

Summary
The solution structure of the potent 95 residue anti-HIV protein scytovirin has been determined and two carbohydrate binding sites have been identified. This unique protein, containing five structurally important disulfide bonds, demonstrates a novel fold with no elements of extended regular secondary structure. Scytovirin contains two 39 residue sequence repeats, differing in only three amino acids and each repeat has primary sequence similarity to chitin binding proteins. Both sequence repeats form similarly structured domains, with the exception of one region. The result is two carbohydrate binding sites with substantially different affinities. The unusual fold clusters aromatic residues in both sites suggesting a binding mechanism similar to other known hevein-like carbohydrate binding proteins but differing in carbohydrate specificity. Scytovirin, originally isolated from the cyanobacterium Scytonema varium, holds potential as an HIV entry inhibitor for both therapeutic and prophylactic anti-HIV applications. The high resolution structural studies reported are an important initial step in unlocking the therapeutic potential of scytovirin. Funded in part by NCI Contract N01-CO-12400.
Keywords: scytovirin, antiviral, HIV, carbohydrate, hevein
Introduction
Interactions of protein with carbohydrate play fundamental roles throughout biology. Bacterial infection, cell growth, inflammation, cell mobility, fertilization, cell to cell adhesion, and viral infection are all influenced by protein/carbohydrate interactions. Therefore protein/carbohydrate interactions are a topic of major interest throughout science. One well studied group of lectins is the hevein-like family. This family is structurally united by the presence of similar chitin binding domains (CBDs) and are members of carbohydrate-binding module family 181. Hevein, isolated from the latex of Hevea brasiliensis, is composed of 43 residues and its structure has been known since 19912. Other hevein-like CBDs vary in length, typically from 38-45 residues, and are often found in multiple repeats within a single protein. A dual CBD-like motif is present in the potent antiviral protein scytovirin, originally isolated from the cyanobacterium Scytonema varium.
Scytovirin is of particular interest due to the fact that it possesses potent and broad spectrum antiviral, including anti-HIV, activity not found in the other hevein-like domain proteins3. Scytovirin exhibits low nanomolar activity against HIV and binds to the HIV envelope glycoproteins gp160, gp120 and gp41, but it does not bind to the T-cell extracellular CD4 receptor or other common cell surface proteins4. The interaction is carbohydrate dependent, with scytovirin most readily binding to glycosylated gp120. Scytovirin's ability to completely inactivate laboratory strains and primary isolates of HIV-14 makes it a promising candidate for anti-HIsV microbicide development.
Scytovirin contains two 39 residue repeats, which exhibit rather low similarity to a subset of diverse CBD containing proteins4, The initial sequence analysis offers little indication of the mechanism of antiviral activity beyond similarity to CBDs. The comparison to hevein-like CBDs was not originally examined; however, the sequence similarity between the scytovirin repeats and the hevein domain is approximately 30%. In addition, several features common to hevein family members5,6 are present in scytovirin: There are two CBD-like domains separated by a proline rich linker. Both scytovirin and hevein-like CBDs have a high percentage of cysteine residues, all participating in disulfide bonds. Each domain of scytovirin has three aromatic residues, similar to the conserved triad of aromatic residues found in other hevein-like CBDs. Thus, it seems plausible that scytovirin is another hevein family member.
However, scytovirin shows distinct differences from the hevein-like proteins. First, scytovirin does not bind chitin4. In fact, scytovirin shows a very restrictive carbohydrate binding specificity, having no measurable affinity for monosaccharides or common trisaccharide cores from higher order m es4; 7, (vide infra). This observation is in stark contrast to the increasing affinity for longer oligosaccharides reported for other hevein-like CBDs8. Second, the disulfide bonds among the conserved cysteines of scytovirin are shuffled compared to the consensus hevein arrangement. Scytovirin has only five cysteine residues per domain. These align exactly with five of the eight cysteines per domain in the classical hevein arrangement. The presence of an odd number of cysteines in each domain of scytovirin causes the disulfide bonds to rearrange, resulting in two disulfides per domain and one inter-domain disulfide4, unlike the four disulfide bonds typically found in hevein domains. Although scytovirin has three aromatic residues per domain, similar to hevein-like CBDs, the sequence arrangement of the aromatic residues does not match the classic hevein domain sequential arrangement, vide infra.
Thus, scytovirin has remained a rather enigmatic, but potent antiviral protein. In order to better understand the antiviral mechanism and enable development of this natural protein, we have determined the three-dimensional solution structure of recombinant scytovirin and found it to have a novel fold, quite different from hevein. We have also examined the carbohydrate binding properties with short oligomannose carbohydrates, which were previously identified7. These structural studies are a vital part of the future development for scytovirin and detail the unique characteristics of this protein, both with respect to its antiviral activity and its relationship to other well-studied carbohydrate binding proteins.
Results
Chemical Shift and NOE Assignments
Chemical shift assignments of recombinant scytovirin (Figure 1a) were complicated by resonance degeneracy. A doubling of most peaks was immediately apparent from the 15N-HSQC, Figure 1b. Duplicity of the amide backbone chemical shifts for a majority of the resonances readily suggested that the sequence repeats of scytovirin have similar backbone folds. There was an indication that each sequence repeat might represent a separate domain of the total protein structure, and we refer to the two sequence repeats as domains, which is borne out in the complete analyses (vide infra). Chemical shift degeneracy was exacerbated for side chains where 13C and 1H resonances were not as well dispersed as 15N chemical shifts. For illustration, strip plots for a stretch of five consecutive residues in each domain, showing the Cα and Cβ resonances and sequential correlations from triple resonance NMR spectra, are shown in Figure 1c.
Figure 1. Scytovirin Sequence, Resonance Assignments, and Chemical Shift Indices.
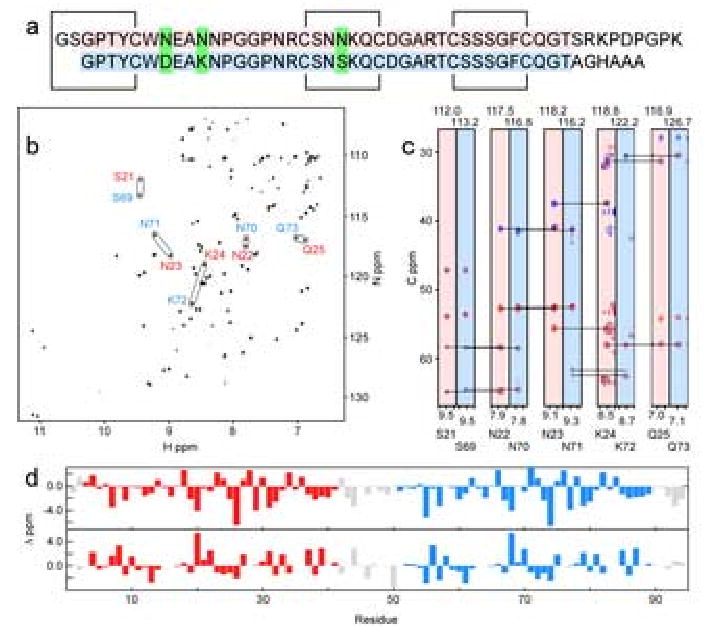
a) Amino acid sequence of scytovirin aligned to show sequence repeats. The background of the first domain (SD1) is shaded in red while the background of the second domain (SD2) is shaded in blue. Black lines indicate disulfide bonds. Green boxes highlight the three natural sequence differences between domains. b) An 15N-HSQC of scytovirin illustrates peak doubling of complementary resonances. Residues S21, N22, N23, K24, and Q25 from SD1 are labeled in red and complementary residues S69, N70, S71, K72, and Q73 from SD2 are labeled in blue. c) Slices from HNCACB and CBCA(CO)NH spectra for the same ten residues as b. HNCACB Cα resonances are shown in red and Cβ resonances in blue. CBCA(CO)NH resonances, from previous residue Cα and Cβ resonances, are shown in magenta. Background shading is red for SD1 and blue for SD2. d) The difference in Cα (top) and Cβ chemical shifts from random coil values. No extended regions of regular secondary structure are expected since the characteristic positive difference for α-helices and negative difference for β-sheets is not observed for Cα chemical shifts.
The three amino acid differences between sequence repeats of scytovirin aided the sequential assignment process; nevertheless, meticulous manual analysis of standard triple resonance spectra had to be supplemented with additional experiments to unambiguously assign backbone resonances. An HMQC-NOESY-HMQC9, twice taking advantage of 15N chemical shift dispersion, greatly aided the resolution of inter- versus intra-domain backbone sequential assignments. Pulse sequences to assign through proline resonances10 were also used to confirm assignments. Information from these experiments was especially useful for the P-G-G-P sequence in each domain and the P-D-P-G-P sequence in the linker. Manual analysis of NOESY data was also utilized to help resolve chemical shift ambiguity. In summary, nearly complete domain specific resonance assignments were obtained for the first 94 residues of scytovirin. Residue 95, the C-terminal alanine, was not observable. Over 98% of backbone and 92% of side chain resonances were accounted for. As expected, each 15N-HSQC peak pair corresponded to complementary resonances of the 39 residue sequence repeats. Chemical shift analysis of backbone Cα and Cβ resonances11; 12 (Fig. 1d) gave the first indication that no extended regions of regular secondary structure were present.
NOESY assignments were also complicated by chemical shift degeneracy, especially for inter- versus intra-domain NOEs. No symmetry for the two domains was assumed and an initial structure was calculated from a sparse set of unambiguously assigned NOEs. A moderate resolution structure was obtained and a degree of two-fold symmetry was immediately apparent. The initial structure allowed for resolution of more inter- versus intra-domain NOE ambiguity. After multiple iterations, 1205 intraresidue, 1006 sequential, and 634 long range NOEs were assigned.
Structural Overview and Backbone Flexibility
The solution structure of scytovirin was determined using NOEs and residual dipolar coupling data13 (RDCs). Initial structures, determined using only NOEs, exhibited a backbone RMSD of 1.4 +/- 0.1 Å to the mean structure. The sequence repeats corresponded to structured domains with residues 3-43 forming structural domain 1 (SD1) and 51-89 forming structural domain 2 (SD2). The disulfide bonding pattern, previously determined from proteolysis and peptide mapping5, was readily apparent in these initial structures. Structure calculations with and without disulfide restraints showed only small differences, and subsequent calculations included these restraints. Backbone RMSDs for the domains, aligned individually, were 1.0 Å for SD1 and 1.3 Å for SD2. More striking than the domain similarity was the complete absence of regular secondary structure. In agreement with chemical shift indexing data, no extended α-helices or β-sheets were observed. Circular dichroism, Supplementary Figure 1, also indicates an atypical fold. The CD spectrum for scytovirin does not exhibit an α-helical, β-sheet, mixed helix/sheet, or unfolded profile. Refinement against J-modulated HαCα and NH RDCs confirmed the unusual fold and total lack of regular secondary structure. Two regions of extended structure are superficially similar to β-sheets. In SD1, residues 28-32 lie antiparallel to residues 38-42. Also, residues 6-12 lie antiparallel to the disjoint sections 17-20 and 26-28. Although reminiscent of β-sheets, neither Phi/Psi angles nor chemical shift indexing strictly indicate the presence of classical β-sheet folds. Furthermore, temperature coefficients of the amide resonances14 are not consistent with hydrogen bonding patterns expected for antiparallel β-sheets. The same holds for complementary residues in SD2. Additionally, there are no Ile, Leu, or Val residues in the sequence, which results in a very minimal hydrophobic core. The limited hydrophobic core within each domain is consistent with the unusual fold. Thus, scytovirin has no recognizable α-helix or β-sheet elements of secondary structure.
The lowest energy 20 refined scytovirin structures are shown in Figure 2a and coordinates are deposited under PDB accession number 2JMV. In the final structures, the backbone RMSD of the entire protein improved to 0.42 +/− 0.11 Å. Backbone RMSDs of the individual domains improved to 0.25 Å for SD1 and 0.40 Å for SD2. In addition to improved precision, RDC refinement helped define the linker and last loop-like structure of each domain. Calculated versus observed RDC plots and cross-validation are included as Supplementary Figure 2. A DALI15 structure search found no similar structures for either SD1, SD2 or full length scytovirin. Thus scytovirin represents a truly novel fold.
Figure 2. Solution Structure of Scytovirin.
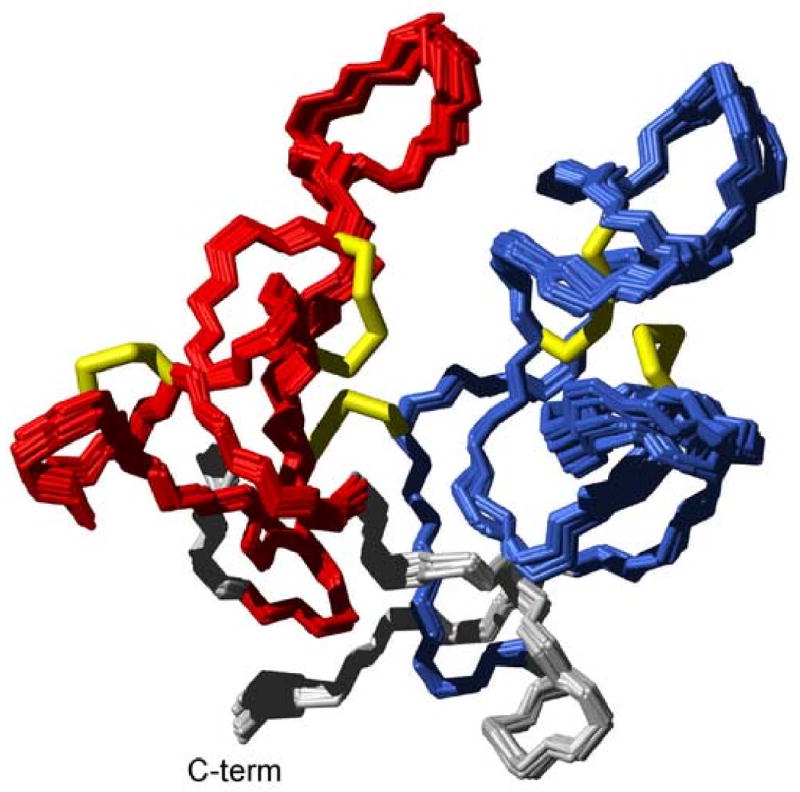
a) Backbone traces of lowest energy 20 scytovirin structures of 200 calculated from refinement against HαCα and NH RDCs. SD1 is colored red, SD2 blue, disulfide bonds in yellow, and the linker and termini gray. b) Stereo view of backbone structure closest to the mean.
Despite the lack of regular secondary structure, scytovirin appears to be quite ordered on NMR measurable timescales. Transverse and longitudinal 15N relaxation rates showed no regions of significant variation except in the C-terminus. The correlation time derived from these data was 5.1 nsec, which corresponds to a monomeric 9.7 kDa protein. Heteronuclear NOEs16 at multiple field strengths resulted in average values (excluding the two C-terminal residues) of 0.80 ± 0.05 and 0.80 ± 0.07 at 600 and 800 MHz, respectively. Data are included as Supplementary Figure 3. No significant change in average NOE is observed when calculated separately for the individual domains. It is apparent that the backbone of scytovirin does not exhibit flexibility, except the last two C-terminal residues. The N-terminus is well ordered, beginning at residue 1, and participates in structuring of SD1. The interactions of residues G1 and S2 with C26, D27, and R30 contribute to the structural difference between domains. Hydrogen/deuterium exchange experiments show that 17 residues in each domain exhibit protected amide protons. All of the protected amides were on the interior of the protein. Some of these amides must participate in hydrogen bonds; however, due to the lack of regular secondary structure it was not possible to unambiguously assign hydrogen bonding partners. Therefore, the structures were calculated without such constraints.
Domain Interface
The interface between the SD1 and SD2 lies in the middle of the protein and helps stabilize the fold of each domain. The structure of each domain was calculated separately, using intra-domain NOEs. The backbone RMSDs worsened due to the loss of inter-domain NOEs, but the same overall folds were recognized. One end of the interface is cross-linked by the disulfide bond between C7 and C55. Multiple unambiguous NOEs, most notable T5 HG to T53 HG, substantiate the presence of the disulfide bond. The remainder of the interface is predominantly hydrophobic. A large number of unambiguous NOEs are observed between residue C20 and residue Q73. The interface is ∼580 Å2 and may help stabilize the domain structures, in agreement with initial studies of the individual domains (data not published). The linker does not appear to contribute significantly to the interface.
Conserved Structural Elements
As expected from the similarity of chemical shifts in each domain, the amino acid sequence repeats have similar overall backbone folds; however, the RMSD between SD1 and SD2 is 5 Å. Close inspection reveals a conformational change that explains the difference between the domains. Before discussion of the differences, it is beneficial to understand the likenesses.
Three discreet, common structural elements are apparent in both domains of scytovirin, Figure 3a. The first conserved structural element, the “top loop,” is formed by residues 12-17(60-65) of SD1(SD2), Figure 3b. The top loop circumnavigates approximately 240° of a complete circle. The first position of the top loop is the site of one of the three sequence differences (N12/K60) between SD1 and SD2, and the rest of the loop is formed by a P-G-G-P sequence. Both top loops exhibit a similar pattern of heteronuclear NOEs at 600 and 800 MHz. The first glycine (G15/G63) in the loop has an average NOE value of 0.77 ± 0.01 whereas the second glycine (G16/G64) has a value of 0.84 ± 0.02. The first glycine is closer to the solvent exposed apex of the loop and therefore it is reasonable that it is slightly less conformationally restricted. The second conserved structural element, denoted the “middle turn,” is formed by residues 22-25(70-73), Figure 3c. A disulfide bond between cysteine residues 20(68) and 26(74) helps demark the boundaries of this conserved structural element. In SD1, a probable hydrogen bond between the backbone carbonyl oxygen of residue 22 and the backbone amide proton of residue 25 produces a likeness to a classical turn. However, unlike a classical turn, the last residue of both middle turns introduces an abrupt change in direction. The change in direction is followed by a kinked extended region leading to the third conserved structural element and most distinguishing feature of scytovirin, denoted the “bottom knot,” which is formed from residues 31-39(79-87), Figure 3d. As with the middle turn, a disulfide bond between residues 32-38 (80-86) helps define the boundaries of this conserved structural element. Refinement against RDCs greatly improves resolution of this element in each domain, revealing a quite unusual fold.
Figure 3. Three Major Sub-structural Elements in Scytovirin.
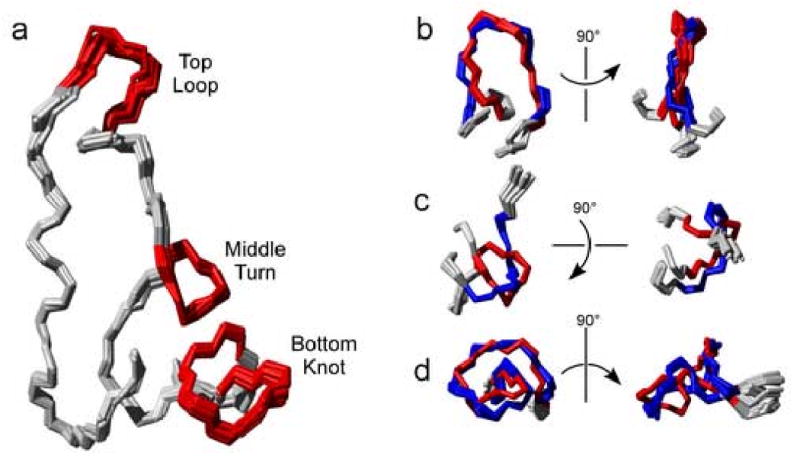
a) Twenty member ensemble of SD1 colored to show the 3 major loop-like elements conserved in both domains in red. See text for discussion. b) The top loop c) middle turn and d) bottom knot of SD1 (red) and SD2 (blue) are aligned to illustrate the structural similarities and differences.
Domain Differences
Both SD1 and SD2 are composed of the same structural elements; however, the relative orientations of the elements within each domain are not the same, resulting in the 5 Å RMSD between SD1 and SD2. The major differences are centered around the middle turn. In SD2, the middle turn is flipped 180° with respect to the rest of the domain when compared to the middle turn of SD1, Figure 3c. Backbone chemical shift differences between SD1 and SD2 are greatest for residues in the middle turn and the extended region directly following, Figure 4. Interactions between residues G1 and C26, D27, R30 of scytovirin are responsible for the difference, since the side chains intimately pack in this region of SD1 and help determine the orientation of the turn. The same does not hold for SD2, since residues P49 and K50 in the linker, equivalent in position to G1 and S2 for SD1, pack and interact differently with the residues near the middle turn of SD2. Residues preceding P49 in the linker, which are absent at the N-terminus, may also contribute to the differences. The final result is a flipped orientation of the middle turn. The structural significance of the two N-terminal residues, G1 and S2, is supported by the decrease in anti-HIV EC50 by greater than a factor of 500 when these two residues are removed17.
Figure 4. Amide Backbone Chemical Shift Difference between Domains.
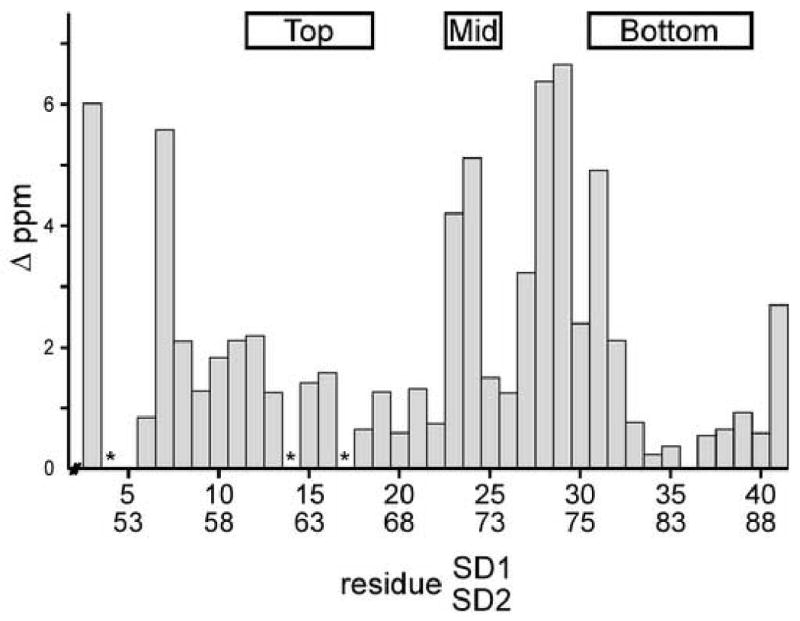
The absolute difference of amide backbone chemical shifts for equivalent residues in SD1 and SD2 are plotted. 1H contributions have been scaled to 15N ppm. Asterisks mark the position of proline residues. At the top, boxes demark residues composing the top loop, middle turn, and bottom knot.
As a consequence of the reversed middle turn, the top loop is also reversed between domains. The right handedness of SD1 is mirrored by the left handedness of SD2. Changes in side chain packing caused by the natural residue differences of N9 to D57 before the top loop and N12 to K60 at the beginning of the top loop contribute to the reversed handedness. Differences also occur in hydrophobic contributions at position A11/A59. For SD1, the domain interface provides some protection for the A11. For A59 of SD2, protection by the interface is not available so the hydrophobic side chain buries in the domain, positioning the residue much differently. These relatively small effects combine to cause the regions between the top loop and middle turn to be different between in SD1 and SD2. These conformations are confirmed by the observed RDCs for these regions, and the difference leads to the large backbone RMSD between the two domains.
The backbone folds of SD1 and SD2 are quite similar, except for the region between the top loop and middle turn. The concerted change in handedness of the top loop and 180° reversal of the middle turn effectively cancel each other, preserving the remainder of the overall fold. The backbone RMSD of the two domains is 2.5 Å, when the top loop through the middle turn are left out of the calculation. The major structural differences are thereby relegated to only part of the protein, and do not dramatically impact residues near the carbohydrate binding site (vide infra). This allows scytovirin to position residues critical for carbohydrate binding similarly between domains. Thus, both domains of scytovirin retain the ability to bind oligosaccharide, albeit with different affinities.
Oligosaccharide Binding
Scytovirin has been shown to bind specific oligosaccharides found on gp41 and gp120.4 In particular, scytovirin was shown to bind to a specific tetrasaccharide substructure of the high mannose oligosaccharide normally found decorating HIV-1 envelope glycoproteins7. The Manα(1→2)Manα(1→6)Manα(1→6)Man tetrasaccharide is pictured in Figure 5a. Chemical shift perturbation mapping of scytovirin was used to monitor titration of this tetrasaccharide, see Figure 5b. Different exchange kinetics were observed for residues in SD1 versus residues in SD2. At 500 MHz, residues in SD1 exhibit intermediate exchange whereas residues in SD2 exhibit fast exchange. These exchange regimes can be shifted by conducting the experiments at 800 MHz, where the intermediate exchange becomes slow and the fast exchange becomes intermediate (data not shown). The results suggest significantly different affinities for binding of the tetrasaccharide to each domain. Chemical shifts as a function of ligand concentration were fit to a two site model (Matlab algorithm kindly provided by Dr. David Fushman, University of Maryland) providing dissociation constant estimates of 30 μM for SD1 and 160 μM for SD2 (Supplementary Figure 4). These measurements agree with data measured from isothermal titration calorimetry (Shenoy & O'Keefe, personal communication.)
Figure 5. Oligosaccharide Titrations Reveal Binding Site.
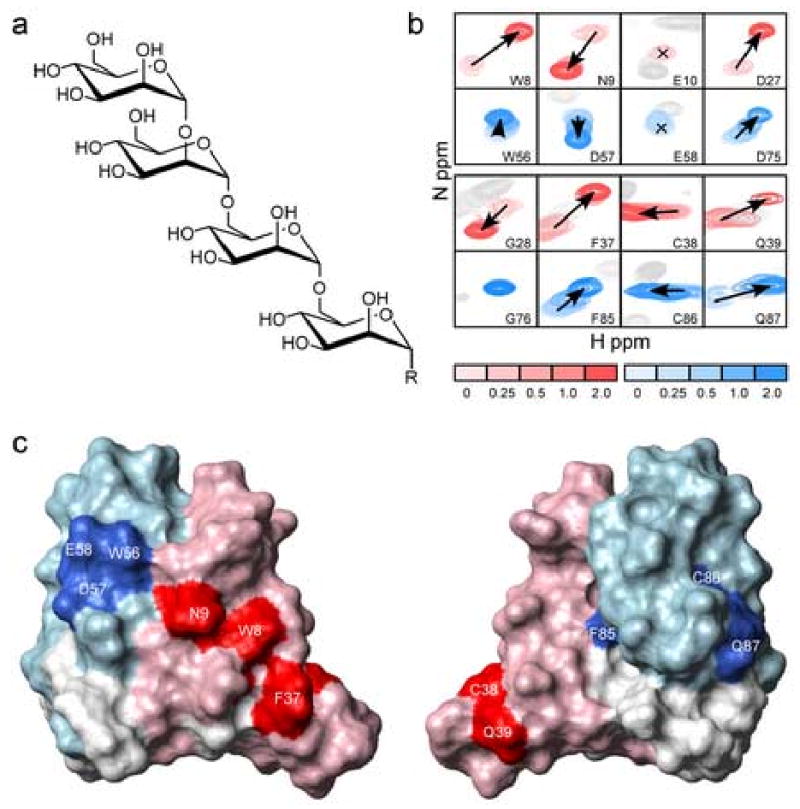
a) Structure of the tetrasaccharide used for NMR binding studies, where R is the same as in Adams et al.7 b) Overlay of sections from 15N-HSQC spectra collected with increasing amounts of tetrasaccharide. Complementary residues showing the largest chemical shift perturbation are shown. Residues from SD1 are in red and residues from SD2 are in blue. Color gradients from light to dark correspond to tetrasaccharide:scytovirin ratios of 0:1, 0.25:0,0.5:1,1:1,2:1. c) Surface representation of scytovirin (SD1 pink, SD2 light blue) colored to show residues having largest change in chemical shift upon tetrasaccharide binding (SD1 red, SD2 blue).
Residues that demonstrate backbone amide chemical shift changes upon addition of tetrasaccharide are almost identical for SD1 and SD2. Mapping those residues onto the scytovirin structure is shown in Figure 5c. It appears that each domain of scytovirin binds oligosaccharide in a similar fashion. This agrees with biochemical studies of the individually expressed domains17. There are three aromatic residues in each domain (SD1: Y6, W8, F37 and SD2: Y54, W56, F85), and, with the exception of F85 in SD2, all the aromatic residues of scytovirin are clustered near the binding sites, Figure 5c. Both Nε1Hε1 of tryptophan W8(SD1) and W56(SD2) demonstrate chemical shift perturbations, thus suggesting involvement in tetrasaccharide binding. The clustering of aromatics agrees well with an existing model in which two aromatic residues, separated by one residue in the amino acid sequence, play a critical role in one class of protein-oligosaccharide interactions.18
Oligosaccharide Binding Site Comparison
The differences between the two oligosaccharide binding sites can partially be explained from the apo structure of scytovirin. The N9 residue in SD1, which is the site of side chain substitution for D57 in SD2, shows large backbone amide chemical shift changes in the presence of tetrasaccharide, implicating involvement in binding. The negative charge of D57 may contribute to decreased oligosaccharide affinity in SD2. Also, the residues between the top loop and middle turn show minor chemical shift perturbations upon addition of tetrasaccharide, and some structural differences between the two domains may contribute to differences in binding. The proximity of G1 and S2 to surface exposed residues with the largest chemical shift perturbations in SD1 indicate the significance of the ordered N-terminus and local structure that contribute to the different oligosaccharide binding affinity. The lack of chemical shift perturbation for G76 in SD2 differs from the significant change observed for the equivalent G28 in SD1. Thus, even though the sites are quite similar, enough difference exists to cause an appreciable difference in oligosaccharide affinity. Furthermore, both sites exhibit considerable specificity. We have confirmed the previous observation that a related Manα(1→6)Manα(1→6)Man trisaccharide7 does not bind to scytovirin based on the absence of observed chemical shift perturbations in NMR titration experiments.
Discussion
At first glance, scytovirin appears to be another member of the hevein-like family since it binds oligosaccharides, has a majority of the signature CBD cysteines2 (Figure 6a), and has a similar aromatic triad involved in carbohydrate binding. The likeness of scytovirin to CBDs, however, does not extend to the backbone fold. The scytovirin structure is quite distinct from that of the hevein domain (Figure 6b). The difference in structure is, in part, due to the different disulfide bond formation. The absence of a cysteine at position 17 in scytovirin, strictly conserved in all hevein family members, causes a dramatic rearrangement of disulfide bonding, see Figure 6a. In scytovirin, the disulfide bonding pattern is C20-C26 and C32-C38 in SD1, C68-C74 and C80-C86 in SD2 with an inter-domain C7-C55 disulfide bond4. This corresponds to a sequential disulfide bonding pattern between the 2nd -3rd and 4th-5th (7th-8th, 9th-10th in SD2) cysteines with one inter-domain disulfide bond, between the 1st-6th cysteines. The disulfide bonds are very important to the structural integrity of the two domains, and the inter-domain disulfide bond assists in bringing the two domains together to form the overall globular structure. In all hevein family members, the pattern of disulfide bonds is not sequential, but intertwined as evidenced from disulfide bonding between the conserved 1st-4th, 2nd -5th, 3rd-6th, and 7th-8th cysteines (Figure 6a). The conserved disulfide pairing makes it reasonable to assume that the hevein domain structure is conserved across all family members, as represented by the B. arreguin hevein domain, Figure 6b. The hevein domain exhibits short α-helical and β-strand secondary structures, contrary to the absence of such structural elements in scytovirin.
Figure 6. Sequence Alignment, Disulfide Bonding Pattern, and Domain Comparison of Scytovirin and Hevein-domains.
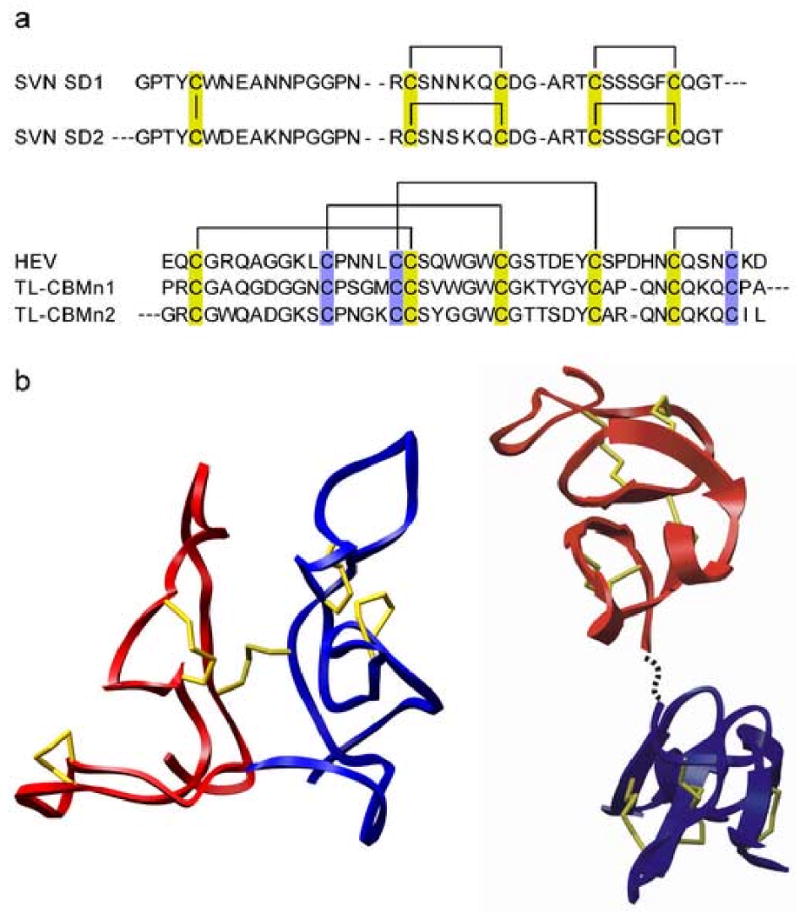
a) An amino acid sequence alignment of both scytovirin domains (SVN SD1 and SD2), hevein (HEV), and the hevein-like tomato lectin N-terminal chitin-binding modules5 (TL-CBMn1 and TL-CBMn2) illustrates the drastically different disulfide bond formation of scytovirin. Disulfide bonds are indicated by lines. Gold boxes indicate cysteines conserved in scytovirin and hevein-like proteins whereas blue boxes indicate cysteines only found in hevein-like CBDs. b) Comparison of the backbone folds of the dual CBDs of scytovirin (left) to a model of two arbitrarily oriented hevein domains (PDB ID: 1HEV) (right). All cysteines side chains are shown, colored in yellow. SVN SD1 is shown in red and SVN SD2 is shown in blue. The model of a dual hevein-domain protein without any inter-domain disulfide bond (right) is formed from two copies of 1HEV and the linker is represented as a dashed line.
The sequence repeat, or dual hevein motif, of scytovirin is also seen in other CBDs. For example, a recently described tomato lectin shows the same duplication of hevein domains joined by proline rich linkers5. The tomato lectin has no inter-domain disulfide bond, hence there is no a priori expectation that the two domains would contact or interact with one another. The dual motif would be anticipated to have a fold comprised of two typical hevein domains connected by a linker, as suggested by the model in Figure 6b. Conversely, the inter-domain disulfide bond of scytovirin is reminiscent of mistletoe lectin where two CBDs are held together by a lone, inter-domain disulfide bond6. The conserved hevein disulfide pairing of the tomato lectin (Figure 6a) would suggest that the individual domains have the hevein fold and that they are simply tethered. It is not known if there is any contact or interaction between the two domains, as seen in scytovirin. This variation in domain structure and interconnection might provide further differentiation in carbohydrate recognition specificity; however, further investigations are required to examine this possibility.
Each domain of scytovirin contains a triad of aromatic residues that is similar to the triad in hevein domains and is known to be responsible for carbohydrate binding in hevein18. Despite the sequence similarity to CBDs, scytovirin lacks measurable binding affinity for chitin, monosaccharides, or common trisaccharides4; 7. Scytovirin does not bind the GlcNAcβ(1→4)GlcNAc structure of chitin but instead shows high specificity for a tetrasaccharide structure of Manα(1→2)Manα(1→6)Manα(1→6)Man. One plausible explanation for these differences is the altered arrangement of key aromatic residues in the binding sites of scytovirin compared to hevein domains. Interestingly, SD1 of scytovirin preserves the structural arrangement of the aromatic triad (Y6, W8, and F37) and has a nearly identical affinity for its tetrasaccharide binding partner as hevein does for (GlcNAc)48. SD2 exhibits a lower apparent binding affinity for the tetrasaccharide, and the aromatic triad (Y54, W56, and F85) is not as tightly clustered. The side chain of F85 is not readily able to participate in binding due to the reversed arrangement of the middle turn. Thus, we predict scytovirin and hevein utilize similar protein:carbohydrate interactions; however a different specificity is achieved as a direct result of the altered disulfide bonding pattern and rearrangement of the aromatic triads in each domain. In binding experiments with the tetrasaccharide, the two binding sites are independent with relatively weak affinity. As noted for other CBDs8, increased affinity is achieved by binding to large carbohydrates or glycoproteins that can delivery carbohydrates to both scytovirin domains, and this multivalency is likely responsible for the high anti-HIV activity of scytovirin.
The presence of two weak carbohydrate binding sites that result in potent antiviral activity towards gp41 and gp120 parallels the observations for another cyanobacterial lectin protein with potent antiviral activity, cyanovirin-N 19. Cyanovirin-N exhibits two binding sites with micromolar affinity, similar to scytovirin. The cyanovirin-N binding sites, however, do not involve aromatic residues, based on both NMR titrations20 and the crystal structure of the cyanovirin-N:Man-9 complex21. Similar to scytovirin, cyanovirin-N exhibits very high affinity toward viral glycoprotein epitopes. A recent study of cyanovirin-N19 observed precipitation of gp41 by cyanovirin-N that could be prevented by mutation of one of the binding sites, suggesting that the precipitation was due to cross linking but concluding that the nanomolar potency was probably not due to cross-linking. Instead, the multi-valency may be related to the spatial specificity of glycoprotein epitopes. Our data suggest that scytovirin may act in a parallel fashion, but the nature of the carbohydrate binding sites are different and lead to a distinct carbohydrate specificity compared to cyanovirin-N7.
The structure of scytovirin introduces a novel fold and new twist to carbohydrate binding proteins. Knowledge of the carbohydrate binding sites provides important opportunities for structural engineering to improve this molecule as an antiviral, and particularly anti-HIV, agent. Further studies are in progress to better understand the protein:carbohydrate complex. A greater understanding of how scytovirin binds to viral glycoprotein carbohydrates will contribute to advanced development of antiviral therapies and potential topical microbicide prophylaxis.
Materials and Methods
Sample Preparation and NMR Data Acquisition
Scytovirin was expressed and purified as described elsewhere.22 The NMR buffer used for all experiments was 20 mM MES, 100 μM EDTA, 10 μM NaN3, pH = 5.5. The concentration of scytovirin was 1 mM for the unbound sample and 0.5 mM for the sample used for carbohydrate titration. All data, unless otherwise noted, were collected at 25° C. The titration, assignment, and 13C-edited NOESY data were collected on Varian Inova spectrometers with cryogenically cooled probes at field strengths of 500, 600 and 800 MHz respectively. 15N-edited NOESY and proline sequential assignment data were collected at 600 MHz on a Bruker Avance spectrometer. 15N-edoted and 13C-edited NOESY mixing times were 100 and 125 milliseconds respectively. Heteronuclear 15N-1H NOEs were collected with a recycle delay of 5.5 sec or recycle delay of 2.5 sec and a saturation time of 3 seconds. Hydrogen/deuterium exchange spectra were collected at times of 20 min, 40 min, 1.5 hrs, 4 hrs, 16 hrs, and 4 days after a lyophilized sample of scytovirin was resuspended in D2O. All data were processed using nmrPipe23 and visualized using SPARKY24.
Structure Calculation and Refinement Against RDCs
Structure calculations were conducted using XPLOR-NIH.25 Due to the lack of extended secondary structure predicted from Cα/Cβ chemical shifts, database calculated dihedral angle restraints were not used. 1206 intraresidue, 1005 sequential, and 634 long range (defined as NOEs between protons of residues five or greater positions apart in the sequence) were used in the calculation. The lowest energy 20 structures of 200 calculated are reported. The precise and consistent suite of J-modulated NH and HαCα RDCs were measured using established pulse sequences.13 Integer multiples of 0.70 and 1.0 milliseconds were used for HαCα and NH coupling delays respectively. Sixty-seven HαCα and 82 NH couplings were used for structural refinement. Ramachandran statistics included 13 dihedral angles in the most favorable region, 32 in the additionally allowed, 19 generously allowed and 5 disallowed.
Oligosaccharide Titration
Oligosaccharides were synthesized as reported elsewhere.26; 27 Titration data were collected at both 500 and 800 MHz in NMR buffer. Ratios of approximately 0:1, 0.5:1, 1:1 and 2:1 were collected for the Manα(1-2)Manα(1-6)Manα(1-6)Man tetrasaccharide. Concentrations of 0:1, 0.5:1, 1:1, 2:1 and 10:1 were collected for the non-binding Manα(1-6)Manα(1-6)Man trisaccharide. At 800 MHz, data were collected at temperatures of 10, 17.5 and 25° C for temperature dependence of oligosaccharide binding and tentative indication of hydrogen bonding.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
NMR and Refinement Statistics for Scytovirin
| Distance Constraints | |
|---|---|
| Total NOE | 2905 |
| Intra-residue | 1247 |
| Inter-residue | 1658 |
| Sequential (|i-j| = 1) | 679 |
| Medium-range (|i-j| < 4) | 324 |
| Long-range (|i-j > 5) | 655 |
| Total RDCs | 148 |
| NH | 82 |
| HαCα | 66 |
| Q (%) | 0.25 |
| Structure Statistics | |
| Violations (mean and s.d.) | |
| Distance constraints > 0.5 Å (Å) | 24 +/- 2 |
| Average violation > 0.5 Å (Å) | 0.69 +/- 0.16 |
| Max. distance constraint violation (Å) | 1.17 |
| Deviations from idealized geometry | |
| Bond lengths (Å) | 0.01 +/- 0.06 |
| Bond angles (°) | 1.59 +/- 2.55 |
| Impropers (°) | 0.08 +/- 2.19 |
| Average r.m.s.d. to mean* (Å) | |
| Heavy | 0.42 +/- 0.11 |
| Backbone | 0.51 +/- 0.11 |
Calculated from 20 refined structures.
Acknowledgments
The authors would like to thank Walter Thompson for protein purification, Jess Li for general laboratory support, and Vadim Gaponenko, Hana Křížová, and Amanda Altieri for useful discussions and review of this manuscript. The authors would also like to thank Charles Schwieters for guidance in utilizing XPLOR-NIH and David Fushman for a Matlab algorithm and advice for analyzing kinetic data. This research is supported by the intramural research program of the NIH, National Cancer Institute, Structural Biophysics Laboratory. This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract N01-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Supplementary Data: Supplementary data associated with this article can be found, in the online version, at XXXXXX.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Boraston AB, Bolam DN, Gilbert HJ, Davies GJ. Carbohydrate-binding modules: fine-tuning polysaccharide recognition. Biochemical Journal. 2004;382:769–81. doi: 10.1042/BJ20040892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rodriguez-Romero A, Ravichandran KG, Soriano-Garcia M. Crystal structure of hevein at 2.8 A resolution. FEBS Letters. 1991;291:307–9. doi: 10.1016/0014-5793(91)81308-u. [DOI] [PubMed] [Google Scholar]
- 3.Charan RD, Munro MH, O'Keefe BR, Sowder R, McKee TC, Currens MJ, Pannell LK, Boyd MR. Isolation and characterization of Myrianthus holstii lectin, a potent HIV-1 inhibitory protein from the plant Myrianthus holstii(1) Journal of Natural Products. 2000;63:1170–4. doi: 10.1021/np000039h. [DOI] [PubMed] [Google Scholar]
- 4.Bokesch HR, O'keefe BR, McKee TC, Pannell LK, Patterson GM, Gardella RS, Sowder RC, 2nd, Turpin J, Watson K, Buckheit RW, Jr, Boyd MR. A potent novel anti-HIV protein from the cultured cyanobacterium Scytonema varium. Biochemistry. 2003;42:2578–84. doi: 10.1021/bi0205698. [DOI] [PubMed] [Google Scholar]
- 5.Peumans WJ, Rouge P, Van Damme EJ. The tomato lectin consists of two homologous chitin-binding modules separated by an extensin-like linker. Biochemical Journal. 2003;376:717–24. doi: 10.1042/BJ20031069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stoeva S, Franz M, Wacker R, Krauspenhaar R, Guthohrlein E, Mikhailov A, Betzel C, Voelter W. Primary structure, isoforms, and molecular modeling of a chitin-binding mistletoe lectin. Archives of Biochemistry & Biophysics. 2001;392:23–31. doi: 10.1006/abbi.2001.2429. [DOI] [PubMed] [Google Scholar]
- 7.Adams EW, Ratner DM, Bokesch HR, McMahon JB, O'Keefe BR, Seeberger PH. Oligosaccharide and glycoprotein microarrays as tools in HIV glycobiology; glycan-dependent gp120/protein interactions. Chemistry & Biology. 2004;11:875–81. doi: 10.1016/j.chembiol.2004.04.010. see comment. [DOI] [PubMed] [Google Scholar]
- 8.Asensio JL, Canada FJ, Siebert HC, Laynez J, Poveda A, Nieto PM, Soedjanaamadja UM, Gabius HJ, Jimenez-Barbero J. Structural basis for chitin recognition by defense proteins: GlcNAc residues are bound in a multivalent fashion by extended binding sites in hevein domains. Chemistry & Biology. 2000;7:529–43. doi: 10.1016/s1074-5521(00)00136-8. [DOI] [PubMed] [Google Scholar]
- 9.Ikura M, Bax A, Clore GM, Gronenborn AM. Detection of nuclear Overhauser effects between degenerate amid proton resonances by heteronuclear three-dimensional NMR spectroscopy. Journal of the American Chemical Society. 1990;112:9020–9022. [Google Scholar]
- 10.Schubert M, Ball LJ, Oschkinat H, Schmieder P. Bridging the gap: A set of selective 1H-15N-correlations to link sequential neighbors of prolines. Journal of Biomolecular NMR. 2000;17:331–5. doi: 10.1023/a:1008362904205. [DOI] [PubMed] [Google Scholar]
- 11.Wishart DS, Sykes BD. The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. Journal of Biomolecular NMR. 1994;4:171–80. doi: 10.1007/BF00175245. [DOI] [PubMed] [Google Scholar]
- 12.Wishart DS, Sykes BD, Richards FM. The chemical shift index: a fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry. 1992;31:1647–51. doi: 10.1021/bi00121a010. [DOI] [PubMed] [Google Scholar]
- 13.McFeeters RL, Fowler CA, Gaponenko VV, Byrd RA. Efficient and precise measurement of H(alpha)-C(alpha), C(alpha)-C', C(alpha)-C(beta) and H(N)-N residual dipolar couplings from 2D H(N)-N correlation spectra. Journal of Biomolecular NMR. 2005;31:35–47. doi: 10.1007/s10858-004-6057-y. [DOI] [PubMed] [Google Scholar]
- 14.Cierpicki T, Zhukov I, Byrd RA, Otlewski J. Hydrogen bonds in human ubiquitin reflected in temperature coefficients of amide protons. Journal of Magnetic Resonance. 2002;157:178–80. doi: 10.1006/jmre.2002.2597. [DOI] [PubMed] [Google Scholar]
- 15.Holm L, Sander C. Protein structure comparison by alignment of distance matrices. Journal of Molecular Biology. 1993;233:123–38. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- 16.Kay LE, Torchia DA, Bax A. Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: applicatio nto staphylococcal nuclease. Biochemistry. 1991;28:8972–8979. doi: 10.1021/bi00449a003. [DOI] [PubMed] [Google Scholar]
- 17.Xiong C, O'Keefe BR, Byrd RA, McMahon JB. Potent anti-HIV activity of scytovirin domain 1 peptide. Peptides. 2006;27:1668–75. doi: 10.1016/j.peptides.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 18.Chavez MI, Andreu C, Vidal P, Aboitiz N, Freire F, Canada FJ, Jimenez-Barbero J. On the Importance of Carbohydrate-Aromatic Interaction for the Molecular Recognition of Oligosaccharides by Proteins: NMR Studies of the Structure and Bidning Affinity of AcAMP2-like Peptides with Non-Natural Naphtyyl and Fluoroaromatic Residues. Chemistry, A European Journal. 2005;11:7060–7074. doi: 10.1002/chem.200500367. [DOI] [PubMed] [Google Scholar]
- 19.Barrientos LG, Matei E, Lasala F, Delgado R, Gronenborn AM. Dissecting carbohydrate-Cyanovirin-N binding by structure-guided mutagenesis: functional implications for viral entry inhibition. Protein Engineering, Design & Selection. 2006;19:525–35. doi: 10.1093/protein/gzl040. [DOI] [PubMed] [Google Scholar]
- 20.Bewley CA, Otero-Quintero S. The potent anti-HIV protein cyanovirin-N contains two novel carbohydrate binding sites that selectively bind to Man(8) D1D3 and Man(9) with nanomolar affinity: implications for binding to the HIV envelope protein gp120. Journal of the American Chemical Society. 2001;123:3892–902. doi: 10.1021/ja004040e. [DOI] [PubMed] [Google Scholar]
- 21.Botos I, O'Keefe BR, Shenoy SR, Cartner LK, Ratner DM, Seeberger PH, Boyd MR, Wlodawer A. Structures of the complexes of a potent anti-HIV protein cyanovirin-N and high mannose oligosaccharides. Journal of Biological Chemistry. 2002;277:34336–42. doi: 10.1074/jbc.M205909200. [DOI] [PubMed] [Google Scholar]
- 22.Xiong C, O'keefe BR, Botos I, Wlodawer A, McMahon JB. Overexpression and purification of scytovirin, a potent, novel anti-HIV protein from the cultured cyanobacterium Scytonema varium. Protein Expression and Purification. 2006;46:233–239. doi: 10.1016/j.pep.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 23.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. Journal of Biomolecular NMR. 1995;6:277–93. doi: 10.1007/BF00197809. see comment. [DOI] [PubMed] [Google Scholar]
- 24.Goddard TD, Kneller DG. SPARKY 3. University of California; San Francisco: [Google Scholar]
- 25.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. Journal of Magnetic Resonance. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 26.Plante OJ, Buchwald SL, Seeberger PH. Halobenzyl Ethers as Protecting Groups for Organic Synthesis. Journal of the American Chemical Society. 2000;122:7148–7149. [Google Scholar]
- 27.Ratner DM, Plante OJ, Seeberger PH. A Linear Synthesis of Branched High-Mannose Oligosaccharides from the HIV-1 Viral Surface Envelope Glycoprotein gp120. European Journal of Organic Chemistry. 2002:826–833. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.