

Abstract
Myelinating cells, oligodendrocytes in the CNS and Schwann cells in the PNS, produce an enormous amount of plasma membrane during the myelination process, making them particularly susceptible to disruptions to the secretory pathway. ER stress, initiated by the accumulation of unfolded or misfolded proteins, activates the unfolded protein response (UPR), which adapts cells to the stress. If this adaptive response is insufficient, the UPR activates an apoptotic program to eliminate the affected cells. Recent observations suggest that ER stress in myelinating cells plays an important role in the pathogenesis of various disorders of myelin, including Charcot-Marie-Tooth disease (CMT), Pelizaeus-Merzbacher disease (PMD), Vanishing White Matter Disease (VWMD), as well as in the most common disorder of myelin, multiples sclerosis (MS). A better understanding of ER stress in myelinating cells has laid the groundwork for the design of novel therapeutic strategies to promote myelinating cell survival in these disorders.
Myelinating cells, oligodendrocytes in the CNS and Schwann cells in the PNS, produce a vast amount of myelin as an extension of their plasma membrane (Figure 1) 1, 2. Myelin, which is a unique, lipid-rich multilamellar sheath that wraps around axons in the CNS and PNS, is not only essential for the fast conduction of the action potential along axons but also for the maintenance of axonal integrity 3. According to estimates from morphometric analyses, the mean surface area of myelin per mature myelinating cell is thousands of times greater than the surface area of a typical mammalian cell 1. Thus, during the active phase of myelination, each myelinating cell must synthesize an enormous amount of myelin membrane proteins, cholesterol, and membrane lipids through the secretory pathway 2. Therefore, it is not surprising that myelinating cells are highly sensitive to the disruption of the secretory pathway, particularly the homeostasis of the endoplasmic reticulum (ER). Recent studies indicate that this increased susceptibility contributes to the pathogenesis of a number of myelin disorders 4-11.
Figure 1. Oligodendrocyte and myelin.
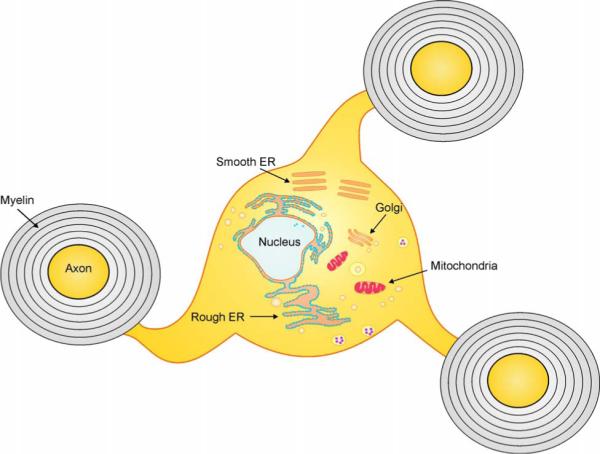
Oligodendrocytes produce as an extension of their plasma membrane vast amounts of myelin; a unique, lipid-rich, multilamellar sheath that wraps axons in the CNS. The secretory pathways for protein and lipid, including rough ER, smooth ER and Glogi apparatus, are well developed in oligodendrocytes. Evidence is accumulating that oligodendrocytes, as well as their PNS counterpart Schwann cells, rank among the cells that are most sensitive to the disruption of the secretory pathway.
The ER has three essential functions 12, 13. Secretory proteins and proteins destined for the cell surface and other intracellular organelles are synthesized by ribosomes on the cytosolic surface of the ER and translocated into the ER lumen through a pore in the ER membrane. Inside the lumen, they are properly modified and folded. Additionally, the biosynthesis of steroids, cholesterol, and other lipids takes place on the cytoplasmic side of the ER membrane. Moreover, cellular calcium is mainly stored in the ER lumen. The concentration of calcium in the cytoplasm is primarily regulated by Ca2+ transport into and out of the ER.
A number of cell stress conditions, such as a perturbed calcium homeostasis or redox status, elevated secretory protein synthesis rates, altered glycosylation levels, and abnormally high cholesterol levels, can interfere with oxidative protein folding and subsequently lead to the accumulation of unfolded or misfolded proteins in the ER lumen. This phenomenon has been referred to as ER stress 14-16. As a consequence, the eukaryotic cell has evolved an adaptive coordinated response, the unfolded protein response (UPR, Figure 2 and Table 1), to limit further accumulation of unfolded proteins in the ER. Three ER-resident transmembrane proteins involved in the UPR have been identified: pancreatic ER kinase (PERK), the kinase encoded by the inositol requiring 1 (IRE1) gene, and activating transcription factor 6 (ATF6). At the onset of ER stress, the most rapidly activated pathway is the PERK pathway, which couples protein folding in the ER with protein synthesis by phosphorylating the α subunit of eukaryotic translation initiation factor 2 (eIF2α) 17, 18. The activation of the PERK-eIF2α pathway attenuates the initiation of translation in response to ER stress and enhances the stress-induced expression of numerous cytoprotective genes 19-21. Activation of IRE1 initiates the splicing of X-box-binding protein (XBP1) mRNA, producing an active transcription factor, spliced XBP-1 (sXBP-1) 22, 23. Additionally, ATF6 becomes an active transcription factor by transiting to the Golgi complex, where it is cleaved by the proteases S1P and S2P, before translocating to the nucleus 24. The activation of IRE1 signaling and the cleavage of ATF6 promote ER expansion and the expression of ER-localized chaperones, which facilitate protein folding in the ER 18, 25. In multicellular organisms, if these adaptive responses are not sufficient to resolve the folding problems in the ER, the UPR will trigger an apoptotic program to eliminate the cells 26, 27.
Figure 2. The UPR pathway in eukaryotic cells.

ER stress triggers PERK dimerization and auto-phosphorylation. Activated PERK attenuates protein biosynthesis by phosphorylating eIF2α. Phosphorylated eIF2α also activates transcription factor ATF4, which enhances the stress-induced expression of numerous cytoprotective genes. Additionally, ATF4 increases the expression of CHOP, which induces GADD34 expression. GAAD34 binds to PP1 and forms the GADD34-PP1 complex that specifically dephosphorylates eIF2α. ER stress also triggers IRE1dimerization and auto-phosphorylation. Activation of IRE1 initiates the splicing of XBP1 mRNA, producing an active transcription factor sXBP-1. Additionally, ATF6 becomes an active transcription factor by transiting to the Golgi complex, where it is cleaved by the proteases S1P and S2P. The activation of IRE1 signaling and the cleavage of ATF6 promote ER expansion and the expression of ER-localized chaperones. In multicellular organisms, if these adaptive responses are not sufficient to resolve the folding problems in the ER, the UPR will trigger an apoptotic program, such as activation of caspase 12 or CHOP, to eliminate the cells.
Table 1.
The functions of major effecter components of the UPR
| UPR Effecter molecule |
Function |
|---|---|
| PERK | At the onset of ER stress, the most rapidly activated ER stress sensor is PERK, which couples protein folding in the ER with protein synthesis by phosphorylating eIF2α 19, 20. |
| eIF2α | eIF2α phosphorylation promotes a stress-resistant state by global attenuation of protein biosynthesis and induction of numerous stress-induced cytoprotective genes 21, 28. Four different kinases are known to phosphorylate eIF2α. They are PERK, general control nonderepressible-2 kinase (GCN2) that is activated by amino acid starvation, double-stranded RNA-dependent protein kinase (PKR) that is activated by viral infection, and heme-regulated inhibitor (HRI) that is activated by iron deficiency 28. |
| ATF4 | Transcription factor ATF4 mRNA contains a unique 5' untranslated region, leading to ATF4 protein expression when eIF2α is phosphorylated. The gene expression program activated by ATF4 is involved in regulating amino acid metabolism and resistance to oxidative stress 29. |
| CHOP | A transcription factor downstream of ATF4, was thought to promote apoptotic cell death, since its deletion diminishes cell death during ER stress. Recent work, however, demonstrated that the deleterious effects of CHOP are associated with the induction of the protein growth arrest and DNA damage 34 (GADD34), rather than the activation of an apoptotic program 25, 30, 31. |
| GADD34 | A regulatory subunit of protein phosphatase 1 (PP1) that specifically dephosphorylates phosphorylated eIF2α. GADD34 blockage increases the levels of phosphorylated eIF2 α in stressed cells and protects cells from the stresses 31, 32. |
| ATF6 | Activation of ATF6 promotes the expression of ER-localized chaperones 18, 25. |
| IRE1 | Activation of IRE1 initiates the splicing of X-box-binding protein (XBP1) mRNA, producing an active transcription factor, spliced XBP-122, 23. |
| XBP-1 | Spliced XBP-1 promotes ER expansion and the expression of ER-localized chaperones 18, 25. |
| Caspase-12 | Activation of Caspase-12, an ER-localized caspase, has been shown to be associated with cell apoptosis induced by ER stress 33, 34. |
It has become increasingly clear that ER stress is an important feature of a number of human diseases, especially those involving cells dedicated to the synthesis of secreted proteins, such as diabetes mellitus, liver diseases, and inflammatory disorders 35-37. In this review, we discuss the current understanding of the contribution of ER stress in myelinating cells to the pathogenesis of inherited myelin disorders, including Charcot-Marie-Tooth disease, Pelizaeus-Merzbacher disease (PMD), Vanishing White Matter Disease, as well as in multiples sclerosis (MS), an autoimmune demyelination disorder of the CNS. We also examine the unique features of ER stress in myelinating cells and how the increased sensitivity of these cells to ER stress might provide selective therapeutic opportunities.
Evidence for abnormalities in the secretory pathway of Schwann cells in Charcot-Marie-Tooth disease
Charcot-Marie-Tooth disease is one of the most common inherited neurological disorders in humans, affecting approximately 1 in 2,500 individuals. Charcot-Marie-Tooth disease is actually a group of disorders of the PNS that are caused by mutations in a number of distinct genes and can be categorized as primarily originating from either the myelinating cell (Charcot-Marie-Tooth 1) or the axons (Charcot-Marie-Tooth 2) 38. Mutations in genes encoding peripheral myelin proteins account for the majority of Charcot-Marie-Tooth 1 cases. Peripheral myelin protein P0, a transmembrane glycoprotein, comprises greater than 50% of the total protein of peripheral myelin, and a number of distinct point mutations in P0 have been shown to be responsible for a dominant form of Charcot-Marie-Tooth 1B 39. There is strong evidence that mutations in the gene encoding P0 can lead to ER stress in myelinating Schwann cells 4, 5. Pennuto et al. have generated an authentic mouse model of Charcot-Marie-Tooth 1B that expresses a human disease-causing form of P0 deleted for S63 (P0S63del) 4. Their studies demonstrate that this mutation causes a perturbed alignment of hydrophobic residues in β strand C of P0 that results in the retention of the mutant protein in the ER of Schwann cells and leads to increased levels of phosphorylated eIF2α (p-eIF2α), ATF4, ATF3 and CAATT enhancer binding protein homologous protein (CHOP) in the nerve, suggesting activation of the PERK pathway 5. The spliced form of XBP-1 is also increased in the nerves of these animals, suggesting activation of the IRE1 pathway. Moreover, cleavage of ATF6 is detected in the P0S63del nerve. Interestingly, the activation of PERK, IRE1 and ATF6 pathways in P0S63del nerves is dose-dependent, which strongly suggests that the retention of P0S63del in the ER results in the activation of the UPR in Schwann cells.
CHOP, a transcription factor downstream of the PERK-eIF2α pathway, has been shown to play an important role in the cell apoptosis that results from ER stress, and CHOP deletion protects various cell types against ER stress-induced apoptosis in cell culture and animal models 25, 30, 31. Pennuto et al. show CHOP is undetectable in Schwann cells of normal nerves, but present in the nuclei of P0S63del Schwann cells 5. To determine the role of the CHOP pathway in the pathogenesis of the myelin abnormalities that occurs in the P0S63del nerve, P0S63del mice were crossed with CHOP null mice 5. In the absence of CHOP activity, the number of demyelinated fibers in P0S63del nerves is reduced, and the behavioral and electrophysiological abnormalities are attenuated. Nevertheless, CHOP inactivation, surprisingly, did not affect the number of apoptotic Schwann cells or the degree of hypomyelination in the P0S63del nerve 5. The mechanism accounting for the detrimental effects of CHOP in Charcot-Marie-Tooth 1B remains to be determined.
Mutations in genes encoding peripheral myelin protein PMP22 are the most common cause for Charcot-Marie-Tooth 1 cases 39. PMP22 is a highly hydrophobic transmembrane protein with four putative transmembrane domains. In humans, the molecular lesion that is responsible for the vast majority of Charcot-Marie-Tooth 1 cases is a duplication of the region of chromosome 17 that contains the PMP22 gene, resulting in increased expression of this membrane protein 40. Similarly, enforced expression of wild type PMP22 in Schwann cells of transgenic mice and rats results in a Charcot-Marie-Tooth like disorder 41. Cumulative evidence indicates that this is a gain of function disorder in which the increased expression of PMP22 has a deleterious effect on myelinating Schwann cells 42. Additionally, spontaneous dominant mouse mutations have been identified in the PMP22 gene, Trembler (Tr) and Trembler-J (Tr-J), that introduce nonconservative amino acid changes into hydrophobic domains of this protein. It has been demonstrated that misfolded PMP22Tr and PMP22Tr-J associate with calnexin, an ER chaperone, and accumulate in the ER of Schwann cells 43, 44. Moreover, Khajavi et al. have shown that treatment with the chemical compound curcumin, which is capable of promoting the transport of misfolded proteins from the ER to the plasma membrane, significantly attenuates Schwann cell apoptosis and improves the neuropathologic phenotype in PMP22Tr-J mice 45. Taken together, these data indicate that the accumulation of mutant PMP22 in the ER causes the apoptosis of Schwann cells, resulting in myelin abnormalities. Nevertheless, this accumulation does not lead to increased expression of the ER stress markers binding immunoglobulin protein (BIP) or CHOP44, 45. These data suggest that the role of the UPR in the pathogenesis of Charcot-Marie-Tooth 1A mutations needs to be examined directly and in greater detail.
Severe ER stress in Pelizaeus-Merzbacher disease
Pelizaeus-Merzbacher disease (PMD) is an X chromosome-linked dysmyelinating disease of the CNS with a broad range of clinical severity that is caused by more than 60 known mutations, including missense and nonsense mutations, in the proteolipid protein (PLP) gene 46-48. In addition, PLP gene duplications are responsible for approximately 50% of PMD cases. PLP and its alternatively spliced isoform, DM20, are believed to contain four membrane-spanning domains and, together, comprise approximately 50% of total CNS myelin protein. It is believed that mutant PLP has a dominant gain-of-toxic function, since PLP deletion does not affect oligodendrocyte development and myelination, and PLP null mice have normal neurological function until late adulthood 46. Moreover, the enforced expression of a wild type PLP gene is not sufficient to rescue the PLP mutant phenotype 46-48. Studies from Gow et al. demonstrate that PLP mutations cause oligodendrocyte death during the myelination process, at the developmental time point when oligodendrocytes are extending extensive processes, making contact with nearby axons, and initiating the synthesis of the myelin proteins 49.
Several studies have shown that the mutant PLP proteins are not properly folded and accumulate in the ER 47, 50, 51. COS-7 cells transfected with a number of distinct PLP mutants show accumulation of mutant proteins in the ER 7, 47. Additionally, immunohistochemical studies show perinuclear accumulation of mutant PLP proteins in the oligodendrocytes of PLP mutant mice 49, 52. This was further supported by electron microscopy analyses, which provided direct evidence that mutant PLP proteins accumulate in the ER of oligodendrocytes, where they disturb the ER ultrastructure 47, 53.
Strong evidence has also recently indicated that the accumulation of mutant PLP in the ER activates the UPR and leads to myelinating oligodendrocyte death 7, 54. Mutant PLP expressed by transfected fibroblasts accumulates in the ER and significantly increases the expression of ER stress markers such as CHOP, BIP and ATF3, markers that are also upregulated in the oligodendrocytes of PLP mutant mice 7. Importantly, CHOP is localized to the nucleus in mutant oligodendrocytes, which is strongly indicative of the ER stress response 7. Similarly, increased CHOP expression is seen in the oligodendrocyte nuclei of PMD patients 7. Lastly, Bauer et al. show that oligodendrocytes from rats transgenically overexpressing PLP are characterized by swollen ER and increased expression of BIP, as well as disulfide isomerase, an ER enzyme involved in protein folding 6.
To determine the involvement of ER stress in oligodendrocyte death in PLP mutant animals, Southwood et al. used a genetic approach, crossing CHOP null mice with PLP mutant mice 7. Surprisingly, they found that the absence of CHOP notably exacerbated the clinical phenotype and increased oligodendrocyte apoptosis in PLP mutant mice. This protective effect of CHOP on oligodendrocytes during ER stress is strikingly contrasted to its proapoptotic effect on other cell types. The mechanisms accounting for the protective effect of CHOP on oligodendrocytes remain a mystery. Interestingly, Southwood et al. show that CHOP induction in PLP mutant mice does not enhance expression of genes that normally act downstream of CHOP, such as DOCs (downstream of CHOP) 1, 4, and 6 7. It is possible that CHOP has a distinct set of target genes depending on cell type, which is reflected in its cell-specific function.
The UPR adapts oligodendrocytes to eIF2B mutations in Vanishing White Matter Disease
Vanishing White Matter Disease is an inherited autosomal-recessive hypomyelinating disorder. The discovery of missense mutations in the ubiquitously-expressed genes encoding the five subunits of eIF2B in Vanishing White Matter Disease patients led to the identification of the genetic causes of Vanishing White Matter Disease 55, 56. This is the first human disease that has been shown to directly affect a protein synthesis factor. eIF2B functions in translation as a guanine nucleotide exchange factor, promoting the release of GDP from eIF2 and the formation of an active eIF2-GTP complex 57. eIF2-GTP then binds aminoacylated initiator methionyl tRNA, forming a ternary complex that is essential for each translation initiation event. p-eIF2α reduces the activity of eIF2B, with the 2 proteins forming a nonproductive p-eIF2–eIF2B complex. eIF2B mutations may impair its response to p-eIF2α, particularly during cell stress 58. Although oligodendrocytes have been described as abnormal and “foamy” in appearance, no evidence has been presented that demonstrates a reduction of oligodendrocyte numbers in Vanishing White Matter Disease 59, 60. Interestingly, van Kollenburg et al. recently presented evidence for the activation of the UPR in oligodendrocytes in Vanishing White Matter Disease patients, including increased levels of phosphorylated PERK, p-eIF2α, ATF4 and CHOP 61, 62. Moreover, it has been shown that primary cultured fibroblasts from Vanishing White Matter Disease patients respond normally to ER stress by reduced global translation rates. Nevertheless, the induction of ATF4 is significantly enhanced in these cells compared to normal controls despite equal levels of cell stress and p-eIF2α 63. Importantly, a recent study has shown that the enforced expression of a Vanishing White Matter Disease-causing eIF2B mutation in an oligodendroglial cell line leads to elevated basal levels of the ER stress markers ATF4, GADD34 and BIP and results in the hyper-induction of these markers in response to a pharmacological stress agent, indicating that the UPR adapts oligodendrocytes to Vanishing White Matter Disease-causing eIF2B mutations 64.
ER stress has beneficial and detrimental effects on oligodendrocyte survival in immune-mediated demyelinating disorders
MS is the most common neurological disorder affecting young adults, with an estimated incidence of approximately one in 500 individuals. It is generally believed that MS and its animal model, experimental autoimmune encephalomyelitis (EAE), are Th1 T cell-mediated autoimmune diseases 65, 66. The pathological hallmarks of MS and EAE include inflammation, demyelination, oligodendrocyte loss, and axonal degeneration. Evidence is emerging that the UPR is involved in the disease pathogenesis of MS and EAE 10, 11, 67. Gene chip analyses have shown upregulation of the ER stress-responsive genes ATF4 and heat shock protein 70 (HSP70) in MS demyelinated lesions 68, 69. Another recent report has shown elevated expression of CHOP, BIP and XBP-1 in multiple cell types within MS demyelinated lesions, including oligodendrocytes, astrocytes, T cells, and microglia 10. Moreover, elevated levels of p-eIF2α have been observed in oligodendrocytes and infiltrating T cells in the CNS during the course of EAE 70, 71.
Importantly, evidence has suggested that activation of the PERK-eIF2α pathway of the UPR in oligodendrocytes protects against EAE-induced oligodendrocyte death and demyelination 72. Interferon-γ (IFN-γ), a T cell-derived pleiotropic cytokine, is believed to play a crucial role in the pathogenesis of MS and EAE 11, 73. CNS delivery of IFN-γ before EAE onset ameliorated disease severity and prevented EAE-induced oligodendrocyte loss, demyelination, and axonal damage 72. Interestingly, the protective effects of this cytokine are accompanied with the activation of the PERK-eIF2α pathway in oligodendrocytes and are abrogated in PERK deficient animals 72. While evidence has shown that IFN-γ is capable of increasing eIF2α phosphorylation through activation of double-stranded RNA-dependent protein kinase (PKR) 74 and the level of phosphorylated PKR is elevated in oligodendrocytes in the course of EAE 70, we have demonstrated that IFN-γ-induced eIF2α phosphorylation in oligodendrocytes is mediated by PERK and the protective effect of IFN-γ in EAE is dependent on PERK 72, 75. Thus, these data provide strong evidence that the UPR-induced by IFN-γ is involved in the pathogenesis of immune-mediated demyelination diseases.
Remyelination is sufficiently robust to repair myelin damage and restore neurological function in some animal models of CNS demyelination. The remyelination process also occurs in the CNS of MS patients. Nevertheless, this remyelination is generally considered to be insufficient 76, 77. Several reports have shown that IFN-γ is a major cytokine that suppresses oligodendrocyte regeneration and inhibits remyelination in MS and EAE demyelinated lesions 78-80. Interestingly, the detrimental effects of IFN-γ on myelin repair are mediated by ER stress in the remyelinating oligodendrocytes 80. Transgenic mice that ectopically express IFN-γ in the CNS during development display a tremoring phenotype, oligodendrocyte loss, and hypomyelination 75, 81, 82. Activation of the PERK-eIF2α pathway is detected in the myelinating oligodendrocytes of these transgenic mice 75. Additionally, decreased activity of the PERK-eIF2α pathway, via PERK inactivation, exacerbates IFN-γ-induced oligodendrocyte apoptosis and hypomyelination 75; whereas, increased activity of the PERK-eIF2α pathway, via GADD34 inactivation, has the opposite effect 83. Moreover, the detrimental effects of IFN-γ on remyelination in demyelinated lesions are associated with ER stress in remyelinating oligodendrocytes, as evidenced by the upregulation of BIP and CHOP and an elevated level of p-eIF2α 80. Importantly, PERK deficiency significantly exacerbates remyelinating oligodendrocyte apoptosis and remyelination failure induced by IFN-γ in demyelinated lesions 80.
Thus, it appears that ER stress has both beneficial and detrimental effects on oligodendrocyte survival in immune-mediated demyelination diseases. ER stress induction in fully myelinated mature oligodendroytes promotes cell survival, but in actively myelinating/remyelinating oligodendrocytes leads to cell death. Although it is largely unknown how the UPR selectively initiates the apoptotic and adaptive pathways 84, we speculate that the outcomes of ER stress in oligodendrocytes are likely determined by the developmental status of the cells 11, 67, 72. During active myelination, the ER in oligodendrocytes is largely occupied with the production of enormous amounts of myelin proteins and lipids 1, 2. Therefore, its adaptive capacity may be limited at this stage. It is likely that the dramatic upregulation of protein synthesis initiated by the inflammatory response overload the ER of myelinating cells, causing severe ER stress and cell death. In contrast, mature oligodendrocytes in adult mice produce just enough myelin proteins and lipids to maintain myelin structure homeostasis 85. The ER in mature oligodendrocytes may have sufficient adaptive capacity to handle the increased protein load. In this situation, the resulting ER stress would be moderate and would not trigger the apoptotic program. Rather, the adaptive response activated by ER stress could act protectively against subsequent insults.
The unique features of ER stress in myelinating cells
As described in the discussion of myelin disorders above, there is evidence to suggest that myelinating cells respond to ER stress in a manner that is somewhat distinct from that observed in other cell types. For example, CHOP, which is a downstream effecter molecule of the PERK-eIF2α pathway, participates in the induction of the apoptotic response in most cell types, but does not appear to contribute to the UPR-induced apoptotic demise of myelinating cells 5, 7, 30. In fact, activation of CHOP promotes oligodendrocyte survival during the ER stress induced by PLP mutations 7. Moreover, it has been demonstrated that CHOP plays an important role in demyelination in the P0 mutant nerve, but is not involved in Schwann cell apoptosis 5.
Caspase-12, an ER-localized caspase, has been shown to play an important role in apoptosis elicited by ER stress in neurodegenerative diseases 33, 34. While activation of caspase-12 is detected in the oligodendrocytes of PLP mutant mice, caspase-12 deletion does not significantly affect the clinical phenotype, oligodendrocyte apoptosis, or myelin abnormalities in these mice 86. Additionally, the upregulation of ATF3 was shown to occur in the oligodendrocytes of PLP mutant mice, but the deletion of ATF3 also fails to have an effect on oligodendrocyte apoptosis in these animals 87. Taken together, these data indicate that the apoptotic process elicited by ER stress in myelinating cells is unique. A more detailed understanding of these unique features could be critical when considering potential therapeutic approaches for myelin disorders.
Perhaps the considerable demand on the secretory pathway of actively myelinating cells has resulted in an alteration to the typical response to ER stress that other cell types display. A more thorough assessment of the UPR in myelinating cells is essential to better understand the response of these cells to ER stress. One difficulty in the study of the UPR in myelinating cells is that they appear most susceptible to secretory pathway perturbations while actively myelinating axons, diminishing the utility of examining their response to stresses in heterologous systems or in isolation. Therefore, the most informative studies have been carried out in vivo using mouse models. Nevertheless, recent advances in the development of in vitro myelination systems, in combination with siRNA technology, should allow for a more rapid characterization of the UPR in myelinating cells 88.
Therapeutic potential and future directions
Considerable progress has now been made towards a detailed understanding the signaling pathways of the ER stress response. Recent studies indicate that manipulating these signaling pathways has the potential to be therapeutic. Chemical chaperones 4-phenylbutyric acid and taurine-conjugated ursodeoxycholic acid, which reduce the phosphorylation of PERK and IRE1 during ER stress, both significantly improve glucose tolerance and insulin sensitivity in insulin-resistant obese mice 89. Another chemical chaperone, the resveratrol tetramer vaticanol B, which suppresses the expression of CHOP and BIP during ER stress, prevents ER stress-induced apoptosis 90. Additionally, treatment with salburinal, a small-molecule inhibitor of eIF2α dephosphorylation, results in sustained eIF2α phosphorylation, and protects cells from ER stress and viral infection 91.
Currently, there is no effective therapy for patients with disorders of myelinating cells. Therapeutic strategies that facilitate the transport of misfolded proteins out of the ER could be beneficial for these patients. It has been shown that treatment with curcumin promotes mutant PMP22 transport from the ER to the plasma membrane and attenuates Schwann cell apoptosis in PMP22Tr-J mice 45. Moreover, our recent data demonstrate that treatment with salburinal promotes the survival of myelinating oligodendrocyte exposed to IFN-γ, implying that therapeutic strategies that enhance the PERK-eIF2α pathway could promote oligodendrocyte survival in immune-mediated demyelination diseases 83. The manipulation of ER stress for therapeutic purposes, without causing severe side effects, is of course a formidable challenge. Nevertheless, just as actively myelinating cells display increased sensitivity to perturbations to the secretory pathway, these cells may also display selective sensitivity and benefit from modulators of the UPR.
Future studies will need to address the many open questions concerning the physiological significance of the various ER-stress signaling pathways during the myelination process, and the pathological significance of these pathways in myelin disorders. For example, considering the relative prevalence of the disorder, it is critical to know if ER stress is an important participant in the pathogenesis of CMT1 in patients that over-express the membrane proteinPMP22. Moreover, the unique aspects of the UPR in myelinating cells need to be better understood. The knowledge gained from such studies will provide a strong foundation for the design of therapeutic strategies for these diseases. In addition, insights gained from these efforts might prove applicable to neurodegenerative disorders that display the selective vulnerability of specific neuronal subtypes. Neuronal populations with excessive secretory requirements might display increased sensitivity to factors, genetic and environmental, that disrupt ER function 92. Future investigations into this possibility might be enlightening.
Acknowledgements
W.L. is supported by a National Multiple Sclerosis Society Career Transition Fellowship Grant (TA 3026-A-1). B.P. is supported by grants from the National Institutes of Health (NS34939 and 027336) and the Myelin Repair Foundation. We thank Darlene Douglas for critical comments on the manuscript.
References
- 1.Pfeiffer SE, Warrington AE, Bansal R. The oligodendrocyte and its many cellular processes. Trends Cell Biol. 1993;3:191–197. doi: 10.1016/0962-8924(93)90213-k. [DOI] [PubMed] [Google Scholar]
- 2.Anitei M, Pfeiffer SE. Myelin biogenesis: sorting out protein trafficking. Curr. Biol. 2006;16:R418–421. doi: 10.1016/j.cub.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 3.Simons M, Trotter J. Wrapping it up: the cell biology of myelination. Curr. Opin. Neurobiol. 2007;17:533–540. doi: 10.1016/j.conb.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 4.Wrabetz L, et al. Different intracellular pathomechanisms produce diverse Myelin Protein Zero neuropathies in transgenic mice. J. Neurosci. 2006;26:2358–2368. doi: 10.1523/JNEUROSCI.3819-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pennuto M, et al. Ablation of the UPR-mediator CHOP restores motor function and reduces demyelination in Charcot-Marie-Tooth 1B mice. Neuron. 2008;57:393–405. doi: 10.1016/j.neuron.2007.12.021. [This paper shows that the UPR is involved in the demyelination process in Charcot-Marie-Tooth 1B mice.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bauer J, et al. Endoplasmic reticulum stress in PLP-overexpressing transgenic rats: gray matter oligodendrocytes are more vulnerable than white matter oligodendrocytes. J. Neuropathol. Exp. Neurol. 2002;61:12–22. doi: 10.1093/jnen/61.1.12. [DOI] [PubMed] [Google Scholar]
- 7.Southwood CM, Garbern J, Jiang W, Gow A. The unfolded protein response modulates disease severity in Pelizaeus-Merzbacher disease. Neuron. 2002;36:585–596. doi: 10.1016/s0896-6273(02)01045-0. [This paper shows that oligodendrocyte apoptosis in PMD is mediated by the UPR.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van der Voorn JP, et al. The unfolded protein response in vanishing white matter disease. J. Neuropathol. Exp. Neurol. 2005;64:770–775. doi: 10.1097/01.jnen.0000178446.41595.3a. [DOI] [PubMed] [Google Scholar]
- 9.van Kollenburg B, et al. Glia-specific activation of all pathways of the unfolded protein response in vanishing white matter disease. J. Neuropathol. Exp. Neurol. 2006;65:707–715. doi: 10.1097/01.jnen.0000228201.27539.50. [DOI] [PubMed] [Google Scholar]
- 10.Mháille AN, et al. Increased expression of endoplasmic reticulum stress-related signaling pathway molecules in multiple sclerosis lesions. J. Neuropathol. Exp. Neurol. 2008;67:200–211. doi: 10.1097/NEN.0b013e318165b239. [This is the first study to show elevated levels of ER stress markers in MS demyelinated lesions.] [DOI] [PubMed] [Google Scholar]
- 11.Lees JR, Cross AH. A little stress is good: IFN-gamma, demyelination, and multiple sclerosis. J. Clin. Invest. 2007;117:297–2999. doi: 10.1172/JCI31254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 1999;13:1211–1233. doi: 10.1101/gad.13.10.1211. [DOI] [PubMed] [Google Scholar]
- 13.Schröder M. Endoplasmic reticulum stress responses. Cell Mol. Life Sci. 2008;65:862–894. doi: 10.1007/s00018-007-7383-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma Y, Hendershot LM. The unfolding tale of the unfolded protein response. Cell. 2001;107:827–830. doi: 10.1016/s0092-8674(01)00623-7. [DOI] [PubMed] [Google Scholar]
- 15.Rutkowski DT, Kaufman RJ. A trip to the ER: coping with stress. Trends Cell Biol. 2004;14:20–28. doi: 10.1016/j.tcb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 16.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 17.Ron D, Harding H. PERK and translational control by stress in the endoplasmic reticulum. In: Hershey J, Mathews M, Sonenberg N, editors. Translational Control. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 2000. pp. 547–560. [Google Scholar]
- 18.Schröder M, Kaufman RJ. Divergent roles of IRE1alpha and PERK in the unfolded protein response. Curr. Mol. Med. 2006;6:5–36. doi: 10.2174/156652406775574569. [DOI] [PubMed] [Google Scholar]
- 19.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 20.Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell. 2000;5:897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- 21.Harding HP, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 22.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 23.Calfon M, et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 24.Ye J, et al. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell. 2000;6:1355–1364. doi: 10.1016/s1097-2765(00)00133-7. [DOI] [PubMed] [Google Scholar]
- 25.Marciniak SJ, Ron D. Endoplasmic reticulum stress signaling in disease. Physiol. Rev. 2006;86:1133–1149. doi: 10.1152/physrev.00015.2006. [DOI] [PubMed] [Google Scholar]
- 26.Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7:880–885. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Faitova J, Krekac D, Hrstka R, Vojtesek B. Endoplasmic reticulum stress and apoptosis. Cell Mol. Biol. Lett. 2006;11:488–505. doi: 10.2478/s11658-006-0040-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Proud CG. eIF2 and the control of cell physiology. Semin. Cell Dev. Biol. 2005;16:3–12. doi: 10.1016/j.semcdb.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 29.Rutkowski DT, Kaufman RJ. All roads lead to ATF4. Dev. Cell. 2003;4:442–444. doi: 10.1016/s1534-5807(03)00100-x. [DOI] [PubMed] [Google Scholar]
- 30.Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- 31.Marciniak SJ, et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18:3066–3077. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J. Cell Biol. 2001;153:1011–1022. doi: 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Szegezdi E, Fitzgerald U, Samali A. Caspase-12 and ER-stress-mediated apoptosis: the story so far. Ann. NY Acad. Sci. 2003;1010:186–194. doi: 10.1196/annals.1299.032. [DOI] [PubMed] [Google Scholar]
- 34.Nakagawa T, et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 35.Scheuner D, Kaufman RJ. The unfolded protein response: a pathway that links insulin demand with beta-cell failure and diabetes. Endocr. Rev. 2008;29:317–333. doi: 10.1210/er.2007-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ji C. Dissection of endoplasmic reticulum stress signaling in alcoholic and non-alcoholic liver injury. J. Gastroenterol. Hepatol. 2008;23:S16–24. doi: 10.1111/j.1440-1746.2007.05276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Todd DJ, Lee AH, Glimcher LH. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat. Rev. Immunol. 2008;8:663–674. doi: 10.1038/nri2359. [DOI] [PubMed] [Google Scholar]
- 38.Skre H. Genetic and clinical aspects of Charcot-Marie-Tooth's disease. Clin. Genet. 1974;6:98–118. doi: 10.1111/j.1399-0004.1974.tb00638.x. [DOI] [PubMed] [Google Scholar]
- 39.Berger P, Niemann A, Suter U. Schwann cells and the pathogenesis of inherited motor and sensory neuropathies (Charcot-Marie-Tooth disease). Glia. 2006;54:243–257. doi: 10.1002/glia.20386. [DOI] [PubMed] [Google Scholar]
- 40.Lupski JR, Chance PF. Hereditary motor and sensory neuropathies involving altered dosage or mutation of PMP22: the CMT1A duplication and HNPP deletion. In: Dyck PJ, Thomas PK, editors. Peripheral neuropathy. Elsevier Saunders; Philadelphia: 2005. pp. 1659–1680. [Google Scholar]
- 41.Sereda MW, Nave KA. Animal models of Charcot-Marie-Tooth disease type 1A. Neuromolecular Med. 2006;8:205–216. doi: 10.1385/nmm:8:1-2:205. [DOI] [PubMed] [Google Scholar]
- 42.Snipes GJ, Orfali W, Fraser A, Dickson K, Colby J. The anatomy and cell biology of peripheral myelin protein-22. Ann. NY Acad. Sci. 1999;883:143–151. doi: 10.1111/j.1749-6632.1999.tb08577.x. [DOI] [PubMed] [Google Scholar]
- 43.Colby J, et al. PMP22 carrying the trembler or trembler-J mutation is intracellularly retained in myelinating Schwann cells. Neurobiol. Dis. 2000;7:561–573. doi: 10.1006/nbdi.2000.0323. [DOI] [PubMed] [Google Scholar]
- 44.Dickson KM, et al. Association of calnexin with mutant peripheral myelin protein-22 ex vivo: a basis for “gain-of-function” ER diseases. Proc. Natl. Acad. Sci. USA. 2002;99:9852–9857. doi: 10.1073/pnas.152621799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khajavi M, et al. Oral curcumin mitigates the clinical and neuropathologic phenotype of the Trembler-J mouse: a potential therapy for inherited neuropathy. Am. J. Hum. Genet. 2007;81:438–453. doi: 10.1086/519926. [This report demonstrates the therapeutic benefit of facilitated ER export in a CMT animal model.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garbern JY. Pelizaeus-Merzbacher disease: Genetic and cellular pathogenesis. Cell Mol. Life Sci. 2007;64:50–65. doi: 10.1007/s00018-006-6182-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Southwood C, Gow A. Molecular pathways of oligodendrocyte apoptosis revealed by mutations in the proteolipid protein gene. Microsc. Res. Tech. 2001;52:700–708. doi: 10.1002/jemt.1054. [DOI] [PubMed] [Google Scholar]
- 48.Gow A, Lazzarini RA. A cellular mechanism governing the severity of Pelizaeus-Merzbacher disease. Nat. Genet. 1996;13:422–428. doi: 10.1038/ng0896-422. [DOI] [PubMed] [Google Scholar]
- 49.Gow A, Southwood CM, Lazzarini RA. Disrupted proteolipid protein trafficking results in oligodendrocyte apoptosis in an animal model of Pelizaeus-Merzbacher disease. J. Cell Biol. 1998;140:925–934. doi: 10.1083/jcb.140.4.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Swanton E, Holland A, High S, Woodman P. Disease-associated mutations cause premature oligomerization of myelin proteolipid protein in the endoplasmic reticulum. Proc Natl Acad Sci USA. 2005;102:4342–4347. doi: 10.1073/pnas.0407287102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dhaunchak AS, Nave KA. A common mechanism of PLP/DM20 misfolding causes cysteine-mediated endoplasmic reticulum retention in oligodendrocytes and Pelizaeus-Merzbacher disease. Proc. Natl. Acad. Sci. USA. 2007;104:17813–17818. doi: 10.1073/pnas.0704975104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Koeppen AH, Barron KD, Csiza CK, Greenfield EA. Comparative immunocytochemistry of Pelizaeus-Merzbacher disease, the jimpy mouse, and the myelin-deficient rat. J. Neurol. Sci. 1988;84:315–327. doi: 10.1016/0022-510x(88)90135-9. [DOI] [PubMed] [Google Scholar]
- 53.Roussel G, Neskovic NM, Trifilieff E, Artault JC, Nussbaum JL. Arrest of proteolipid transport through the Golgi apparatus in Jimpy brain. J. Neurocytol. 1987;16:195–204. doi: 10.1007/BF01795303. [DOI] [PubMed] [Google Scholar]
- 54.McLaughlin M, et al. Genetic background influences UPR but not PLP processing in the rumpshaker model of PMD/SPG2. Neurochem. Res. 2007;32:167–176. doi: 10.1007/s11064-006-9122-y. [DOI] [PubMed] [Google Scholar]
- 55.Schiffmann R, Elroy-Stein O. Childhood ataxia with CNS hypomyelination/vanishing white matter disease--a common leukodystrophy caused by abnormal control of protein synthesis. Mol. Genet. Metab. 2006;88:7–15. doi: 10.1016/j.ymgme.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 56.Leegwater PA, et al. Subunits of the translation initiation factor eIF2B are mutant in leukoencephalopathy with vanishing white matter. Nat. Genet. 2001;29:383–8. doi: 10.1038/ng764. [DOI] [PubMed] [Google Scholar]
- 57.Mohammad-Qureshi SS, Jennings MD, Pavitt GD. Clues to the mechanism of action of eIF2B, the guanine-nucleotide-exchange factor for translation initiation. Biochem. Soc. Trans. 2008;36:658–664. doi: 10.1042/BST0360658. [DOI] [PubMed] [Google Scholar]
- 58.van der Knaap MS, Pronk JC, Scheper GC. Vanishing white matter disease. Lancet Neurol. 2006;5:413–423. doi: 10.1016/S1474-4422(06)70440-9. [DOI] [PubMed] [Google Scholar]
- 59.Wong K, et al. Foamy cells with oligodendroglial phenotype in childhood ataxia with diffuse central nervous system hypomyelination syndrome. Acta. Neuropathol. 2000;100:635–646. doi: 10.1007/s004010000234. [DOI] [PubMed] [Google Scholar]
- 60.Van Haren K, et al. The life and death of oligodendrocytes in vanishing white matter disease. J. Neuropathol. Exp. Neurol. 2004;63:618–630. doi: 10.1093/jnen/63.6.618. [DOI] [PubMed] [Google Scholar]
- 61.van der Voorn JP, et al. The unfolded protein response in vanishing white matter disease. J. Neuropathol. Exp. Neurol. 2005;64:770–775. doi: 10.1097/01.jnen.0000178446.41595.3a. [DOI] [PubMed] [Google Scholar]
- 62.van Kollenburg B, et al. Glia-specific activation of all pathways of the unfolded protein response in vanishing white matter disease. J. Neuropathol. Exp. Neurol. 2006;65:707–715. doi: 10.1097/01.jnen.0000228201.27539.50. [DOI] [PubMed] [Google Scholar]
- 63.Kantor L, et al. Heightened stress response in primary fibroblasts expressing mutant eIF2B genes from CACH/VWM leukodystrophy patients. Hum. Genet. 2005;118:99–106. doi: 10.1007/s00439-005-0024-x. [DOI] [PubMed] [Google Scholar]
- 64.Kantor L, et al. A point mutation in translation initiation factor 2B leads to a continuous hyper stress state in oligodendroglial-derived cells. PLoS ONE. 2008;3:e3783. doi: 10.1371/journal.pone.0003783. [This paper provides evidence that the UPR adapts oligodendrocytes to eIF2B mutations in Vanishing White Matter Disease] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Frohman EM, Racke MK, Raine CS. Multiple sclerosis--the plaque and its pathogenesis. N. Engl. J. Med. 2006;354:942–955. doi: 10.1056/NEJMra052130. [DOI] [PubMed] [Google Scholar]
- 66.Trapp BD, Nave KA. Multiple sclerosis: an immune or neurodegenerative disorder? Annu. Rev. Neurosci. 2008;31:247–69. doi: 10.1146/annurev.neuro.30.051606.094313. [DOI] [PubMed] [Google Scholar]
- 67.Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454:455–462. doi: 10.1038/nature07203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mycko MP, Papoian R, Boschert U, Raine CS, Selmaj KW. Microarray gene expression profiling of chronic active and inactive lesions in multiple sclerosis. Clin. Neurol. Neurosurg. 2004;106:223–229. doi: 10.1016/j.clineuro.2004.02.019. [DOI] [PubMed] [Google Scholar]
- 69.Cwiklinska H, et al. Heat shock protein 70 associations with myelin basic protein and proteolipid protein in multiple sclerosis brains. Int. Immunol. 2003;15:241–249. doi: 10.1093/intimm/dxg022. [DOI] [PubMed] [Google Scholar]
- 70.Chakrabarty A, Danley MM, LeVine SM. Immunohistochemical localization of phosphorylated protein kinase R and phosphorylated eukaryotic initiation factor-2 alpha in the central nervous system of SJL mice with experimental allergic encephalomyelitis. J. Neurosci. Res. 2004;76:822–833. doi: 10.1002/jnr.20125. [DOI] [PubMed] [Google Scholar]
- 71.Chakrabarty A, Fleming KK, Marquis JG, LeVine SM. Quantifying immunohistochemical staining of phospho-eIF2alpha, heme oxygenase-2 and NADPH cytochrome P450 reductase in oligodendrocytes during experimental autoimmune encephalomyelitis. J. Neurosci. Methods. 2005;144:227–234. doi: 10.1016/j.jneumeth.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 72.Lin W, et al. The integrated stress response prevents demyelination by protecting oligodendrocytes against immune-mediated damage. J. Clin. Invest. 2007;117:448–456. doi: 10.1172/JCI29571. [This paper shows the beneficial effects of the UPR in an immune-mediated demyelinating disease.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Imitola J, Chitnis T, Khoury SJ. Cytokines in multiple sclerosis: from bench to bedside. Pharmacol. Ther. 2005;106:163–177. doi: 10.1016/j.pharmthera.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 74.Su Q, et al. Interferons induce tyrosine phosphorylation of the eIF2alpha kinase PKR through activation of Jak1 and Tyk2. EMBO Rep. 2007;8:265–270. doi: 10.1038/sj.embor.7400891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lin W, Harding HP, Ron D, Popko B. Endoplasmic reticulum stress modulates the response of myelinating oligodendrocytes to the immune cytokine interferon-gamma. J. Cell Biol. 2005;169:603–612. doi: 10.1083/jcb.200502086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bruck W, Kuhlmann T, Stadelmann C. Remyelination in multiple sclerosis. J. Neurol. Sci. 2003;206:181–185. doi: 10.1016/s0022-510x(02)00191-0. [DOI] [PubMed] [Google Scholar]
- 77.Franklin RJ. Why does remyelination fail in multiple sclerosis? Nat. Rev. Neurosci. 2002;3:705–714. doi: 10.1038/nrn917. [DOI] [PubMed] [Google Scholar]
- 78.Panitch HS, Hirsch RL, Schindler J, Johnson KP. Treatment of multiple sclerosis with gamma interferon: exacerbations associated with activation of the immune system. Neurology. 1987;37:1097–1102. doi: 10.1212/wnl.37.7.1097. [DOI] [PubMed] [Google Scholar]
- 79.Renno T, et al. Interferon-gamma in progression to chronic demyelination and neurological deficit following acute EAE. Mol. Cell. Neurosci. 1998;12:376–389. doi: 10.1006/mcne.1998.0725. [DOI] [PubMed] [Google Scholar]
- 80.Lin W, et al. Interferon-gamma inhibits central nervous system remyelination through a process modulated by endoplasmic reticulum stress. Brain. 2006;129:1306–1318. doi: 10.1093/brain/awl044. [This paper show ER stress induction in actively remyelinating oligodendrocytes leads to cell death and remyelination failure in an immune-mediated demyelinating diseases.] [DOI] [PubMed] [Google Scholar]
- 81.Corbin JG, et al. Targeted CNS expression of interferon-gamma in transgenic mice leads to hypomyelination, reactive gliosis, and abnormal cerebellar development. Mol. Cell. Neurosci. 1996;7:354–370. doi: 10.1006/mcne.1996.0026. [DOI] [PubMed] [Google Scholar]
- 82.LaFerla FM, Sugarman MC, Lane TE, Leissring MA. Regional hypomyelination and dysplasia in transgenic mice with astrocyte-directed expression of interferon-gamma. J. Mol. Neurosci. 2000;15:45–59. doi: 10.1385/JMN:15:1:45. [DOI] [PubMed] [Google Scholar]
- 83.Lin W, et al. Enhanced Integrated Stress Response Promotes Myelinating Oligodendrocyte Survival in Response to Interferon-{gamma} Am. J. Pathol. 2008;173:1508–1517. doi: 10.2353/ajpath.2008.080449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Boyce M, Yuan J. Cellular response to endoplasmic reticulum stress: a matter of life or death. Cell Death Differ. 2006;13:363–373. doi: 10.1038/sj.cdd.4401817. [DOI] [PubMed] [Google Scholar]
- 85.Morell P, Quarles RH. Myelin formation, structure and biochemistry. In: Siegel GJ, Agranoff BW, Albers RW, Fisher SK, Uhler MD, editors. Basic Neurochemistry: Molecular, Cellular, and Medical Aspects. Lippincott-Raven Publishers; Philadelphia: 1999. pp. 69–93. [Google Scholar]
- 86.Sharma R, Gow A. Minimal role for caspase 12 in the unfolded protein response in oligodendrocytes in vivo. J. Neurochem. 2007;101:889–897. doi: 10.1111/j.1471-4159.2007.04541.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sharma R, Jiang H, Zhong L, Tseng J, Gow A. Minimal role for activating transcription factor 3 in the oligodendrocyte unfolded protein response in vivo. J. Neurochem. 2007;102:1703–1712. doi: 10.1111/j.1471-4159.2007.04646.x. [DOI] [PubMed] [Google Scholar]
- 88.Watkins TA, Emery B, Mulinyawe S, Barres BA. Distinct stages of myelination regulated by gamma-secretase and astrocytes in a rapidly myelinating CNS coculture system. Neuron. 2008;60:555–569. doi: 10.1016/j.neuron.2008.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ozcan U, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Takano K, et al. A dibenzoylmethane derivative protects dopaminergic neurons against both oxidative stress and endoplasmic reticulum stress. Am. J. Physiol. Cell Physiol. 2007;293:C1884–1894. doi: 10.1152/ajpcell.00305.2007. [DOI] [PubMed] [Google Scholar]
- 91.Boyce M, et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science. 2005;307:935–939. doi: 10.1126/science.1101902. [This paper demonstrates the potential of small molecules to protect cells from ER stress.] [DOI] [PubMed] [Google Scholar]
- 92.Lindholm D, Wootz H, Korhonen L. ER stress and neurodegenerative diseases. Cell Death Differ. 2006;13:385–392. doi: 10.1038/sj.cdd.4401778. [DOI] [PubMed] [Google Scholar]