

Summary
RND (Resistance-Nodulation-Division) family transporters are widespread especially among Gram-negative bacteria, and catalyze the active efflux of many antibiotics and chemotherapeutic agents. They have very large periplasmic domains, and form tripartite complexes with outer membrane channels and periplasmic adaptor proteins. AcrAB-TolC complex of Escherichia coli, which pumps out a very wide range of drugs, has been studied most intensively. Early studies showed that the transporter captures even those substrates that cannot permeate across the cytoplasmic membrane, such as dianionic β-lactams, suggesting that the capture can occur from the periplasm. It was also suggested that the capture occurs from the cytoplasmic membrane/periplasm interface, because most substrates contain a sizable hydrophobic domain; however, this may simply be a reflection of the nature of the binding site within AcrB. Genetic studies of chimeric transporters showed that much of the substrate specificity is determined by their periplasmic domains. Biochemical studies with intact cells recently led to the determination of the kinetic constants of AcrB for some β-lactams, and the result confirms the old prediction that AcrB is a rather slow pump. Reconstitution of purified AcrB and its relatives showed that the pump is a drug/proton antiporter, that AcrA strongly stimulates the activity of the pump, and that AcrB seems to have a highest affinity for conjugated bile salts. Structural study with mutants of the network of charged residues in the transmembrane domain showed that protonation here produced a far-reaching conformational change, which was found to be present in one of the protomers in the asymmetric crystal structure of the wild-type AcrB. The functional rotatory hypothesis then predicts that the drug bound in the periplasmic domain is extruded through this conformational change initiated by the protonation of one of the residues in the aforementioned network, an idea that was recently supported by disulfide cross-linking as well as by the behavior of linked AcrB protomers.
Keywords: AcrB, AcrD, TolC, AcrA, reconstitution, disulfide cross-linking, proton relay network
INTRODUCTION
Efflux pumps of the RND (Resistance-Nodulation- Division) superfamily (such as AcrB of Escherichia coli and MexB of Pseudomonas aeruginosa) play an important role in producing multidrug resistance (both intrinsic and elevated) in Gram-negative bacteria. This is because these pumps become associated with two other classes of proteins, the outer membrane channel such as TolC of E. coli and OprM of P. aeruginosa, belonging to the OMF (outer membrane factor) family of proteins [1], and the periplasmic “adaptor” protein such as AcrA of E. coli and MexA of P. aeruginosa, classified into the MFP (membrane fusion protein) family [2]. Importantly, each of these three component proteins is essential for drug efflux, and the absence of even one component makes the entire complex totally nonfunctional [3, 4]. The construction of this tripartite complex suggested that the drugs are here exported directly into the external medium, rather than into the periplasm [5]. This is a huge advantage for bacterial cells, because once exported into the external space, drug molecules must traverse the outer membrane barrier to reenter the cells. (An early conceptual diagram is shown in Fig. 1). Thus these pumps work synergistically with the outer membrane barrier. Wild-type strains of most Gram-negative bacteria are resistant to most lipophilic antibiotics (for E. coli they include penicillin G, oxacillin, cloxacillin, nafcillin, macrolides, novobiocin, linezolid, and fusidic acid), and this “intrinsic resistance” was often thought to be caused by the exclusion of drugs by the outer membrane barrier. Indeed breaching the outer membrane barrier does sensitize E. coli cells to the drugs just mentioned [6]. However, the inactivation of the major and constitutively expressed RND pump AcrB makes the bacteria almost completely susceptible to these agents (the minimal inhibitory concentration [MIC] of a lipophilic penicillin, cloxacillin, goes down from 512 μg/ml in the wild type to only 2 μg/ml [7]) even in the presence of the intact outer membrane barrier. Thus the characteristic intrinsic resistance of gram-negative bacteria owes as much to the RND pumps as to the outer membrane barrier.
Fig. 1.

An early schematic view of the tripartite pump complex. Note that amphiphilic substrates (empty and filled-in rectangles represent hydrophobic and hydrophilic parts of the molecule) are hypothesized to be captured either from the periplasm (or the periplasm-plasma membrane interface) or from the cytosol (or the cytosol-membrane interface). For the latter process, two possible pathways are envisaged: either the substrate is flipped over to the outer surface of the membrane first and then follows the regular periplasmic capture pathway, or it follows a different capture pathway from the cytosol. From [5].
SUBSTRATE RANGE OF AcrB AND THE PERIPLASMIC SUBSTRATE CAPTURE
We noted early [5] that AcrB of E. coli can handle a very wide range of compounds. These include cationic dyes such as acriflavine, crystal violet, ethidium bromide, and rhodamine 6G; antibiotics such as penicillins, cephalosporins, fluoroquinolones, macrolides, chloramphenicol, tetracyclines, novobiocin, fusidic acid, oxazolidinones, and rifampicin; detergents such as Triton X-100, sodium dodecylsulfate, and bile acids; and even simple organic solvents. It was obvious that there is no structural similarity between most of these compounds. One feature that was common was that the substrates were either lipophilic or at least contained lipophilic domains [5]. This led to the hypothesis that the substrates first interact with the membrane lipid bilayer [5], and become captured either from within the bilayer or from the bilayer-aqueous interface [5](Fig. 1). However, now that the structure of AcrB is known [8], we can interpret these observations in an alternative manner as the result of interaction between the substrate and the hydrophobic interior of the substrate-binding site(s) within AcrB.
Another question that attracted our attention in the early days of research was the possibility of the capture of substrates from the periplasm or a location that is in rapid equilibrium with periplasm. This question arose because the overexpression of the main RND pump in Pseudomonas aeruginosa (later identified as MexB [9]) produced resistance to β-lactams [10], which were believed to remain in the periplasm because their targets, penicillin-binding proteins, are located in this compartment. We actually measured the distribution of β-lactams within P. aeruginosa cells [10]. We found that monoanionic penicillins and cephalosporins, such as benzylpenicillin and cephalothin, slowly traverse the cytoplasmic membrane and enter the cytosol. However, dianionic compounds such as carbenicillin or ceftriaxone could not cross the cytoplasmic membrane, and stayed completely in the periplasm. Yet carbenicillin was a good substrate for this RND pump, an observation that forced us to conclude that the pump unexpectedly could capture its substrates from the periplasm, or from the periplasm-cytoplasmic membrane interface (Fig. 1).
This issue was revisited several years later by the use of Salmonella typhimurium ( S. enterica serovar Typhimurium) [7]. Again, it was confirmed that dianionic β-lactams cannot cross the cytoplasmic membrane. Furthermore, a correlation was observed between the lipophilicity of the side-chain and the efficiency at which the compounds were pumped out by AcrB. The latter observation, however, is not easily interpreted, as mentioned earlier.
Finally, a strong evidence supporting the periplasmic capture of aminoglycosides was obtained by the in vitro reconstitution experiment of AcrD [11], which will be described later. Thus it seems clear that periplasmic capture occurs. It is not clear, however, at this point whether this is the predominant mode of capture, or how and to what extent the substrates may be captured from the cytosol.
In this connection, it is interesting that the general inhibitor of P. aeruginosa and E. coli RND pumps, MC-207,110 (phenylalanyl-arginyl-β-naphthylamine)[12], which apparently is a favored substrate of the pumps, affected the carbenicillin MIC only to a small extent [12]. Thus the addition of this inhibitor decreased the MICs of drugs such as erythromycin and levofloxacin to almost exactly the same degree as the genetic inactivation of the MexB-MexA-OprM pump. In contrast the inhibitor produced a decrease of only fourfold in carbenicillin MIC, although the genetic inactivation decreased it 512-fold. A similar discrepancy was also found for the MICs of tetracycline and ethidium bromide [12]. Since we know that carbenicillin must be captured exclusively from periplasm, these results might suggest that this particular inhibitor acts more effectively for drugs that are predominantly captured in the cytosol. However, the results may be caused by the binding of various substrates to different subdomains of the large binding pocket of the transporter.
The RND pumps have been shown to act in synergy with the “simple” pumps that extrude drugs only into the periplasm [13]. For example, in P. aeruginosa, the expression of a simple tetracycline pump TetA produces only a very modest degree of resistance (MIC = 32 μg/ml) in the absence of the main tripartite pump MexB-MexA-OprM, but MIC is raised very strongly to 512 μg/ml if the tripartite pump in addition is expressed at the normal, constitutive level. That the high resistance is not due to the pumping of tetracycline by the tripartite pump alone is seen from the fact that the tripartite pump, in the absence of TetA, produces an MIC of only 4 μg/ml. Thus these two types of pumps appear to act truly in a synergistic manner. The most likely molecular explanation of these results is that the tetracycline pumped out into the periplasm by the TetA pump is then captured by the MexB pump, and is extruded into the outside medium through the tripartite structure [13]. This concept then explains why the simple, or the single-component transporters such as TetA [14] or S. aureus QacA [15] confer significant resistance to E. coli cells. It also emphasizes the importance of periplasmic substrate capture mechanism in the function of RND pumps.
KINETIC CONSTANTS OF THE AcrB PUMP
In order to understand the behavior of the pump in intact cells, it is obviously essential to know its kinetic constants. The first hint on the binding affinity of various substrates was obtained by using them as competitive inhibitors in the reconstitution assay of AcrB [16], which will be described in detail later in this review. In this assay, which depended on the export of fluorescent phospholipids by the purified AcrB protein, taurocholate, a conjugated bile salt, inhibited the reaction most strongly (presumably by competing as substrates), the 50% inhibition occurring at 15 μM. In contrast, antibiotics were less efficient inhibitors, and cloxacillin and erythromycin caused 50% inhibition only around 100 μM. These results are important in showing that the conjugated bile salts are the preferred, and most likely the natural, substrates of the AcrB pump of E. coli, which must live in an environment full of these membrane-disrupting detergents. However, this is not a direct assay of substrate binding, and we do not know the true affinity of each substrate to the pump. (We should also mention that running similar assays in intact cells is difficult because both the substrate and the competitive inhibitor must diffuse across the outer membrane before binding to the pump.)
There was a recent attempt to measure the binding of substrates to purified AcrB protein by the use of fluorescence polarization [17]. Because binding of fluorescent substrates to a large AcrB trimer is expected to slow the tumbling of these molecules, this ingenious method offers advantages. The apparent dissociation constants reported ranged from 5.5 μM for rhodamine 6G to 74 μM for ciprofloxacin. However, the method does not give reliable values of binding stoichiometry, and because the authors did not use mutant AcrB proteins that are altered in substrate-binding sites, it is unclear whether the binding occurred to the true binding sites, or to some extraneous pockets that bind lipophilic molecules in a non-specific manner.
Several laboratories followed the efflux of fluorescent dyes from cells after their energization. Typically, cells are preloaded with dyes that become fluorescent only within the membrane. The preloading requires inactivation of the RND pump by proton-conductor such as CCCP (carbonyl cyanide m-chlorophenylhydrazone), and re-energization of the cells is done by adding compounds such as glucose or formate. If everything goes perfectly, the time-dependent decrease in fluorescence intensity should follow the integrated form of the Michaelis-Menten equation, which should give us the kinetic constants Km and Vmax. In the first paper reporting on the use of these probes [18], the results were not convincing because the efflux rates from P. aeruginosa strains either producing or not producing the MexB-MexA-OrpM efflux complex showed little difference, presumably because deenergization with cyanide could not be reversed rapidly. However, efflux assays were successfully performed since then as a qualitative assay, for example with NPN (N-phenylnaphthylamine) and E. coli [12], and with DASPEI (2-[4-dimethylamino]styryl- N-ethylpyridinium iodide) and E. coli [19]. No effort to use the efflux curve for quantitative analysis of the transport process was reported. We have tried to do this quantitative analysis with a fluorescent dye, Nile Red using E. coli (J. Bohnert and H. Nikaido, unpublished results). However, the system is extremely complex, because CCCP used in the dye-loading period may still persist in the cells at least in the beginning, and the full membrane energization with carbon sources will take a few seconds, thus making the analysis of the early phase of the efflux curve quite difficult.
Very recently we had the first success with the estimation of kinetic constants of the AcrB pump (K. Nagano and H. Nikaido, manuscript in preparation). With attempts using intact cells, the difficulty is always with the estimation of the substrate concentration within periplasm, where it is interacting with the pump. We solved this problem by using β-lactams as substrates. Since β-lactams are hydrolyzed by the periplasmic β-lactamase, and since we know the Km and Vmax of the enzyme, we can calculate the periplasmic concentration of β-lactams from the Michaelis-Menten equation if we follow the hydrolysis of β-lactams by intact cells. Once the periplasmic concentration is known, the rate at which the β-lactams cross the outer membrane by diffusion through porin channels (Vin) can be calculated because it is proportional to the concentration difference of the β-lactam between the external medium and the periplasm. The efflux rate is the difference between the influx rate Vin and the hydrolysis rate Vhyd, or Vin-Vhyd. This approach was successfully applied to the influx, efflux, and hydrolysis of a cephalosporin, nitrocefin, and we found that the efflux occurs with the Michaelis-Menten saturation kinetics, with the apparent Km of 3–5 μM. There was little sign of positive cooperativity, a result that is interesting in view of the functional rotating mechanism of the pump and an earlier report on the reconstituted CzcA pump [20], described below. A turnover number of about 10 s−1 was calculated. The low turnover number was as expected, because the tripartite pumps have to deal only with the small number of drug molecules that have trickled through the effective outer membrane barrier [5].
Very recently, binding of 24-, 25-, or 27-hydroxysterols to Niemann-Pick C1 protein (NPC1), a member of the RND family involved in cholesterol trafficking in vertebrate cells [21], was reported [22]. The binding took place in the presence of a high concentration (1 %) of the detergent Nonidet P40, and the half-saturation was reached with about 0.1 μM of 25-hydroxysterol. 7-, 19, 20-Hydroxysterols did not bind to this protein. In comparison with bacterial RND pumps, the NPC1 protein has an N-terminal extension consisting of an extramembranous domain of about 240 residues and an additional transmembrane segment. This extramembranous domain is secreted from cultured cells as a soluble dimer, which bound 25-hydroxysterol with a strong affinity (KD = 10 nM)[23]. Since this domain is uniquely present in NPC1, it is unclear whether these beautiful results would help us in our effort to understand the ligand binding process in efflux transporters.
SUGGESTIONS FROM GENETIC STUDIES
Genetic approaches have been able to provide valuable insights on the reaction mechanism of RND pumps, as described below.
a. Use of chimeric genes
Replacing large segments of genes with sequences coming from another homolog has been most useful in identifying the domains responsible for functions of these pumps. This was first done in 2002 by Elkins and Nikaido [24] as well as Tikhonova, Wang, and Zgurskaya [25]. The first group used E. coli AcrB, which gives a strong resistance to ciprofloxacin, novobiocin, fusidic acid, erythromycin, and taurocholate, and AcrD, which generates no resistance to these agents except a modest one against taurocholate. Replacing both of the large periplasmic loops of AcrD with those from AcrB created a transporter pumping out all of the tested AcrB substrates efficiently. Replacing loops of AcrB with those from AcrD created a transporter that behaved like AcrD in terms of its substrate specificity. Finally, replacing the transmembrane regions of AcrD with corresponding sequences from AcrB did not alter the substrate specificity of the transporter. These results suggest that the substrate specificity, and presumably the critical binding of substrates, occur in the periplasmic domain, not in the transmembrane domain. The latter group used AcrB and P. aeruginosa MexB. The results suggested that the substrate specificity was determined largely by the second external loop of these transporters, a conclusion consistent with the location of the substrate-binding pocket identified in the asymmetric trimer crystal of AcrB (see Inference from Structure, below). They also showed that replacing the region coding for the transmembrane helices 8 through 12 of AcrB with the MexB sequence produced a somewhat MexB-like pattern for a few drugs (lower efflux of ethidium bromide and lincomycin, and higher efflux of cinnoxacin); it is difficult to explain these results in terms of structure. Finally, this group showed that the N-terminal periplasmic loop and the first part of the C-terminal loop essentially determine the interaction of the RND pumps with their cognate periplasmic adaptor proteins. In 2003, a study replacing the transmembrane strands of P. aeruginosa MexB with those of MexY, an aminoglycoside pump [26], was published. There was no change in substrate specificity. Although these are negative results, they are at least consistent with the hypothesis that the periplasmic domain plays a large role in determining the substrate specificity.
b. Use of random mutants
In this approach pioneered by Lomovskaya’s laboratory, spontaneous mutants of P. aeruginosa MexD that acquired the ability to extrude carbenicillin, which is not handled by the wild-type MexD, were isolated [27]. All mutants mapped to the periplasmic domain, none to the transmembrane domain, further supporting the conclusion mentioned above. Among the residues identified, Phe608 (corresponding to Phe610 of AcrB) is a part of the substrate-binding pocket more recently identified (see below). Some others (Gln34, Gln89 [corresponding to Glu 89 of AcrB], Asn673[Thr676]) are on the walls of the large lateral cleft of the periplasmic domain, which will be discussed later as a potential site of periplasmic entry for substrates.
Bohnert et al. [28] used an E. coli strain that lacked both AcrB and AcrF and instead overproduced another RND pump, YhiV. A spontaneous mutant obtained after repeated exposure to levofloxacin was shown to owe its increased resistance to a change of an aliphatic Val610 into an aromatic Phe in YhiV. This mutant pump has an altered specificity. It produces a stronger resistance to relatively small, aromatic compounds such as levofloxacin, linezolid and tetracycline, but the resistance to a non-aromatic, bulky macrolides becomes weaker than in the parent protein. This residue, which corresponds to Val612 of AcrB, is seen to be a part of the substrate-binding pocket in the asymmetric AcrB trimer structure that will be discussed later.
In a different approach, Middlemiss and Poole [29] carried out an in vitro random mutagenesis of the mexB gene from P. aeruginosa. This is a more comprehensive approach, but is expected to generate more “noise.” Indeed the group of mutants that decreased the level of resistance to most of drugs included the presumed proton relay mutants in Asp407 and Asp408 of the transmembrane domain (see below), or Gly220 mutant in the “peg” that is inserted into the periplasmic domain of the neighboring protomer. The mutants that are significantly altered in the substrate specificity, in contrast, often contained alterations in the periplasmic domain, as expected. Among these, alterations of Ala618 and Arg716 (corresponding to Arg717 of AcrB) occur on the opposing walls of the large lateral cleft. However, there were several mutants in the transmembrane domain, and their properties remain to be explained.
c. Site-directed mutagenesis
In 1999, inspection of amino acid sequence of an RND-family toxic cation efflux pump CzcA of Ralstonia sp. and other RND pumps showed that there are several conserved charged residues in the transmembrane domain, including Asp402, Asp408, and Glu415 [20]. Changing these residues into non-acidic residues abolished the cation pumping activity. This was followed up a few years later by the site-directed mutagenesis study of MexB [30]. In MexB, the Asp402 and Asp408 of CzcA are replaced by a tandem pair of aspartate residues, Asp407 and Asp408 on transmembrane segment (TMS) 4, and both of them were found to be essential for the pump function. This study also identified an essential Lys939 located in TMS 10, and suggested that these three residues produce a proton-relay network. Finally, we have identified Thr978 of AcrB on TMS 11 as another, functionally essential component of this network [31]. (This study also identified Arg971, located close to the cytosolic end of TMS11, as an essential residue [31], although it is far away from the network of other charged residues.)
Site-directed mutagenesis was also applied to residues assumed to be involved in the binding or passage of substrates. When we reported on the crystal structures of AcrB with ligands in the central cavity of the transmembrane domain [32], we were aware of the problem that the ligand might be binding to any hydrophobic pocket of the protein, which has nothing to do with the normal pathway of the exported ligand. Thus we changed residues that appeared to be involved in the binding through site-directed mutagenesis, and we obtained an assuring result that conversion of Phe386 to alanine nearly totally abolished the resistance to ethidium, rhodamine 6G, and dequalinium [32]. However, this result was obtained by using a very high copy number vector of the pUC series, and we now are aware that this approach may produce misleading results possibly by making the cell membrane leaky through a strong overproduction of intrinsic membrane proteins. Indeed, Phe386Ala mutation hardly affected susceptibility of E. coli to most drugs, when tested by expression with the vector pSPORT1 [33]. Nevertheless, Hearn and coworkers [34], using a Pseudomonas fluorescens homolog of AcrB, EmhB, expressed from a medium copy number plasmid, found that decreased efflux of dequalinium was produced by the same change (Phe386Ala) as well as the change of Asn99 to alanine. Furthermore, changing Asp101, which is located at the “ceiling” of the central cavity, into alanine decreased the efflux of most drugs as well as of polycyclic hydrocarbons.
With AcrB mutant Asn101Ala, which will be described later in the section Inference from Structure, changing into alanine the residues Glu673, Phe664, or Phe666, all located in the right (looking into the center of molecule with the periplasmic domain up) wall or the bottom of the large periplasmic cleft, close to its entrance, decreased the efflux activity [33]. With the Cu+ and Ag+ efflux pump CusA of E. coli, changing into leucine the methionine residues 573, 623, and 672, all close to the entrance of the periplasmic large cleft in the AcrB model, strongly reduced its activity [35]. Together with the binding of substrates to this cleft observed in the Asn101Ala mutant AcrB [33], these results favor the assumption that the periplasmic uptake of substrates by these pumps involves the entry from the large periplasmic cleft.
In an interesting study, Dastidar et al. [36] introduced cysteine residues to various positions of Haemophilus influenzae AcrB, chosen on the basis of their effects on substrate specificity in MexD [27] (described above), and measured their accessibility to a relatively hydrophilic probe, fluorescein maleimide in the presence and absence of efflux substrates. Most substrates, except ethidium, inhibited strongly the modification of cysteine at position 288, which corresponds to Gly290 of AcrB that is close to the bottom of the substrate-binding pocket in the asymmetric trimer structure (described later in the review). Interestingly, the modification of cysteine at position 601, corresponding to Phe617 of AcrB, one of the residues lining the substrate-binding pocket, is inhibited by Triton X-100, but not by other substrates. These data suggest the large size (or the flexibility) of the binding pocket that would accommodate different ligands in different ways.
Murakami et al. [19] used cysteine scanning mutagenesis to examine the role of the central “pore,” which appears to be closed in the crystal structure of AcrB. Although alterations of many residues, including Asp101 just mentioned, were found to decrease the extrusion of drugs, the interpretation is complicated by the fact that the pore is made by a three-stranded coiled-coil structure composed of all three protomers of AcrB, so that inter-subunit disulfide bonds are formed in some cases.
RECONSTITUTION STUDIES
In the biochemical studies of most bacterial transporters, membrane vesicles, either right-side-out or inverted, have been most useful. However, with RND family multidrug efflux pumps, this approach was not successful, although it was vigorously pursued in several laboratories including our own. The likely reasons for this failure include the inherently slow kinetics of the pump, described above, and the hydrophobic nature of most substrates which allows a spontaneous equilibration of transported substrates across the membrane layer. Another possibility may be that the periplasmic adaptor protein, which seems to be necessary to activate the transporter (see below), becomes stripped off during the preparation of the membranes.
The first successful functional reconstitution of an RND pump was achieved by Zgurskaya and Nikaido [16] in 1999. Cells containing acrAB genes on a high-copy-number pUC plasmid were grown without induction, and the membrane proteins were solubilized with 5% Triton X-100 overnight in the cold. The native AcrB protein contains four histidine residues among the eight residues at its C-terminus, and the protein could be purified by adsorption to a Cu2+-chelate matrix followed by elution with imidazole, always in the presence of Triton X-100. Just before the reconstitution, AcrB was adsorbed again to the Cu2+-chelate matrix, and Triton X-100 was exchanged with octyl-β-D-glucoside by elution with a buffer containing the latter detergent as well as imidazole. Since AcrB formed visible aggregates in octylglucoside within a few hours, it was immediately reconstituted into proteoliposomes by the octylglucoside dilution method.
Innovative approaches were required for assays of the transport activity of AcrB. Unlike many transporters that move hydrophilic ligands (see below), most of the substrates of AcrB are quite hydrophobic [5], and are expected to traverse across membrane bilayers through spontaneous diffusion. Thus it was impossible to rely upon the quantitation of ligands accumulated within the intravesicular space. Inspired by the report that mammalian P-glycoprotein, which also transports hydrophobic compounds, has activity as a phospholipid flippase [37], we used a 7-nitrobenz-2-oxa-1,3-diazol-4-yl (NBD)-labeled fluorescent phospholipid as the substrate. Phospholipids, however, are likely to become reinserted into the original membrane even when they are expelled from it by the AcrB pump. In order to minimize this possibility, an excess of “acceptor” liposomes, which did not contain AcrB, were used to “trap” the fluorescent phospholipids extruded. Finally, the amount of the NBD-labeled phospholipids remaining in the AcrB-containing “donor” proteoliposomes was estimated by initially quenching the NBD fluorescence through fluorescence energy transfer to rhodamine-labeled phospholipids (Fig. 2A).
Fig. 2.
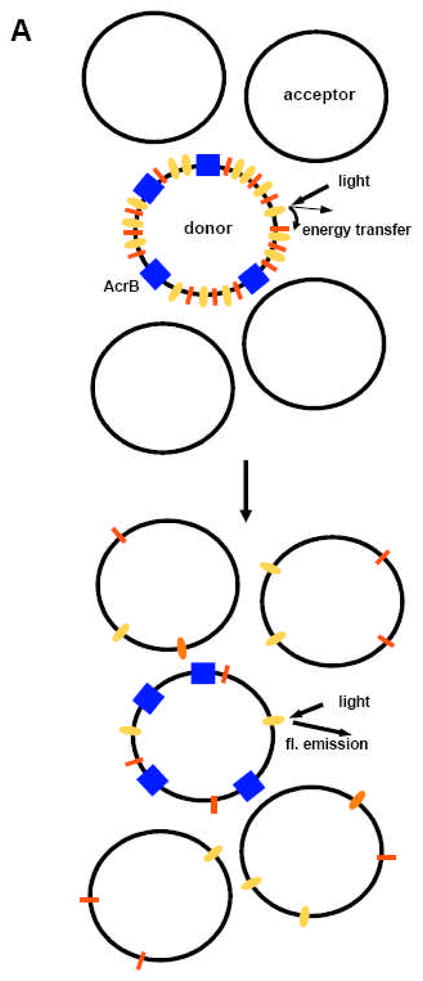
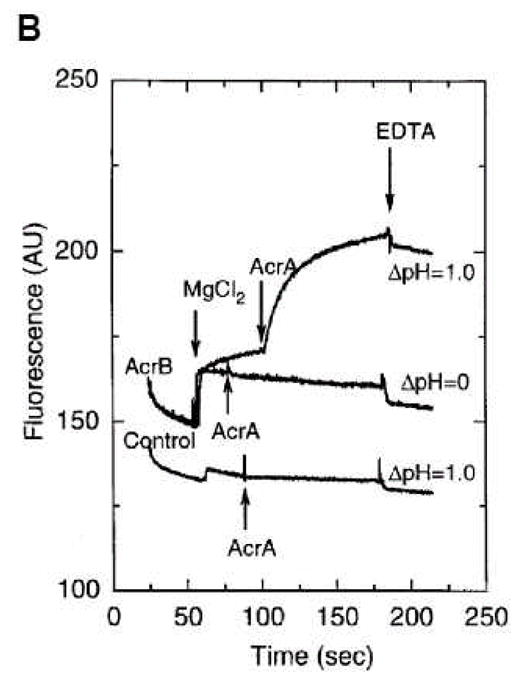
Phospholipid extrusion assay of reconstituted AcrB. A. Principle of the assay. Donor proteoliposomes containing AcrB (blue rectangles) were mixed with acceptor liposomes not containing any protein. The NBD fluorescence from the NBD-labeled phosphatidylethanolamine (orange) in the donor vesicles is initially quenched by fluorescent emission energy transfer to the rhodamine-labeled phosphatidylethanolamine (red). However, following the export of phospholipids to the acceptor vesicles, the surface density of labeled phospholipids decreases, and the fluorescent emission from NBD increases. B. An example of data. When a transmembrane pH gradient was generated by diluting the vesicles (internal pH, 7.0) into a buffer of pH 6.0, donor vesicles not containing any AcrB (bottom trace) do not show much change in fluorescence. Nor do the AcrB-containing vesicles in the absence of pH gradient (middle trace). However, when AcrB-containing vesicles are exposed to pH gradient, there is the expected gradual increase in NBD fluorescence in the presence of Mg2+. Note that the rate of fluorescence increase is strongly accelerated if AcrA (15 μg/ml) is also added. From [16].
Fig. 2B shows a representative result. Addition of Mg2+ activated the transport process, which is consistent with the observation that in intact cells, another RND-family multidrug efflux pump MexXY requires the presence of Mg2+ in the medium for its function [38]. The pumping was further accelerated by the addition of lipid-free AcrA. The latter result was interpreted as the result of AcrA protein connecting the donor and acceptor vesicles [16]. Alternatively, however, AcrA might have activated the pumping activity of AcrB directly, in view of the fact that AcrD, a close homolog of AcrB, is activated in a reconstituted system by AcrA [11](see below).
To show more directly that AcrB functions as a proton/drug antiporter, the system was energized by using the valinomycin-induced flux of K+, which was converted into a proton gradient in the presence of KCl. By measuring the intravesicular pH with a fluorescent, membrane-impermeable pH indicator pyranine, we confirmed that proton efflux occurred accompanying the pH-gradient-driven influx of drugs (Fig. 3). When the number of protons moved per vesicle is calculated, it appears that the AcrB pump is functioning extremely slowly, with a turnover rate of less than one per minute. We note that the assay was carried out without the addition of Mg2+ and AcrA, which were needed for the activation of AcrB according to the fluorescent phospholipid extrusion assay. We also do not know if a fraction of the pump was inactivated during the purification and reconstitution.
Fig. 3.
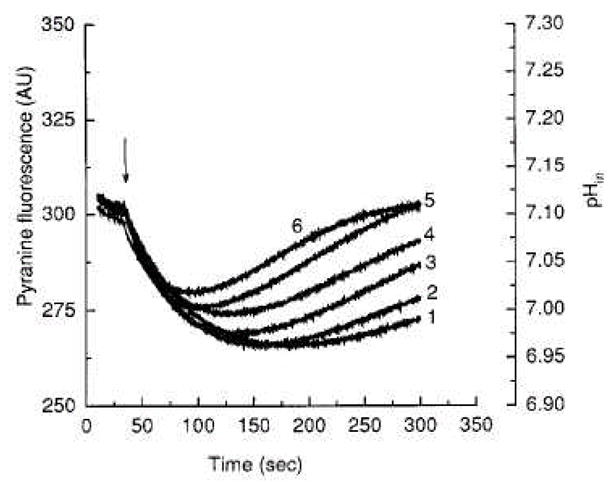
Proton flux assay of reconstituted AcrB. AcrB-containing vesicles were made in the presence of 1 mM pyranine, a fluorescent pH indicator, and the extravesicular pyranine was removed by gel filtration. The interior-acidic pH gradient was generated by diluting the vesicles containing 0.1 M KCl into 0.1 M NaCl and then by adding valinomycin (arrow). The acidic internal pH was maintained for several minutes in the absence of drug (curve 1). When erythromycin (curve 2), chloramphenicol (curve 3), cloxacillin (curve 4), glycocholate (curve 5), or taurocholate (curve 6) were present at 0.2 mM, the interior pH returned to its initial value more rapidly, presumably due to the proton efflux accompanying the influx of drugs by the AcrB antiporter. From [16].
The affinity of substrates for AcrB was estimated in two ways. First, the substrates were added as potential competitors in the fluorescent phospholipid efflux assay. This showed that conjugated bile acids, such as taurocholate, were the most effective inhibitors of phospholipid efflux (Fig. 4). Second, the potential substrates were added in the proton efflux assay, and again taurocholate was the most effective among the compounds tested (Fig. 3). These results fit the notion that the natural substrate for the AcrB pump are the bile salts, most of which exist in the conjugated form in the intestinal tract of higher animals.
Fig. 4.
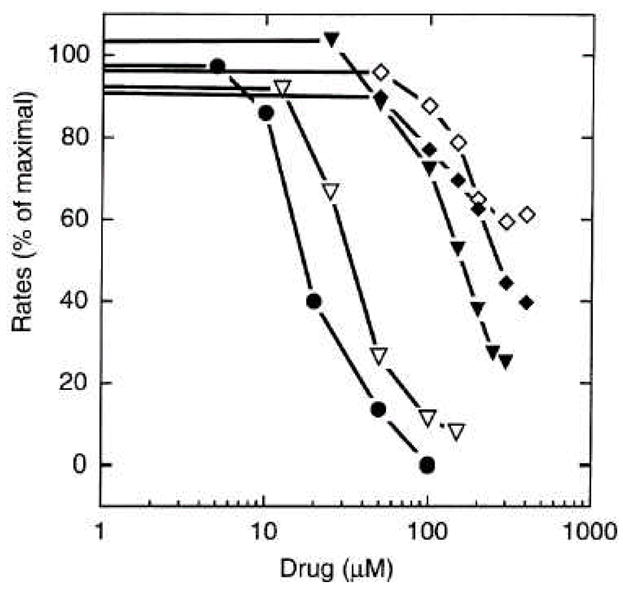
Inhibition of the phospholipid extrusion activity of reconstituted AcrB by known substrates. The initial rates of the intermembrane lipid transfer were measured in a setup similar to Fig. 2 in the presence of erythromycin (◇), cloxacillin (◆), glycochenodeoxycholate (▼;), glycocholate ( ▽ ), and taurocholate (●). The vesicles were first equilibrated with the drug. Initial rates were measured after the addition of AcrA to 25 μg/ml. From [16].
Soon afterward, the successful reconstitution of another RND pump, CzcA protein of Ralstonia, was reported [20]. CzcA catalyzes the export of toxic divalent cations such as Zn2+, Co2+, and Cd2+. The PCR-amplified gene was inserted into a commercial vector pASK-IBA3, which supplies a C-terminal, eight-residue tag. The protein was purified by affinity chromatography with a modified streptavidin matrix that binds the C-terminal tag [39]. It may be significant that solubilization and purification were performed in the presence of phospholipids, as specified by [40]. The purified CzcA was mixed with Triton X-100-treated liposomes, and the proteoliposomes were formed by removal of the detergent with Bio-Beads. A pH gradient was imposed across the membrane by diluting proteoliposomes containing 100 mM Tris-Cl at pH 5.0 into 100 mM Tris-Cl at pH 7.0. Since the substrates do not diffuse spontaneously across the membrane bilayer, the pumping activity can be assessed by traditional methods, and indeed vesicles were shown to accumulate divalent cations such as Zn2+. Better results were obtained by acidifying the vesicle interior by the diffusion of NH3 from proteoliposomes loaded with 0.5 M NH4Cl. The data with Zn2+ were reported to show a sigmoidal kinetics with a Hill coefficient of 2, although only four concentrations of the substrate were used. The half-maximal rate was achieved at a very high concentration of 6.6 mM, which casts doubt on the physiological relevance of these data. We now know that a similar cation pump, CusA captures ions from periplasm (see Coda below); yet these reconstitution experiments were done to detect the capture of extravesicular (corresponding to cytosolic) capture of cations, although we cannot exclude the possibility that the two systems are very different.
Reconstitution of another multidrug efflux RND pump AcrD was achieved in 2005 [11]. AcrD was of special interest, as some of its substrates, aminoglycosides, are very hydrophilic and are not expected to cross membrane bilayers spontaneously. AcrD, in a form with a C-terminal hexahistidine tag, was expressed from a high copy number plasmid with isopropyl-β-D-thiogalactoside (IPTG) induction, and was purified in dodecylmaltoside. The AcrD preparations in dodecylmaltoside were used directly for proteoliposome reconstitution by octylglucoside dilution method, without the prior removal of dodecylmaltoside. Routine assays relied on the proton eflux determined with the intravesicular pH indicator pyranine, as described earlier. It was soon discovered that no acceleration of proton flux occurred, with aminoglycosides (70 μM) either outside or inside vesicles, unless AcrA (with the lipidation site removed) was added at the time of reconstitution, and thus to the interior of the vesicles (Fig. 5A). It was known that AcrA was required for the activity of AcrD in intact cells [24], but this was expected because AcrA (or one of its homologs) would be needed for the construction of a tripartite efflux machinery. In the proteoliposomes containing AcrD, a tripartite structure is not needed for efflux, and the result strongly suggested that AcrA and its homologs of the MFP family had another important function in directly activating the pumping function of the cognate RND efflux transporter. A similar situation was found more recently with the MFP MacA for the activation of an ABC efflux transporter MacB [41]. When the aminoglycosides were added to the external medium, the drugs are occupying a compartment that is similar to the cytosol in intact cells. Under such a setup, we could show that kanamycin, tobramycin, gentamicin, and amikacin produced an increased efflux of proton (Figs. 5B and 5C), presumably because the aminoglycosides were being pumped into the vesicles by AcrD. Indeed, the intravesicular accumulation of an aminoglycoside could be demonstrated by using [3H]gentamicin [11]. When aminoglycosides were added into the intravesicular space by performing reconstitution in their presence, and the vesicle interior is acidified, we get a situation in which the drugs occupy an acidic space comparable to the periplasmic space. Under such conditions, gentamicin and amikacin both strongly accelerated the proton efflux, showing directly that these substrates of the RND pump can be captured from the periplasm (Fig. 6). Another interesting finding was that one aminoglycoside with the structure very different from all others, streptomycin, stimulated proton movement only when it was incorporated into the intravesicular space (corresponding to periplasm in intact cells), and not when it was present in extravesicular medium (corresponding to cytosol) [11]. This suggests that there are two separate ways for capturing substrates in AcrD.
Fig. 5.
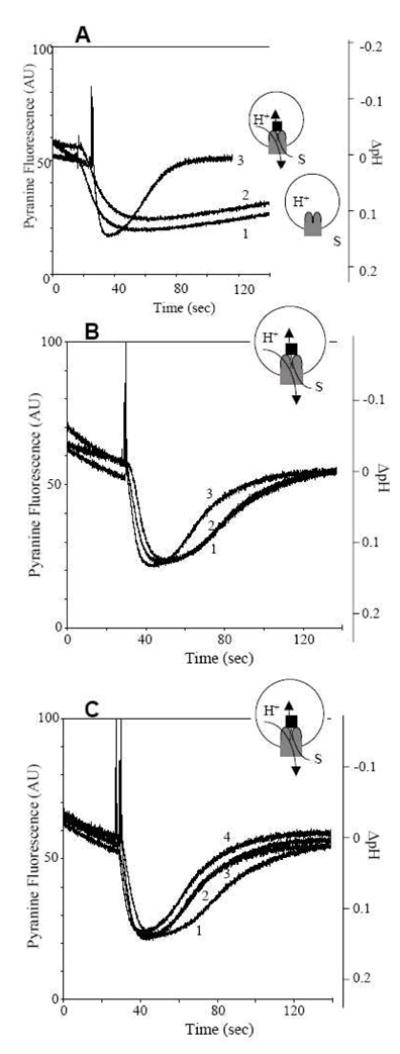
Proton flux assay of reconstituted AcrD with extravesicular substrates. AcrD-containing proteoliposomes were made in KCl, and an interior-acidic pH gradient was generated by diluting them into NaCl and by adding valinomycin at 30 s. The insets show the experimental set-up. The gray object denotes AcrD, with its periplasmic domain shown as ellipses. The black rectangle denotes AcrA, and the substrates are shown as “S”. A. The requirement for AcrA. Curve 2 shows that, even when gentamicin (70 μM) was present in the extravesicular space, no increase in proton efflux was seen in comparison with the control vesicles not containing AcrD (curve 1). However, when AcrA (plus Mg2+) was included within the vesicles (curve 3), a strong stimulation of proton efflux (presumably accompanying gentamicin transport) was seen. B. The effect of gentamicin addition in a single batch of AcrD-containing proteoliposomes with AcrA inside the vesicles. Curve 1, no antibiotic added. Curve 3, gentamicin added to the external medium. Curve 2, gentamicin and anti-AcrD polyclonal antiserum added to the external medium. C. The effect of various aminoglycosides on proton efflux. To a single batch of AcrD-containing proteoliposomes with AcrA inside, 70 μM amikacin (curve 2), tobramycin (curve 3), or gentamicin (curve 4) was added to the extravesicular space. Curve 1 shows no drug addition control. From [11].
Fig. 6.
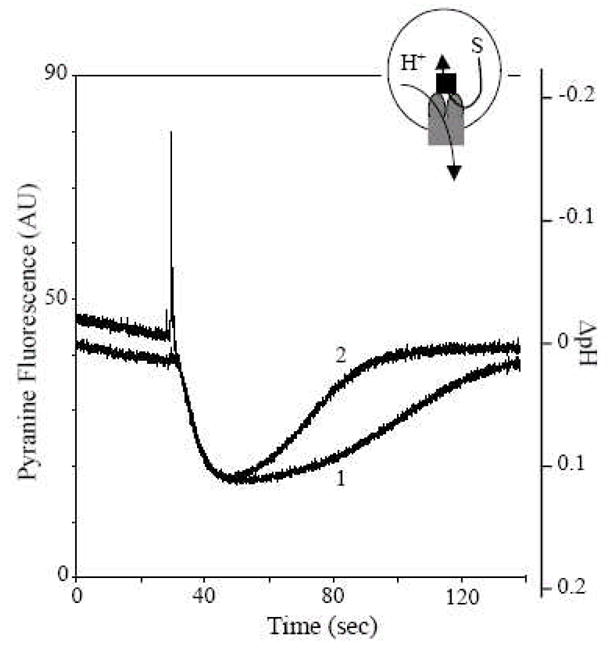
Proton flux assay of reconstituted AcrD with an intravesicular substrate. Two AcrD-containing proteoliposome preparations were made side-by-side with entrapped AcrA. One preparation did not contain any substrate (curve 1), but the other was made with entrapped gentamicin (curve 2). After the generation of pH gradient (inside acidic), the vesicles with entrapped gentamicin are seen to show an enhanced proton efflux, presumably because the substrate is captured by AcrD and is released into the same intravesicular space. From [11].
INFERENCE FROM STRUCTURE
Very important pieces of knowledge on the mechanisms of RND pump complex came from crystallography. Thus the structure of the outer membrane channel TolC was determined in 2000 [42](shown in red in Fig. 7A), followed by the structure of its P. aeruginosa homolog, OprM [43]. These proteins exist as a tightly woven trimer, containing a single 12-stranded β-barrel traversing the outer membrane, and a remarkably long (close to 100 Å) periplasmic extension of the channel as a long a-helical bundle. Murakami et al. solved the structure of AcrB as a homotrimer with a three-fold rotational symmetry in 2002 [8], the first crystallographic structure for a proton-motive-force-driven transporter solved (green in Fig. 7A; more details are shown in Fig. 7B). The periplasmic portion is at least as large as the transmembrane portion as was predicted from the amino acid sequence, and the top of the periplasmic domain, TolC-binding domain, has the dimension that is similar to the tip of the a-helical bundle of TolC. This was followed by the crystallographic elucidation of the structures of the central portion (about two-thirds) of the adaptor proteins MexA [44, 45] and AcrA [46](blue in Fig. 7A). These are elongated proteins as shown earlier [47], and in crystals show a strong tendency to become packed side-by-side to form hexamers and heptamers. Thus the structures of all three component classes of the tripartite assembly are known, and it is possible to propose how these proteins are assembled together [48](Fig. 7A). In such models, the coiled coil domain of the adaptor is assumed to interact with the outer membrane channel protein, a hypothesis that is supported by biochemical data [49, 50]. Studies with chimeric constructs of AcrA homologs showed that the C-terminal domain, whose structure is not known from crystallography, is essential for interaction with AcrB [51]. Mutant studies also suggest that the domain close to this end of AcrA, containing the a+β structure, is the main site of its interaction with the periplasmic domain of RND pumps [52].
Fig. 7.
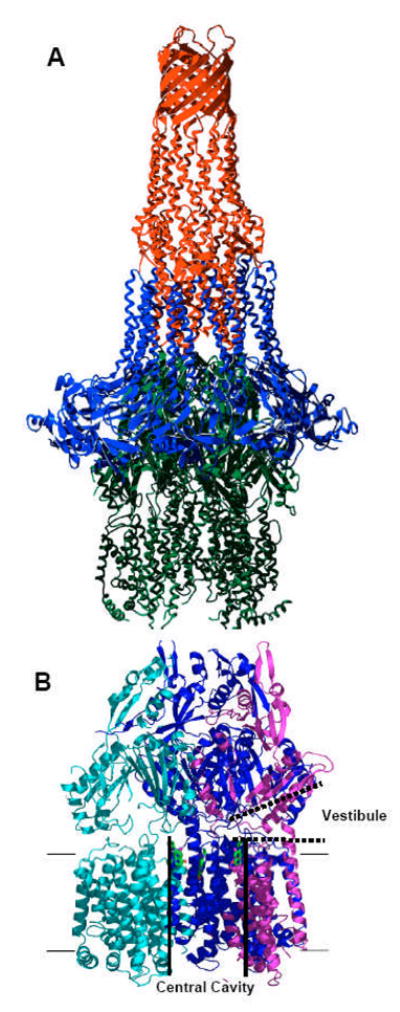
Crystal structure of TolC, AcrA, and AcrB. A. A hypothetical arrangement of the tripartite TolC (red)-AcrA (blue)-AcrB (green) structure based on x-ray crystal structures. From [45], with permission from Elsevier. B. AcrB trimer. Each protomer is shown in cyan, mauve, and blue. The large central cavity (thick black lines) is connected to the periplasm through vestibules (thick dotted lines) between protomers. Substrate molecules (ciprofloxacin) bound to the ceiling of the central cavity are shown in green stick models. Proximal portion of the structure was cut away to reveal the presence of vestibule. Drawn by using PyMol with Protein Data Bank coordinate 1OYE.
Earlier we mentioned that AcrB likely captures some of its substrates from the periplasm, and the prediction made in the early days that drugs may first partition into the membrane-periplasm interface (Fig. 1). Indeed the AcrB trimer structure showed that there was a small opening (vestibule) between subunits at the bottom of the periplasmic domain, close to the external surface of the membrane bilayer [8], and this led to the top of the large central cavity in the transmembrane domain (Fig. 7B). It was thus hypothesized that the drugs diffused through the vestibule into the central cavity, where it was captured. Indeed, co-crystallization of AcrB with various drugs produced crystals with drug molecules within this central cavity [53]. However, there was no proof that the bound drugs were on their correct path to extrusion, and the extrusion pathways were not clear in the crystal structure. Nevertheless it must be stated that in the AcrB-YajC cocrystal obtained accidentally from E. coli grown in ampicillin-containing medium [54] contains two ampicillin molecules per AcrB protomer at this position.
We obtained another set of data with one site-directed mutant of AcrB, Asn109Ala [33]. In the simplest hypothesis for drug extrusion, the drug molecules are captured at the ceiling of the central cavity, enter the central pore bordered by three helices (roughly corresponding to Asp99 through Leu117) from the three protomers, and pass through this pore to reach the top of the periplasmic domain. However, the pore in the crystal structure appears closed. Since the side-chain of Asn109 appears to extend deeply into the center of the pore, we naively thought that changing it to alanine might weaken the interaction of helices from the three subunits, and “loosen up” the pore. The mutant AcrB functions almost normally as judged from the resistance levels to several inhibitors. Interestingly, when the mutant protein was crystallized in the presence of several ligands (ethidium, rhodamine 6G, ciprofloxacin, nafcillin, and the inhibitor Phe-Arg-β-naphthylamide [12]), ligands were found to be bound not only to the ceiling of the central cavity (as seen earlier [53]) but also to the periplasmic domain, to one side of the large, deep, external cleft. Again, there was the problem that the ligands could have become bound to any lipophilic pocket which may not be on the proper pathway for export. Changing residues that appear to be interacting with the ligands into alanine through site-directed mutagenesis produced a drastic decrease in resistance to most drugs in the case of Glu673, and more modest yet significant decreases with Phe666 and Phe664. Even these data are not decisive, however, because they may inactivate the transporter by a mechanism unrelated to ligand binding. Nevertheless the more recent asymmetric AcrB structures (discussed later in this review) are consistent with the hypothesis that this is the portal for the periplasmic entry of substrates.
Because of these difficulties, we tried a different approach. We crystallized AcrB mutants in which one of the residues in the transmembrane domain putatively involved in proton translocation (see “site-directed mutagenesis” above) was altered [31, 55]. We assumed that during the normal function cycle of AcrB, a proton antiporter, one of these residues will be altered in its protonation state, and this may cause conformational changes of the whole protein which would be coupled to ligand export. If so, our mutant proteins would mimic this transient state, and would assume a conformation similar to that of a transient intermediate during the process of drug extrusion. In the mutants examined, Asp407, Asp408, or Lys940 was changed into Ala. Since inspection of the AcrB crystal structure showed that the side-chain of Thr978 also interacts tightly with the three amino acid residues just mentioned [31], we also included this residue for analysis. When the crystal structures (without ligands) of these mutant proteins were examined [55], we found an extensive conformational alteration in the transmembrane domain (Fig. 8). It was quite reassuring that the conformational alterations were similar in all four mutants, confirming that they were due to the disruption of the salt bridge/H-bonding network among these four residues, rather than to specific effects of individual mutations. Much of the changes involve the shortening or melting of helices, except that helix 8, one of the helices connecting the transmembrane domain with the periplasmic domain, becomes extended into the periplasmic domain (Fig. 8). When the structure of the periplasmic domain was examined, however, there were only very small changes [55], a disappointing result because we expected major conformational alterations in the periplasmic domain during substrate binding and extrusion (see above). The reason for this failure was suggested from the studies describing the asymmetric trimer structure of AcrB, described below.
Fig. 8.
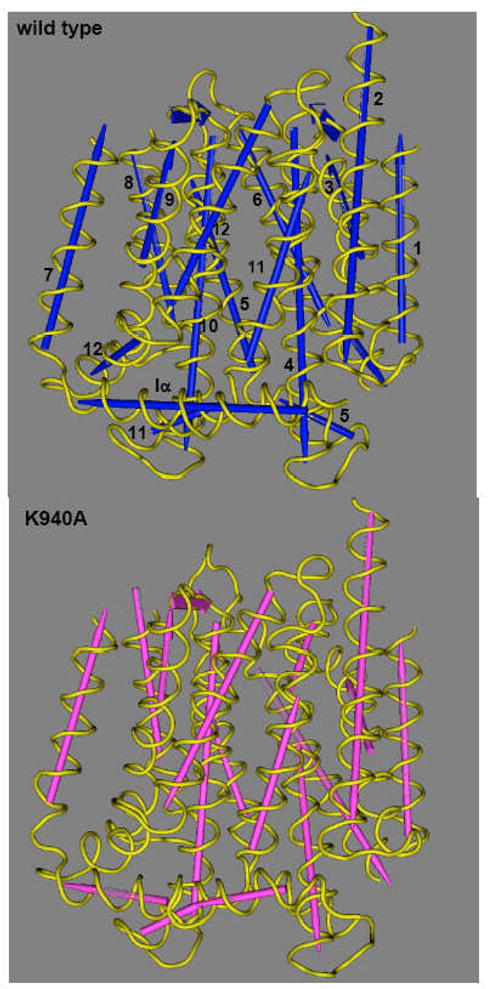
Structure of transmembrane domains of AcrB proton-relay mutants. The figure was drawn by using the Cn3D program (National Center for Biotechnology Information) after alignment of the three-dimensional structures of the wild type (PDB file 1IWG) and the Lys940Ala mutant (this study) through the Vector Alignment Search Tool, available at the National Center for Biotechnology Information website. Note that the assignment of the secondary structure was done in a uniform manner by the Vector Alignment Search Tool program. The TM helices are numbered in the wild-type structure. Although not shown here, other mutants of the network residues (Asp407Ala, Asp408Ala, Thr978Ala) show very similar conformational alterations. From [53].
At about the same time as our study of the proton-relay network mutants, three papers appeared describing the asymmetric trimer structure of AcrB [56–58]. In contrast to the symmetric structures seen before, in these structures each protomer takes a unique conformation. All groups identified one protomer in which a cavity is opened up in the periplasmic domain, a likely substrate-binding site, and in the study by the Murakami group [56], this cavity was indeed found with a bound substrate molecule (minocycline or doxorubicin)(Fig. 9). This protomer is called the “binding” protomer in the nomenclature of the Murakami group. Other protomers are called the “access” and “extrusion” protomers. In the extrusion protomer, which shows the largest conformational alteration, the binding pocket becomes collapsed, and there appears to be an opening of the pathway from the binding pocket to the top (central funnel) of the periplasmic domain. Inspection of the structures of the transmembrane domains shows that the tight association of Asp407-Asp408-Lys940-Thr978 becomes disrupted in the extrusion protomer, likely as a consequence of the protonation of Asp407 [56–58]; indeed the arrangement of these side-chains here is nearly identical with that found in our mutant protein [55]. This is consistent with our hypothesis that the disruption of the network among the four residues mentioned alters the conformation of the periplasmic domain. However, the asymmetric structure also shows that extensive conformational changes in the periplasmic domain require complementary accommodation by the neighboring protomers [56–58]. This was not possible in our construct, in which all protomers corresponded to the mutant protein.
Fig. 9.
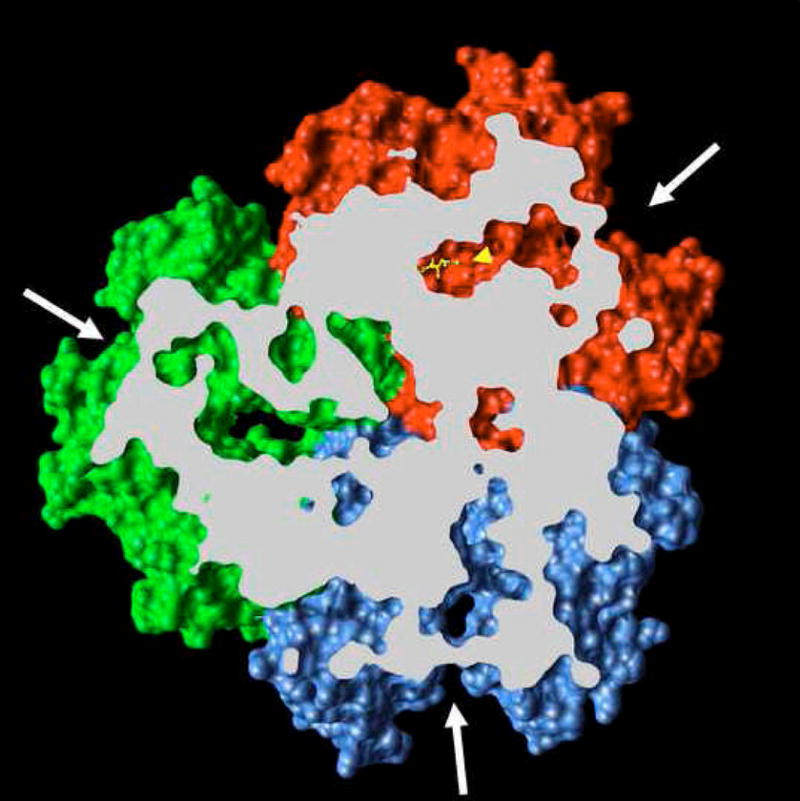
Periplasmic domain of the asymmetric crystal structure of AcrB. The view is from the top (periplasm), and the proximal portion is removed as identified by the white slab plain. The binding protomer (red) contains bound minocycline (ball-and-stick representation in yellow, identified by a yellow arrowhead). The large external cleft (white arrows) is open in the binding protomer as well as in the access protomer (blue), but is closed in the extrusion protomer (green). The figure is based on the PDB file 2DRD, and was drawn by using UCSF Chimera package [69] from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco.
As mentioned already, the extrusion protomer is the subunit where the conformation of the periplasmic domain is altered most strongly from the conformation seen in the symmetric trimer. This conformational alteration is presumably driven by the extension of helix 8 toward the periplasm, this change in turn is caused by the disruption of the salt bridge/H-bonding network among Asp407, Asp408, Lys940, and Thr978, as seen in the structures of both the wild-type protein in the asymmetric model [56–58] and the structure of the proton-relay network mutants [55].
The possible route of substrate binding and extrusion is suggested by the structure of the asymmetric trimer [56–58]. In the binding protomer, the periplasmic domain contains an expanded binding pocket containing several aromatic and hydrophobic residues (Phe136, 178, 610, 615, 617, and 628; Val139 and 612; Ile277 and 626; and Tyr327 [57]). This pocket was indeed found to contain drugs minocycline and doxorubicin [56], and its role is also supported by the previously described mutation data with the AcrB homolog YhiV [28]. The significance of this binding is also suggested by the observation that the pocket becomes collapsed in the extrusion protomer. Finally, its physiological significance is further supported by the comparison of its surface in AcrB and AcrD (Fig. 10): although the surface is entirely hydrophobic in AcrB, in the aminoglycoside pump AcrD it contains many oxygen atoms that may function in the binding of the hydrophilic, basic substrates. How do the substrates reach the binding pocket and how do they leave the pocket? All three groups agree that there is an open pathway between the binding pocket and the external cleft of the periplasmic domain [56–58] (see Fig. 11A). Although this cleft was earlier thought by some investigators to be the site of interaction between AcrA and AcrB, at this point the possibility of substrate entry from the lateral cleft is also supported by the results of random mutagenesis data and the results of site-directed mutagenesis already described. (The entry of the substrate from the cleft was advocated by Lomovskaya and Totrov [59], before the appearance of the asymmetric crystal structures.) The Murakami group suggests in addition that the pathway to the cleft is connected to the vestibule. If this is the case, it keeps open the possibility that the substrate may go through the initial part of the vestibule, and then go into the binding pocket. In the extrusion protomer, the binding pocket becomes narrower, and the pathway to the vestibule becomes closed (Fig. 11B). At the same time, a new pathway from the binding pocket to the funnel-like structure at top becomes open. These crystallographic observations suggest that the protomers within the trimeric AcrB goes through a cyclic conformational change, from the open, “access” conformation through the ligand-bound, “binding” one to finally the “extrusion” conformer, whose transmembrane domain shows signs of disrupted network among proton-translocating residues [56–58].
Fig. 10.
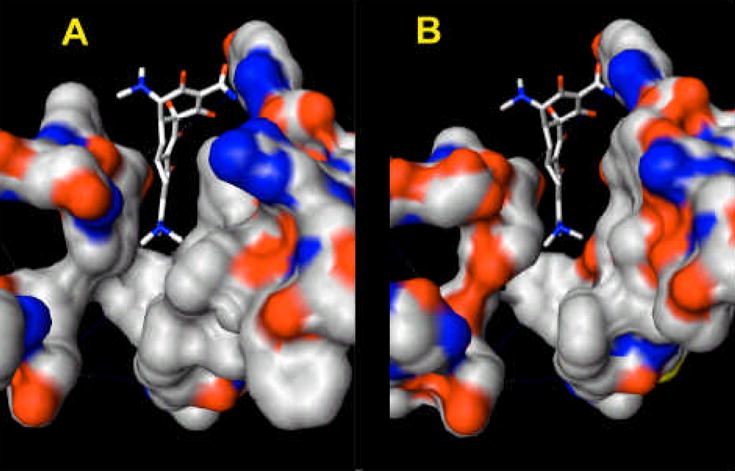
The substrate-binding pocket of the binding protomer in AcrB (left) and AcrD (AcrD). The residues that are close to the surface of the pocket (136–139, 176–178, 275–278, 327, and 610–620) are shown as solid surfaces, with coloring based on the element (oxygen, red; nitrogen, blue; and carbon, white). The bound minocycline is shown in the stick representation at the center. As seen, in AcrB (based on the PDB file 2DRD) the cavity surface is dominated by hydrophobic residues such as F178, V139, F610, F628, F615, V612, I278, and I277. In the hypothetical structure of AcrD (right), the surface has become much more hydrophilic as many of these residues are replaced with hydrophilic amino acids. The direction of view is similar to the Fig. 11, although here the subject was turned clockwise approximately by 30°. The figure was drawn by using UCSF Chimera package [69].
Fig. 11.
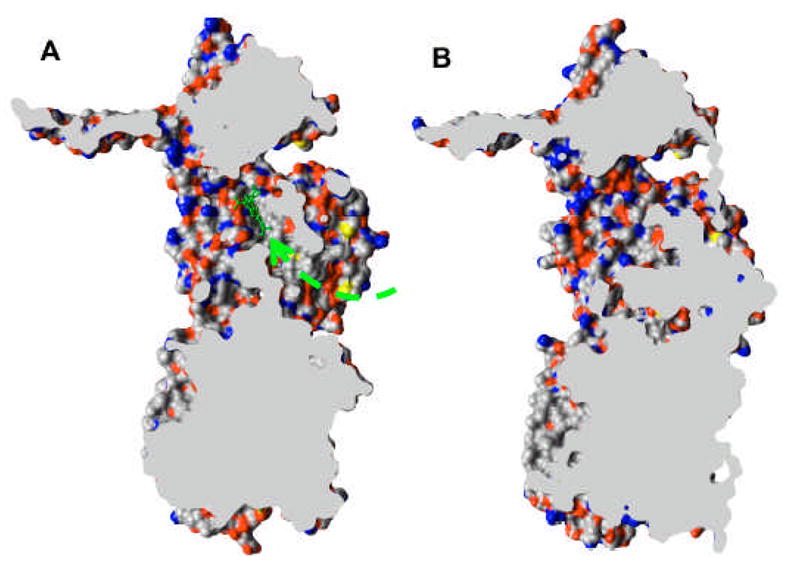
Cut-out view of the binding protomer with the bound minocycline (in a ball-and-stick representation) (A) and the extruding protomer (B), both from PDB file 2DRD, drawn by using UCSF Chimera package [69]. The wide passageway from the external cleft to the binding site (dashed arrow) seen in A appears to be completely closed in B.
BACK TO BIOCHEMISTRY
Although the structure of the asymmetric AcrB trimer suggests strongly the mechanism of ligand extrusion coupled to proton influx, this mechanism still remains a hypothesis. We set out to prove this hypothesis by carrying out biochemical studies of the pump. We note that the proposed cyclic change involves the opening and closing of the large external cleft in the periplasmic domain (Fig. 9). Thus if the opening of the cleft is prevented, we can predict that the pump will cease to function. We introduced pairs of cysteine residues at various positions on the opposing walls of the cleft [60]. Although single cysteine mutations produced little inhibition of transport, proteins containing double mutations (Asp566Cys and Thr678Cys; Phe666Cys and Thr678Cys; Phe666Cys and Gln830Cys) were strongly compromised in function. This is likely to be due to the forced closing of the cleft by disulfide bonds, but we cannot rule out the possibility that the trimeric assembly failed because of the premature formation of disulfide bonds between protomers. Indeed with cysteine pairs at other positions, there were data suggesting this interpretation. To avoid this problem, we expressed mutated AcrB in a host strain with a defective DsbA, a periplasmic disulfide oxidoreductase that plays a major role in the formation of disulfide bonds in this compartment. Indeed in this strain, the AcrB protein containing Phe666Cys and Thr678Cys (or Gln830Cys) largely retained its transport activity, presumably because disulfide bond formation did not occur in the absence of DsbA enzyme activity. We tried to see, by using these mutants, if the cross-linking and inactivation of AcrB can be observed in “real time” by using a fast-acting disulfide cross-linking agents based on the methanethiosulfonate groups. Indeed, addition of such cross-linkers to cells that are pumping out a fluorescent dye, ethidium, was shown to stop the function of the pump instantaneously (Fig. 12), thus providing biochemical support for the rotating mechanism of drug transport. A similar result was obtained with a somewhat different approach [61].
Fig. 12.
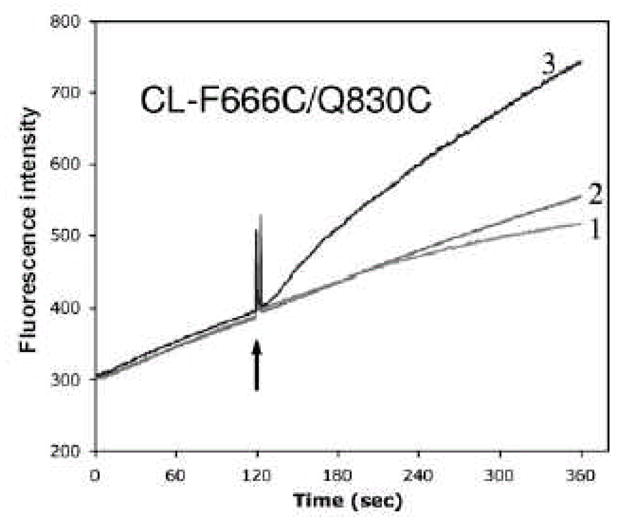
Real-time inhibition of pump function through closure of the periplasmic cleft. After the conversion of AcrB protein into the cysteineless (CL) form by substituting native Cys with Ser, two residues close to each other in the cleft, Phe666 and Gln830 were converted into cysteine by site-directed mutagenesis, generating CL-F666C/Q830C. A plasmid construct expressing this mutant AcrB was introduced into an E. coli ΔacrB::spc strain also containing dsbA::kan mutation, which prevents the formation of disulfide bonds in the periplasm. The AcrB efflux pump was active as it was able to prevent the rapid entry of the ethidium dye (curve 1, where only the solvent was added at the arrow). It remained active also when both Cys residues were modified by a methanethiosulfonate SH reagent 5-MTS (curve 2). However, the pump stopped functioning when the two Cys residues were cross-linked by the addition of a cross-linking methanethiosulfonate agent MTS-2-MTS (curve 3). For other control experiments see [60].
BACK TO GENETICS
The functional rotation mechanism of AcrB function [56–58] predicts that if one of the three protomers is defective in the proton relay network, then the pumping action by the entire trimer comes to a halt. This hypothesis could not be tested by the disulfide cross-linking experiment above, because all protomers had the same mutated sequence. We devised a way to test this hypothesis by creating a giant gene, in which three acrB sequences were connected together through short linker sequences (Y. Takatsuka and H. Nikaido, unpublished). This linked trimer is expressed well as a single giant protein, and produces drug resistance sometimes even slightly higher than that produced by the monomeric acrB gene. When only one of the three component acrB sequences in the giant gene was changed to include mutations in the proton-relay network, then the entire trimer became inactive. When only one of the three acrB sequences was made to contain the Phe666Cys and Gln830Cys, then cross-linking of these two cysteine residues by a fast-acting methanethiosulfonate cross-linker instantaneously inactivated the entire trimer, regardless of the position of the altered protomer in the giant sequence. These results again strongly support the notion that the trimer acts by a functional rotating mechanism.
CODA
A plausible outline on the mechanism of substrate capture and extrusion can now be proposed thanks mainly to the elucidation of crystallographic structures of the asymmetric AcrB trimer [56–58]. Furthermore, this functional rotatory mechanism has been confirmed by subsequent biochemical [60, 61] and genetic (Y. Takatsuka and H. Nikaido, unpublished results) studies, as described above. However, there are still many questions that remain. For example, the pathway of cytosolic capture of substrates has not been established, although one group finds a pathway connecting the binding pocket and the external leaflet of the bilayer [58]. Another issue is the precise way the substrate binds to the very large substrate-binding cavity. Already the two substrates that were found in the asymmetric trimers [56], minocycline and doxorubicin, are occupying the pocket in significantly different ways. Perhaps the large size of the cavity and many potential binding modes of substrates might explain the observation that Phe-Arg-β-naphthylamide inhibits the efflux of various substrates with very different efficiency. The rotatory mechanism shows that the large external cleft of the periplasmic domain of AcrB becomes closed in the extrusion protomer, but this cleft is where AcrA is generally thought to bind to AcrB, and it is unknown what happens to the bound AcrA in this protomer. Nor do we have any idea on how AcrA stimulates the function of AcrB. Finally, some RND transporters, such as MdtBC of E. coli [62, 63], contain two protomers of different sequences, and it is unknown how the functionally rotating mechanism would work in such a situation.
The functions of the MFP, AcrA and its homologs, are described by others in this series. However, it should be mentioned that the MFP of the Cus system of E. coli, CusB, is known to bind the substrate Cu+ [64], and most excitingly, the direct transfer of metal ion from the periplasmic binding protein CusF to CusB was recently demonstrated [65]. Thus it seems quite possible that in this system the substrate ion flows from the periplasmic binding protein to the periplasmic adaptor protein, and then finally to the periplasmic domain of an RND transporter, CusA.
There are also unresolved questions on the function of tripartite RND pumps in intact cells. For example, P. aeruginosa strains overexpressing a tripartite pump MexCD-OprJ were often found to have reduced function of other tripartite pumps MexAB-OprM and MexXY-OprM although the latter pumps are expressed at the normal level [66]. Another curious observation is that TonB, a protein that functions in energizing the outer membrane siderophore receptors, is needed in P. aeruginosa for the function of the MexAB-OprM pump [67, 68]. As far as the authors are aware, there is no explanation of this effect at the molecular level, but this reminds us that a small protein YajC was accidentally co-crystallized with AcrB [54]. Although the deletion of gene yajC had only a small effect on drug susceptibility, this observation certainly requires further follow-up study, especially because this co-crystal was obtained from cells expressing both YajC and AcrB at normal constitutive levels, without the lopsided, plasmid-driven overexpression of only AcrB.
Acknowledgments
The studies in the authors’ laboratory are supported by a U.S. Public Health Service grant, AI 09644. We thank V. Koronakis for supplying the high resolution version of Fig. 7A.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
LITERATURE CITED
- 1.Paulsen IT, Park JH, Choi PS, Saier MH., Jr A family of gram-negative bacterial outer membrane factors that function in the export of proteins, carbohydrates, drugs and heavy metals from gram-negative bacteria. FEMS Microbiol Lett. 1997;156:1–8. doi: 10.1111/j.1574-6968.1997.tb12697.x. [DOI] [PubMed] [Google Scholar]
- 2.Dinh T, Paulsen IT, Saier MH., Jr A family of extracytoplasmic proteins that allow transport of large molecules across the outer membranes of gram-negative bacteria. J Bacteriol. 1994;176:3825–31. doi: 10.1128/jb.176.13.3825-3831.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ma D, Cook DN, Alberti M, Pon NG, Nikaido H, Hearst JE. Molecular cloning and characterization of acrA and acrE genes of Escherichia coli. J Bacteriol. 1993;175:6299–313. doi: 10.1128/jb.175.19.6299-6313.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma D, Cook DN, Alberti M, Pon NG, Nikaido H, Hearst JE. Genes acrA and acrB encode a stress-induced efflux system of Escherichia coli. Mol Microbiol. 1995;16:45–55. doi: 10.1111/j.1365-2958.1995.tb02390.x. [DOI] [PubMed] [Google Scholar]
- 5.Nikaido H. Multidrug efflux pumps of gram-negative bacteria. J Bacteriol. 1996;178:5853–9. doi: 10.1128/jb.178.20.5853-5859.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vaara M. Antibiotic-supersusceptible mutants of Escherichia coli and Salmonella typhimurium. Antimicrob Agents Chemother. 1993;37:2255–60. doi: 10.1128/aac.37.11.2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nikaido H, Basina M, Nguyen V, Rosenberg EY. Multidrug efflux pump AcrAB of Salmonella typhimurium excretes only those beta-lactam antibiotics containing lipophilic side chains. J Bacteriol. 1998;180:4686–92. doi: 10.1128/jb.180.17.4686-4692.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murakami S, Nakashima R, Yamashita E, Yamaguchi A. Crystal structure of bacterial multidrug efflux transporter AcrB. Nature. 2002;419:587–93. doi: 10.1038/nature01050. [DOI] [PubMed] [Google Scholar]
- 9.Li XZ, Nikaido H, Poole K. Role of mexA-mexB-oprM in antibiotic efflux in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 1995;39:1948–53. doi: 10.1128/aac.39.9.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li XZ, Ma D, Livermore DM, Nikaido H. Role of efflux pump(s) in intrinsic resistance of Pseudomonas aeruginosa: active efflux as a contributing factor to beta-lactam resistance. Antimicrob Agents Chemother. 1994;38:1742–52. doi: 10.1128/aac.38.8.1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aires JR, Nikaido H. Aminoglycosides are captured from both periplasm and cytoplasm by the AcrD multidrug efflux transporter of Escherichia coli. J Bacteriol. 2005;187:1923–9. doi: 10.1128/JB.187.6.1923-1929.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lomovskaya O, Warren MS, Lee A, Galazzo J, Fronko R, Lee M, Blais J, Cho D, Chamberland S, Renau T, Leger R, Hecker S, Watkins W, Hoshino K, Ishida H, Lee VJ. Identification and characterization of inhibitors of multidrug resistance efflux pumps in Pseudomonas aeruginosa: novel agents for combination therapy. Antimicrob Agents Chemother. 2001;45:105–16. doi: 10.1128/AAC.45.1.105-116.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee A, Mao W, Warren MS, Mistry A, Hoshino K, Okumura R, Ishida H, Lomovskaya O. Interplay between efflux pumps may provide either additive or multiplicative effects on drug resistance. J Bacteriol. 2000;182:3142–50. doi: 10.1128/jb.182.11.3142-3150.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tamura N, Konishi S, Yamaguchi A. Mechanisms of drug/H+ antiport: complete cysteine-scanning mutagenesis and the protein engineering approach. Curr Opin Chem Biol. 2003;7:570–9. doi: 10.1016/j.cbpa.2003.08.014. [DOI] [PubMed] [Google Scholar]
- 15.Paulsen IT, Brown MH, Littlejohn TG, Mitchell BA, Skurray RA. Multidrug resistance proteins QacA and QacB from Staphylococcus aureus: membrane topology and identification of residues involved in substrate specificity. Proc Natl Acad Sci USA. 1996;93:3630–5. doi: 10.1073/pnas.93.8.3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zgurskaya HI, Nikaido H. Bypassing the periplasm: reconstitution of the AcrAB multidrug efflux pump of Escherichia coli. Proc Natl Acad Sci USA. 1999;96:7190–5. doi: 10.1073/pnas.96.13.7190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Su CC, Yu EW. Ligand-transporter interaction in the AcrB multidrug efflux pump determined by fluorescence polarization assay. FEBS Lett. 2007;581:4972–6. doi: 10.1016/j.febslet.2007.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ocaktan A, Yoneyama H, Nakae T. Use of fluorescence probes to monitor function of the subunit proteins of the MexA-MexB-oprM drug extrusion machinery in Pseudomonas aeruginosa. J Biol Chem. 1997;272:21964–9. doi: 10.1074/jbc.272.35.21964. [DOI] [PubMed] [Google Scholar]
- 19.Murakami S, Tamura N, Saito A, Hirata T, Yamaguchi A. Extramembrane central pore of multidrug exporter AcrB in Escherichia coli plays an important role in drug transport. J Biol Chem. 2004;279:3743–8. doi: 10.1074/jbc.M308893200. [DOI] [PubMed] [Google Scholar]
- 20.Goldberg M, Pribyl T, Juhnke S, Nies DH. Energetics and topology of CzcA, a cation/proton antiporter of the resistance-nodulation-cell division protein family. J Biol Chem. 1999;274:26065–70. doi: 10.1074/jbc.274.37.26065. [DOI] [PubMed] [Google Scholar]
- 21.Chang TY, Chang CC, Ohgami N, Yamauchi Y. Cholesterol sensing, trafficking, and esterification. Annu Rev Cell Dev Biol. 2006;22:129–57. doi: 10.1146/annurev.cellbio.22.010305.104656. [DOI] [PubMed] [Google Scholar]
- 22.Infante RE, Abi-Mosleh L, Radhakrishnan A, Dale JD, Brown MS, Goldstein JL. Purified NPC1 protein. I. Binding of cholesterol and oxysterols to a 1278-amino acid membrane protein. J Biol Chem. 2008;283:1052–63. doi: 10.1074/jbc.M707943200. [DOI] [PubMed] [Google Scholar]
- 23.Infante RE, Radhakrishnan A, Abi-Mosleh L, Kinch LN, Wang ML, Grishin NV, Goldstein JL, Brown MS. Purified NPC1 protein: II. Localization of sterol binding to a 240-amino acid soluble luminal loop. J Biol Chem. 2008;283:1064–75. doi: 10.1074/jbc.M707944200. [DOI] [PubMed] [Google Scholar]
- 24.Elkins CA, Nikaido H. Substrate specificity of the RND-type multidrug efflux pumps AcrB and AcrD of Escherichia coli is determined predominantly by two large periplasmic loops. J Bacteriol. 2002;184:6490–8. doi: 10.1128/JB.184.23.6490-6498.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tikhonova EB, Wang Q, Zgruskaya H. Chimeric analysis of the multicomponent multidrug efflux transporters from Gram-negative bacteria. J Bacteriol. 2002;184:6499–507. doi: 10.1128/JB.184.23.6499-6507.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eda S, Maseda H, Nakae T. An elegant means of self-protection in gram-negative bacteria by recognizing and extruding xenobiotics from the periplasmic space. J Biol Chem. 2003;278:2085–8. doi: 10.1074/jbc.C200661200. [DOI] [PubMed] [Google Scholar]
- 27.Mao W, Warren MS, Black DS, Satou T, Murata T, Nishino T, Gotoh N, Lomovskaya O. On the mechanism of substrate specificity by resistance nodulation division (RND)-type multidrug resistance pumps: the large periplasmic loops of MexD from Pseudomonas aeruginosa are involved in substrate recognition. Mol Microbiol. 2002;46:889–901. doi: 10.1046/j.1365-2958.2002.03223.x. [DOI] [PubMed] [Google Scholar]
- 28.Bohnert JA, Schuster S, Fähnrich E, Trittler R, Kern WV. Altered spectrum of multidrug resistance associated with a single point mutation in the Escherichia coli RND-type MDR efflux pump YhiV (MdtF) J Antimicrob Chemother. 2007;59:1216–22. doi: 10.1093/jac/dkl426. [DOI] [PubMed] [Google Scholar]
- 29.Middlemiss JK, Poole K. Differential impact of MexB mutations on substrate selectivity of the MexAB-OprM multidrug efflux pump of Pseudomonas aeruginosa. J Bacteriol. 2004;186:1258–69. doi: 10.1128/JB.186.5.1258-1269.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guan L, Nakae T. Identification of essential charged residues in transmembrane segments of the multidrug transporter MexB of Pseudomonas aeruginosa. J Bacteriol. 2001;183:1734–9. doi: 10.1128/JB.183.5.1734-1739.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takatsuka Y, Nikaido H. Threonine-978 in the transmembrane segment of the multidrug efflux pump AcrB of Escherichia coli is crucial for drug transport as a probable component of the proton relay network. J Bacteriol. 2006;188:7284–9. doi: 10.1128/JB.00683-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu EW, Aires JR, Nikaido H. AcrB multidrug efflux pump of Escherichia coli: composite substrate-binding cavity of exceptional flexibility generates its extremely wide substrate specificity. J Bacteriol. 2003;185:5657–64. doi: 10.1128/JB.185.19.5657-5664.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu EW, Aires JR, McDermott G, Nikaido H. A periplasmic drug-binding site of the AcrB multidrug efflux pump: a crystallographic and site-directed mutagenesis study. J Bacteriol. 2005;187:6804–15. doi: 10.1128/JB.187.19.6804-6815.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hearn EM, Gray MR, Foght JM. Mutations in the central cavity and periplasmic domain affect efflux activity of the resistance-nodulation-division pump EmhB from Pseudomonas fluorescens cLP6a. J Bacteriol. 2006;188:115–23. doi: 10.1128/JB.188.1.115-123.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Franke S, Grass G, Rensing C, Nies DH. Molecular analysis of the copper-transporting efflux system CusCFBA of Escherichia coli. J Bacteriol. 2003;185:3804–12. doi: 10.1128/JB.185.13.3804-3812.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dastidar V, Mao W, Lomovskaya O, Zgurskaya HI. Drug-induced conformational changes in multidrug efflux transporter AcrB from Haemophilus influenzae. J Bacteriol. 2007;189:5550–8. doi: 10.1128/JB.00471-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ruetz S, Gros P. Phosphatidylcholine translocase: a physiological role for the mdr2 gene. Cell. 1994;77:1071–81. doi: 10.1016/0092-8674(94)90446-4. [DOI] [PubMed] [Google Scholar]
- 38.Mao W, Warren MS, Lee A, Mistry A, Lomovskaya O. MexXY-OprM efflux pump is required for antagonism of aminoglycosides by divalent cations in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2001;45:2001–7. doi: 10.1128/AAC.45.7.2001-2007.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schmidt TG, Skerra A. The Strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nature Protocols. 2007;2:1528–35. doi: 10.1038/nprot.2007.209. [DOI] [PubMed] [Google Scholar]
- 40.Hanada K, Yamato I, Anraku Y. Purification and reconstitution of Escherichia coli proline carrier using a site specifically cleavable fusion protein. J Biol Chem. 1988;263:7181–5. [PubMed] [Google Scholar]
- 41.Tikhonova EB, Devroy VK, Lau SY, Zgurskaya HI. Reconstitution of the Escherichia coli macrolide transporter: the periplasmic membrane fusion protein MacA stimulates the ATPase activity of MacB. Mol Microbiol. 2007;63:895–910. doi: 10.1111/j.1365-2958.2006.05549.x. [DOI] [PubMed] [Google Scholar]
- 42.Koronakis V, Sharff A, Koronakis E, Luisi B, Hughes C. Crystal structure of the bacterial membrane protein TolC central to multidrug efflux and protein export. Nature. 2000;405:914–9. doi: 10.1038/35016007. [DOI] [PubMed] [Google Scholar]
- 43.Akama H, Kanemaki M, Yoshimura M, Tsukihara T, Kashiwagi T, Yoneyama H, Narita S, Nakagawa A, Nakae T. Crystal structure of the drug discharge outer membrane protein, OprM, of Pseudomonas aeruginosa: dual modes of membrane anchoring and occluded cavity end. J Biol Chem. 2004;279:52816–9. doi: 10.1074/jbc.C400445200. [DOI] [PubMed] [Google Scholar]
- 44.Akama H, Matsuura T, Kashiwagi S, Yoneyama H, Narita S, Tsukihara T, Nakagawa A, Nakae T. Crystal structure of the membrane fusion protein, MexA, of the multidrug transporter in Pseudomonas aeruginosa. J Biol Chem. 2004;279:25939–42. doi: 10.1074/jbc.C400164200. [DOI] [PubMed] [Google Scholar]
- 45.Higgins MK, Bokma E, Koronakis E, Hughes C, Koronakis V. Structure of the periplasmic component of a bacterial drug efflux pump. Proc Natl Acad Sci USA. 2004;101:9994–9. doi: 10.1073/pnas.0400375101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mikolosko J, Bobyk K, Zgurskaya HI, Ghosh P. Conformational flexibility in the multidrug efflux system protein AcrA. Structure. 2006;14:577–87. doi: 10.1016/j.str.2005.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zgurskaya HI, Nikaido H. AcrA is a highly asymmetric protein capable of spanning the periplasm. J Mol Biol. 1999;285:409–20. doi: 10.1006/jmbi.1998.2313. [DOI] [PubMed] [Google Scholar]
- 48.Eswaran J, Koronakis E, Higgins MK, Hughes C, Koronakis V. Three’s company: component structures bring a closer view of tripartite drug efflux pumps. Curr Opin Struct Biol. 2004;14:741–7. doi: 10.1016/j.sbi.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 49.Stegmeier JF, Polleichtner G, Brandes N, Hotz C, Andersen C. Importance of the adaptor (membrane fusion) protein hairpin domain for the functionality of multidrug efflux pumps. Biochemistry. 2006;45:10303–12. doi: 10.1021/bi060320g. [DOI] [PubMed] [Google Scholar]
- 50.Lobedanz S, Bokma E, Symmons MF, Koronakis E, Hughes C, Koronakis V. A periplasmic coiled-coil interface underlying TolC recruitment and the assembly of bacterial drug efflux pumps. Proc Natl Acad Sci USA. 2007;104:4612–7. doi: 10.1073/pnas.0610160104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Elkins CA, Nikaido H. Chimeric analysis of AcrA function reveals the importance of its C-terminal domain in its interaction with the AcrB multidrug efflux pump. J Bacteriol. 2003;185:5349–56. doi: 10.1128/JB.185.18.5349-5356.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krishnamoorthy G, Tikhonova EB, Zgurskaya HI. Fitting periplasmic membrane fusion proteins to inner membrane transporters: mutations that enable Escherichia coli AcrA to function with Pseudomonas aeruginosa MexB. J Bacteriol. 2007;190:691–8. doi: 10.1128/JB.01276-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu EW, McDermott G, Zgurskaya HI, Nikaido H, Koshland DE., Jr Structural basis of multiple drug-binding capacity of the AcrB multidrug efflux pump. Science. 2003;300:976–80. doi: 10.1126/science.1083137. [DOI] [PubMed] [Google Scholar]
- 54.Törnroth-Horsefield S, Gourdon P, Horsefield R, Brive L, Yamamoto N, Mori H, Snijder A, Neutze R. Crystal structure of AcrB in complex with a single transmembrane subunit reveals another twist. Structure. 2007;15:1663–73. doi: 10.1016/j.str.2007.09.023. [DOI] [PubMed] [Google Scholar]
- 55.Su CC, Li M, Gu R, Takatsuka Y, McDermott G, Nikaido H, Yu EW. Conformation of the AcrB multidrug efflux pump in mutants of the putative proton relay pathway. J Bacteriol. 2006;188:7290–6. doi: 10.1128/JB.00684-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Murakami S, Nakashima R, Yamashita E, Matsumoto T, Yamaguchi A. Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. Nature. 2006;443:173–9. doi: 10.1038/nature05076. [DOI] [PubMed] [Google Scholar]
- 57.Seeger MA, Schiefner A, Eicher T, Verrey F, Diederichs K, Pos KM. Structural asymmetry of AcrB trimer suggests a peristaltic pump mechanism. Science. 2006;313:1295–8. doi: 10.1126/science.1131542. [DOI] [PubMed] [Google Scholar]
- 58.Sennhauser G, Amstutz P, Briand C, Storchenegger O, Grutter MG. Drug export pathway of multidrug exporter AcrB revealed by DARPin inhibitors. PLoS Biol. 2007;5:e7. doi: 10.1371/journal.pbio.0050007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lomovskaya O, Totrov M. Vacuuming the periplasm. J Bacteriol. 2005;187:1879–83. doi: 10.1128/JB.187.6.1879-1883.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Takatsuka Y, Nikaido H. Site-directed disulfide cross-linking shows that cleft flexibility in the periplasmic domain is needed for the multidrug efflux pump AcrB of Escherichia coli. J Bacteriol. 2007;189:8677–8684. doi: 10.1128/JB.01127-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Seeger MA, von Ballmoos C, Eicher T, Brandstatter L, Verrey F, Diederichs K, Pos KM. Engineered disulfide bonds support the functional rotation mechanism of multidrug efflux pump AcrB. Nat Struct Mol Biol. 2008;15:199–205. doi: 10.1038/nsmb.1379. [DOI] [PubMed] [Google Scholar]
- 62.Baranova N, Nikaido H. The BaeSR two-component regulatory system activates transcription of the yegMNOB (mdtABCD) transporter gene cluster in Escherichia coli and increases its resistance to novobiocin and deoxycholate. J Bacteriol. 2002;184:4168–76. doi: 10.1128/JB.184.15.4168-4176.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nagakubo S, Nishino K, Hirata T, Yamaguchi A. The putative response regulator BaeR stimulates multidrug resistance of Escherichia coli via a novel multidrug exporter system, MdtABC. J Bacteriol. 2002;184:4161–7. doi: 10.1128/JB.184.15.4161-4167.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bagai I, Liu W, Rensing C, Blackburn NJ, McEvoy MM. Substrate-linked conformational change in the periplasmic component of a Cu(I)/Ag(I) efflux system. J Biol Chem. 2007;282:35695–35702. doi: 10.1074/jbc.M703937200. [DOI] [PubMed] [Google Scholar]
- 65.Bagai I, Liu W, Rensing C, Blackburn NJ, McEvoy MM. Direct metal transfer between periplasmic proteins identifies a bacterial copper chaperone. Biochemistry. 2008 doi: 10.1021/bi801638m. e-published 10/11/2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jeannot K, Elsen S, Kohler T, Attree I, van Delden C, Plesiat P. Resistance and virulence of Pseudomonas aeruginosa clinical strains overproducing the MexCD-OprJ efflux pump. Antimicrob Agents Chemother. 2008;52:2455–62. doi: 10.1128/AAC.01107-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhao Q, Li XZ, Mistry A, Srikumar R, Zhang L, Lomovskaya O, Poole K. Influence of the TonB energy-coupling protein on efflux-mediated multidrug resistance in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 1998;42:2225–31. doi: 10.1128/aac.42.9.2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhao Q, Poole K. Differential effects of mutations in tonB1 on intrinsic multidrug resistance and iron acquisition in Pseudomonas aeruginosa. J Bacteriol. 2002;184:2045–9. doi: 10.1128/JB.184.7.2045-2049.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]