

Abstract
Axonal degeneration is a key component of a variety of neurological diseases. Studies using wlds mutant mice have demonstrated that delaying axonal degeneration slows disease course and prolongs survival in neurodegenerative disease models. The Wlds protein is normally localized to the nucleus, and contains the N terminus of ubiquitination factor Ube4b fused to full-length Nmnat1, an NAD biosynthetic enzyme. While Nmnat enzymatic activity is necessary for Wlds-mediated axonal protection, several important questions remain including whether the Ube4b component of Wlds also plays a role, and in which cellular compartment (nucleus vs cytosol) the axonal protective effects of Nmnat activity are mediated. While Nmnat alone is clearly sufficient to delay axonal degeneration in cultured neurons, we sought to determine whether it was also sufficient to promote axonal protection in vivo. Using cytNmnat1, an engineered mutant of Nmnat1 localized only to the cytoplasm and axon, that provides more potent axonal protection than that afforded by Wlds or Nmnat1, we generated transgenic mice using the prion protein promoter (PrP). The sciatic nerve of these cytNmnat1 transgenic mice was transected, and microscopic analysis of the distal nerve segment 7 d later revealed no evidence of axonal loss or myelin debris, indicating that Nmnat alone, without any other Wlds sequences, is all that is required to delay axonal degeneration in vivo. These results highlight the importance of understanding the mechanism of Nmnat-mediated axonal protection for the development of new treatment strategies for neurological disorders.
Introduction
Axonal degeneration is involved in many neurological disorders and often precedes neuronal cell death (Raff et al., 2002; Coleman, 2005). Insight into the axonal degeneration process has been gained through studies of Wallerian degeneration slow (wlds) mice in which axonal degeneration after injury is delayed (Lunn et al., 1989; Perry et al., 1991). The wlds mutation results in the overexpression of a fusion protein (Wlds) composed of N-terminal 70 aa of Ube4b linked to full-length Nmnat1, an enzyme that catalyzes the formation of NAD from the substrate NMN (Coleman et al., 1998). This Wlds fusion protein protects axons from degeneration initiated by a variety of insults both in vitro and in vivo (Mack et al., 2001; Conforti et al., 2007).
The molecular mechanism of axonal protection by Wlds was initially investigated using an in vitro DRG culture system, where overexpression of the Nmnat1 portion of Wlds alone was sufficient to prevent axonal degeneration after injury (Araki et al., 2004). Nmnat1 or Wlds mutants lacking NAD synthetic capability do not delay axonal degeneration, indicating that Nmnat enzymatic activity is essential for axonal protection (Araki et al., 2004; Jia et al., 2007). Further evidence that Nmnat1 is the key component for Wlds-mediated axonal protection was obtained from experiments showing that Nmnat enzymes localized to multiple cellular compartments or derived from species ranging from Archaebacterium Methanococcus jannaschii to Saccharomyces cerevisiae to Mus musculus all provide robust axonal protection (Sasaki et al., 2006; Press and Milbrandt, 2008; Sasaki et al., 2009). In apparent contrast to these findings, one study in Drosophila found that Drosophila melanogaster Nmnat mutants with diminished enzymatic activity prevent neuronal death through a chaperone-like mechanism (Zhai et al., 2008).
The axonal protective activity of Nmnat1 or its product NAD has been reproduced in a number of laboratories (Wang et al., 2005; MacDonald et al., 2006); however, others have reported that Nmnat1 is ineffective or less protective than Wlds protein (Conforti et al., 2007; Watanabe et al., 2007). The conflicting results obtained in these studies regarding Wlds- versus Nmnat1-mediated protection could emanate from differences in the cellular distribution of these proteins, or perhaps, from differing levels of transgene expression. Interestingly, studies of Wlds transgenic mice revealed a clear gene dosage effect, as axons in mice with high levels of Wlds expression showed maximum axonal protection while those with low Wlds levels showed minimal axonal protection after sciatic nerve transection (Mack et al., 2001). Most importantly, these authors found that similarly generated transgenic mice expressing Nmnat1 alone showed no evidence of axonal protection (Conforti et al., 2007), giving rise to the idea that non-Nmnat1 portions of the Wlds are crucial for delayed axonal degeneration.
In this study, we designed experiments to address these apparently conflicting results to better define the molecular mechanism of Wlds-mediated axonal protection. We first performed a rigorous comparison of axonal protection mediated by different Nmnat/Wlds proteins in vitro, and found the most potent protection from cytNmnat1, an Nmnat1 mutant localized to the cytosolic and axonal compartments. Next we generated transgenic mice overexpressing the most potent axonal protectant, cytNmnat1, in neurons via the prion protein promoter. We found that both axonal degeneration and functional decline in axonal and synaptic physiology were dramatically delayed in cytNmnat1 transgenic mice after sciatic nerve transection, demonstrating that Nmnat1 is the active axonal protective moiety of the Wlds fusion protein in vivo.
Materials and Methods
Plasmids and generation of transgenic animals.
Plasmids encoding 6xHis epitope-tagged mouse Nmnat1, Wlds, and cytNmnat1 were described previously (Araki et al., 2004; Sasaki et al., 2006). The epitope-tagged cytNmnat1 cDNA was subcloned into the prion protein promoter (PrP)-based transgenic vector MoPrP.Xho (ATCC; JHU-2). Pronuclear injection of the vector to produce cytNmnat1 transgenic mice was performed using C57BL/CBA F1 strain in the Mouse Genetics Core at Washington University. The seven founders identified by PCR genotyping were mated with C57BL/6 mice and four of them transmitted the transgene through the germline and produced progeny that expressed the cytNmnat1 transgene. All experiments were conducted using mice derived from these four lines in which the founders were mated (F2).
DRG culture and in vitro axotomy.
DRG drop cultures were performed based on a previously described method (Sasaki et al., 2009). Mouse DRGs were collected from embryonic day 13.5 CD1 or transgenic mice expressing cytNmnat1 embryos. The DRGs from 5 embryos (∼200 total DRGs) from CD1 mice were dissociated and resuspended in 50 μl of Neurobasal media (Invitrogen) containing 2% B27 (Invitrogen) and 50 ng/ml NGF (2.5S; Harlan Bioproducts) per dissected embryo. For transgenic DRG experiments, DRGs were collected separately from each embryo (transgenic or littermate wild type), dissociated, and resuspended in 18 μl of media. The suspended DRG neurons (2 μl) were placed as a drop in the center of the well (24-well plate) and incubated at 37°C with 5% CO2 for 15 min. After cell attachment, 500 μl of Neurobasal media containing 2% B27, 50 ng/ml NGF, 1 μm 5-fluoro-2′-deoxyuridine (Sigma), and 1 μm uridine (Sigma) was added to each well. Axotomy was performed at 14 DIV (days in vitro) by transecting axons away from the cell body cluster with a microscalpel. For vincristine-induced axonal degeneration assay, 14 DIV cultures were subjected to 4 μm vincristine treatment. Axonal degeneration was monitored for 3 d and assessed using the ImageJ (NIH) based programs described previously (Sasaki et al., 2009). Cultures from 10 transgenic and 10 wild-type embryos derived from three independent litters were analyzed.
Adenovirus and lentivirus production and infection.
Adenoviruses expressing Wlds, Nmnat1, cytNmnat1, or EGFP were prepared as previously described (Le et al., 2005). For adenovirus infection of DRG neurons, 103 to 107 CFU were added per well. The virus-containing medium was replaced with fresh media after 24 h of the addition of viruses. Lentiviruses expressing Nmnat1 or cytNmnat1 were prepared and used as described previously (Sasaki et al., 2006).
Quantification of relative protein levels in adenovirus infected DRG neurons.
The viral dose–response experiments required an evaluation of protein expression levels over a wide range. To achieve this goal, we used an adenovirus expression system with dual CMV promoters; one to drive an EGFP reporter and the other to independently drive expression of the gene of interest (e.g., Nmnat1). In this system, the fluorescent reporter expression is well correlated with the expression level of the gene of interest (Mitsuuchi et al., 2006). To confirm this tight correlation, we examined the relationship between EGFP fluorescence intensity (FI) and expression of each of the Nmnat/Wlds proteins we studied. First, we compared the expression level of the protein (e.g., Nmnat1, Wlds, or cytNmnat1) and the fluorescence intensity of EGFP (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). We infected HEK293T cells (1 × 105 in 24-well) with different amounts of adenovirus expressing either Nmnat1, Wlds, or cytNmnat1. Three days after infection, the fluorescence intensity from each well was determined using an inverted microscope with 10× objective lens and constant emission light intensity obtained using a metal halide lamp (X-cite 120; EXFO) with a 0.5 s exposure time. The images were captured using a CCD camera (Cool SNAP ES; Nikon) and fluorescence intensity was calculated as the total summation of pixel intensity (after background subtraction) using MetaMorph software (Molecular Devices).
To assess transgene protein expression, lysates were prepared from each 24-well and centrifuged at 10,000 × g for 10 min. The supernatants (10 μl) were diluted 10-fold with PBS and applied to a slot blot manifold (Minifold II, Schleiger). The filter was incubated with 6xHis antibody (R&D Systems) to detect tagged Nmnat or Wlds proteins, the signals were visualized by chemiluminescence (EpiChemiII, Bioimaging Systems) and quantified using ImageJ (NIH). This signal was normalized to total protein amount (P) determined by Ponceau S staining of the same blot.
As shown in supplemental Figure S1 (available at www.jneurosci.org as supplemental material), the relationship between average EGFP fluorescence intensity and Nmnat/Wlds protein levels was linear (r2 = 0.92 for Nmnat1, 0.99 for Wlds, and 0.98 for cytNmnat1) (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). However, a given fluorescence intensity corresponded to a different absolute protein expression level for each protein (e.g., Nmnat1, Wlds, and cytNmnat1), presumably due to slight differences in the synthesis and/or degradation of the individual proteins. This is clearly demonstrated by comparing the plots of fluorescence intensity versus Nmnat1 levels with that of fluorescence intensity versus Wlds levels (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). For example, while a fluorescence intensity of 1 × 107 corresponds to 1.0 arbitrary unit (a.u.) of Nmnat1, this intensity corresponds to only 0.6 a.u. of Wlds. Values were plotted of multiple infections using different amounts of virus, and the slope of the P versus FI for each construct was determined. These equations were then used to calculate the protein concentration of each experiment. The equations used were as follows: P = FI(4 × 10−8) + 0.65 for Nmnat1, P = FI(2 × 10−8) + 0.41 for Wlds, and P = FI(8 × 10−8) + 0.20 for cytNmnat1. For dosage experiments using DRG neurons, the average fluorescence intensity was determined using three fields per well. Through this approach, we used fluorescence intensity to derive the relative expression from the Nmnat1/Wlds transgenes.
Subcellular localization studies.
DRG neurons infected with lentivirus expressing Nmnat1 or cytNmnat1 were examined by immunofluorescence to determine the subcellular distribution of these proteins. Five days after infection, the neurons were fixed with 4% paraformaldehyde, permeabilized with PBS containing 0.1% Tween 20 (PBS-T), and incubated with PBS-T containing 5% BSA for 1 h. The neurons were incubated with 6xHis tag antibody (1:1000, R&D Systems) diluted in PBS-T containing 5% BSA for 16 h at 4°C. After washing in PBS-T, the neurons were incubated with Alexa Fluor 594-conjugated secondary antibody (Invitrogen) for 1 h, and the proteins were visualized by fluorescence microscopy (Nikon).
Immunohistochemistry.
Animals were perfused with 4% paraformaldehyde and the brain and DRGs were dissected. The tissues were incubated in 4% paraformaldehyde for 16 h at 4°C and embedded in O.C.T. compound (Tissue-Tek). Sections (25 μm) were cut from the block with a cryostat and postfixed with 4% paraformaldehyde on the glass slides, permeabilized with PBS-T, and incubated with PBS-T containing 5% BSA for 1 h. Sections were then incubated with 6xHis tag antibody (1:500, R&D Systems) diluted in PBS-T containing 5% BSA for 16 h at 4°C. After washing in PBS-T, the sections were incubated with Alexa Fluor 594-conjugated secondary antibody (1:1000, Invitrogen) for 1 h and cytNmnat1 proteins were visualized by fluorescence microscopy (Nikon).
Surgical procedures and histology.
Mice were anesthetized by intraperitoneal injection of 2-2-2 tribromoethanol at a dose of 500 mg/kg. The sciatic nerve was exposed at the hip and transected using a small scissors. The distal stump was deflected onto an adjacent muscle to prevent regeneration. Nerve lesions were produced on the right side and the contralateral nerve was left intact and served as the uninjured control. To analyze the nerve structure, the distal segments of the sciatic nerves (injured and control) were fixed with 4% paraformaldehyde for 16 h, rinsed with PBS, and incubated with 1% osmium tetroxide, 1.5% potassium ferricyanide, and 0.5 m phosphate buffer, pH 7.0, for 1 h at 25°C. The nerves were embedded in paraffin and tissue sections (4 μm) were prepared using a microtome (Leica RM2135) and stained with toluidine blue. For the cytoskeletal protein analysis, distal sciatic nerve segments as well as uninjured control nerves were harvested 7 d after sciatic nerve transection and homogenized in 10 mm phosphate buffer, pH 8.0, containing 0.3 m NaCl, and protease inhibitors and analyzed by Western blotting using anti-Neurofilament 165 kDa component (1:1000, DSHB; 2H3). Equal amounts of protein loading were confirmed by Ponceau S staining of the blot. Electron microscopy was performed using thin sections stained with uranyl acetate and lead citrate, and photographed with a JEOL 1200 electron microscope as described previously (Ryu et al., 2007). g-ratios were calculated by dividing the axon diameter by the total diameter of the myelinated axons as described before (Ryu et al., 2007). One hundred g-ratios of myelinated axons over 2 μm total diameter from two digital electron micrographs of each genotype were used to calculate an average g-ratio.
Nmnat enzymatic assay from the brain lysate.
Brain hemispheres were dissected from the mice and homogenized in 2 ml of 10 mm HEPES, pH 7.4, 0.3 m NaCl, and protease inhibitors (Roche). Nmnat enzymatic activity was measured by incubating 25 μg of brain lysate at 37°C for 1 h in buffer containing 10 mm Tris-HCl, pH 8.0, 0.2 m KCl, 1 mm MgCl2, 1 mm ATP, and 1 mm NMN. The reactions were stopped by addition of an equal volume of 1 m HClO4, the reactants were spun at 14,000 × g in a microcentrifuge, and neutralized with K2CO3. The neutralized solution was centrifuged for 10 min, the supernatant was mixed with an equal volume of phosphate buffer, pH 7.0, and analyzed by HPLC using LC-18T reverse-phase column at a flow rate of 1 ml/min. The NAD concentration was determined by measuring the absorbance at 254 nm compared with NAD standards (obtained from Sigma).
Nerve electrophysiology.
Compound motor action potential (CMAP) measurements were performed as previously described (Baloh et al., 2009). The tibial nerve was stimulated at the ankle (distal to the transection site), and recording electrodes were placed in the foot. CMAP measurements were performed on the side of sciatic nerve transection at 0, 2, 4, and 7 d after axotomy.
Results
Cytoplasmic Nmnat1 (cytNmnat1) mutant protein provides robust axonal protection
In transgenic mice overexpressing Wlds protein, the extent of axonal protection is directly correlated with transgene expression level, with complete loss of protection occurring at a level ∼20% of that present in wlds homozygote mice (Mack et al., 2001). Thus, the varying results obtained with respect to Nmnat versus Wlds-mediated axonal protection (Araki et al., 2004; Wang et al., 2005; MacDonald et al., 2006; Conforti et al., 2007), may reflect different levels of Nmnat1 and Wlds expression. Alternatively, they may result from different threshold amounts required for protection, different culture conditions, or from the semiquantitative nature of most axonal degeneration assays.
To address the genesis of these discrepant results and directly determine whether Wlds and Nmnat proteins had different levels of axonal protective activity, we used a quantitative, automated axonal degeneration assay that we recently developed (Sasaki et al., 2009). An adenovirus system in which the levels of Nmnat/Wlds protein could be defined over a wide range using by measuring fluorescence intensity of a coexpressed marker (see Materials and Methods) was also used. DRG neurons were infected with varying amounts of adenovirus (103 to 107 CFU per well) expressing Nmnat1, Wlds, cytNmnat1 (a cytoplasmic Nmnat1 mutant), or EGFP. Three days after infection, the axons were transected and, after another 3 d, axonal degeneration was assessed using quantitative image analysis (Sasaki et al., 2009).
We found that at high protein levels, Nmnat1 (16 a.u. of protein), Wlds (4.4 a.u.), and cytNmnat1 (15 a.u.) all provided significant axonal protection, with the degeneration index (DI) remaining in the range observed before axotomy (i.e., axonal degeneration after axotomy was delayed) (Fig. 1B). No protection was provided by adenovirus expressing EGFP alone (Fig. 1A). Interestingly, at a lower dose of adenovirus (106 CFU), where transgene expression was decreased ∼10-fold (Nmnat1: 1.6 a.u., Wlds: 0.4 a.u., cytNmnat1: 1.3 a.u.), only cytNmnat1 showed significant axonal protection (Fig. 1A,B).
Figure 1.

Cytoplasmic Nmnat1 (cytNmnat1) provides more robust axonal protection than Wlds protein or wild-type Nmnat1. A, Representative images of EGFP fluorescence from the DRG cell bodies and phase-contrast images of axons at 0 and 72 h after transection are shown. DRG neurons were infected with relatively low amounts of adenovirus (106 CFU per well) expressing the indicated proteins. Only cytNmnat1-expressing neurons have intact axons 3 d after axotomy. B, The axonal degeneration indices (±SD) were plotted against time after axotomy. The number indicated to the right of each plot is the relative expression level of each protein calculated from EGFP fluorescent images (see Materials and Methods). Note that axons expressing cytNmnat1 were protected at lower expression levels than those expressing Nmnat1 or Wlds. C–E, To determine a wider dose–response curve, DRG neurons were infected with the indicated amount of adenovirus (103 to 107 CFU per well) and analyzed as above. The relative transgene expression was plotted against the axonal degeneration index 3 d after axotomy. DRG cultures from three independent litters of mice (17–19 wells total) were analyzed for each protein. Axons from Nmnat1- and Wlds-expressing DRG neurons remained intact at relative transgene expression levels above 16 a.u. and 1.6 a.u., whereas axons from cytNmnat1-expressing DRG neurons showed no axonal degeneration until transgene expression levels dropped below 0.3 a.u. Importantly, at expression levels between 0.3 and 1 a.u., axons from cytNmnat1-expressing neurons were significantly more intact than those of Nmnat1-, or Wlds-expressing DRGs (p < 0.01 by ANOVA and Tukey's post hoc test). Note that DI > 0.2 corresponds to visibly degenerated axons. Scale bar, 100 μm.
This difference in potency encouraged us to conduct experiments using a wider range of Nmnat/Wlds protein levels to examine the dose-dependent nature of Nmnat-mediated axonal protection. We determined the DI of DRG cultures infected with different levels of adenovirus expressing the various Nmnat/Wlds proteins (from 103 to 107 CFU per well) (Fig. 1C–E). For Nmnat1-expressing DRGs, no axonal degeneration (DI < 0.2) was detected until protein levels dropped below 16 a.u. (Fig. 1C). This result strongly indicated that Nmnat1 levels are critical for axonal protection and demonstrated a threshold, below which, Nmnat1 could no longer delay axonal degeneration. Similar experiments using Wlds protein demonstrated that Wlds was ∼10-fold more potent than native Nmnat1 at promoting axonal protection (Fig. 1D). This indicates there is a dosage range where Wlds protein is protective but native Nmnat1 is not, providing a possible explanation for why some studies have not found protection by native Nmnat1 compared with Wlds (Conforti et al., 2007). Intriguingly, cytNmnat1-expressing DRG neurons showed essentially no axonal degeneration even at very low expression levels (∼50-fold below the threshold of Nmnat1) (Fig. 1E). This result indicated that cytNmnat1 protects axons more efficiently than Nmnat1, with Wlds providing an intermediate level of protection.
Nmnat1 and cytNmnat1 have similar levels of enzymatic activity, suggesting that other parameters must be responsible for the differing levels of axonal protection afforded by these proteins (Araki et al., 2004; Sasaki et al., 2006). Given that cytNmnat1 differs from Nmnat1 only by the mutation of its nuclear localization signal, we looked carefully at the subcellular localization of these two proteins. DRG neurons were infected with lentiviruses expressing either wild-type Nmnat1 or cytNmnat1, and immunocytochemistry was performed (Fig. 2). In accord with previous reports, Nmnat1 is predominantly found in the nucleus but not in the axon (Berger et al., 2005). Conversely, cytNmnat1 was found predominantly in the cytoplasm and at high levels within axons. Overall, these experiments indicate that cytNmnat1, which is abundant in axons, is more protective than either wild-type Nmnat1 or Wlds, which are both located primarily in the nucleus, and suggest that the axonal localization of cytNmnat1 contributes to its enhanced axonal protective activity.
Figure 2.
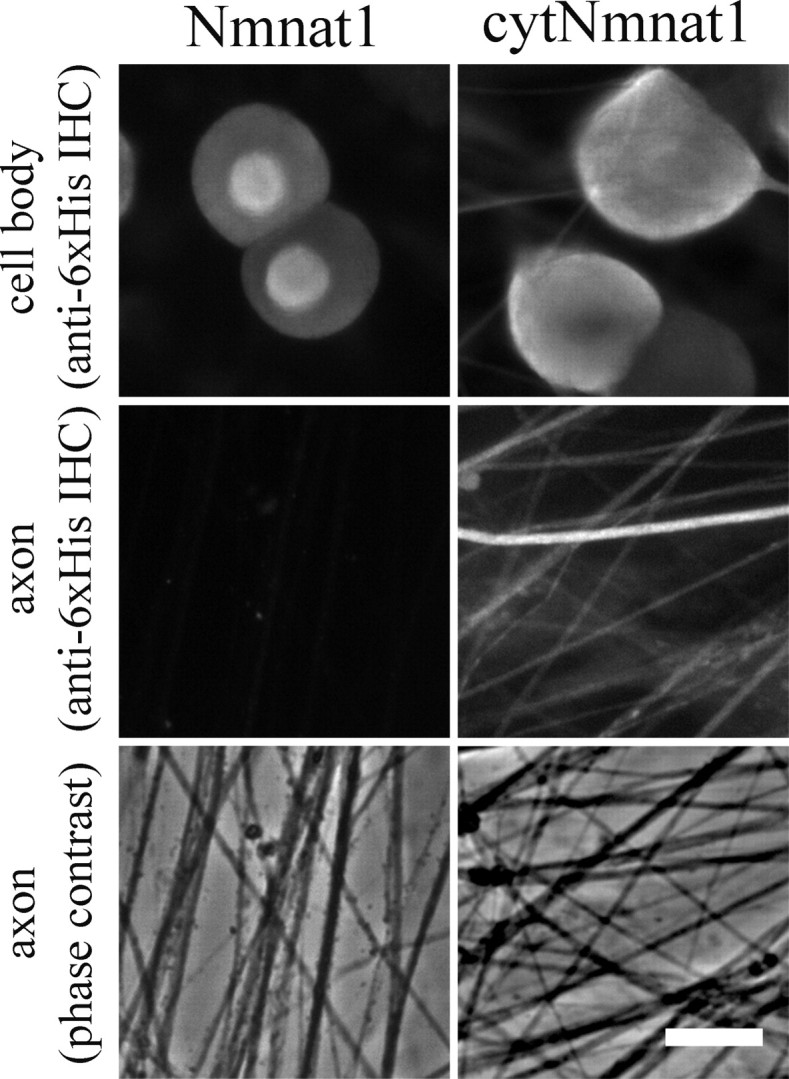
cytNmnat1 is present in axons. DRG neurons were infected with lentivirus expressing 6xHis epitope-tagged Nmnat1 or cytNmnat1. The infected neurons were stained with anti-6xHis antibody. Nmnat1 was predominantly expressed in the nucleus and cytNmnat1 is expressed diffusely in the cytosol (top row). cytNmnat1 was detected in axons at high levels, whereas no Nmnat1 was detected within the axon (middle row). Phase-contrast pictures corresponding to the axonal area stained with anti-6xHis antibody are shown (bottom row). Scale bar, 25 μm.
Generation of transgenic mice expressing cyt-Nmnat1 in neurons
In contrast to the clear demonstration of Nmnat-mediated axonal protection in vitro, its effectiveness in vivo has been controversial (Conforti et al., 2007). The demonstration that higher levels of Nmnat1 are required to protect axons compared with Wlds or cytNmnat1, suggested that the failure to observe protection in vivo could be due to insufficient expression of Nmnat1 in neurons of the reported transgenic animals (Conforti et al., 2007). To address this issue and examine whether Nmnat1 alone, in the absence of all other sequences present in the Wlds protein, can provide axonal protection in vivo, we generated cytNmnat1 transgenic mice. We used the prion protein promoter (PrP) to drive strong neuronal expression of cytNmnat1, a mutant form that protects axons at 50-fold lower concentrations than Nmnat1. Four PrP-cytNmnat1 transgenic (Tg) lines (A–D) were obtained. No overt phenotypic abnormalities were observed in any of these mice up to 10 months of age.
To examine cytNmnat1 expression in these Tg lines, we performed Western blot analysis using anti-His tag antibody on brain lysates prepared from 6-week-old heterozygous PrP-cytNmnat1 Tg mice from each line (Fig. 3A). The expression of the cytNmnat1 transgene varied ∼17-fold, with line A expressing the lowest level and lines C and D the highest levels of cytNmnat1. To ensure that the cytNmnat1 expressed in these Tg mice was active, we performed Nmnat enzymatic assays using brain lysates from 6-week-old animals of each Tg line as well as wlds and wild-type mice. We found that the level of expression by Western blot correlated well with Nmnat enzymatic activity, with brain lysates from PrP-cytNmnat1(A–D) containing ∼15-fold (line A), 100-fold (line B), and 300-fold (lines C, D) greater activity than lysates from wild-type brain (Fig. 3B). We found that wlds brain lysates had sevenfold greater activity than wild-type brain, consistent with previous reports (Mack et al., 2001). We also measured NAD+ levels in these brain lysates; however, in accord with previous work on Nmnat transfected cells and wlds mice (Mack et al., 2001), no changes in steady state NAD+ levels were observed in the PrP-cytNmnat1 Tg mice (data not shown).
Figure 3.
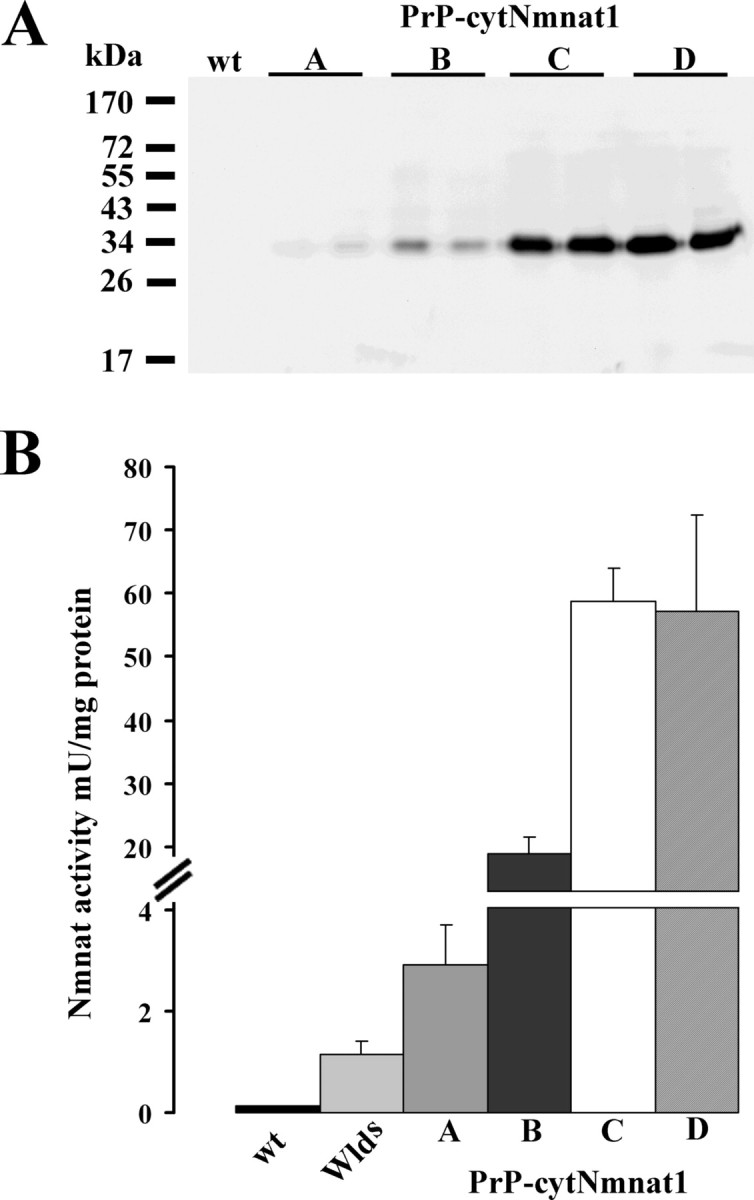
Nmnat enzymatic activity is increased in Prp-cytNmnat1 Tg mouse brain. A, cytNmnat1 expression levels in lysates prepared from the 6-week-old brains of two independent PrP-cytNmnat1 Tg mice of all four (A–D) lines and wild-type (wt) mice were analyzed by Western blotting using anti-6xHis tag antibody. Transgene expression was observed in all four lines, with the lowest level in line A and the highest level in lines C and D. B, Nmnat enzymatic activity was measured in 6-week-old brain lysates prepared from each of the PrP-cytNmnat1 Tg lines, wlds, and wild-type mice. Each of the four Tg lines showed significant increases in Nmnat enzymatic activity that was well correlated with the protein expression levels observed in A. Three mice of each genotype were analyzed and the data are displayed as mean ± SD.
To further examine the pattern of cytNmnat1 expression in the nervous system, we performed immunohistochemistry with anti-His antibody using tissues from Prp-cytNmnat1(D) Tg mice. High expression of the cytNmnat1 transgene was detected in the hippocampus, in particular in the CA1 region and in the granule cells of the dentate gyrus (Fig. 4A,B,E). In addition, cytNmnat1 was expressed in cortical neurons, cerebellar granule cells, spinal motor neurons, and DRG sensory neurons (Fig. 4C,F; data not shown). We did not observe strong staining in the Purkinje cells of the cerebellum. Consistent with our results in cultured cells, we found that cytNmnat1 is present outside of the neuronal nucleus and is found in the neuronal projections (Fig. 4B,C,E, arrows). These results demonstrate that PrP-cytNmnat1 Tg mice express high levels of enzymatically active cytNmnat1 in multiple neuronal populations.
Figure 4.
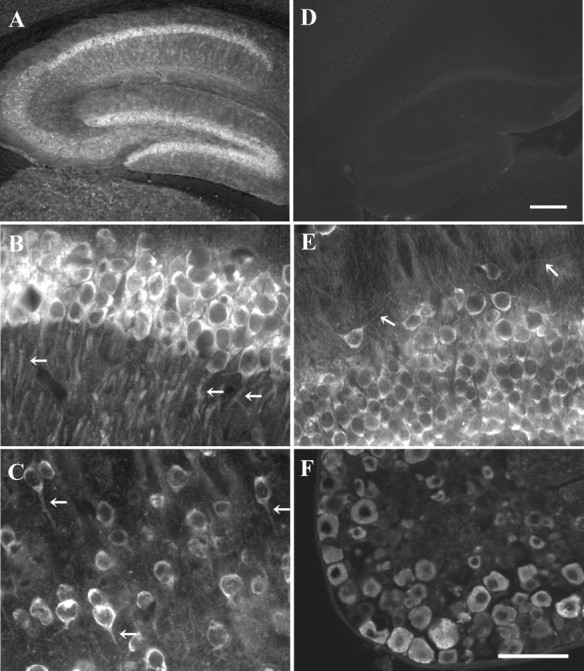
Neuronal expression of the cytNmnat1 transgene. Immunofluorescence with the anti-6xHis tag antibody was used to examine the expression of the cytNmnat1 transgene in 6-week-old PrP-cytNmnat1(D) Tg brain (A, B, C, E) and DRG (F). Wild-type brain is shown in D. The highest cytNmnat1 expression was detected in the hippocampus (A). Higher magnification revealed cytNmnat1 expression in the cytosol and neuronal projections in the CA1 pyramidal cells, cortical neurons, and neurons in the dentate gyrus (B, C, E; arrows indicating representative neuronal projections). High levels of cytNmnat1 were also detected in DRG neurons (F). Scale bars: (in D) A, D, 250 μm; (in F) B, C, E, F, 50 μm.
Axonal degeneration in PrP-cytNmnat1 Tg mice is delayed after sciatic nerve transection
The high neuronal expression of the cytNmnat1 transgene in the DRG sensory and spinal motor neurons in these mice prompted us to examine its effects on axonal degeneration after nerve injury. Initially, we cultured DRG neurons from E13.5 PrP-cytNmnat1(A) embryos (the lowest expressing line) and tested whether their axons were protected after injury in vitro. As shown in Figure 5A, axons from Tg DRG neurons showed no degeneration even after 72 h, whereas wild-type axons had completely degenerated within 24 h. We also treated cultured Tg DRG neurons with vincristine, a chemotherapeutic agent that causes axonal degeneration. Again, axons from Tg DRG neurons were well preserved, whereas axons from wild-type neurons were completely degenerated within 24 h (Fig. 5B). These results confirmed that the expression of the cytNmnat1 transgene was sufficient to delay in vitro axonal degeneration caused by either axotomy or vincristine.
Figure 5.
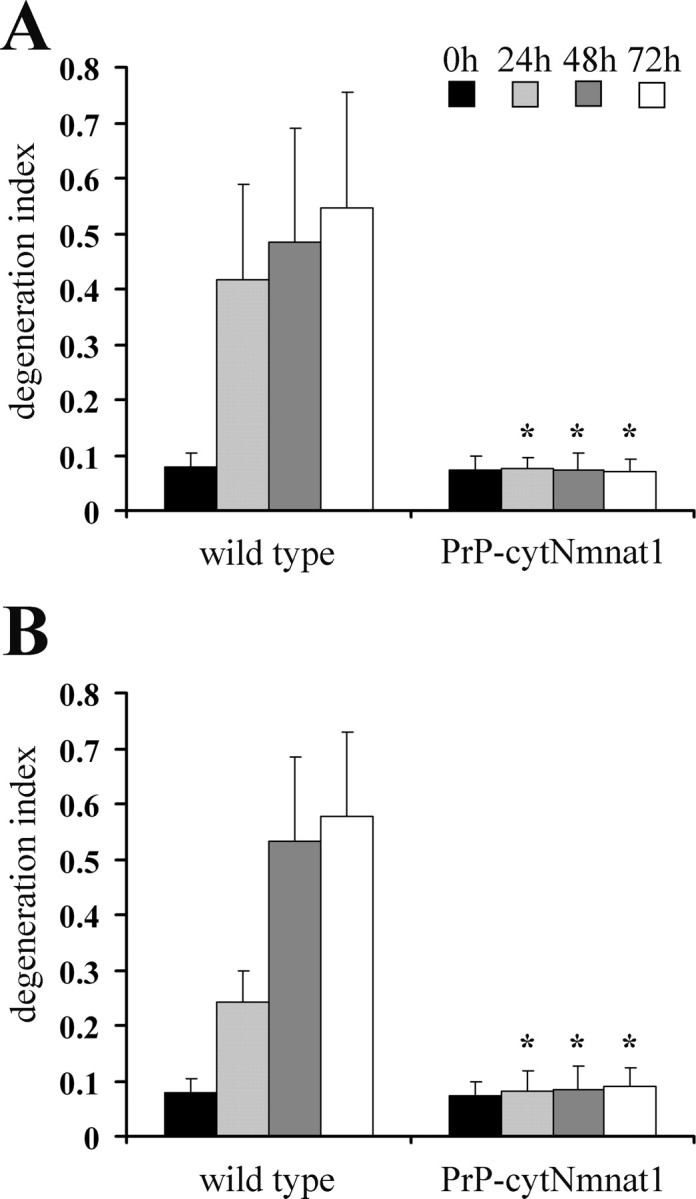
Delayed axonal degeneration in cultured DRG neurons from PrP-cytNmnat1(A) Tg mice. A, Axons from wild-type and PrP-cytNmnat1(A) Tg DRG neurons were transected after 14 d in culture. Degeneration indices were calculated 0, 1, 2, and 3 d later. Wild-type axons were completely degenerated after 1 d whereas transgenic axons were intact even 3 d after injury (n = 10). B, DRG neurons from PrP-cytNmnat1(A) Tg mice were cultured for 14 d, then treated with 4 μm vincristine. Degeneration indices were calculated at 2 and 3 d after vincristine addition. Wild-type axons were completely degenerated after 2 d whereas transgenic axons were intact 3 d after vincristine treatment (n = 10). *Significant difference (p < 0.001, Student's t test) between neurons from PrP-cytNmnat1 and wild type of the indicated time.
To test whether overexpression of cytNmnat1 alone can delay axonal degeneration after nerve injury in vivo, we examined Wallerian degeneration of the distal segment after sciatic nerve transection. The sciatic nerves of the PrP-cytNmnat1 Tg lines A–D as well as wild-type mice were transected and the distal nerve segments (from both the injured and uninjured sides) were harvested 7 d later. Lysates were prepared and the amount of neurofilament remaining was measured using Western blotting to assess the degree of axonal degeneration. In wild-type mice, there was no detectable neurofilament in the transected nerve, whereas the amount of neurofilament remaining in the distal nerve segments of all four PrP-cytNmnat1 Tg mice was similar to that observed in the uninjured nerve control (Fig. 6A; data not shown). The distal segment from the wlds mouse had an intermediate level of neurofilament 7 d after transection.
Figure 6.
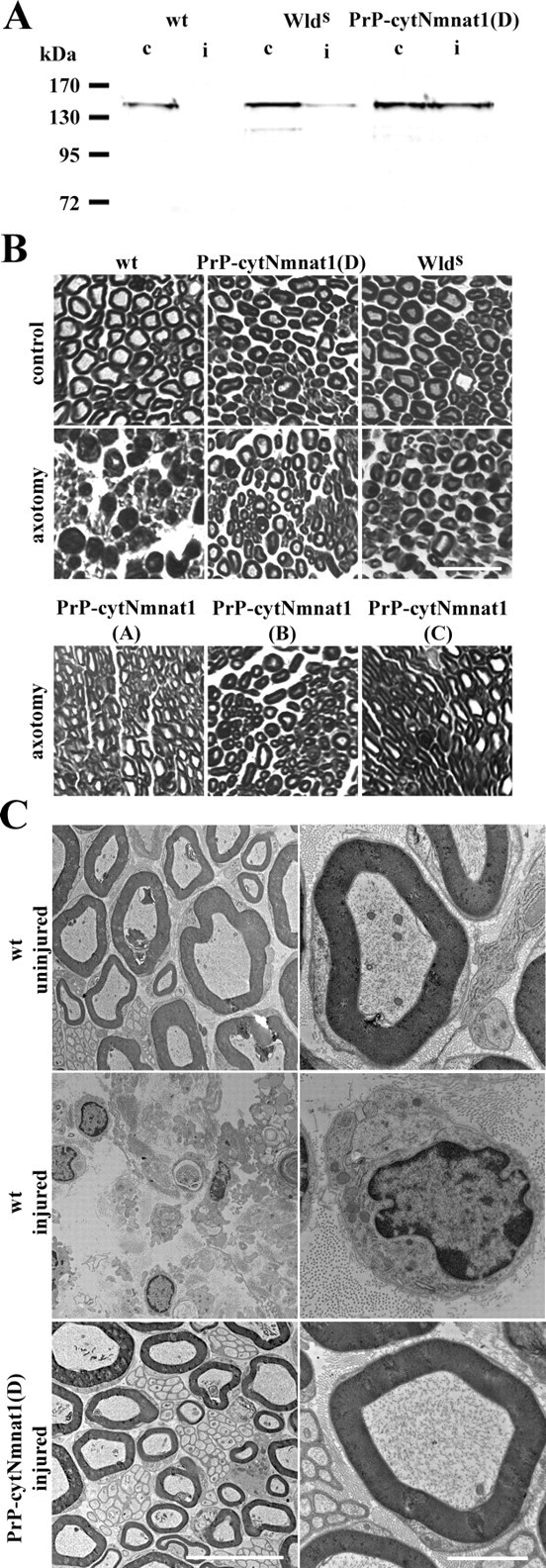
Axonal degeneration after sciatic nerve transection is delayed in PrP-cytNmnat1 Tg mice. A, Neurofilament (165 kDa component) levels remaining in the distal sciatic nerve segment 7 d after transection were measured by Western blot using anti-neurofilament antibody to examine axonal loss. Neurofilament levels in transected distal segment (i) of PrP-cytNmnat1(D) Tg mice were equivalent to those in the uninjured control (c) nerve. No neurofilament remained in the wild-type distal segment after injury. Transected distal segment from wlds mice also contained neurofilament. B, Plastic thin sections (4 μm) of distal sciatic nerve taken from PrP-Nmnat1(A–D) Tg, wlds, or wild-type mice 7 d after transection were stained with toluidine blue. Axons and the myelin structures of PrP-cytNmnat1 Tg and wlds mice were well preserved, whereas the wild-type nerve structure was completely disorganized with massive axonal loss and large amounts of myelin debris. C, Electron microscopic evaluation of these nerves revealed that the wild-type axonal structure was completely degenerated 7 d after axotomy (middle) compared with the uninjured nerve control (top). In contrast, the distal nerve from Prp-cytNmnat1(D) was well preserved (bottom). Higher magnification clearly showed an intact nerve structure including microtubules and mitochondria inside the axon in the transected transgenic nerve. Scale bars: B, 50 μm; C, left column, 10 μm; C, right column, 2 μm.
To further characterize the in vivo axonal protection, thin sections of the distal nerve segments were stained with toluidine blue and examined microscopically. We detected no differences in the control sciatic nerves isolated from wild-type, PrP-cytNmnat1(A–D), or wlds mice (Fig. 6B; data not shown). However, an examination of the distal segments of the injured sciatic nerves 7 d after axotomy revealed dramatic differences. In wild-type nerves, the myelin structure was completely destroyed and there were few, if any, intact axons (Fig. 6B). In contrast, there was no evidence of axon loss or myelin debris in the distal segments from the PrP-cytNmnat1(A–D) or wlds mice 7 d after injury (Fig. 6B).
To examine the distal sciatic nerve segments from wild-type and PrP-cytNmnat1(D) Tg mice in more detail, we performed electron microscopy. No normal myelinated or unmyelinated fibers were observed in injured wild-type nerve (Fig. 6C). In contrast, analysis of the distal segment of the cytNmnat1 transgenic mice showed complete preservation of both myelinated and small unmyelinated fibers (Fig. 6C). Furthermore, higher magnification revealed an intact myelin sheath and the presence of mitochondria and microtubular structures within the ensheathed cytNmnat1 transgenic axons. To quantify the extent of the preservation in the PrP-cytNmnat1 Tg mice, we compared the g-ratios of the injured (0.61 ± 0.11) and uninjured (0.63 ± 0.10) sciatic nerves as well as that of the uninjured wild-type nerve (0.60 ± 0.11). No g-ratios could be determined for the transected wild-type nerve because no intact axons were present. These experiments clearly show that the distal segments of PrP-cytNmnat1 Tg sciatic nerves are structurally intact after transection.
Electrophysiologic preservation of axons and neuromuscular transmission in PrP-cytNmnat1 mice after nerve transection
In previous studies of wlds mice, the ability to conduct action potentials, in addition to axonal structural integrity, is preserved after nerve transection (Mack et al., 2001; Gillingwater et al., 2002). To determine whether cytNmnat1 overexpression also preserved the functional integrity of the distal segment, we performed compound motor action potential (CMAP) measurements from the foot muscles after stimulation of the tibial nerve (a branch of the sciatic nerve distal to the transection site). For the distal CMAP response to be preserved, there must be (1) intact ionic gradients and channel function in axons to generate an action potential, (2) neuromuscular synaptic transmission, and (3) muscle contraction (the CMAP response). The distal CMAP was measured before (day 0) and after (at 2, 4, and 7 d) sciatic nerve transection in 10-week-old wild-type, wlds, and PrP-cytNmnat1(D) Tg mice. In wild-type mice, CMAP was completely absent by 2 d after axotomy (Fig. 7). In PrP-cytNmnat1 Tg mice, the CMAP was maintained at ∼80% of its original value at 2 d after transection, but gradually decreased to ∼10% by 7 d after transection, indicating significant functional protection of the distal axons after nerve transection by cytNmnat1. This was similar to that observed in wlds mice, indicating that the degree of axonal protection in PrP-cytNmnat1 Tg mice is comparable.
Figure 7.

Axonal and synaptic integrity are both protected in PrP-cytNmnat1(D) Tg mice after axotomy. The CMAP amplitude is shown as a percentage of the initial value (0 h) after stimulation distal to site of sciatic transaction in wild-type, PrP-cytNmnat1(D) Tg, and wlds mice. At 10 weeks of age, wild-type mice had no response 2 d after axotomy, whereas PrP-cytNmnat1 and wlds mice retained ∼80% of initial CMAP amplitude. This response declined to ∼10% in both PrP-cytNmnat1 Tg and wlds mice at 7 d after axotomy. At 17 weeks, PrP-cytNmnat1 Tg mice had an ∼90% decline in the CMAP response 7 d after axotomy, similar to that observed in 10-week-old Tg mice. * and ** indicate significant difference (p < 0.001 for * and p < 0.05 for **, Student's t test) between relative CMAP at indicated time and 0 h. *** indicates no significant difference (p > 0.1, Student's t test) between 10-week-old and 17-week-old PrP-cytNmnat1 of CMAP at indicated time.
The basis of the loss of CMAPs is presumably due to synaptic withdrawal and loss of neuromuscular transmission, which is known to occur before structural axonal degeneration in 2-month-old Wlds mice (Gillingwater et al., 2002). Furthermore, synaptic withdrawal occurs more rapidly in older wlds mice, such that no electrophysiologic protection is apparent 7 d after transection at advanced ages. To investigate whether PrP-cytNmnat1 Tg mice show a similar age dependency, we also measured distal CMAPs in 4-month-old wild-type and PrP-cyt-Nmnat1 Tg mice. In wild-type mice, the CMAP response disappeared within 2 d; however, the older cytNmnat1 transgenic mice showed a similar time course of CMAP loss as the 2-month-old mice, indicating the lack of an age-dependent decrement in protection. These results indicate that Nmnat overexpression preserves both axonal and synaptic integrity after axotomy. Furthermore, and similar to wlds mice, synaptic integrity appears to be lost before structural axonal degeneration in Prp-cytNmnat1 Tg mice. In total, these studies prove that neuronal overexpression of Nmnat alone, in the absence of all other Wlds protein sequences, is sufficient to protect both axonal and synaptic structure and function after axotomy in vivo.
Discussion
Molecular dissection of the Wlds fusion protein responsible for the delayed axonal degeneration in wlds mice revealed that the Nmnat1 moiety alone can provide axonal protection in vitro. However, other studies reported a lack of protective activity by Nmnat1 in vivo, leading to the conclusion that other sequences within the Wlds protein are also required. In this study, we used transgenic mice in which cytNmnat1, which is present in neuronal cell bodies and axons and has potent axonal protective activity in vitro, was expressed in neurons using the PrP promoter (Borchelt et al., 1996). These mice express cytNmnat1 in multiple neuronal populations, and degeneration of axons emanating from DRG neurons cultured from these transgenic mice is significantly delayed. Morphological as well as functional analysis of sciatic nerves in these PrP-cytNmnat1 Tg mice demonstrated dramatically delayed axonal degeneration after transection. These experiments clearly demonstrated that overexpression of the Nmnat enzyme alone is sufficient to delay axonal degeneration in vivo to a similar, if not greater extent, than reported for the original wlds mice.
Why do the PrP-cytNmnat1 Tg mice reported in this study demonstrate robust axonal protection, whereas previous work showed that axonal degeneration in transgenic mice expressing Nmnat1 was not delayed after sciatic nerve transection (Conforti et al., 2007)? The lack of axonal protection by nuclear Nmnat1 in the previous study was surprising in light of multiple reports demonstrating that Nmnat1 effectively protects axons in vitro (Araki et al., 2004; Wang et al., 2005) and in vivo using a Drosophila model (MacDonald et al., 2006). One potential explanation for these results stems from the observation that axonal protection by the Wlds protein is dose dependent and demonstrates a clear threshold effect (Mack et al., 2001).
To address this issue, we developed a quantitative assay for axonal degeneration and used it to establish an axonal protection dose–response curve for Nmnat1, Wlds, and cytNmnat1. We found that cytNmnat1 provided protection at very low levels, whereas Wlds and Nmnat1 were only effective at fivefold and 50-fold higher levels than cytNmnat1. This implies that the previously reported Nmnat1 transgenic mice could have expressed subthreshold amounts of the enzyme at levels that were insufficient to prevent axonal degeneration in response to injury. Indeed, the total enzymatic activity in the brain of these animals would appear to be insufficient to provide axonal protection (Conforti et al., 2007). Furthermore, although expression levels in DRG neurons were not reported in that study, they were presumably very low as DRG neurons from these mice failed to show delayed axonal degeneration even when cultured in vitro.
Detailed studies of nerve structure and function after injury in wlds mice clearly showed that there is a delay in axonal and synaptic degeneration. Synaptic withdrawal followed by synaptic degeneration causes deficiencies in neuromuscular transmission in wlds mice (Gillingwater et al., 2002). For example, in 2-month-old wlds mice, synaptic withdrawal is the major cause of functional deficits in neuromuscular integrity, which occur within 10 d after injury. However, in older wlds mice (4-month-old), synaptic degeneration predominates and causes an earlier physiological dysfunction after injury (7 d) than is observed in young animals (Gillingwater et al., 2002). Interestingly, the time course of CMAP decline is similar in young PrP-cytNmnat1 and wlds mice; however, in contrast to the more rapid decline observed in older wlds mice, the CMAP measurements in older PrP-cytNmnat1 Tg mice were equivalent to those observed in 2-month-old animals. Overall, we found that maintenance of functional integrity after injury by cytNmnat1, like that promoted by Wlds protein, involves both axonal and synaptic protection. In both PrP-cytNmnat1 Tg and wlds mice, the duration of axonal protection is longer than synaptic protection, suggesting that the fundamental mechanisms of these processes are different.
The molecular mechanism of Nmnat-mediated axonal protection remains enigmatic; however, we now know that it is operant in vivo in an evolutionarily conserved manner, ranging from Drosophila to mouse (MacDonald et al., 2006). In previous work, we showed that enzymes located in the nucleus as well as in mitochondria or in the cytoplasm could effectively preserve injured axons (Sasaki et al., 2006). These results suggest that diffusible molecules modified by Nmnat enzymes are responsible for axonal protection. Indeed, the fact that Nmnat enzymes from Drosophila, Saccharomyces cerevisiae, and the Archaebacterium Methanococcus jannaschii can all promote axonal protection, and that mutants with reduced NAD synthetic activity do not, strongly suggests that enzymatic activity rather than interactions with other cellular proteins underlies Nmnat axonal protective activity (Sasaki et al., 2009). In this case, one would expect that steady-state NAD levels would be altered in cells where Nmnat or Wlds protein is overexpressed; however, multiple laboratories have been unable to detect such differences (Mack et al., 2001; Araki et al., 2004; Wang et al., 2005). In addition, we investigated the role of intracellular steady-state NAD elevations in axonal protection using mice lacking CD38, an NAD glycohydrolase, and found that twofold increases in NAD do not promote axonal protection either in vitro or in vivo (Sasaki et al., 2009). Furthermore, reducing NAD steady-state levels using pharmacologic or genetic inhibitors of NAD biosynthesis did not inhibit Nmnat-mediated axonal protection. In contrast, a recent study showed that treatment with FK866, a chemical inhibitor of Nampt, could weaken Wlds-mediated axonal protection (Conforti et al., 2009). The differences in these results could reflect the less potent protection mediated by Wlds compared with cytNmnat1. While mammalian Wlds and Nmnat enzymes mediate protection in both mammalian and Drosophila axonal degeneration models, another enzyme that produces NAD+, NAD synthetase, failed to preserve injured axons in either mammalian (Sasaki et al., 2006) or Drosophila neurons (Avery et al., 2009). It is puzzling that axonal protection requires Nmnat enzymatic activity, yet no changes in steady-state NAD levels are observed (Araki et al., 2004; Avery et al., 2009; Conforti et al., 2009; Sasaki et al., 2009). Equally surprising are results showing that alternative manipulations to substantially increase NAD levels do not in themselves provide robust axonal protection, nor do interventions that dramatically decrease NAD levels abolish Nmnat-mediated protection. Together, these observations suggest that Nmnat has additional activities and perhaps additional substrates that are crucial for delaying axonal degeneration.
In examining the dose dependency of Nmnat axonal protection, we found that cytNmnat1 was active at much lower levels than either Nmnat1 or Wlds protein. What is the mechanism of this increased potency? It is not due to differences in enzymatic activity, as the kcat of both Nmnat1 and cytNmnat1 are equivalent (data not shown). Instead, we believe that the presence of the protein in the cytoplasm, particularly the localization of cytNmnat1 within the axon, where Nmnat1 is never found, is likely to underlie the increased axonal protection. In this regard, the moderately stronger protection provided by Wlds protein compared with Nmnat1 may be due to its higher accumulation outside of the nucleus via its enhanced translocation into the cytosol through its interaction with VCP (Laser et al., 2006). This hypothesis is supported by a recent study in Drosophila demonstrating that Wlds lacking the N-terminal VCP-interaction domain provided a level of axonal protection that was equivalent to that promoted by Nmnat1 in vivo (Avery et al., 2009). In previous work, we examined the importance of Nmnat subcellular localization for effective axonal protection and found that Nmnat1, cytNmnat1, and even Nmnat3, which localizes to mitochondria, can all promote axonal protection (Sasaki et al., 2006). These results have now been replicated in vivo (Avery et al., 2009; Beirowski et al., 2009), and effectively demonstrate that cytosolic localization of Nmnat is important for robust axonal protection. Because the minimum amount of cytNmnat1 required to delay axonal degeneration is very low, it is likely that axonal protection mediated by Nmnat1, Nmnat3, or Wlds protein results from a small proportion of these proteins residing in the cytoplasm. This implies that molecules abundant in the cytosol or axon are the critical targets of Nmnat for promoting axonal protection.
In summary, we have shown that Nmnat1 alone, in the absence of all other Wlds sequences, provides robust axonal protection after nerve injury in vivo. These results indicate that focusing on Nmnat as the key component responsible for the delayed axonal degeneration first discovered in the wlds mouse mutant will likely lead to new mechanistic insights. Understanding the molecular basis of Nmnat-mediated axonal protection will potentially lead to the development of new therapeutic agents for neurodegenerative diseases.
Footnotes
This work was supported by National Institutes of Health Neuroscience Blueprint Center Core Grant P30 NS057105 to Washington University, the Hope Center for Neurological Disorders, National Institutes of Health Grants NS040745 (J.M.), AG13730 (J.M.), and 1K08NS055980 (R.H.B.), Ministry of Health, Labour, and Welfare of Japan (SHA4431), and Muscular Dystrophy Association Grants 10040 (J.M.) and 4152 (R.H.B.). R.H.B. holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund and is supported by the Children's Discovery Institute. We thank Kelli Simburger, Amy Strickland, Amber Neilson, and Tim Fahrner for experimental assistance and members of the Milbrandt laboratory for their comments on the manuscript and helpful discussions.
The authors and Washington University may derive benefit from a licensing agreement with Sirtris Pharmaceuticals, which did not provide any support for this work.
References
- Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305:1010–1013. doi: 10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- Avery MA, Sheehan AE, Kerr KS, Wang J, Freeman MR. Wld S requires Nmnat1 enzymatic activity and N16-VCP interactions to suppress Wallerian degeneration. J Cell Biol. 2009;184:501–513. doi: 10.1083/jcb.200808042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baloh RH, Strickland A, Ryu E, Le N, Fahrner T, Yang M, Nagarajan R, Milbrandt J. Congenital hypomyelinating neuropathy with lethal conduction failure in mice carrying the Egr2 I268N mutation. J Neurosci. 2009;29:2312–2321. doi: 10.1523/JNEUROSCI.2168-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beirowski B, Babetto E, Gilley J, Mazzola F, Conforti L, Janeckova L, Magni G, Ribchester RR, Coleman MP. Non-nuclear Wld(S) determines its neuroprotective efficacy for axons and synapses in vivo. J Neurosci. 2009;29:653–668. doi: 10.1523/JNEUROSCI.3814-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger F, Lau C, Dahlmann M, Ziegler M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J Biol Chem. 2005;280:36334–36341. doi: 10.1074/jbc.M508660200. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Davis J, Fischer M, Lee MK, Slunt HH, Ratovitsky T, Regard J, Copeland NG, Jenkins NA, Sisodia SS, Price DL. A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet Anal. 1996;13:159–163. doi: 10.1016/s1050-3862(96)00167-2. [DOI] [PubMed] [Google Scholar]
- Coleman M. Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci. 2005;6:889–898. doi: 10.1038/nrn1788. [DOI] [PubMed] [Google Scholar]
- Coleman MP, Conforti L, Buckmaster EA, Tarlton A, Ewing RM, Brown MC, Lyon MF, Perry VH. An 85-kb tandem triplication in the slow Wallerian degeneration (Wlds) mouse. Proc Natl Acad Sci U S A. 1998;95:9985–9990. doi: 10.1073/pnas.95.17.9985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti L, Fang G, Beirowski B, Wang MS, Sorci L, Asress S, Adalbert R, Silva A, Bridge K, Huang XP, Magni G, Glass JD, Coleman MP. NAD(+) and axon degeneration revisited: Nmnat1 cannot substitute for Wld(S) to delay Wallerian degeneration. Cell Death Differ. 2007;14:116–127. doi: 10.1038/sj.cdd.4401944. [DOI] [PubMed] [Google Scholar]
- Conforti L, Wilbrey A, Morreale G, Janeckova L, Beirowski B, Adalbert R, Mazzola F, Di Stefano M, Hartley R, Babetto E, Smith T, Gilley J, Billington RA, Genazzani AA, Ribchester RR, Magni G, Coleman M. Wld(S) protein requires Nmnat activity and a short N-terminal sequence to protect axons in mice. J Cell Biol. 2009;184:491–500. doi: 10.1083/jcb.200807175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillingwater TH, Thomson D, Mack TG, Soffin EM, Mattison RJ, Coleman MP, Ribchester RR. Age-dependent synapse withdrawal at axotomised neuromuscular junctions in Wld(s) mutant and Ube4b/Nmnat transgenic mice. J Physiol. 2002;543:739–755. doi: 10.1113/jphysiol.2002.022343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia H, Yan T, Feng Y, Zeng C, Shi X, Zhai Q. Identification of a critical site in Wld(s): essential for Nmnat enzyme activity and axon-protective function. Neurosci Lett. 2007;413:46–51. doi: 10.1016/j.neulet.2006.11.067. [DOI] [PubMed] [Google Scholar]
- Laser H, Conforti L, Morreale G, Mack TG, Heyer M, Haley JE, Wishart TM, Beirowski B, Walker SA, Haase G, Celik A, Adalbert R, Wagner D, Grumme D, Ribchester RR, Plomann M, Coleman MP. The slow Wallerian degeneration protein, WldS, binds directly to VCP/p97 and partially redistributes it within the nucleus. Mol Biol Cell. 2006;17:1075–1084. doi: 10.1091/mbc.E05-04-0375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le N, Nagarajan R, Wang JY, Svaren J, LaPash C, Araki T, Schmidt RE, Milbrandt J. Nab proteins are essential for peripheral nervous system myelination. Nat Neurosci. 2005;8:932–940. doi: 10.1038/nn1490. [DOI] [PubMed] [Google Scholar]
- Lunn ER, Perry VH, Brown MC, Rosen H, Gordon S. Absence of Wallerian degeneration does not hinder regeneration in peripheral nerve. Eur J Neurosci. 1989;1:27–33. doi: 10.1111/j.1460-9568.1989.tb00771.x. [DOI] [PubMed] [Google Scholar]
- MacDonald JM, Beach MG, Porpiglia E, Sheehan AE, Watts RJ, Freeman MR. The Drosophila cell corpse engulfment receptor draper mediates glial clearance of severed axons. Neuron. 2006;50:869–881. doi: 10.1016/j.neuron.2006.04.028. [DOI] [PubMed] [Google Scholar]
- Mack TG, Reiner M, Beirowski B, Mi W, Emanuelli M, Wagner D, Thomson D, Gillingwater T, Court F, Conforti L, Fernando FS, Tarlton A, Andressen C, Addicks K, Magni G, Ribchester RR, Perry VH, Coleman MP. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat Neurosci. 2001;4:1199–1206. doi: 10.1038/nn770. [DOI] [PubMed] [Google Scholar]
- Mitsuuchi Y, Powell DR, Gallo JM. Adenoviral modification of mouse brain derived endothelial cells, bEnd3, to induce apoptosis by vascular endothelial growth factor. Oncogene. 2006;25:954–958. doi: 10.1038/sj.onc.1209142. [DOI] [PubMed] [Google Scholar]
- Perry VH, Brown MC, Lunn ER. Very slow retrograde and Wallerian degeneration in the CNS of C57BL/Ola mice. Eur J Neurosci. 1991;3:102–105. doi: 10.1111/j.1460-9568.1991.tb00815.x. [DOI] [PubMed] [Google Scholar]
- Press C, Milbrandt J. Nmnat delays axonal degeneration caused by mitochondrial and oxidative stress. J Neurosci. 2008;28:4861–4871. doi: 10.1523/JNEUROSCI.0525-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raff MC, Whitmore AV, Finn JT. Axonal self-destruction and neurodegeneration. Science. 2002;296:868–871. doi: 10.1126/science.1068613. [DOI] [PubMed] [Google Scholar]
- Ryu EJ, Wang JY, Le N, Baloh RH, Gustin JA, Schmidt RE, Milbrandt J. Misexpression of Pou3f1 results in peripheral nerve hypomyelination and axonal loss. J Neurosci. 2007;27:11552–11559. doi: 10.1523/JNEUROSCI.5497-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Araki T, Milbrandt J. Stimulation of nicotinamide adenine dinucleotide biosynthetic pathways delays axonal degeneration after axotomy. J Neurosci. 2006;26:8484–8491. doi: 10.1523/JNEUROSCI.2320-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Bhupinder PSV, Milbrandt J. Nmnat-mediated axonal protection requires enzymatic activity but not increased levels of neuronal NAD+ J Neurosci. 2009;29:5525–5535. doi: 10.1523/JNEUROSCI.5469-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Zhai Q, Chen Y, Lin E, Gu W, McBurney MW, He Z. A local mechanism mediates NAD-dependent protection of axon degeneration. J Cell Biol. 2005;170:349–355. doi: 10.1083/jcb.200504028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Tsukiyama T, Hatakeyama S. Protection of vincristine-induced neuropathy by WldS expression and the independence of the activity of Nmnat1. Neurosci Lett. 2007;411:228–232. doi: 10.1016/j.neulet.2006.09.068. [DOI] [PubMed] [Google Scholar]
- Zhai RG, Zhang F, Hiesinger PR, Cao Y, Haueter CM, Bellen HJ. NAD synthase NMNAT acts as a chaperone to protect against neurodegeneration. Nature. 2008;452:887–891. doi: 10.1038/nature06721. [DOI] [PMC free article] [PubMed] [Google Scholar]