynactin subunit p150glued | Motor neuron disease with spinal and bulbar muscular atrophy, possibly due to abnormal movement of autophagosomes. | Puls et al., 2005 |
| CLN3a | Juvenile onset neuronal lipofuscinosis (Batten disease), possibly due to reduced autophagosome/lysosome fusion. | Vesa and Peltonen, 2002 |
| LAMP-2a | Familial X-linked cardiomyopathy (Danon disease), possibly due to reduced autophagosome/lysosome fusion. | Nishino et al., 2000 |
|
| ||
| Examples of Gene Products that Regulate Autophagy and Other Cellular Processes | ||
| AKT, PI3K, PTEN (somatic) | Cancer. Gain-of-function mutations or (somatic) amplifications in the oncogenes PI3K and Akt and loss-of-function mutations in the tumor suppressor gene PTEN confer autonomy from growth factors and inhibit autophagy. | Cully et al., 2006 |
| TSC1, TSC2 (germline and somatic) | Tuberous sclerosis complex (autosomal dominant disorder with benign hamartomatous growths in multiple organs). Mutations in TSC1 or TSC2 abolish their inhibition of mTOR, leading to decreased autophagy. | Schwartz et al., 2007 |
| LKB1/STK11 (germline and somatic) | Peutz-Jeghers syndrome (autosomal dominant syndrome with benign hamartomatous polyps in gastrointestinal tract and increased incidence of epithelial cancers). Somatic mutations in non-small cell lung cancer. LKB1 activates AMPK and stimulates autophagy. | Ji et al., 2007; Liang et al., 2007 |
| p53 (somatic) | Cancer. Mutations in p53 mutations are found in >50% of all human tumors. p53 may activate autophagy after genotoxic stress. | Feng et al., 2005; Crighton et al., 2006 |
| Bcl-2 (somatic) | Follicular lymphoma. Amplification of Bcl-2 may inhibit autophagy by targeting Beclin 1. | Pattingre et al., 2005 |
| IRGM1 (germline) | Crohn’s diease (a type of inflammatory bowel disease). IRGM1 is an immunity-related GTPase that stimulates autophagy. | Massey and Parkes, 2007 |
Indicates that mice harboring identical mutations have been shown to have impaired autophagy and display similar disease phenotype (see Table 1 for details).
The role of autophagy in protection against neurodegenerative diseases is established in animal models but not yet in patients. Nonetheless, preclinical animal data provide a strong rationale for proceeding with clinical trials with autophagy-stimulatory agents; this is especially true as agents shown to be beneficial in reducing neurotoxicity of mutant aggregate-prone proteins are already in clinical use to treat other diseases. Rapamycin analogs, which are approved for the use of preventing organ transplant rejection and postangioplasty coronary artery restenosis and are in phase II oncology trials, protect against neurodegeneration seen in Drosophila and mouse polyQ disease models (Rubinsztein et al., 2007). However, because TOR also affects protein synthesis, cell proliferation, cell growth, cell death, and immune function, its inhibition has some adverse effects; perhaps intermittent rather than continuous stimulation of autophagy with rapamycin may reduce such side effects while maintaining therapeutic efficacy. Lithium chloride, a drug used for the treatment of bipolar disorder, induces autophagy by reducing IP3 levels and enhances the clearance of aggregate-prone proteins (Rubinsztein et al., 2007). Another group of new agents, small molecule enhancers of rapamycin (SMERs), enhance the clearance of mutant huntingtin and α-synuclein and protect against neurodegeneration in a Drosophila Huntington’s disease model (Sarkar et al., 2007). Given that lithium and SMERs both act independently of TOR, it is possible that they may be used therapeutically in combination with rapamycin analogs. Of long-term interest, both for the treatment of neurodegenerative diseases and, potentially, the prevention of aging, would be drugs that can reverse age-dependent declines that occur in CNS autophagy protein expression and lysosomal clearance of autophagosomes.
Autophagy and Liver Disease
Tissue-specific knockout studies in mice (discussed above) indicate an important role for basal hepatocyte autophagy in intracellular protein and organelle quality control. The protein quality-control function may be important in the pathogenesis of the most common genetic cause of human liver disease, α1-antitrypsin deficiency, which is associated with chronic inflammation and carcinogenesis (Perlmutter, 2006). Perhaps, similar to neurodegenerative disorders caused by aggregate-prone proteins, pharmacological activation of autophagy may be helpful in this setting.
A toxic gain-of-function point mutation in α1-antitrypsin Z (α1-ATZ) impairs proper protein folding and renders a normally secreted hepatic protein prone to form aggregated polymers within the hepatocyte ER. Whereas wild-type α1-antitrypsin is degraded primarily by the proteasome, mutant α1-ATZ is thought to be degraded primarily by autophagy (Yorimitsu and Klionsky, 2007). In cell lines depleted of atg5, there is decreased degradation of the mutant α1-ATZ, especially insoluble forms, and increased accumulation of cytoplasmic inclusion bodies (Kamimoto et al., 2006). Moreover, transgenic expression of α1-ATZ is sufficient to induce mouse hepatocyte autophagy in vivo. The precise role of α1-ATZ aggregates in hepatotoxicity is not yet clear; one interesting question is whether such aggregates sequester autophagy proteins, leading to a reduction in hepatocyte autophagy (and its cytoprotective and tumor suppressor effects). A broader question of biomedical relevance is whether the protein quality-control function of autophagy plays a more general role in protecting the liver against alcohol and other hepatotoxic agents.
Autophagy and Muscle Disease
Similar to neurodegenerative diseases, the pathogenesis of myodegenerative diseases may involve either the failure of autophagosomes to fuse with lysosomes or the aggregation of misfolded proteins that exceed the autophagic clearance capacity of the myocyte. Danon disease, a genetic disease characterized by cardiomyopathy, myopathy, and variable mental retardation, results from a mutation in the lysosomal protein LAMP-2 and is associated with extensive accumulation of autophagosomes in the muscles of LAMP-2-deficient mice and patients (Tables 1 and 2). The concept that failure of lysosomes to fuse with autophagosomes contributes to myopathy is further supported by evidence that pharmacological inhibition of this fusion step (e.g., with chloroquine or hydroxychloroquine) causes severe vacuolar myopathies in rats and humans (Bolanos-Meade et al., 2005). There are several other histologically related diseases, such as X-linked myopathy with excessive autophagy, infantile autophagic vacuolar myopathy, adult-onset vacuolar myopathy with multiorgan involvement, and X-linked congenital autophagic vacuolar myopathy, that have unclear molecular defects (Nishino, 2006); however, the prediction is that these disorders may be due, at least in part, to an impairment in autophagosome-lysosome fusion. Another severe inherited disorder of skeletal and cardiac muscle, Pompe disease, which is caused by the deficiency of glycogen-degrading lysosomal enzyme acid α-glucosidase (GAA), is often resistant to GAA enzyme replacement in skeletal muscle but not in heart (Fukuda et al., 2006). Endocytic trafficking of recombinant GAA is abnormal in skeletal muscle; it accumulates primarily in autophagosomes and fails to reach the lysosomal compartment, providing an example of how the “traffic jam” provoked by defective lysosomal function may be pathogenic.
Muscle diseases in which autophagy may promote the clearance of disease-causing proteins include sporadic inclusion body myositis, limb girdle muscular dystrophy type 2B, and Miyoshi myopathy. In sporadic inclusion body myositis, the most common acquired muscle disease in patients above 50 years of age, overexpression of amyloid precursor protein (APP) and accumulation of its proteolytic fragment β-amyloid in vacuolated cells is thought to be a central pathogenetic mechanism (Askanas and Engel, 2006). Both APP and β-amyloid colocalize with LC3 in cultured human muscle cells and in degenerating muscle fibers in human biopsy tissue, suggesting that these proteins are cleared by autophagy (Lunemann et al., 2007). Limb girdle muscular dystrophy type 2B and Miyoshi myopathy are both caused by mutations of the gene encoding dysferlin, a type II transmembrane protein expressed primarily in muscle sarcolemma. Although wild-type dysferlin in the ER is degraded primarily by the ubiquitin-proteasome system, mutant dysferlin, which spontaneously aggregates in the ER, is degraded primarily by the autophagy/lysosomal system (Askanas and Engel, 2006). In cells expressing mutant dysferlin, autophagy inhibition due to atg5 depletion or a mutation in the ER-stress-induced eIF2α-mediated autophagy pathway increases aggregation whereas rapamycin activation of ER-stress-induced autophagy decreases aggregation. Thus, a general theme seems to be emerging in diseases associated with aggregate-prone proteins not only in the brain but also in the liver and muscle; the autophagy pathway plays a central role in the clearance of such proteins, and pharmacologic upregulation of autophagy may protect against aggregate accumulation.
Autophagy and Cardiac Disease
As noted, defective autophagy (due to impaired autophagosome-lysosome fusion) may play a role in relatively rare forms of inherited diseases of the heart (e.g., Danon disease, Pompe disease). Of greater medical significance is the possibility that autophagy may constitute an important physiological or pathophysiological response to cardiac stresses such as ischemia or pressure overload, which are frequently encountered in patients with coronary artery disease, hypertension, aortic valvular disease, and congestive heart failure. The accumulation of autophagosomes has been noted in cardiac biopsy tissues of patients with these disorders, rodent models of these cardiac diseases, and isolated stressed cardiomyocytes (Terman and Brunk, 2005). Prior to genetic studies, it was largely assumed that autophagy invariably contributed to myocyte degeneration in such settings. However, more recent data challenge this view; the cytoprotective effects of autophagy (either via ATP production, protein and organelle quality control, or other mechanisms) may predominate in certain settings.
The cardiomyocyte, similar to the neuron, is a postmitotic cell in which basal autophagy may be important in protein and organelle quality control. Heart-specific knockout of atg5 in adult mice results in cardiac hypertrophy and contractile dysfunction that is accompanied by increased levels of ubiquitinated proteins and ultrastructural evidence of sarcomere and mitochondrial structural abnormalities (Table 1). Beyond this need for autophagy under basal conditions, the heart may uniquely rely on stress-induced upregulation of autophagy to ensure the availability of energy substrates and to promote cellular remodeling. The heart consumes more energy per gram than any other organ in the body, and common cardiac disorders (e.g., cardiac ischemia and heart failure) are characterized by a reduction in the availability of energy substrates, a factor that contributes to transient or sustained impairment of cardiac function. Furthermore, when cardiac stresses are sustained for long periods of time, myocytes remodel their cellular architecture (e.g., undergo elongation and hypertrophy) to adapt to stress. The needs of the stressed heart for more energy substrates and for cellular remodeling may be met in part through the autophagy pathway; cardiac-specific deficiency of atg5 early in cardiogenesis does not result in any phenotypic abnormalities under basal conditions but results in more severe cardiac dysfunction following treatment with pressure overload or β-adrenergic stress (Table 1). These data suggest that upregulation of autophagy in failing hearts is an adaptive response that protects against hemodynamic or neurohormonal stresses.
Although this is an attractive model, not all studies support an adaptive role for autophagy during cardiac stress. In a pressure overload model, heterozygous disruption of beclin 1 led to preservation not deterioration of contractile function (Table 1). These results differ from those obtained in atg5 knockout mice, which may reflect different contributions of atg5 versus beclin 1 or different effects of homozygous versus heterozygous disruption of an ATG gene. Heterozygous disruption of beclin 1 also decreases the size of the myocardial infarction after ischemia/reperfusion (Table 1). However, in an in vitro model of cardiac ischemia/reperfusion, beclin 1 overexpression decreased cell injury and dominant-negative Atg5 overexpression increased cell injury, suggesting a protective effect for both ATG genes in ischemia/reperfusion (Hamacher-Brady et al., 2006). Additional studies are needed to clarify whether autophagy activation serves as an adaptive or maladaptive cardiac response to hemodynamic and ischemic stress. It is noteworthy, however, that in patients, interventions known to exacerbate ischemia or heart failure (e.g., beta-adrenergic receptor agonists) act to reduce autophagy in the heart whereas interventions known to ameliorate ischemia or heart failure (e.g., beta-adrenergic receptor antagonists) tend to enhance autophagy in the heart (Bahro and Pfeifer, 1987).
Autophagy and Cancer
In the past decade, several genetic links have emerged between autophagy defects and cancer, providing increasing support for the concept that autophagy is a bona fide tumor suppressor pathway (Levine, 2007; Mathew et al., 2007a).
The regulation of autophagy overlaps closely with signaling pathways that regulate tumorigenesis. Several tumor suppressor genes involved in the upstream inhibition of TOR signaling, including PTEN, TSC1, and TSC2, stimulate autophagy and, conversely, TOR-activating oncogene products such as class I PI3K and Akt inhibit autophagy (Table 2). P53, the most commonly mutated tumor suppressor gene in human cancers, positively regulates autophagy in DNA-damaged cells, perhaps through AMPK activation of the TSC1/TSC2 complex and subsequent TOR inhibition or perhaps via upregulation of DRAM (damage-regulated autophagy modulator), a lysosomal protein that may induce autophagy (Crighton et al., 2006; Feng et al., 2005). The death-associated protein kinase, DAPk, also induces autophagy and apoptosis, is commonly silenced in human cancers by methylation, and has tumor and metastasis suppressor properties (Gozuacik and Kimchi, 2006). The cellular proto-oncoproteins, Bcl-2 and Bcl-XL, which are often overexpressed in human cancers, are generally thought to mediate oncogenesis by suppressing mitochondrial membrane permeabilization, one of the rate-limiting steps of apoptosis. In addition, ER-localized Bcl-2 and Bcl-XL inhibit autophagy by binding to the Beclin 1 autophagy protein (Pattingre et al., 2005; Maiuri et al., 2007b). Thus, there is a strong correlation between molecules that are involved in autophagy induction and tumor suppression and between molecules that are involved in autophagy inhibition and oncogenesis. It will be important to determine whether this autophagy modulation is mechanistically important in the action of these tumor suppressor genes and oncogenes. Such a finding would suggest a broad involvement of autophagy in most human cancers.
The first specific link between the autophagy machinery and human cancer was established in 1999, when it was discovered that the ATG gene beclin 1 was a candidate tumor suppressor (Liang et al., 1999). Beclin 1 maps to a tumor susceptibility locus that is monoallelically deleted in a high percentage of human breast, ovarian, and prostate cancers (Table 2), and decreased expression of Beclin 1 has been reported in human breast, ovarian, and brain tumors (Liang et al., 1999; Miracco et al., 2007). Beclin 1 gene transfer inhibits growth of tumor cell lines in vitro and the tumorigenicity of human breast carcinoma cells in mouse xenograft models (Liang et al., 1999). The monoallelic deletions of beclin 1 in human cancer are likely mechanistically important in tumorigenesis, given that targeted mutant mice with heterozygous disruption of beclin 1 have decreased autophagy, are more prone to the development of spontaneous tumors including lymphomas, lung carcinomas, hepato-cellular carcinomas, and mammary precancerous lesions, and undergo accelerated hepatitis B virus-induced carcinogenesis (Table 1). Further, immortalized kidney and mammary epithelial cells derived from beclin 1 heterozygous-deficient mice are more tumorigenic than those derived from wild-type mice (Karantza-Wadsworth et al., 2007; Mathew et al., 2007b).
The Mittleman Breakpoint Database reveals frequent chromosomal aberrations of several other components of the autophagic machinery in human cancers, although it is not yet known if these components are biallelically mutated or function as haploinsufficient tumor suppressors. Atg5, a component of the ubiquitin-like protein conjugation system, has tumor suppressor effects in a mouse xenograft model (Yousefi et al., 2006), and knockout of atg4c, a cysteine protease involved in processing LC3, increases chemically induced fibrosarcomas in mice (Table 1). This suggests that tumor suppression may be a shared property of autophagy proteins that act at different steps in the pathway. Other proteins that form part of the Beclin 1/class III PI3K complex and are required for its autophagy activity may also play a role in cell growth control and/or tumor suppression. UVRAG, originally identified through its ability to complement UV-radiation sensitivity in tumor cells, is monoallelically deleted at a high frequency in human colon cancers and suppresses the proliferation and tumorigenicity of human colon cancer cells (Liang et al., 2006). Bif-1 interacts with Beclin 1 through UVRAG and activates the Beclin 1/class III PI3K complex, and its deletion in mice results in the development of spontaneous tumors (Table 1). Another component of the complex that positively regulates autophagy, Ambra1, has only been studied in the context of CNS development, but ambra1 deficiency in mouse embryos leads to severe neural tube defects associated with autophagy impairment and uncontrolled cell proliferation (Table 1), further supporting the concept that the Beclin 1/class III PI3K complex regulates cell growth.
The genetic links between deficiencies in the autophagy machinery and tumor susceptibility highlight the likely importance of autophagy in tumor suppression. However, the molecular mechanisms by which autophagy functions in tumor suppression are poorly defined. Increasing evidence suggests that the tumor suppressor functions of autophagy may be independent of both potential prodeath and prosurvival effects (Mathew et al., 2007a). Epithelial tumors that arise from cells with monoallelic or biallelic deletions of beclin 1 or atg5, respectively, do not display decreased cell death; in fact, in regions of metabolic stress, they display increased cell death, as would be expected with loss of autophagy-dependent survival (Mathew et al., 2007b). The association between increased cell death and increased tumorigenic potential in the setting of ATG gene deficiency suggests that autophagy-dependent survival, at least in certain experimental models, does not promote tumorigenesis. White and colleagues have proposed two hypotheses to explain how loss of autophagy, a cell survival pathway, may stimulate oncogenesis. First, when tumor cells cannot die by apoptosis upon exposure to metabolic stress, autophagy may prevent death from necrosis, a process that might exacerbate local inflammation and thereby increase tumor growth rate (Degenhardt et al., 2006). Second, ATG gene deletion may promote genomic instability in metabolically stressed cells, leading to oncogene activation and tumor progression (Mathew et al., 2007a). Indeed, immortalized mouse epithelial cells with ATG gene deficiency (monoallelic loss of beclin 1 or biallelic loss of atg5) display increased DNA damage, centrosome abnormalities, aneuploidy, numerical and structural chromosomal abnormalities, and gene amplification, especially during ischemic stress that is associated with increased tumorigenicity. These cells are engineered to have concurrent defects in cell-cycle checkpoints and apoptosis (e.g., p53 and Rb inactivation, Bcl-2 overexpression); thus, it is not yet known whether autophagy prevents genomic instability in normal cells and thereby plays a role in preventing tumor initiation.
Another possibility is that autophagy plays a more direct role in negative growth control, perhaps by degrading specific organelles or proteins essential for cell growth regulation. In support of this theory, enforced Beclin 1 expression slows the proliferation of tumor cell lines (without affecting cell death) and causes a decrease in expression of cyclin E and phosphorylated Rb (Koneri et al., 2007; Liang et al., 1999). Also, mice with a monoallelic deletion of beclin 1 display hyperproliferation of both mammary epithelial cells and splenic lymphocytes (Qu et al., 2003). In Drosophila, overexpression of Atg1, which causes the hyperactivation of autophagy, directly inhibits cell growth and induces cell death (Scott et al., 2007). In the nematode, caloric restriction and insulin-signaling mutations that increase autophagic activity reduce tumor cell division, although it is not yet known whether these antiproliferative effects are mediated by autophagy (Pinkston et al., 2006). One hypothesis is that, despite its prosurvival effects, autophagy activation in tumor cells by metabolic stress prevents inappropriate cell division. The spontaneous proliferation of normally quiescent mammary epithelial cells in beclin 1+/− mice suggests that autophagy (similar to TOR inhibition) plays a role in coordinating environmental cues and entry or maintenance of the G0 state.
Although it is presently unclear whether cell survival/cell death effects are relevant to the tumor suppressor role of autophagy, such effects are likely important in cancer therapeutics. A large series of clinically approved and experimental anticancer therapies induce the accumulation of autophagosomes in tumor cell lines in vitro (Maiuri et al., 2007a). For many years, it was thought that these therapies kill cells through autophagy (i.e., induce “autophagic cell death”). However, specific inhibition of autophagy with siRNAs targeted against ATG genes usually accelerates, rather than prevents, cell death in these settings (Maiuri et al., 2007a), indicating that autophagy activation represents a cellular attempt to cope with stress induced by cytotoxic agents. This suggests that inhibition of autophagy (rather than stimulation of autophagy) might be beneficial in cancer treatment. Indeed, in mice harboring c-Myc-induced lymphomas, the drug chloroquine, an alkalinizing lysosomotropic drug that impairs autophagic degradation, enhanced the ability of either p53 or a DNA alkylating agent to induce tumor cell death and tumor regression (Amaravadi et al., 2007), indicating a potential prosurvival and protumorigenic role for autophagy during cancer chemotherapy. Because chloroquine has pleiotropic effects, both on tumor cells (for instance on the multidrug pump) and on the host immune system (where it stimulates antitumor responses; Apetoh et al., 2007), it may be premature to explain its antitumor effects solely via its autophagy inhibitory action. Further in vivo studies are needed with more specific inhibitors of autophagy to determine whether the beneficial effects of blocking a tumor cell survival pathway outweigh the potential detrimental effects of blocking a tumor suppressor pathway.
Autophagy and Aging
The notion that intermittent fasting promotes longevity is not merely a cultural belief shared by diverse civilizations throughout history; it is a scientific truth that extends across eukaryotic organisms, including yeast, flies, worms, and rodents (Guarente and Kenyon, 2000). Dietary restriction is a potent inducer of autophagy in virtually all species (Levine and Klionsky, 2004). In the model organism, C. elegans, autophagy is required for the life-extending effects of dietary restriction; feeding-defective worms do not live longer if treated with siRNA against atg genes (Jia and Levine, 2007). A similar requirement exists for atg genes in the longevity phenotype of worms with a loss-of-function mutation in the insulin/IGF-1 signaling pathway (Melendez et al., 2003), a hormonal pathway that negatively regulates autophagy and life span in diverse species. Interestingly, dietary-restricted or long-lived insulin/IGF-1 mutants are resistant to many age-related diseases, including Huntington’s disease and cancer in C. elegans disease models, sarcopenia in worms, heart failure in Drosophila, and cancer in rodents (Kenyon, 2005). In view of the protective role of autophagy in certain age-related diseases such as neurodegeneration, cardiomyopathy, and cancer, a critical question is whether autophagy activation in long-lived mutant animals is mechanistically responsible for resistance to age-related disease.
A corollary question is whether autophagy function declines with age, and if so, whether such a decline contributes to aging and susceptibility to age-related diseases. Indeed, both classical autophagy (macroautophagy) and chaperone-mediated autophagy decline with aging in rodents and probably in humans (Martinez-Vicente and Cuervo, 2007). The mechanism for the decline in autophagy with aging is unknown but, at least in the rodent liver, is thought to involve alterations both in responses to hormonal regulation of autophagy (e.g., glucagon, insulin) and in the degradation of autophagosomes (Del Roso et al., 2003). It is possible that the accumulation of undigested material inside secondary lysosomes—a characteristic feature of aged, postmitotic cells—interferes with the ability of lysosomes to fuse with autophagosomes and degrade their cargo, thus creating a vicious cycle leading to a progressive defect in autophagosome degradation. However, it is equally plausible that this feature of aging results from, rather than causes, autophagy impairment. It will be important to further determine what age-related changes occur in the expression or function of proteins directly involved in autophagy regulation and execution or in lysosomal function.
A cardinal feature of aging postmitotic cells is the accumulation of damaged proteins and organelles—especially functionally disabled mitochondria and lipofuscin-loaded lysosomes. Mutations that decrease mitochondrial metabolism or reduce levels of reactive oxygen species extend life span in C. elegans and rodents, presumably by decreasing the generation of damaged proteins and organelles (Levine and Klionsky, 2004). In parallel, the activation or inhibition of autophagy likely prevent or promote aging, respectively, due to alterations in the removal of damaged proteins and organelles. As noted above, tissue-specific deletion of ATG genes in postmitotic cells, including neurons, hepatocytes, and cardiomyocytes, reveals a critical role for basal autophagy in protein and organelle turnover and prevention of cellular degeneration. Therefore, it is easy to imagine how either the cumulative effects of incomplete autophagic clearance over prolonged periods and/or age-related declines in autophagy function could contribute to aging. Caloric restriction can extend life span in diverse species and reverse the age-related decline in autophagy in rodent liver (Kenyon, 2005). Yet, despite the practice of fasting to promote longevity throughout human history, alternative approaches should be sought that mimic the beneficial effects of caloric restriction on autophagy while avoiding the potential adverse effects of caloric restriction on other aspects of human health. One such candidate is an antilipolytic drug that has been used successfully to increase autophagic activity in rodent liver and to extend life span (Bergamini, 2005). The life-span effects of other clinically used autophagy inducers such as rapamycin and lithium chloride have not been investigated. Ideally, the molecular basis for the age-related decline in autophagy function should be identified and pharmacologically targeted.
Autophagy in Infection, Immunity, and Inflammatory Diseases
The autophagic machinery is used in a multipronged defense against microbes, including the selective delivery of microorganisms to degradative lysosomes (a process referred to as xenophagy) and the delivery of microbial nucleic acids and antigens to endo/lysosomal compartments for activation of innate and adaptive immunity (see Levine and Deretic, 2007; Schmid and Munz, 2007 for detailed reviews) (Figure 4). Numerous medically important pathogens are degraded in vitro by xenophagy, including bacteria such as group A Streptococcus, Mycobacterium tubercuolosis, Shigella flexneri, Salmonella enterica, Listeria monocytogenes, Francisella tularensis; viruses such as herpes simplex virus type I (HSV-1); and parasites such as Toxoplasma gondii. It is predicted that xenophagy participates in pathogen protection in vivo, but data supporting this are limited to certain viral diseases such as tobacco mosaic virus in plants and HSV-1 and Sindbis virus in mice (Levine and Deretic, 2007). With the availability of tissue-specific ATG gene knockout mice, it should be possible to more broadly evaluate the role of xenophagy in microbial pathogenesis.
Figure 4. Autophagy in Innate and Adaptive Immunity.

In xenophagy, intracellular pathogens (bacteria, protozoans, and viruses) that are either inside the cytosol or in pathogen-containing vacuoles are surrounded by isolation membranes, engulfed into autophagosomes, and degraded inside autolysosomes. Autophagy may be involved in the activation of innate immunity by delivering viral nucleic acids to endosomal compartments containing Toll-like receptor 7 (TLR7), which signals the induction of type 1 interferon (IFN) production. Autophagy may be involved in adaptive immunity by delivering endogenously synthesized microbial antigens and self-antigens to late endosomes, where they are loaded onto MHC class II molecules for presentation to CD4+ T cells.
Recent studies also indicate that autophagy participates in trafficking events that activate innate and adaptive immunity (Levine and Deretic, 2007; Schmid and Munz, 2007). With respect to innate immunity, atg5 is required for the delivery of viral nucleic acids from Sendai virus and vesicular stomatitis virus to the endosomal toll-like receptor (TLR), TLR7, and subsequent activation of type I interferon signaling in plasmacytoid dendritic cells (Table 1). With respect to adaptive immunity, autophagy is involved in the delivery of certain endogenously synthesized microbial antigens (e.g., Epstein Barr viral antigens) to MHC class II antigen-presenting molecules, leading to the activation of CD4+ T lymphocytes. The selective targeting of other viral antigens, such as the influenza virus matrix protein, to the autophagosome by fusion with LC3 results in dramatic enhancement of CD4+ T lymphocyte responses. Although it is not yet known how universally important the contribution of autophagic delivery of microbial antigens is in adaptive immunity during natural infections, this latter finding has immediate implications for vaccine development; targeting proteins for autophagic delivery to MHC class II loading compartments may be an effective means to improve T helper cell responses and thereby increase vaccine efficacy. Not only does the autophagic machinery function in innate and adaptive immunity, but several innate and adaptive immune mediators involved in intracellular pathogen control stimulate autophagy, including the interferon-inducible antiviral molecule, PKR; CD40-CD40 ligand interactions; IFNγ and its downstream immunity-related GTPases; TNFα; T-helper type 1 lymphocytes; and the cell-surface receptor, TLR4 (Levine and Deretic, 2007).
Given the likely importance of autophagy in host defense against intracellular pathogens, microbial virulence may be partly determined by the ability of such pathogens to successfully antagonize host autophagy. Moreover, for organisms such as bacteria that can replicate outside of host cells, the ability to antagonize autophagy may influence whether or not the organism is an extracellular or intracellular pathogen. For example, group A Streptococcus is an extracellular pathogen that invades into the cytoplasm of epithelial cells but is rapidly degraded by an autophagy-dependent pathway (Nakagawa et al., 2004). Successful intracellular microbes may antagonize both the signaling pathways that activate autophagy as well as the membrane trafficking events required for lysosomal delivery and degradation, either indirectly or directly by inhibitory interactions with autophagy proteins (see Levine and Deretic, 2007 for detailed review). Perhaps the most compelling evidence to date that microbial evasion of autophagy is important for disease is a recent study with HSV-1; the ability of the virus to cause fatal encephalitis in mice is severely compromised by a mutation in a virally encoded neurovirulence factor that abrogates its ability to bind to Beclin 1 and inhibit host autophagy (Orvedahl et al., 2007). Thus, whereas host-related factors that contribute to autophagy impairment (e.g., mutations in genes involved in autophagy or lysosomal function, epigenetic changes that affect autophagolysomsomal trafficking) may contribute to susceptibility to certain other diseases (e.g., neuro-and myodegenerative disorders and aging), microbes may be able to successfully disarm even an optimally functioning host autophagy system to cause pathology. The selective disruption of interactions between microbial virulence factors and their targeted host autophagy proteins represents an attractive new antimicrobial therapeutic strategy.
Autophagy has additional effects on immunity that are not directly relevant to pathogen control (reviewed in Levine and Deretic, 2007). The role of autophagy in cellular homeostasis and life and death decisions may extend to T lymphocyte development and to T lymphocyte depletion during HIV infection. CD4+ and CD8+ T cells from atg5−/− mice fail to undergo efficient proliferation after T cell receptor stimulation. Upon transplantation into lethally irradiated mice, atg5−/− lymphocytes develop normally in the thymus but fail to repopulate the periphery due to overwhelming cell death (Table 1). One interpretation of this finding is that T cells, upon exit from the thymus, become exposed to nutritional stress due to limitations in trophic factor support and require autophagy for survival. In contrast, another study found that TH2 cells become more resistant to cell death induced by growth-factor withdrawal when autophagy is blocked using pharmacological or genetic methods (Li et al., 2006). This cell death process may be exploited by viruses such as HIV, as the HIV envelope glycoprotein induces autophagic cell death by binding to CXC-chemokine receptor 4 in uninfected bystander CD4+ T cells (Espert et al., 2006). Although only studied so far in the context of T cells, autophagy may also be involved in the homeostasis, differentiation, and function of other populations of immune cells. A particularly interesting question is whether autophagy plays a role in thymic selection and central tolerance. The role of autophagy in MHC class II presentation of self-antigens has not yet been directly examined, but the high levels of autophagic activity in the thymic epithelial cells of newborn mice (Mizushima et al., 2004) suggests that autophagy may enable thymic epithelial cells to present self-antigens to lymphocytes during positive and negative selection.
The recently discovered function of autophagy in ATP-dependent generation of engulfment signals and heterophagic removal of apoptotic corpses (Qu et al., 2007) hints at a potential role for autophagy in the prevention of inflammation and autoimmunity. The rapid removal of apoptotic corpses is critical for the prevention of tissue inflammation (Grossmayer et al., 2005), and indeed, autophagy-deficient atg5−/− embryos have increased inflammation in tissues that have impaired clearance of apoptotic cells (Qu et al., 2007). Moreover, the lack of efficient apoptotic cell clearance may overcome tolerance to self-antigens and lead to autoimmune diseases such as systemic lupus erythematosis (SLE) (Grossmayer et al., 2005). It is therefore possible that defective autophagy may contribute to the pathogenesis of SLE or other autoimmune diseases.
Several recent genome-wide scans have uncovered strong genetic associations between two genes involved in autophagy, the autophagy-stimulatory immunity-related GTPase, IRGM1, and the autophagy execution gene, Atg16L, and susceptibility to Crohn’s disease, a chronic inflammatory disease of the intestine (Table 2). These studies suggest a potential role for autophagy deregulation in the pathogenesis of Crohn’s disease. However, as of yet, it is not known whether the ATG16L variant (T300A) is defective in autophagy function and whether this genetic link is indicative of an underlying mechanistic role of autophagy impairment in Crohn’s disease pathogenesis. The pathogenic mechanisms of Crohn’s disease are incompletely understood but are postulated to involve a dysregulated immune response to commensal intestinal bacteria, altered mucosal barrier function, and/or defects in bacterial clearance (Baumgart and Carding, 2007). It is speculated that autophagy deficiency might contribute to one or more of these pathological mechanisms in Crohn’s disease (Levine and Deretic, 2007). Studies in targeted mutant mice with conditional deletion of atg16 and other ATG genes should help elucidate potential interrelationships between alterations in autophagy and the pathogenesis of Crohn’s disease.
Conclusion
Based on new understandings of the physiological functions of autophagy we now know that both basal levels of autophagy and stress-induced increases in autophagy are likely important in promoting mammalian health. Autophagy maintains nutrient and energy homeostasis in the face of a limited food supply; it clears intracellular proteins and damaged organelles that may lead to tissue degeneration, genomic instability, cancer, and aging; and it defends mammalian cells against microbial attack. Perhaps this is why so many cultures have empirically incorporated periodic fasting, a practice that activates autophagy, into their healing traditions.
The activation of autophagy, however, is not without potential risks. Autophagy may help keep alive those cells that should die, such as chemotherapy-treated tumor cells, or, if present in excess, kill cells that should live. If the lysosomal clearance of autophagosomes fails, the activation of autophagy results in a cellular traffic jam that may lead to increased pathology—a scenario that may in fact occur with aging, Parkinson’s disease, and other neuro-and myodegenerative disorders. Yet, in most diseases, we still do not fully understand how the potential risks of autophagy are weighed against its numerous adaptive physiological functions. The preponderance of currently available evidence from Drosophila, C. elegans, and mouse models does suggest that the primary function of autophagy may be to promote health and longevity. Nonetheless, further research is needed to define the precise determinants of whether autophagy is beneficial or pathological in more specific disease contexts.
Clearly, we have learned and will continue to learn a great deal from autophagy-deficient model organisms. The critical question is whether these lessons translate into predicted insights regarding the roles of autophagy in human disease. This seems likely given the evolutionarily conserved nature of autophagy, the regulation of its signaling pathways, its molecular machinery, and its physiological functions, but formal proof is lacking. If our predictions from animal models are correct, we will need more specific ways to stimulate autophagy and the clearance of autophagosomes (for the prevention/treatment of aging, infection, cancer, and degenerative diseases) as well as to inhibit autophagy (perhaps for cancer therapy). To accomplish this goal, we need a better understanding of the signaling control of autophagy, the molecular actions of the Atg proteins, the regulation of autophagosome/lysosome fusion, and the mechanistic basis for age-related and disease-specific defects in autophagy activation and autophagolysosomal maturation. Perhaps then, we will be able to live longer and healthier lives—without having to fast.
Acknowledgments
B.L. is supported by the National Institutes of Health, American Cancer Society, and Ellison Medical Foundation. G.K. is supported by Ligue Nationale contre le Cancer, Agence Nationale de Recherche, Institut National contre le Cancer, Cancéropôle Ile-de France, and European Union (Active p53, ApoSys, ChemoRes, DeathTrain, RIGHT, TransDeath). We thank R. Talley for administrative assistance and M. Packer for helpful comments.
References
- Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A, Thompson CB. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest. 2007;117:326–336. doi: 10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apetoh L, Ghriringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, et al. Toll-like receptor 4-dependent contribution of the immune system to anti-cancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–1059. doi: 10.1038/nm1622. [DOI] [PubMed] [Google Scholar]
- Askanas V, Engel WK. Inclusion-body myositis: a myodegenerative conformational disorder associated with Aβ, protein misfolding, and proteasome inhibition. Neurology. 2006;66:S39–S48. doi: 10.1212/01.wnl.0000192128.13875.1e. [DOI] [PubMed] [Google Scholar]
- Bahro M, Pfeifer U. Short-term stimulation by propanolol and verapamil of cardiac cellular autophagy. J Mol Cell Cardiol. 1987;19:1169–1178. doi: 10.1016/s0022-2828(87)80527-8. [DOI] [PubMed] [Google Scholar]
- Bassham DC, Laporte M, Marty F, Moriyasu Y, Ohsumi Y, Olsen LJ, Yoshimoto K. Autophagy in development and stress responses of plants. Autophagy. 2006;2:2–11. doi: 10.4161/auto.2092. [DOI] [PubMed] [Google Scholar]
- Baumgart DC, Carding SR. Inflammatory bowel disease: cause and immunobiology. Lancet. 2007;369:1627–1647. doi: 10.1016/S0140-6736(07)60750-8. [DOI] [PubMed] [Google Scholar]
- Bergamini E. Targets for antiageing drugs. Expert Opin Ther Targets. 2005;9:77–82. doi: 10.1517/14728222.9.1.77. [DOI] [PubMed] [Google Scholar]
- Bolanos-Meade J, Zhou L, Hoke A, Corse A, Vogelsang G, Wagner KR. Hydroxychloroquine causes severe vacuolar myopathy in a patient with chronic graft-versus-host disease. Am J Hematol. 2005;78:306–309. doi: 10.1002/ajh.20294. [DOI] [PubMed] [Google Scholar]
- Cao Y, Espinola JA, Fossale E, Massey AC, Cuervo AM, MacDonald ME, Cotman SL. Autophagy is disrupted in a knock-in mouse model of juvenile neuronal ceroid lipofuscinosis. J Biol Chem. 2006;281:20483–20493. doi: 10.1074/jbc.M602180200. [DOI] [PubMed] [Google Scholar]
- Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–134. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- Criollo A, Maiuri MC, Tasdemir E, Vitale I, Fiebig AA, Andrews D, Molgo J, Diaz J, Lavandero S, Harper F, et al. Regulation of autophagy by the inositol trisphosphate receptor. Cell Death Differ. 2007;14:1029–1039. doi: 10.1038/sj.cdd.4402099. [DOI] [PubMed] [Google Scholar]
- Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer. 2006;6:184–192. doi: 10.1038/nrc1819. [DOI] [PubMed] [Google Scholar]
- de Duve C, Wattiaux R. The lysosome. Annu Rev Physiol. 1966;28:435–492. doi: 10.1146/annurev.ph.28.030166.002251. [DOI] [PubMed] [Google Scholar]
- Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gelinas C, Fan Y, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Roso A, Vittorini S, Cavallini G, Donati A, Gori Z, Masini M, Pollera M, Bergamini E. Ageing-related changes in the in vivo function of rat liver macroautophagy and proteolysis. Exp Gerontol. 2003;38:519–527. doi: 10.1016/s0531-5565(03)00002-0. [DOI] [PubMed] [Google Scholar]
- Espert L, Denizot M, Grimaldi M, Robert-Hebmann V, Gay B, Varbanov M, Codogno P, Biard-Piechaczyk M. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J Clin Invest. 2006;116:2161–2172. doi: 10.1172/JCI26185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felbor U, Kessler B, Mothes W, Goebel HH, Ploegh HL, Bronson RT, Olsen BR. Neuronal loss and brain atrophy in mice lacking cathepsins B and L. Proc Natl Acad Sci USA. 2002;99:7883–7888. doi: 10.1073/pnas.112632299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci USA. 2005;102:8204–8209. doi: 10.1073/pnas.0502857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, Corazzari M, Fuoco C, Ucar A, Schwartz P, et al. Ambra1 regulates autophagy and development of the nervous system. Nature. 2007;447:1121–1125. doi: 10.1038/nature05925. [DOI] [PubMed] [Google Scholar]
- Fukuda T, Ahearn M, Roberts A, Mattaliano RJ, Zaal K, Ralston E, Plotz PH, Raben N. Autophagy and mistargeting of therapeutic enzyme in skeletal muscle in Pompe disease. Mol Ther. 2006;14:831–839. doi: 10.1016/j.ymthe.2006.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozuacik D, Kimchi A. DAPk protein family and cancer. Autophagy. 2006;2:74–79. doi: 10.4161/auto.2.2.2459. [DOI] [PubMed] [Google Scholar]
- Grossmayer GE, Munoz LE, Gaipl US, Franz S, Sheriff A, Voll RE, Kalden JR, Hermann M. Removal of dying cells and systemic lupus erythematous. Mod Rheumatol. 2005;15:383–390. doi: 10.1007/s10165-005-0430-x. [DOI] [PubMed] [Google Scholar]
- Guarente L, Kenyon C. Genetic pathways that regulate ageing in model organisms. Nature. 2000;408:255–262. doi: 10.1038/35041700. [DOI] [PubMed] [Google Scholar]
- Hamacher-Brady A, Brady NR, Gottlieb RA. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J Biol Chem. 2006;281:29776–29787. doi: 10.1074/jbc.M603783200. [DOI] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Iwata J, Ezaki J, Komatsu M, Yokota S, Ueno T, Tanida I, Chiba T, Tanaka K, Kominami E. Excess peroxisomes are degraded by autophagic machinery in mammals. J Biol Chem. 2006;281:4035–4041. doi: 10.1074/jbc.M512283200. [DOI] [PubMed] [Google Scholar]
- Ji H, Ramsey MR, Hayes DN, Fan C, McNamara K, Kozlowski P, Torrice C, Wu MC, Shimamura T, Perea SA, et al. LKB1 modulates lung cancer differentiation and metastastis. Nat Rev Cancer. 2007;448:807–811. doi: 10.1038/nature06030. [DOI] [PubMed] [Google Scholar]
- Jia K, Levine B. Autophagy is required for dietary restriction-mediated lifespan extension in C. elegans. Autophagy. 2007;3:597–599. doi: 10.4161/auto.4989. [DOI] [PubMed] [Google Scholar]
- Jia K, Hart AC, Levine B. Autophagy genes protect against disease caused by polyglutamine expansion proteins in Caenorhabditis elegans. Autophagy. 2007;3:21–25. doi: 10.4161/auto.3528. [DOI] [PubMed] [Google Scholar]
- Jin S, White E. Role of autophagy in cancer: management of metabolic stress. Autophagy. 2007;3:28–31. doi: 10.4161/auto.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamimoto T, Shoji S, Hidvegi T, Mizushima N, Umebayashi K, Perlmutter DH, Yoshimori T. Intracellular inclusions containing mutant α-1 antitrypsin Z are propagated in the absence of autophagic activity. J Biol Chem. 2006;281:4467–4476. doi: 10.1074/jbc.M509409200. [DOI] [PubMed] [Google Scholar]
- Kang C, You YJ, Avery L. Dual roles of autophagy in the survival of Caenorhabditis elegans during starvation. Genes Dev. 2007;21:2161–2171. doi: 10.1101/gad.1573107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S, White E. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 2007;21:1621–1635. doi: 10.1101/gad.1565707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon C. The plasticity of aging: insights from long-lived mutants. Cell. 2005;120:449–460. doi: 10.1016/j.cell.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Kim I, Rodriquez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007;462:245–253. doi: 10.1016/j.abb.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8:931–937. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- Koneri K, Goi T, Hirono Y, Katayama K, Yamaguchi A. Beclin 1 gene inhibits tumor growth in colon cancer cell lines. Anticancer Res. 2007;27:1453–1457. [PubMed] [Google Scholar]
- Koike M, Shibata M, Waguri S, Yoshimura K, Tanida I, Kominami E, Gotow T, Peters C, von Figura K, Mizushima N, et al. Participation of autophagy in storage of lysosomes in neurons from mouse models of neuronal ceroid-lipofuscinoses (Batten disease) Am J Pathol. 2005;167:1713–1728. doi: 10.1016/S0002-9440(10)61253-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science. 2007;315:1398–1401. doi: 10.1126/science.1136880. [DOI] [PubMed] [Google Scholar]
- Levine B. Cell biology: autophagy and cancer. Nature. 2007;446:745–747. doi: 10.1038/446745a. [DOI] [PubMed] [Google Scholar]
- Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol. 2007;7:767–777. doi: 10.1038/nri2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- Li C, Capan E, Zhao Y, Zhao J, Stolz D, Watkins SC, Jin S, Lu B. Autophagy is induced in CD4+ T cells and important for the growth-factor withdrawal cell death. J Immunol. 2006;177:5163–5168. doi: 10.4049/jimmunol.177.8.5163. [DOI] [PubMed] [Google Scholar]
- Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, Jung JU. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol. 2006;8:688–699. doi: 10.1038/ncb1426. [DOI] [PubMed] [Google Scholar]
- Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, Kondo S, Dumont DJ, Gutterman JU, Walker CL, et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007;9:218–224. doi: 10.1038/ncb1537. [DOI] [PubMed] [Google Scholar]
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- Lucas GJ, Daroscewska A, Ralston SH. Contribution of genetic factors to the pathogenesis of Paget’s disease of bone and related disorders. J Bone Miner Res. 2006;21:31–37. doi: 10.1359/jbmr.06s206. [DOI] [PubMed] [Google Scholar]
- Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–248. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- Lunemann JD, Schmidt J, Schmid D, Barthel K, Wrede A, Dalakas MC, Munz C. β-Amyloid is a substrate of autophagy in sporadic inclusion body myositis. Ann Neurol. 2007;61:476–483. doi: 10.1002/ana.21115. [DOI] [PubMed] [Google Scholar]
- Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007a;8:741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K, Tavernarakis N, et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007b;26:2527–2539. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino G, Salvador-Montoliu N, Fueyo A, Knecht E, Mizushima N, Lopez-Otin C. Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3. J Biol Chem. 2007;282:18573–18583. doi: 10.1074/jbc.M701194200. [DOI] [PubMed] [Google Scholar]
- Martinez-Vicente M, Cuervo AM. Autophagy and neurodegeneration: when the cleaning crew goes on strike. Lancet Neurol. 2007;6:352–361. doi: 10.1016/S1474-4422(07)70076-5. [DOI] [PubMed] [Google Scholar]
- Massey DC, Parkes M. Genome-wide association scanning highlights two autophagy genes, ATG16L and IRGM, as being significantly associated with Crohn’s disease. Autophagy. 2007;3:649–651. doi: 10.4161/auto.5075. [DOI] [PubMed] [Google Scholar]
- Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007a;7:961–967. doi: 10.1038/nrc2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, Chen G, Jin S, White E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007b;21:1367–1381. doi: 10.1101/gad.1545107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion. Circ Res. 2007;100:914–922. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- Meijer AJ, Codogno P. Signalling and autophagy regulation in health, aging, and disease. Mol Aspects Med. 2006;27:411–425. doi: 10.1016/j.mam.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Melendez A, Talloczy Z, Seaman M, Eskelinen EL, Hall DH, Levine B. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science. 2003;301:1387–1391. doi: 10.1126/science.1087782. [DOI] [PubMed] [Google Scholar]
- Miracco C, Cosci E, Oliveri G, Luzi P, Pacenti L, Monciatti I, Mannucci S, De Nisi MC, Toscano M, Malagnino V, et al. Protein and mRNA expression of autophagy gene Beclin 1 in human brain tumors. Int J Oncol. 2007;30:429–436. [PubMed] [Google Scholar]
- Mizushima N, Klionsky D. Protein turnover via autophagy: implications for metabolism. Annu Rev Nutr. 2007;27:19–40. doi: 10.1146/annurev.nutr.27.061406.093749. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in responose to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair U, Klionsky DJ. Molecular mechanisms and regulation of specific and nonspecific autophagy pathways in yeast. J Biol Chem. 2005;280:41785–41788. doi: 10.1074/jbc.R500016200. [DOI] [PubMed] [Google Scholar]
- Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T, Nara A, Funao J, Nakata M, Tsuda K, et al. Autophagy defends cells against invading group A Streptococcus. Science. 2004;306:1037–1040. doi: 10.1126/science.1103966. [DOI] [PubMed] [Google Scholar]
- Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- Nishino I. Autophagic vacuolar myopathy. Semin Pediatr Neurol. 2006;13:90–95. doi: 10.1016/j.spen.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Nishino I, Fu J, Tanji K, Yamada T, Shimojo S, Koori T, Mora M, Riggs JE, Oh SJ, Koga Y, et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease) Nature. 2000;406:906–910. doi: 10.1038/35022604. [DOI] [PubMed] [Google Scholar]
- Orvedahl A, Alexander D, Talloczy Z, Sun Q, Wei Y, Zhang W, Burns D, Leib DA, Levine B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe. 2007;1:23–35. doi: 10.1016/j.chom.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–863. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- Pankiv S, Hoyvarade Clausen T, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. p62/SGSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24135. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Perlmutter DH. The role of autophagy in alpha-1-antitrypsin deficiency. Autophagy. 2006;2:258–263. doi: 10.4161/auto.2882. [DOI] [PubMed] [Google Scholar]
- Pinkston JM, Garigan D, Hansen M, Kenyon C. Mutations that increase the life span of C. elegans inhibit tumor growth. Science. 2006;313:971–975. doi: 10.1126/science.1121908. [DOI] [PubMed] [Google Scholar]
- Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med. 2007;204:25–31. doi: 10.1084/jem.20061303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puls I, Oh SJ, Summer CJ, Wallace KE, Floeter MK, Mann EA, Kennedy WR, Wendelschafer-Crabb G, Vortmeyer A, Powers R, et al. Distal spinal and bulbar muscular atrophy caused by dynactin mutation. Ann Neurol. 2005;57:687–694. doi: 10.1002/ana.20468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–1820. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X, Zou Z, Sun Q, Luby-Phelps K, Cheng P, Hogan RN, Gilpin C, Levine B. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell. 2007;128:931–946. doi: 10.1016/j.cell.2006.12.044. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Acevedo-Arozena A, Imarisio S, Berger Z, Vacher C, O’Kane CJ, Brown SD, Rubinsztein DC. Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nat Genet. 2005;37:771–776. doi: 10.1038/ng1591. [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Potential therapeutic applications of autophagy. Nat Rev Drug Discov. 2007;6:304–312. doi: 10.1038/nrd2272. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Perlstein EO, Imarisio S, Pineau S, Cordenier A, Maglathlin RL, Webster JA, Lewis TA, O’Kane CJ, Schreiber SL, Rubinsztein DC. Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nat Chem Biol. 2007;3:331–338. doi: 10.1038/nchembio883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid D, Munz C. Innate and adaptive immunity through autophagy. Immunity. 2007;27:11–21. doi: 10.1016/j.immuni.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz RA, Fernandez G, Kotulska K, Jozwiak S. Tuberous sclerosis complex: advances in diagnosis, genetics, and management. J Am Acad Dermatol. 2007;57:189–202. doi: 10.1016/j.jaad.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Scott RC, Juhasz G, Neufeld TP. Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr Biol. 2007;17:1–11. doi: 10.1016/j.cub.2006.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shacka JJ, Klocke BJ, Young C, Shibata M, Olney JW, Uchiyama Y, Saftig P, Roth KA. Cathepsin D deficiency induces persistent neurodegeneration in the absence of Bax-dependent apoptosis. J Neurosci. 2007;27:2081–2090. doi: 10.1523/JNEUROSCI.5577-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata M, Lu T, Furuya T, Degterev A, Mizushima N, Yoshimori T, Mac-Donald M, Yankner B, Yuan J. Regulation of intracellular accumulation of mutant Huntingtin by Beclin 1. J Biol Chem. 2006;281:14474–14485. doi: 10.1074/jbc.M600364200. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JQ, Mul JJ, et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol. 2007;10:1142–1151. doi: 10.1038/ncb1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takacs-Vellai K, Vellai T, Puoti A, Passannante M, Wicky C, Streit A, Kovacs AL, Muller F. Inactivation of the autophagy gene bec-1 triggers apoptotic cell death in C. elegans. Curr Biol. 2005;15:1513–1517. doi: 10.1016/j.cub.2005.07.035. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Gudhe G, Suter A, Eskelinen EL, Hartmann D, Lullmann-Rauch R, Janssen PM, Blanz J, von Figura K, Saftig P. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature. 2000;406:902–906. doi: 10.1038/35022595. [DOI] [PubMed] [Google Scholar]
- Terman A, Brunk UT. Autophagy in cardiac myocyte homeostasis, aging, and pathology. Cardiovasc Res. 2005;68:355–365. doi: 10.1016/j.cardiores.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Vesa J, Peltonen L. Mutated genes in juvenile and variant late infantile neuronal ceroid lipofuscinosis. Curr Mol Med. 2002;2:439–444. doi: 10.2174/1566524023362311. [DOI] [PubMed] [Google Scholar]
- Williams A, Jahreiss L, Sarkar S, Saiki S, Menzies FM, Ravikumar B, Rubinsztein DC. Aggregate-prone proteins are cleared from the cytosol by autophagy: therapeutic implications. Curr Top Dev Biol. 2006;76:89–101. doi: 10.1016/S0070-2153(06)76003-3. [DOI] [PubMed] [Google Scholar]
- Yorimitsu T, Klionsky DJ. Eating the endoplasmic reticulum: quality control by autophagy. Trends Cell Biol. 2007;17:279–285. doi: 10.1016/j.tcb.2007.04.005. [DOI] [PubMed] [Google Scholar]
- Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, Brunner T, Simon HU. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. 2006;8:1124–1132. doi: 10.1038/ncb1482. [DOI] [PubMed] [Google Scholar]
- Yu WH, Cuervo AM, Kumar A, Peterhoff CM, Schmidt SD, Lee JH, Mohan PS, Mercken M, Farmery MR, Tjernberg LO, et al. Macroautophagy - a novel β-amyloid peptide-generating pathway activated in Alzheimer’s disease. J Cell Biol. 2005;171:87–98. doi: 10.1083/jcb.200505082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Z, Horton A, Bravin M, DeJager PL, Selimi F, Heintz N. A novel protein complex linking the delta 2 glutamate receptor and autophagy: implications for neurodegeneration in lurcher mice. Neuron. 2002;35:921–933. doi: 10.1016/s0896-6273(02)00861-9. [DOI] [PubMed] [Google Scholar]
- Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci USA. 2003;100:15077–15082. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Qi H, Taylor R, Xu W, Liu LF, Jin S. The role of autophagy in mitochondria maintenance: Characterization of mitochondrial functions in autophagy-deficient S. cerevisiae strains. Autophagy. 2007;3:337–346. doi: 10.4161/auto.4127. [DOI] [PubMed] [Google Scholar]
- Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA, Hill JA. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest. 2007;117:1782–1793. doi: 10.1172/JCI27523. [DOI] [PMC free article] [PubMed] [Google Scholar]