

Abstract
Prostaglandin H synthase isoforms 1 and -2 (PGHS-1 and -2) react with peroxide to form a radical on Tyr385 that initiates the cyclooxygenase catalysis. The tyrosyl radical EPR signals of PGHS-1 and -2 changes over time and are altered by cyclooxygenase inhibitor binding. We characterized the tyrosyl radical dynamics using wild type human PGHS-1 (hPGHS-1) and its Y504F, Y385F and Y385F/Y504F mutats to determine whether the radical EPR signal changes involve Tyr504 radical formation, Tyr385 radical phenyl ring rotation, or both. Reaction of hPGHS-1 with peroxide produced a wide singlet, whereas its Y504F mutant produced only a wide doublet signal, assigned to the Tyr385 radical. The cyclooxygenase specific activity and KM for arachidonate of hPGHS-1 were not affected by the Y504F mutation, but the peroxidase specific activity and the KM for peroxide were increased. The Y385F and Y385F/Y504F mutants retained only a small fraction of the peroxidase activity; the former had a much-reduced yield of peroxide-induced radical and the latter essentially none. After binding of indomethacin, a cyclooxygenase inhibitor, hPGHS-1 produced a narrow singlet but the Y504F mutant did not show much radical. These results indicate that peroxide-induced radicals form on Tyr385 and Tyr504 of hPGHS-1, with radical primarily on Tyr504 in the wild type protein; indomethacin binding prevented radical formation on Tyr385 but still on Tyr504. Thus, hPGHS-1 and -2 have different distributions of peroxide-derived radical between Tyr385 and Tyr504. Y504F mutants in both hPGHS-1 and -2 significantly decreased the cyclooxygenase activation efficiency, indicating that formation of the Tyr504 radical is functionally important for both isoforms.
Introduction
Prostaglandin H synthase (PGHS)-1 and -2 are bifunctional, membrane bound hemoproteins that catalyze the first committed steps in prostanoid biosynthesis, the conversion of arachidonic acid (AA) to prostaglandin G2 (PGG2) via cyclooxygenase catalysis, and the subsequent reduction of PGG2 to PGH2 through a classic heme peroxidase mechanism [1-3]. These two activities are linked by an essential tyrosine radical generated at Tyr385, which is located near the heme (Fig. 1). The first intermediate in the peroxidase cycle (Intermediate I) contains a ferryl-oxo porphyrin radical cation that can undergo intramolecular electron transfer to form Intermediate II, which contains a ferryl-oxo heme and a Tyr385 radical [4, 5].
Figure 1.
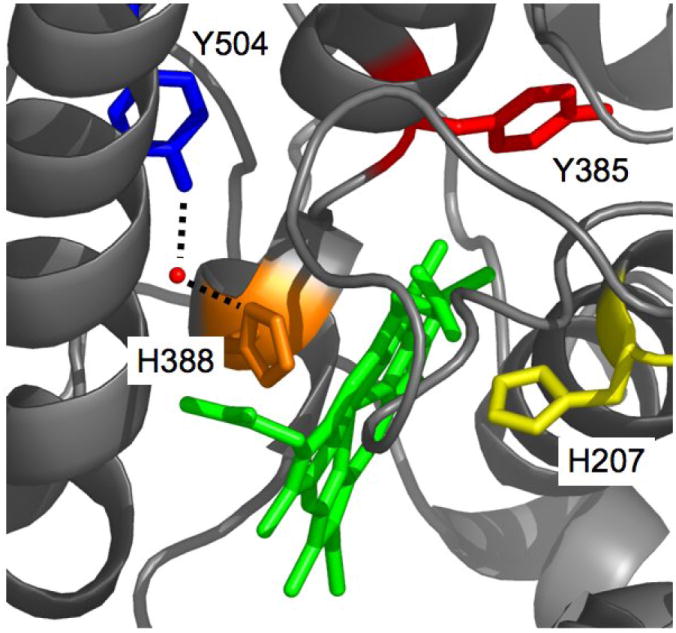
Depiction of oPGHS-1 structure near the peroxidase pocket (PDB entry 2AYL) showing Tyr385 (red stick), Tyr504 (blue stick), Mn-PPIX (green stick), the proximal ligand, His388 (orange stick), His207 (yellow stick) [64]. The red sphere is a structural water H-bonded to Tyr504 and His388.
Tyrosyl radicals have been encountered as cofactors in enzymes as diverse as ribonucleotide reductase, KatG catalase/peroxidase, and photosystem II [6-11]. However, the Tyr385 radical in PGHS is so far the only case of a tyrosyl radical interacting directly with substrate [12]. This unique catalytic role and the physiological and pharmacological importance of cyclooxygenase activity have increased interest in the PGHS-1 and -2 tyrosyl radicals. The majority of studies have been carried out with oPGHS-1, due to the protein’s abundance in sheep seminal vesicles.
Participation of a tyrosyl radical in the PGHS catalytic cycle has been the subject of extensive EPR studies. Interestingly, the EPR signal produced by reaction with peroxide undergoes variations in lineshape and intensity depending upon reaction time, cyclooxygenase inhibitor binding, and self-inactivation [13]. This complexity of tyrosyl radical EPR lineshape led to initial misinterpretation of the wide-singlet tyrosyl radical found in oPGHS-1 as a discrete species [14, 15] and even raised doubt about whether the tyrosyl radical was relevant to catalysis or just a marker of self-inactivation [13, 16]. Thus, it is fundamentally important to understand the origin and role of each tyrosyl radical intermediate observed during PGHS catalysis.
oPGHS-1 and hPGHS-2 display initial wide doublet (WD) signals that convert to wide singlet (WS) signals within 50 s (oPGHS-1) or 50 ms (hPGHS-2) [17]. In oPGHS-1, longer reaction times and self-inactivation result in a transition of the WS to a narrow singlet (NS1b) [15, 16, 18]. In both oPGHS-1 and hPGHS-2, pretreatment with some cyclooxygenase inhibitors results in a second type of NS (NS1a or NS2) [19, 20]; the NS1a signal seen in inhibitor-bound oPGHS-1 has more pronounced hyperfine features than does NS1b [14, 15, 21]. In hPGHS-2, Tyr504 (also shown in Fig. 1) serves as an alternate radical site, accounting for the NS2 spectrum; the WD → WS transition in hPGHS-2 is caused by changing proportions of radical at Tyr385 and Tyr504, with the mixture of WD and NS2 signals adding to the WS signal [17, 22]. In oPGHS-1, however, NO trapping of AA-induced tyrosyl radical identified only Tyr385 as a radical site, seeming to argue against formation of a Tyr504 radical in PGHS-1 [23]. The observation of two NS radical species in oPGHS-1 and one NS in hPGHS-2 and the NO-trapping data with oPGHS-1 raise the question of whether one or both of the NS species seen in PGHS-1 are due to formation of a Tyr504 radical in that isoform, or whether the PGHS-1 NS signals result from changes in the phenyl ring conformation of the Tyr385 radical, as proposed earlier [24].
The primary goal of the present study was to determine whether the tyrosyl radical dynamics and their functional impacts differ between PGHS-1 and -2. We have examined the catalytic and spectroscopic characteristics of recombinant wild type hPGHS-1 and the corresponding Y385F, Y504F, and Y385F/Y504F mutants. The results indicate that the Tyr385 radical in hPGHS-1 does not undergo ring rotation and that Tyr504 is an alternate site for peroxide-generated radical, with the Tyr504 and Tyr385 radicals forming in parallel. Thus, hPGHS-1 and -2 behave similarly in the location(s) of tyrosyl radical formation, although the radical dynamics show clear differences.
A second goal of these studies was to determine whether formation of radical at Tyr504 in hPGHS-1 or -2 affected the regulation of cyclooxygenase catalysis by peroxide level. The results of peroxidase competition experiments in Y504F mutants revealed marked decreases in cyclooxygenase activation efficiency, indicating that the alternative radical site at Tyr504 has a functional role in both isoforms. This study, which is the first examination of tyrosyl radical in hPGHS-1, revealed important similarities and differences in tyrosyl radical dynamics between hPGHS-1 and -2, significantly adding to our previous site-directed mutation study with hPGHS-2 [22].
EXPERIMENTAL PROCEDURES
Materials
AA was from NuChek Preps (Elysian, MN). The EtOOH solution was a gift from Dr. G. Barney Ellison, University of Colorado; the peroxide identity was confirmed by NMR and mass spectrometry. Tween-20 was from Anatrace (Maumee, OH), trans-5-phenyl-4-pentenyl-1-hydroperoxide (PPHP) and the PGHS-1 polyclonal antiserum was from Cayman Chemical (Ann Arbor, MI). All other reagents were obtained from Sigma-Aldrich (St. Louis, MO).
Construction of plasmids for hPGHS-1 mutants
The plasmid coding for hPGHS-1 with a Y504F point mutation was constructed with the QuikChange site-directed mutagenesis kit (Stratagene, Garden Grove, CA) using the pSG5 plasmid containing the wild type hPGHS-1 cDNA [25] as template. This wild type cDNA codes for methionine at the polymorphic site at position 237 [26]. The primer pairs for the Y504F mutation were (base changes are underlined):
Y504F-f: 5’-GATGCGTTGGAGTTCTTCCCTGGACTGCTTCTTG-3’
Y504F-r: 5’-CAAGAAGCAGTCCAGGGAAGAACTCCAACGCATC-3’
The Y385F single mutant and the Y385F/Y504F double mutants were constructed similarly; using the Y504F mutant plasmid as the template and the following primer pairs (base changes are underlined):
Y385F-f: 5’-GAGTTCAACCATCTCTTCCACTGGCACCCC-3’
Y385F-r: 5’-GGGGTGCCAGTGGAAGAGATGGTTGAACTC-3’
Introduction of cDNA coding for histidine tag
The 6-his tag sequence was introduced after the signal peptide cleavage site near the amino terminus of hPGHS-1, as previously reported [27]. Nucleotides coding for the six histidine residues were inserted into the wild type hPGHS-1/pSG5 vector [25] and each of the vectors for the hPGHS-1 mutants using PCR with PfuTurbo DNA polymerase (Stratagene, CA) and appropriate primers:
The sense primer introduced a CACCATCAC sequence (coding for three histidines) before Asp26. The antisense primer introduced an ATGGTGATG sequence (coding for another three histidines) after Ala25. The template was denatured at 95 °C for 1 min, annealed at 60 °C for 50 s, and the extension reaction was at 68 °C for 12 min; there were 18 amplification cycles. The PCR mixture was digested with DpnI and the reaction products were separated by agarose gel electrophoresis; the 6 kb band was extracted and ligated with T4 DNA ligase (New England Biolabs, Ipswich, MA). Following transformation into E. coli strain DH5α, the ampicillin resistant colonies were analyzed by restriction enzyme digest and DNA sequencing.
Baculovirus generation and expression of recombinant proteins
The plasmids containing the histidine-tagged PGHS-1 constructs were each digested with BamHI and XbaI and cloned into the pVL1393 transfer vector (BD Biosciences, San Jose, CA) with T4 DNA ligase. The integrity of each resulting transfer vector was verified by restriction digestion and DNA sequencing. Generation, amplification, and titer determination of the recombinant baculoviruses containing the desired hPGHS-1 cDNA, and expression of the recombinant proteins used previously published techniques [28]; some expression runs were performed in a 5 L bioreactor by the Recombinant Protein Expression/Proteomics Core at the Baylor College of Medicine (Houston, Texas). Additional expression runs of the Y385F mutant of hPGHS-1 were conducted in medium supplemented 12 hr after infection with ibuprofen (0.1 mM), or heme (5 μM) / ALA (0.5 mM), or heme (5 μM) / ALA (0.5 mM) / ibuprofen (0.1 mM).
Purification of recombinant proteins
The purification procedure was modified from a published protocol [29]. After disruption of the Sf9 cells expressing the recombinant protein, the homogenate was centrifuged at 150,000 × g at 4 °C for 1 hr, and the pellet was resuspended with a Dounce homogenizer in 100 mL of 25 mM NaPi, 100 mM NaCl, 20 mM imidazole, pH 7.4, 0.1 mM phenol, 1% w/v Tween-20 and stirred for 1 hr at 4 °C. Unextracted membrane residue was removed by another round of centrifugation. The rest of the purification proceeded as described previously, except that 0.1 mM phenol was included in all buffers [29]. Holoenzyme was reconstituted by addition of heme in the presence of 50 μM phenol [17].
Protein Characterization
The concentrations of recombinant hPGHS-1 apoenzymes were determined by dot blot [28] or by a modified Lowry method [30]; holoenzyme concentrations were calculated from the absorbance at 411 nm (165 mM-1cm-1). hPGHS-1 mutants were analyzed by electrophoresis under denaturing conditions on 10% polyacrylamide gels and visualized either by Coomassie blue staining or by immunoblotting [28]; densitometry of Coomassie stained gels was used to assess homogeneity.
Cyclooxygenase activity
Oxygen uptake was assayed polarographically at 30 °C in 100 mM KPi, pH 7.2 with 1.0 mM phenol, 1.0 μM heme and 0.1% Tween-20 as described previously. One unit of cyclooxygenase activity has an optimal velocity of 1 nmol O2/min. Cyclooxygenase KM values were determined by measuring the activity with 1-100 μM AA and fitting the values to the Michaelis-Menten equation using SigmaPlot 8.0 (SPSS Inc). Cyclooxygenase self-inactivation rates were calculated by dividing the optimal velocity (nmol O2/min) by the reaction extent at complete self-inactivation (nmol O2 consumed) [31].
Assessment of cyclooxygenase activation efficiency
The efficiency of feedback activation by peroxide was assessed by measuring the cyclooxygenase activity of a fixed number of units of enzyme in the presence of varying amounts of GPx-1, using oPGHS-1 as a standard [28]. The cyclooxygenase activation index was calculated by dividing the slope of the activity vs. GPx-1 units/Cox units plot for oPGHS-1 by the corresponding slope for each hPGHS-1 construct.
Peroxidase activity
Peroxidase activity was calculated from the velocity of oxidation of guaiacol (5 mM; 6.7 A436 (mM-1cm-1) [32] in stopped-flow reactions at 25 °C with 1-60 μM PPHP in 0.1 M Tris, pH 8.0 [33]. One unit represents 1 nmol peroxide reduced/min. Vmax and KM values were determined by fitting the data to the Michaelis-Menten equation using SigmaPlot 8.0.
EPR and RFQ experiments
Initial EPR analysis of the hPGHS-1 tyrosyl radicals was done by hand-mixing enzyme with 5 eq of EtOOH in an EPR tube on ice for ~8 s and then plunging the tube in an ethanol/dry ice bath to freeze it rapidly. For freeze-thaw experiments, the EPR samples were thawed in an ice bath (0 °C) for the specified period of time, and then refrozen in an ethanol/dry ice bath. For complexes with indomethacin or meclofenamic acid, enzyme was preincubated with inhibitor (2 eq) at room temperature for 30 min (<10% of control activity) before reacting with EtOOH. RFQ experiments were performed with an Update Instruments (Madison, WI) System 1000 chemical/freeze quench apparatus with a Model 1019 syringe ram, a Model 715 ram controller, and a 0.008 inch nozzle [34]. The ram velocity was 2.0 cm/s, and the dead time was 4-5 ms.
EPR spectra were recorded with a Bruker EMX spectrometer at 9.2-9.3 GHz with a modulation amplitude of 2.0 G, a modulation frequency of 100 kHz, a time constant of 327 ms, a microwave power of 1 mW, and a temperature of 113 K. Radical concentrations were determined by double integration of the EPR signals with reference to a copper standard [15]; the calculations for RFQ-EPR samples used a packing correction factor of 0.45. Simulations of spectra used the Bruker Simfonia package. The modified McConnell relationship, Aiso = ρC1(βo + β2cos2θ), where Aiso is the isotropic hydrogen hyperfine coupling arising from interactions of a β hydrogen, ρC1 is the spin density at C1 of the phenyl ring (set at 0.38 [14]), and βo and β2 are constants with values of 0 and 162.4 mHz, respectively [35], permits calculation of θ, the dihedral angle between the β hydrogen and the pz orbital of the phenyl ring [36]. The most “satisfactory” simulation, when adjusted to the observed total spin concentration, was that with the smallest fractional standard error of estimate (S.E.E.) value:
| [1] |
Here yi’ is the observed EPR signal amplitude, yi is the simulated amplitude at the same magnetic field and n is the number of data points. The term in the denominator normalizes the calculated standard error of estimate for variations in the absolute EPR signal amplitude.
RESULTS
Electrophoretic analysis of hPGHS-1 proteins
Polyacrylamide gel electrophoresis with subsequent visualization by Coomasie staining or immunoblotting of the purified recombinant proteins revealed a major band at ~70 kDa for wild type hPGHS-1 and each of the mutants (data not shown). This confirms that all proteins were expressed as detergent-soluble, full-length proteins. The electrophoretic analysis indicated that the purified proteins were 80-90% homogeneous, except for the Y385F/Y504F double mutant, which was ~50% pure.
Cyclooxygenase activity
The specific activity, KM for AA, and cyclooxygenase inactivation rates were measured for each of the recombinant proteins (Table 1). The specific activity and KM values for wild type hPGHS-1 fell within the range of values reported previously for the purified recombinant protein [27, 37, 38]. The KM value for hPGHS-1 is about four-fold higher than that observed for oPGHS-1 [39]. This is probably due to the higher level of detergent used in the present assays, as increasing detergent concentration is known to increase the cyclooxygenase KM value [40]. Mutation of Tyr504 to phenylalanine resulted in a 24% decrease in cyclooxygenase specific activity, a smaller change than seen for the analogous mutation in hPGHS-2 [22], but did not change significantly the KM value for arachidonate (Table 1). The Y504F mutation also did not significantly change the cyclooxygenase inactivation rate, indicating that Tyr504 is not involved in the self-inactivation process, consistent with our recent finding that the ferryl-oxo heme is the major initiator of self-inactivation [41]. As expected, both Y385F and Y385F/Y504F mutants that lack the key Tyr385 residue completely lost cyclooxygenase activity (Table 1).
Table 1.
Cyclooxygenase and peroxidase kinetic parameters of hPGHS-1 proteins
| Cyclooxygenase | Peroxidase | ||||
|---|---|---|---|---|---|
| hPGHS-1 construct | Specific activity (u/μg) | KM (μM AA) | kinact(min-1) | Specific activity (u/μg) | KM (μM PPHP) |
| Wild type | 21 ± 8 | 12 ± 1 | 1.8 ± 0.1 | 160 ±40a | 32±11a |
| Y504F | 16 ± 3 | 11 ± 1 | 2.2 ± 0.3 | 310±100a | 260±50a |
| Y385F | Not detected | 6.5b | Not determined | ||
| Y385F/Y504F | Not detected | 22c | 28c | ||
Values for wild type and Y504F mutant represent the average ± standard deviation from at least two separate enzyme preparations. Specific activity calculated from fitted Vmax values.
Specific activity estimated from the average peroxidase velocity in quadruplicate assays (SD: ±0.2 u/μg) of one enzyme preparation at 100 μM PPHP. Three other preparations of the Y385F mutant expressed in Sf9 cells with special supplementation with ALA, heme, and/or ibuprofen also had very low peroxidase activities (data not shown).
Vmax and KM values determined from substrate dependence for one enzyme preparation. The specific activity was calculated from the fitted Vmax value. The standard deviation of the fit was ±5 u/μg for specific activity and ±11 μM for KM.
Peroxidase activity
Introduction of the Y504F mutation increased the peroxidase Vmax and KM values compared to wild type hPGHS-1 (Table 1). The fact that saturation required a higher peroxide level in the Y504F mutant indicates that this substitution makes oxidized enzyme intermediates more easily reduced by cosubstrate or impairs the interaction of enzyme with peroxide. Neither possibility can be ruled out from the position of Tyr504 in the crystallographic model (Fig. 1). The Y385F single mutation and Y385F/Y504F double mutation were very detrimental to peroxidase activity, retaining less than 15% of the wild type activity in the purified proteins (Table 1). Several chemical chaperone strategies were tried to improve expression of active protein for the Y385F mutant (Experimental Procedures). Each of these approaches gave similar, moderate amounts of recombinant protein that could be detergent-solubilized and purified without difficulty but nonetheless had a low peroxidase activity that precluded full kinetic analysis.
Cyclooxygenase activation efficiency in wild type and mutant hPGHS-1 and -2
Cyclooxygenase activation efficiency was assessed by titration with the peroxide scavenger, GPx-1[28]. The observed cyclooxygenase activity declined linearly with added GPx-1 for each of the enzymes tested, and cyclooxygenase activation index values were calculated from the ratio of the slope of the fitted line for the oPGHS-1 reference to the fitted line for each test protein (Table 2). The activation efficiency index for hPGHS-1 was almost one, indicating that the human and sheep enzymes were essentially identical. The index value for the hPGHS-1 Y504F mutant was only 0.67, indicating that mutation of Tyr504 markedly impaired the cyclooxygenase activation process.
Table 2.
Cyclooxygenase activation efficiencies of hPGHS-1, hPGHS-2 and their Y504F mutants. The activation efficiency values were determined by titrations with GPx-1 and are relative to an oPGHS-1 standard. Each value represents the average from two or three separate batches of recombinant protein. Details are described in Experimental Procedures.
| Protein | Cyclooxygenase Activation Efficiencya |
|---|---|
| hPGHS-1 | 0.98 ± 0.01 |
| hPGHS-1 Y504F | 0.67 ± 0.02b |
| hPGHS-2 | 5.1 ± 0.6 |
| hPGHS-2 Y504F | 2.6 ± 0.3c |
Relative to oPGHS-1 (= 1.00)
p = 0.012 compared to wild type hPGHS-1
p = 0.003 compared to wild type hPGHS-2
Tyrosyl radical formation kinetics in wild type and mutant hPGHS-1
Reaction of hPGHS-1 with excess EtOOH in RFQ experiments produced a 31 G (peak to trough) WS signal (Fig. 2A) that increased in intensity at a rate of 80 s-1 over the first 50 ms, and then leveled off (Fig. 2B). Minimal lineshape changes occurred during the first 100 ms, but later a 17 G hyperfine splitting began to form and became fully developed by 3-9 s (Fig. 2A). Reaction of hPGHS-1 with 5x EtOOH in hand-mixed experiments at 0 °C produced a 29 G (peak to trough) WS species centered at g = 2.004 with prominent 17 G hyperfine splitting similar to the 9s spectrum shown in Fig. 2A. The yield of the WS tyrosyl radical in hand-mixed samples was 0.2-0.5 spin/heme. Repeated cycles of thawing and re-freezing to allow further reaction caused a decrease in radical intensity without lineshape changes (data not shown). The WS observed in the RFQ samples reacted less than 100 ms (termed WS1) is distinct from the WS observed in the 9s RFQ sample and in hand-mixed samples (termed WS2). Compared to WS2, WS1 has a slightly wider signal (30-31 G in WS1 vs. 27-29 G in WS2; peak to trough), lacks the 17 G hyperfine splitting and has a higher half-saturation power, or P1/2, value (4.5 mW vs. 2.6 mW). No wide doublet signal was seen with hPGHS-1, even at the earliest time point (4 ms), in sharp contrast to oPGHS-1 but somewhat similar to that for hPGHS-2 [17].
Figure 2.
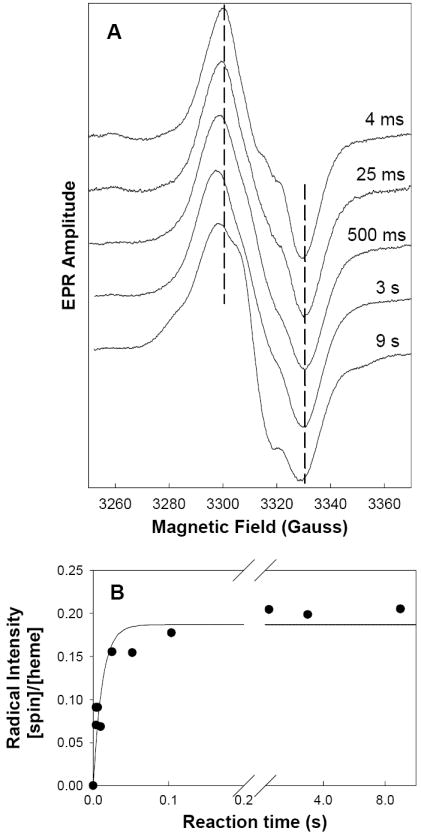
Radicals EPR signals during reaction of hPGHS-1 with EtOOH. (A) hPGHS-1 (47 μM heme) in 100 mM KPi, pH 7.2, 0.1% Tween-20, 10% glycerol was reacted at room temperature with 5 equiv of EtOOH for the indicated times before the sample was freeze-quenched and the EPR spectrum recorded. Vertical dashed lines indicate the peak and trough positions in the 4 ms spectrum. (B) The radical intensity in each sample was determined by double integration and normalized to the hPGHS-1 heme concentration. The line represents the first-order fit to the data.
Reaction of the Y504F mutant of hPGHS-1 with excess EtOOH in hand-mixed experiments formed a WD species, 30-33 G wide with ~16 G hyperfine splitting, centered at g = 2.003 (Fig. 3). The yield of this WD radical was only 0.01-0.04 spin/heme, roughly one tenth of the yield of WS1 radical in wild type hPGHS-1. Freeze-thawing the Y504F mutant samples to allow further reaction caused a small decrease in radical concentration, with no lineshape changes (data not shown). The P1/2 value of the Y504F WD was 9.5 mW at 110K. The low radical yield in reactions of the hPGHS-1 Y504F mutant with peroxide precluded RFQ studies.
Figure 3.
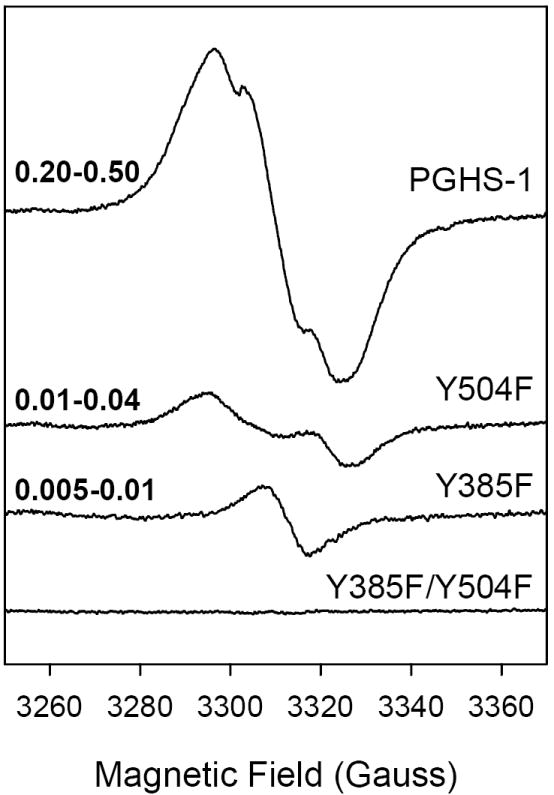
EPR spectra of radicals formed upon reaction of wild type or mutant hPGHS-1 with EtOOH. The samples (21 μM heme) in 100 mM KPi, pH 7.2, 0.1% Tween-20, 10% glycerol were reacted on ice with 5 equiv of EtOOH for ~10 s. The range of radical intensity (in spin/heme) obtained in replicate experiments is indicated at the left side of each spectrum.
Reaction of the Y385F single mutant with EtOOH by manual mixing gave a low yield (0.01 -0.03 spin/heme) 10-G radical centered at g = 2.0028 with P1/2 <1 mW (Fig. 3). This narrow radical species is very different from the WD, WS1 and WS2 radical species (Fig. 3), though the Y385F radical presumably resides on Tyr504. The Y385F/Y504F double mutant showed essentially no radical EPR upon reaction with EtOOH (Fig. 3). This suggests that Tyr385 and Tyr504 are the only significant sites of peroxide-induced radicals in hPGHS-1, though the decreased peroxidase activity in the double mutant (Table 1) indicates a decreased capacity to produce oxidized enzyme intermediates in general.
Tyrosyl radical in inhibitor complexes of wild type and mutant hPGHS-1
Indomethacin (2 fold)-treated hPGHS-1 reacted with EtOOH produced a NS EPR species, 24 G wide, centered at g = 2.004 with ~14 G hyperfine features and a P1/2 = 2.3 mW (Fig. 4). This radical signal was produced in yields of 0.18-0.50 spin/heme, essentially the same as for hPGHS-1 without inhibitor (Fig. 3). Longer reaction times did not change the EPR signal lineshape, but did result in decreased radical intensity (data not shown). Reaction of the indomethacin complex of the hPGHS-1 Y504F mutant with EtOOH produced only a very small amount (0.001-0.004 spin/heme) of a 9 G NS species with a P1/2 = 1.1 mW (Fig. 4). This EPR feature is very similar to that of EtOOH-induced radical in the Y385F mutant in both the presence or absence of indomethacin (Figs. 3 and 4). Inhibitor pre-treated Y385F/Y504F failed to show any detectable radical signal when reacted with peroxide.
Figure 4.
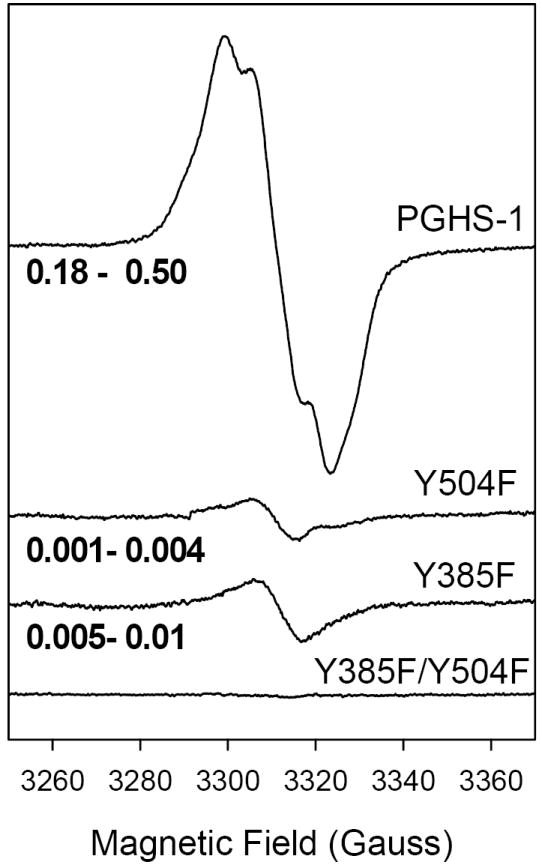
EPR spectra of radicals formed by indomethacin complexes of wild type and mutant hPGHS-1 upon reaction with EtOOH. Each sample (21 μM heme) in 100 mM KPi, pH 7.2, 0.1% Tween-20, was preincubated with 2 equiv of indomethacin before reaction on ice with 5 equiv of EtOOH for ~10 s. The range of radical intensity (in spin/heme) obtained in replicate experiments is indicated at the left side of each spectrum.
In contrast with indomethacin, meclofenamate has been found to inhibit oPGHS-1 cyclooxygenase activity without altering the EPR lineshape of the peroxide-induced tyrosyl radical [42]. For hPGHS-1, preincubation with meclofenamate inhibited the cyclooxygenase activity by 92%, confirming binding of the inhibitor. Reaction of the meclofenamate-hPGHS-1 complex with excess EtOOH produced a ~30 G WS EPR signal (Fig. 5) similar to that observed with inhibitor-free hPGHS-1 (Fig. 2). Thus, meclofenamate and indomethacin have divergent effects on tyrosyl radical distribution in hPGHS-1 and oPGHS-1 despite sharing cyclooxygenase inhibitory actions in both proteins.
Figure 5.
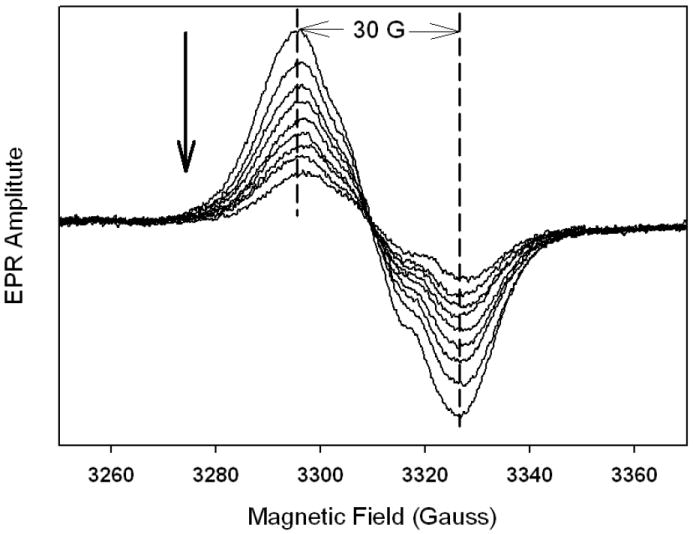
Tyrosyl radical EPR spectra during reaction of the meclofenamate complex of hPGHS-1 with EtOOH. hPGHS-1 (29 μM heme) pre-treated with 2 equivalents of meclofenamate in 100 mM KPi, pH 7.2, 0.1% Tween-20, was reacted on ice with 5 equiv of EtOOH for ~10 s, frozen, and the first EPR spectrum was recorded. For recording subsequent spectra, the sample was thawed and allowed to react at 0 °C for ~10 s before being re-frozen. The arrow indicates the direction of amplitude changes.
Analysis of tyrosyl radical spectra observed with hPGHS-1 and Y504F PGHS-1
The WD spectrum from Y504F hPGHS-1, the NS spectrum from indomethacin/hPGHS-1 and the WS1 spectrum from hPGHS-1 itself seemed the most likely to arise primarily from single radical species. Accordingly, the Bruker Simfonia program was used to simulate EPR powder pattern lineshapes to determine if a single tyrosyl radical could account for all these spectra, and to predict the sidechain conformation and spin distribution of the tyrosine radical(s) involved. The modified McConnell relationship, Aiso = ρC1(βo + β2cos2θ), where Aiso is the isotropic hydrogen hyperfine coupling arising from interactions of a β hydrogen, ρC1 is the spin density at C1 of the phenyl ring (set at 0.38 [14]), and βo and β2 are constants with values of 0 and 162.4 MHz, respectively [35], permits calculation of θ, the dihedral angle between the β hydrogen and the pz orbital at C1 of the phenyl ring [36]. It should be cautioned that these parameter values obtained by simulation of X-band EPR data may not be unique, and the use of other methods such as high-field EPR and ENDOR will be required for precise determination.
The Y504F hPGHS-1 WD can be simulated by a single tyrosyl radical with EPR parameters (Table 2) (Fig. 6A) that resemble those determined for Y504F hPGHS-2 [43], although the β hydrogen coupling is more isotropic in the former than in the latter. Due to the asymmetry of the EPR peak and trough, the S.E.E. is the largest, 0.32, in all the simulations (Fig. 6B). The calculated Cβ proton dihedral angles for the hPGHS-1 Y504F WD tyrosyl radical are 30° and -90°. This result presumably provides an estimate of the structure of the Tyr385 radical in hPGHS-1.
Figure 6.
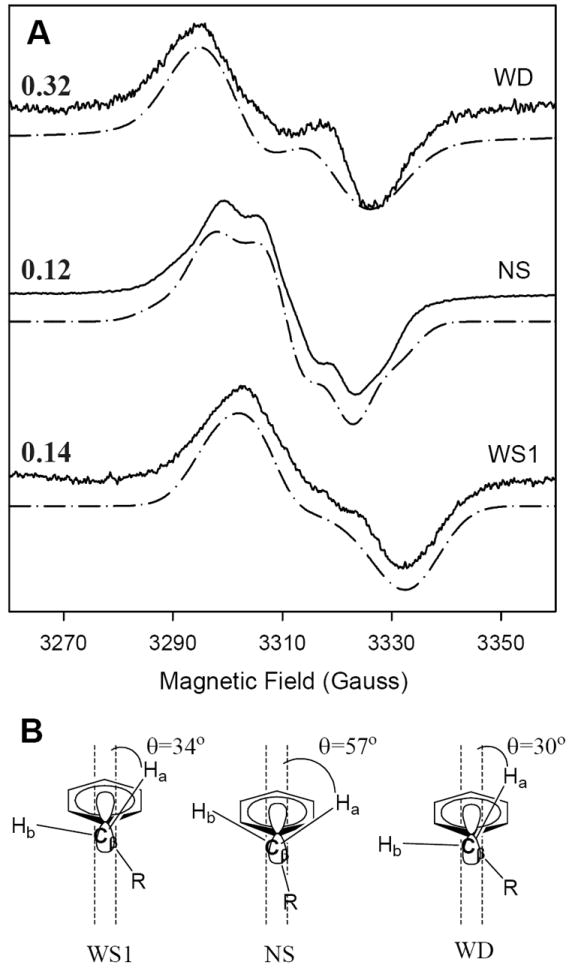
Simulation of peroxide-induced tyrosyl radical EPR signals from wild type and mutant hPGHS-1. (A) Experimental (––––) and simulated (– • –) EPR spectra of the wide doublet (WD; Y504F hPGHS-1, 9 s at 0 °C), narrow singlet (NS; indomethacin-hPGHS-1, 9 s at 0 °C), and early wide singlet (WS1; hPGHS-1, 4 ms at room temperature) EPR signals. Parameters for the simulations are given in Table 3. The S.E.E. values for the agreement between observed and simulated spectra are indicated at the left side. (B) Structural diagrams of the proposed conformations of the beta-methylene groups of tyrosyl radicals for the simulated spectra.
The NS species formed by the hPGHS-1/indomethacin complex upon reaction with EtOOH is well simulated (S.E.E. 0.12) by a single tyrosyl radical with the EPR parameters in Table 2 (Fig. 6A). These parameters are similar to those reported for oPGHS-1 NS [14] except that inclusion of two β hydrogens was required for a satisfactory fit. The EPR parameters for the indomethacin-hPGHS-1 NS signal correspond to Cβ proton dihedral angles of 57° and -63° (Fig. 6B).
The hPGHS-1 WS1 could be well simulated by a single tyrosyl radical with the parameters in Table 3 and S.E.E. value of 0.14 (Fig. 6A). The optimal value of the fitting parameters are similar to those for oPGHS-1 WS with more isotropic H3,5 hyperfine splitting values and dihedral angles for the Cβ protons of 34° and -86° [14]. The hPGHS-1 WS1 spectra could not be well reproduced by taking a weighted sum of the hPGHS-1 Y504F WD and the hPGHS-1 /indomethacin NS signals. However, the experience with oPGHS-1 and hPGHS-2 [13] cautions that the hPGHS-1 WS1 may well represent a combination of the WD signal and an NS signal that is distinct from the NS observed in inhibitor-treated hPGHS-1.
Table 3.
EPR parametersa used for simulation of WD, NS and WS1 signals from Y504F hPGHS-1, the indomethacin complex of hPGHS-1 and hPGHS-1 itself.
| Parameter | Y504F hPGHS-1 wide doublet (WD) | hPGHS-1/ indomethacin narrow singlet (NS) | hPGHS-1 wide singlet 1 (WS1) |
|---|---|---|---|
| gx | 2.0089 | 2.0059 | 2.0074 |
| gy | 2.0044 | 2.0040 | 2.0040 |
| gz | 2.0023 | 2.023 | 2.0030 |
| H3,5Ax | 6.2 | 9.7 | 8.5 |
| H3,5Ay | 6.2 | 2.6 | 7.4 |
| H3,5Az | 6.2 | 7.3 | 6.7 |
| Hβ1Ax | 13.3 | 12.5 | 13.0 |
| Hβ1Ay | 13.3 | 9.4 | 12.2 |
| Hβ1Az | 23.0 | 10.4 | 21.5 |
| Hβ2Ax | b--- | 11.4 | b--- |
| Hβ2Ay | --- | 9.4 | --- |
| Hβ2Az | --- | 9.4 | --- |
Hyperfine coupling values are given in gauss.
Values for only one beta proton are given as the hyperconjugation with the other beta proton is insignificant.
The WS2 has distinct hyperfine features with 17 G splitting, and very broad wings and most likely not a single radical species. As expected, the WS2 spectrum could not be adequately reproduced by weighted addition of the WD and NS spectra. Subtraction of the WS1 from WS2 signal after normalized to the same total spin yielded a symmetric 9-10 G EPR signal which is very similar to the radical observed for Y385F reaction with EtOOH (Fig. 3) (data not shown).
DISCUSSION
Structural basis of WD and WS in hPGHS-1 tyrosyl radical EPR
The early EPR observations of purified oPGHS-1 reacted with peroxide revealed an initial WD signal that converted over time to a WS signal [15, 19]. The WD was ascribed to a torsionally strained radical on Tyr385, and subsequent ring rotation in the Tyr385 radical to a relaxed conformation was suggested to give rise to a NS EPR signal, which mixed with the remaining WD to account for the transition to a WS signal [14, 18]. This explanation can be termed the “ring rotation” mechanism. A second possible mechanism for the WD → WS transition (the “alternate radical site” mechanism) postulates supplementation or replacement of the torsionally-strained Tyr385 radical with a torsionally-relaxed radical located on another tyrosine residue. The two radicals might form on parallel paths with differing kinetics, without direct electron transfer between the other tyrosine and the Tyr385 radical.
The “alternative radical site” mechanism has been seen in other enzyme systems including cytochrome c peroxidase [44], cytochrome c oxidase [45, 46], and ribonucleotide reductase [47, 48]. Formation of the alternate radical is often related to the redox function of the radical intermediate(s) and the alternative site residue need not have the same side chain structure. In PGHS, the observed alternative radical site at Tyr504 is the only other tyrosyl radical formed despite the fact that Trp387 is as close to the heme as either Tyr385 or Tyr504. Furthermore, there are 27 tyrosines in oPGHS-1 and 26 tyrosines in mouse PGHS-2. Tyr348, 404 and 148 have as good a predicted electron coupling efficiency as Tyr385 and Tyr504 but only the latter two residues were observed as sites of radical formation [22]. This is a strong indication of the non-random nature of the alternate radical site in PGHS and suggests the possibility of functional significance. As for examples of tyrosyl radical conformational changes, rotations of tyrosine radical side chains around Cα-Cβ and Cβ-C1 were found in two Class I ribonucleotide reductases by single-crystal HFepr [49, 50]. Rotation of these two bonds was proposed to have a direct impact on the proton-coupled electron transfer between the tyrosyl radical and the di-iron center [49]. Thus, distinguishing between the two possible types of tyrosyl radical dynamics in PGHS is necessary to understand the observed differences in tyrosyl radical species regarding their EPR spectral lineshape and their time-dependent changes, and to link them to the functional differences between hPGHS-1, oPGHS-1 and hPGHS-2.
The ring rotation mechanism has the virtue of simplicity, as it requires only one site for tyrosyl radical in the protein. A single tyrosyl radical site is supported by the results of nitric oxide trapping experiments, which identified nitric oxide adduct only at Tyr385 in oPGHS-1 [23]. Further, the ring rotation mechanism is easy to reconcile with the peroxide-induced NS EPR observed in complexes of oPGHS-1 and hPGHS-2 with some cyclooxygenase inhibitors [42, 51, 52]. These inhibitors abut Tyr385 in crystallographic models [53-55] and could quite plausibly alter the ring conformation of this tyrosine directly or indirectly.
Early evidence for the alternate radical site mechanism came from mutagenesis studies showing that an Y385F mutant of oPGHS-1 could still form a NS tyrosyl radical EPR signal [56]. Direct tests for alternate site radical formation and tyrosine ring rotation were later carried out on hPGHS-2 using tyrosine mutagenesis and EPR studies [22]. The results indicated that changes in the proportions of radical on Tyr385 and radical on Tyr504 accounted for the sequence of EPR signal changes observed in hPGHS-2. In this interpretation, the initial WD in hPGHS-2 originates from the Tyr385 radical, with subsequent formation of a Tyr504 radical contributing a NS signal that mixes with the WD signal to produce the observed WS spectrum.
Ring rotation of the Tyr385 radical was not observed with wild type hPGHS-2, but was seen with the Y348F/Y504F double mutant, where alternate site radical formation on Tyr504 was blocked and Tyr348, the hydrogen bonding partner of Tyr385, was mutated [22, 43]. The lack of this hydrogen bond allowed the Tyr385 radical to rotate slowly, giving rise to WD → NS signal changes similar to those observed in oPGHS-1 [43]. The hPGHS-2 results suggested that both ring rotation and alternate site radical formation might contribute to the signal changes in PGHS-1.
In the present study, the Y504F mutant of hPGHS-1 retained most cyclooxygenase and peroxidase activity, indicating that the substitution did not lead to nonspecific structural perturbation. In particular, formation of the key catalytic radical at Tyr385 was obviously not blocked by the Y504F mutation of hPGHS-1. However, the presence of phenylalanine, which is not readily oxidized, at position 504 of hPGHS-1 did result in production of a persistent WD EPR signal instead of the WS observed with the wild type (Figs. 2 and 3. The WD from hPGHS-1 Y504F can be accounted for by a single tyrosyl radical species (Table 3) and is similar to the WD observed in the Y504F mutant of hPGHS-2 and ascribed to the Tyr385 radical [22]. Thus, the present results with Y504F mutations of hPGHS-1 provide strong evidence that Tyr504 is an alternate tyrosyl radical site in hPGHS-1.
Interestingly, the yield of WD radical in the Y504 mutant is quite different for the two human isoforms. In hPGHS-2 the Y504F protein forms WD tyrosyl radical in yields close to that of the native enzyme [22], whereas in hPGHS-1 the WD radical yields from the Y504 mutant are significantly lower, only 4-10% that of the wild type (Fig. 4). This suggests that the radical accumulation on Tyr385 is lower in hPGHS-1 than in either hPGHS-2 or oPGHS-1.
With Tyr504 established as an alternate site for peroxide-derived radicals in hPGHS-1, the WS1 observed in wild type hPGHS-1 reasonably can be attributed to a mixture of a small portion of WD from the Tyr385 radical and a large portion of the signal from the Tyr504 radical. The observation that the WS1 spectrum can be simulated as a single radical species (Fig. 6 and Table 3) is consistent with a low WD contribution. The Y385F mutant of hPGHS-1 was constructed to isolate the Tyr504 radical for EPR characterization. Unfortunately, insertion of phenylalanine at residue 385 appears to lead to nonspecific structural perturbation in hPGHS-1, as evidenced by the large decrease in peroxidase activity in both the Y385F single mutant and the Y385F/Y504F double mutant (Table 1). This leads to some concern that the narrow and weak EPR signal observed in reaction of the Y385F mutant was reacted with peroxide (Fig. 3) does not represent a “normal” Tyr504 radical. The probable nonspecific disruption accompanying the Y385F substitution also means that the absence of EPR signal in peroxide-treated Y385F/Y504F double mutant (Fig. 3) is not absolute evidence against formation of radical at a third site besides Tyr385 and Tyr504.
The lack of lineshape change over time of the WD signals in Y504F mutants of both isoforms indicates that when the tyrosyl radical is confined to Tyr385, the tyrosyl sidechain does not undergo ring rotation despite being in a strained, energetically unfavorable conformation. Hydrogen bonding between Tyr385 and Tyr348 appears to be a factor in constraining ring rotation in hPGHS-2 [43], and the same may be true for hPGHS-1. The functional role, if any, of such constraints on Tyr385 ring rotation in PGHS-1 and -2, remains unclear, though Y348F mutations in oPGHS-1, hPGHS-2 and mPGHS-2 do result in modest decreases in cyclooxygenase activity [43, 57, 58].
Effects of Tyr385 mutations in hPGHS-1
The results of the present study reveal distinctions between hPGHS-1 and hPGHS-2 in the effects of mutation at Tyr385. The Y385F mutant of hPGHS-2 retains full peroxidase activity and produces an intense 24-26G NS EPR corresponding to the Tyr504 radical [22]. In addition, in hPGHS-2 summation of the NS from the Y385F mutant and the WD from the Y504F mutant can account for the lineshape and intensity of the wild type EPR. In contrast, the Y385F mutant of hPGHS-1 gave a peroxide-induced EPR signal that is too narrow and too weak to combine with the WD of the Y504F mutant to account for either the WS1 or WS2 signals observed with wild type (Fig. 3). Also, the peroxidase activity of hPGHS-1 Y385F is only a small fraction of the wild type activity (Table 1). These large differences were not a result of low protein expression, as the recombinant protein levels and electrophoretic purity of the hPGHS-1 Y385F mutant were close to those for the wild type. Similar radical yield and EPR lineshape were observed for hPGHS-1 Y385F that was expressed by Sf9 cells grown in medium supplemented with heme or flurbiprofen as chemical chaperones in attempts to reduce proteolytic susceptibility and improve folding of the recombinant protein [59] (data not shown). It thus appears that the hPGHS-1 protein is particularly susceptible to some structural perturbation from Tyr → Phe substitution at position 385 that is not confined to the cyclooxygenase pocket, leading to low radical production or conversion to a radical species other than the tyrosyl radical during peroxidase catalysis.
The radical with 10 G line width produced by the hPGHS-1 Y385F mutant (Fig. 3) is too narrow to be a tyrosyl radical or other well-known amino acid radicals [60] and the exact radical location is unresolved. Mutation induced protein structural changes could change the stability of the target radical, as observed with the KatG catalase/peroxidase amino acid radical [61, 62]. Perturbation of the heme geometry by the Y385F mutation and thus the nature of its peroxide-induced radical cannot be excluded. It is possible that this 10 G radical may be associated with peroxidase inactivation at longer reaction times as its EPR lineshape is similar to that of the difference EPR between WS2 and WS1.
Tyr504 and peroxidase kinetics
The evaluations of peroxidase kinetics in hPGHS-1 and -2 with Y504F mutations (Table 1 and [22]) provide an initial glimpse into the possible role of Tyr504 in peroxidase catalysis, either as the site of peroxide-derived oxidant or in some other capacity. PGHS peroxidase follows a sequential bisubstrate kinetic mechanism [4]. Consequently, the increased KM for peroxide observed with Y504F mutants of both hPGHS-1 and -2 (Table 1 and [22]) could reflect either altered binding and reaction with peroxide or altered binding and reaction with the reducing cosubstrate [63] or a combination of the two.
Tyr504 is located near the heme in both PGHS isoforms (Fig. 1 and [54, 64, 65]). In oPGHS-1 Tyr504 interacts with the proximal heme ligand, His388, via H-bonding with a structured water [64]. Accordingly, a Tyr504 mutation might indirectly perturb the heme structure and impair the binding of peroxide or the reactivity of heme, thus increasing the KM value. In this connection, mutation of a tryptophan radical site ~ 7 Å away from the heme propionate of KatG catalase/peroxidase was able to modulate the heme structure via an H-bonding network [61, 62]. Due to low expression yield of the Y504F hPGHS-1, it is difficult to examine directly the effect of mutation on the heme by EPR, as was done with KatG catalase/peroxidase [62].
An alternative explanation for the altered peroxidase kinetics in Y504F mutants involves changes in interactions with cosubstrate. Tyr504 is one of three sites for peroxide-derived oxidant in both isoforms. An Y504F mutation restricts oxidant to the heme and Tyr385, both of which appear to be more exposed than Tyr504 to small, water soluble reductants in crystallographic models [54, 64, 65]. This would increase the proportion of oxidant susceptible to reduction and raise the level of peroxide needed for saturation, thus increasing the KM value for peroxide indirectly. Distinguishing between the two possible explanations for the effects of Y504F mutations on peroxidase kinetics will require more detailed analysis of peroxidase kinetics to identify the step(s) affected by the Y504F mutations.
Tyr504 and cyclooxygenase catalysis
Peroxide-derived oxidant has a central role in cyclooxygenase catalysis [13, 66]. The discovery that Tyr504 can accumulate a considerable portion of peroxide-derived oxidant in both hPGHS-1 and -2 indicates that Tyr504 may have a significant role in cyclooxygenase catalysis. The Y504F substitution results in only modest decreases in hPGHS-1 and -2 cyclooxygenase activity under Vmax assay conditions (Table 1 and [22]), demonstrating that Tyr504 does not have a direct role in cyclooxygenase catalysis in either isoform. This is not surprising given the distance between Tyr504 and the cyclooxygenase pocket in crystallographic models [54, 64, 65]. However, assays under conditions where peroxide levels are limiting reveal that an Y504F mutation significantly decreases the cyclooxygenase activation efficiency in both hPGHS-1 and -2 (Table 2). This indicates that the ability to form a Tyr504 radical actually is important to regulation of cyclooxygenase catalysis by peroxide in both human isoforms. Crystallographic data places Tyr504 at some distance from bound fatty acid in both PGHS-1 and -2 [54, 64, 65], so the effect of the Tyr504 radical on cyclooxygenase catalysis is likely to have an indirect mechanism.
If formation of a Tyr504 radical simply diverted oxidant from the catalytically active radical at Tyr385, a Y504F mutation should increase the concentration of the Tyr385 radical and boost cyclooxygenase activation efficiency. The fact that Y504F mutation actually decreases activation efficiency in both isoforms (Table 2) rules out a simple reciprocal relationship between oxidant at Tyr385 and Tyr504 and requires a mechanism providing a positive effect of radical formation at Tyr504 on cyclooxygenase activation. Cyclooxygenase activation involves generation and retention of oxidized protein intermediates for use in cyclooxygenase catalysis [66, 67]. As noted above, the elevated peroxidase KM values observed in Y504F mutants of both hPGHS-1 and hPGHS-2 suggest that Tyr504 has a role in sustaining levels of peroxide-derived oxidant. Both oxidant generation and oxidant retention will need to be examined in the context of Tyr504 mutants to resolve this question fully.
Redistribution of oxidant for cyclooxygenase catalysis
Radical on Tyr504 is quite some distance from the position of bound fatty acid and so the oxidant would have to be redistributed to Tyr385 (presumably via the heme) for cyclooxygenase catalysis. In both PGHS isoforms, binding of competitive cyclooxygenase inhibitors can change the distribution of protein oxidant upon subsequent reaction with peroxide (Figs. 3 and 4; [22]), so it is plausible that fatty acid binding in the cyclooxygenase site triggers conformational changes that promote redistribution of radical from Tyr504 to Tyr385. There is evidence from oPGHS-1 for long range conformational linkages between the cyclooxygenase and peroxidase regions that might lead to redistribution of oxidant. Binding of ligand (inhibitor) to the cyclooxygenase site changes the conformation of the Arg277 loop near the peroxidase site, rendering it resistant to protease attack [59]. Cyclooxygenase inhibitor binding also increases the rate of tyrosyl radical formation, presumably at Tyr504 [17]. In addition, binding of heme at the oPGHS-1 peroxidase site increases the affinity for inhibitors at the cyclooxygenase site [68].
Redistribution of peroxide-derived oxidant by cyclooxygenase inhibitors
The effects of cyclooxygenase inhibitors provide a useful window into the management of peroxide-derived oxidant in the PGHS isoforms. Complexation of hPGHS-1 with the cyclooxygenase inhibitor indomethacin resulted in production of a NS signal, whereas the indomethacin complex of Y504F hPGHS-1 was essentially radical-free upon reaction with peroxide (Fig. 4). Thus, binding of indomethacin determined the distribution of peroxide-derived oxidant in hPGHS-1, blocking formation of the Tyr385 radical and promoting formation of the Tyr504 radical. Similar behavior was seen with hPGHS-2, where nimesulide binding blocked subsequent peroxide-induced formation of the Tyr385 radical and enhanced formation of the Tyr504 radical (23). Note that this effect of cyclooxygenase inhibitors on oxidant distribution (favoring radical on Tyr504) is opposite to that proposed above for fatty acids (favoring radical on Tyr385). Such distinct actions would not be totally surprising given the large structural differences between anti-cyclooxygenase agents, which typically contain rigid structural elements, and fatty acids, which have much more conformational flexibility. Thus, the conformational perturbations caused by AA and cyclooxygenase inhibitor should be quite different. Crystallographic studies have thus far been unable to identify the structural changes involved [53, 54, 65, 69-72].
For oPGHS-1 the ability to change the distribution of oxidant is known to be dependent on the structure of the inhibitor, with fenamates lacking the effect seen with other inhibitors [42]. The contrast between indomethacin and meclofenamate in their effects on the peroxide-induced radical EPR in hPGHS-1 (Figs. 4 and 5) and previously in oPGHS-1 [42] suggests a similar dependence of oxidant redistribution on inhibitor structure. Clearly, the two contrasting groups of cyclooxygenase inhibitors will be valuable probes in experiments to define which steps in the formation and dissipation of peroxide-derived oxidants on heme, Tyr385 and Tyr504 are affected by ligands in the cyclooxygenase site.
Tyrosyl radical dynamics
In hPGHS-1, radical appears to accumulate faster and to a greater extent at Tyr504 than at Tyr385, as revealed by the lack of observable WD formation even early in the wild type reaction and the lower radical yield in the Y504F mutant (Figs. 2 and 3). In hPGHS-2, radical accumulation at the two tyrosines is more closely balanced, with a Tyr385 WD dominating initially, but then being joined by comparable levels of a Tyr504 NS within about 50 ms [17, 22]. In oPGHS-1, the initial WD forms more slowly than in hPGHS-2, and the transition between WD and WS is even slower, taking many seconds to complete [17]. Presumably formation of both the Tyr385 and the Tyr504 radicals are slower in oPGHS-1 than in hPGHS-2. Thus, there are many differences in the behavior of the Tyr385 and Tyr504 radicals between hPGHS-1 and -2, and even between hPGHS-1 and oPGHS-1.
hPGHS-1 and oPGHS-1 are 93% identical in protein sequence, but examination of the oPGHS-1 crystallographic structure [64] reveals several differences in the vicinity of Tyr504 (Fig. 7). In oPGHS-1, residue 440 (~5 Å from 504) is an isoleucine, whereas hPGHS-1 has methionine at that position. Residue 525 is methionine in oPGHS-1 but isoleucine in hPGHS-1; and residue 383, which lies directly over Tyr504, is glutamine in oPGHS-1 and histidine in hPGHS-1. The characteristics of the residues surrounding a tyrosyl radical can influence its stability [62, 73], and the variations in the Tyr504 environment among the PGHS proteins may account for much of the observed differences in their Tyr504 radicals. It is now becoming clear that the wide variety of tyrosyl radical EPR signals observed in the two PGHS isoforms originates primarily from variations in the kinetics of radical generation at Tyr385 and Tyr504, both residues that are conserved in all known PGHS proteins. Characterizing the distribution of peroxide-derived oxidant among the heme and these two tyrosine residues and its relationship to control of cyclooxygenase activation will advance understanding of catalytic regulation in these important enzymes.
Figure 7.
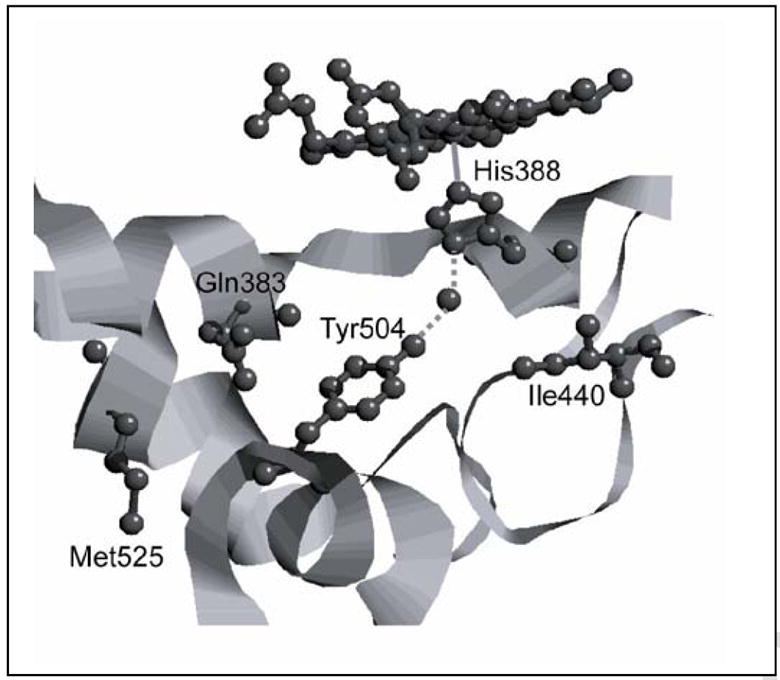
Structure of the vicinity of Tyr504 in oPGHS-1 showing Ile440, Gln383, Met525, His388 and Mn-PPIX (PDB entry 2AYL: Gupta K et al. (2006) Acta Cryst D 62, 151). The corresponding amino acid residues in hPGHS-1 are indicated after the position number.
Summary
Tyr385 and Tyr504 are conserved in all known PGHS proteins, from mammals to coelenterates. The present results with hPGHS-1 make it clear that the wide variety of tyrosyl radical EPR signals observed in the two PGHS isoforms originates primarily from variations in the kinetics of radical generation at Tyr385 and Tyr504. hPGHS-1 is structurally more sensitive to the Y385F mutation than either hPGHS-2 or oPGHS-1. The tyrosyl radical dynamics of hPGHS-1 are quite different from those of hPGHS-2 or oPGHS-1, with two WS (and no WD) tyrosyl radical intermediates observed during reaction with peroxide. The present results also show that radical distribution between Tyr385 and Tyr504 is also very different for hPGHS-1 and hPGHS-2 (or oPGHS-1). Tyr504 appears to be functionally important to both PGHS-1 and -2. Thus, the way is opened for experiments to determine the factors controlling oxidant distribution and stability and their relation to cyclooxygenase catalytic control in these important enzymes.
Acknowledgments
We thank Dr. G. Barney Ellison at the University of Colorado for the generous gift of EtOOH. We also thank Dr. Gang Wu for help with evaluation of the EPR simulations. This work was supported by United States Public Service Grants GM44911 (to A.-L.T.) and GM52170 (to R.J.K).
ABBREVIATIONS
- PGHS-1
prostaglandin H synthase-1
- oPGHS-1
ovine PGHS-1
- hPGHS-1
human PGHS-1
- PGHS-2
prostaglandin H synthase-2
- ALA
delta-amino levulinic acid
- WD
wide doublet tyrosyl radical
- WS
wide singlet tyrosyl radical
- NS
narrow singlet tyrosyl radical
- ENDOR
electron nuclear double resonance spectroscopy
- AA
arachidonic acid
- TMPD
N,N,N’,N’-tetramethyl-p-phenylenediamine
- RFQ
rapid freeze quench
- PPIX
protoporphyrin IX
- PPHP
trans-5-phenyl-4-pentenyl-1-hydroperoxide
- GPx-1
cytosolic glutathione peroxidase
- S.E.E
standard error of estimation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Smith WL, Garavito RM, DeWitt DL. J Biol Chem. 1996;271:33157–33160. doi: 10.1074/jbc.271.52.33157. [DOI] [PubMed] [Google Scholar]
- 2.Herschman HR. Biochim Biophys Acta. 1996;1299:125–140. doi: 10.1016/0005-2760(95)00194-8. [DOI] [PubMed] [Google Scholar]
- 3.Smith WL, Eling TE, Kulmacz RJ, Marnett LJ, Tsai A-L. Biochemistry. 1992;31:3–7. doi: 10.1021/bi00116a001. [DOI] [PubMed] [Google Scholar]
- 4.Lambeir AM, Markey CM, Dunford HB, Marnett LJ. J Biol Chem. 1985;260:14894–14896. [PubMed] [Google Scholar]
- 5.Dietz R, Nastainczyk W, Ruf HH. Eur J Biochem. 1988;171:321–328. doi: 10.1111/j.1432-1033.1988.tb13793.x. [DOI] [PubMed] [Google Scholar]
- 6.Sjoberg BM, Reichard P, Graslund A, Ehrenberg A. J Biol Chem. 1978;253:6863–6865. [PubMed] [Google Scholar]
- 7.Ivancich A, Jouve HM, Sartor B, Gaillard J. Biochemistry. 1997;36:9356–9364. doi: 10.1021/bi970886s. [DOI] [PubMed] [Google Scholar]
- 8.Debus RJ, Barry BA, Babcock GT, McIntosh L. Biophys J. 1988;53:A270–A270. [Google Scholar]
- 9.Debus RJ, Barry BA, Sithole I, Babcock GT, McIntosh L. Biochemistry. 1988;27:9071–9074. doi: 10.1021/bi00426a001. [DOI] [PubMed] [Google Scholar]
- 10.Rogers MS, Dooley DM. Cur Op Chem Biol. 2003;7:189–196. doi: 10.1016/s1367-5931(03)00024-3. [DOI] [PubMed] [Google Scholar]
- 11.Chouchane S, Girotto S, Yu SW, Magliozzo RS. J Biol Chem. 2002;277:42633–42638. doi: 10.1074/jbc.M207916200. [DOI] [PubMed] [Google Scholar]
- 12.Pesavento RP, Van Der Donk WA. Adv Prot Chem. 2001;58:317–385. doi: 10.1016/s0065-3233(01)58008-0. [DOI] [PubMed] [Google Scholar]
- 13.Tsai A-L, Kulmacz RJ. Prostagl & Oth Lip Med. 2000;62:231–254. doi: 10.1016/s0090-6980(00)00083-6. [DOI] [PubMed] [Google Scholar]
- 14.Shi W, Hoganson CW, Espe M, Bender CJ, Babcock GT, Palmer G, Kulmacz RJ, Tsai A-L. Biochemistry. 2000;39:4112–4121. doi: 10.1021/bi992561c. [DOI] [PubMed] [Google Scholar]
- 15.Tsai A-L, Palmer G, Kulmacz RJ. J Biol Chem. 1992;267:17753–17759. [PubMed] [Google Scholar]
- 16.Lassmann G, Odenwaller R, Curtis JF, DeGray JA, Mason RP, Marnett LJ, Eling TE. J Biol Chem. 1991;266:20045–20055. [PubMed] [Google Scholar]
- 17.Tsai A-L, Wu G, Palmer G, Bambai B, Koehn JA, Marshall PJ, Kulmacz RJ. J Biol Chem. 1999;274:21695–21700. doi: 10.1074/jbc.274.31.21695. [DOI] [PubMed] [Google Scholar]
- 18.DeGray JA, Lassmann G, Curtis JF, Kennedy TA, Marnett LJ, Eling TE, Mason RP. J Biol Chem. 1992;267:23583–23588. [PubMed] [Google Scholar]
- 19.Kulmacz RJ, Ren Y, Tsai A-L, Palmer G. Biochemistry. 1990;29:8760–8771. doi: 10.1021/bi00489a037. [DOI] [PubMed] [Google Scholar]
- 20.Xiao G, Tsai A-L, Palmer G, Boyar WC, Marshall PJ, Kulmacz RJ. Biochemistry. 1997;36:1836–1845. doi: 10.1021/bi962476u. [DOI] [PubMed] [Google Scholar]
- 21.Dorlet P, Seibold SA, Babcock GT, Gerfen GJ, Smith WL, Tsai A-L, Un S. Biochemistry. 2002;41:6107–6114. doi: 10.1021/bi015871f. [DOI] [PubMed] [Google Scholar]
- 22.Rogge CE, Liu W, Wu G, Wang LH, Kulmacz RJ, Tsai AL. Biochemistry. 2004;43:1560–1568. doi: 10.1021/bi035717o. [DOI] [PubMed] [Google Scholar]
- 23.Goodwin DC, Gunther MR, Hsi LC, Crews BC, Eling TE, Mason RP, Marnett LJ. J Biol Chem. 1998;273:8903–8909. doi: 10.1074/jbc.273.15.8903. [DOI] [PubMed] [Google Scholar]
- 24.Barry BA, el-Deeb MK, Sandusky PO, Babcock GT. J Biol Chem. 1990;265:20139–20143. [PubMed] [Google Scholar]
- 25.Ren Y, Ruan KH, Walker C, Kulmacz RJ. Adv Prostagl Thromb Leuk Res. 1995;23:113–115. [PubMed] [Google Scholar]
- 26.Ulrich CM, Bigler J, Sibert J, Greene EA, Sparks R, Carlson CS, Potter JD. Hum Mut. 2002;20:409–410. doi: 10.1002/humu.9080. [DOI] [PubMed] [Google Scholar]
- 27.Smith T, Leipprandt J, DeWitt D. Arch Biochem Biophys. 2000;375:195–200. doi: 10.1006/abbi.1999.1659. [DOI] [PubMed] [Google Scholar]
- 28.Bambai B, Rogge CE, Stec B, Kulmacz RJ. J Biol Chem. 2004;279:4084–4092. doi: 10.1074/jbc.M304762200. [DOI] [PubMed] [Google Scholar]
- 29.MirAfzali Z, Leipprandt JR, McCracken JL, DeWitt DL. Arch Biochem Biophys. 2005;443:60–65. doi: 10.1016/j.abb.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 30.Peterson GL. Meth Enzymol. 1983;91:95–119. doi: 10.1016/s0076-6879(83)91014-5. [DOI] [PubMed] [Google Scholar]
- 31.Smith WL, Lands WE. J Biol Chem. 1972;247:1038–1047. [PubMed] [Google Scholar]
- 32.Maehly AC, Chance B. Meth Enzymol. 1955;2:764–775. [Google Scholar]
- 33.Shimokawa T, Smith WL. J Biol Chem. 1991;266:6168–6173. [PubMed] [Google Scholar]
- 34.Tsai A-L, Berka V, Kulmacz RJ, Wu G, Palmer G. Anal Biochem. 1998;264:165–171. doi: 10.1006/abio.1998.2774. [DOI] [PubMed] [Google Scholar]
- 35.Bender CJ, Sahlin M, Babcock GT, Barry BA, Chandrashekar TK, Salowe SP, Stubbe J, Lindstrom B, Petersson L, Ehrenberg A, Sjoberg BM. J Am Chem Soc. 1989;111:8076–8083. [Google Scholar]
- 36.Fessenden RW, Schuler RH. J Chem Phys. 1963;39:2147–2195. [Google Scholar]
- 37.Barnett J, Chow J, Ives D, Chiou M, MacKenzie R, Osen E, Nguyen B, Tsing S, Bach C, Freire J, Chan H, Sigal E, Ramesha C. Biochim Biophys Acta. 1994;1209:130–139. doi: 10.1016/0167-4838(94)90148-1. [DOI] [PubMed] [Google Scholar]
- 38.Gierse JK, Hauser SD, Creely DP, Koboldt C, Rangwala SH, Isakson PC, Seibert K. Biochem J. 1995;305:479–484. doi: 10.1042/bj3050479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu W, Cao D, Oh SF, Serhan CN, Kulmacz RJ. FASEB J. 2006;20:1097–1108. doi: 10.1096/fj.05-5273com. [DOI] [PubMed] [Google Scholar]
- 40.Rand Doyen J, Yucer N, Lichtenberger LM, Kulmacz RJ. Prostagl Oth Lip Med. 2008;85:134–143. doi: 10.1016/j.prostaglandins.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu G, Rogge CE, Wang JS, Kulmacz RJ, Palmer G, Tsai AL. Biochemistry. 2007;46:534–542. doi: 10.1021/bi061859h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kulmacz RJ, Palmer G, Tsai A-L. Mol Pharmacol. 1991;40:833–837. [PubMed] [Google Scholar]
- 43.Rogge CE, Ho B, Liu W, Kulmacz RJ, Tsai AL. Biochemistry. 2006;45:523–532. doi: 10.1021/bi051235w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barrows TP, Bhaskar B, Poulos TL. Biochemistry. 2004;43:8826–8834. doi: 10.1021/bi049531g. [DOI] [PubMed] [Google Scholar]
- 45.Lemma-Gray P, Weintraub ST, Carroll CA, Musatov A, Robinson NC. FEBS Lett. 2007;581:437–442. doi: 10.1016/j.febslet.2006.12.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Svistunenko DA, Wilson MT, Cooper CE. Biochim Biophys Acta. 2004;1655:372–380. doi: 10.1016/j.bbabio.2003.06.006. [DOI] [PubMed] [Google Scholar]
- 47.Potsch S, Lendzian F, Ingemarson R, Hornberg A, Thelander L, Lubitz W, Lassmann G, Graslund A. J Biol Chem. 1999;274:17696–17704. doi: 10.1074/jbc.274.25.17696. [DOI] [PubMed] [Google Scholar]
- 48.Wiertz FG, Richter OM, Cherepanov AV, MacMillan F, Ludwig B, de Vries S. FEBS Lett. 2004;575:127–130. doi: 10.1016/j.febslet.2004.08.048. [DOI] [PubMed] [Google Scholar]
- 49.Galander M, Uppsten M, Uhlin U, Lendzian F. J Biol Chem. 2006;281:31743–31752. doi: 10.1074/jbc.M605089200. [DOI] [PubMed] [Google Scholar]
- 50.Hogbom M, Galander M, Andersson M, Kolberg M, Hofbauer W, Lassmann G, Nordlund P, Lendzian F. Proc Natl Acad Sci U S A. 2003;100:3209–3214. doi: 10.1073/pnas.0536684100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goodwin DC, Rowlinson SW, Marnett LJ. Biochemistry. 2000;39:5422–5432. doi: 10.1021/bi992752f. [DOI] [PubMed] [Google Scholar]
- 52.Tsai A-L, Palmer G, Xiao G, Swinney DC, Kulmacz RJ. J Biol Chem. 1998;273:3888–3894. doi: 10.1074/jbc.273.7.3888. [DOI] [PubMed] [Google Scholar]
- 53.Loll PJ, Picot D, Garavito RM. Nat Struc Biol. 1995;2:637–643. doi: 10.1038/nsb0895-637. [DOI] [PubMed] [Google Scholar]
- 54.Luong C, Miller A, Barnett J, Chow J, Ramesha C, Browner MF. Nat Struc Biol. 1996;3:927–933. doi: 10.1038/nsb1196-927. [DOI] [PubMed] [Google Scholar]
- 55.Picot D, Loll PJ, Garavito RM. Nature. 1994;367:243–249. doi: 10.1038/367243a0. [DOI] [PubMed] [Google Scholar]
- 56.Tsai A-L, Hsi LC, Kulmacz RJ, Palmer G, Smith WL. J Biol Chem. 1994;269:5085–5091. [PubMed] [Google Scholar]
- 57.Hsi LC, Hoganson CW, Babcock GT, Garavito RM, Smith WL. Biochem Biophys Res Commun. 1995;207:652–660. doi: 10.1006/bbrc.1995.1237. [DOI] [PubMed] [Google Scholar]
- 58.Hochgesang GP, Rowlinson SW, Marnett LJ. J Am Chem Soc. 2000;122:6514–6515. [Google Scholar]
- 59.Kulmacz RJ. J Biol Chem. 1989;264:14136–14144. [PubMed] [Google Scholar]
- 60.Stubbe J, Van der Donk WA. Chem Rev. 1998;98:705–762. doi: 10.1021/cr9400875. [DOI] [PubMed] [Google Scholar]
- 61.Ivancich A, Jakopitsch C, Auer M, Un S, Obinger C. J Am Chem Soc. 2003;125:14093–14102. doi: 10.1021/ja035582+. [DOI] [PubMed] [Google Scholar]
- 62.Jakopitsch C, Obinger C, Un S, Ivancich A. J Inorg Biochem. 2006;100:1091–1099. doi: 10.1016/j.jinorgbio.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 63.Kulmacz RJ. Arch Biochem Biophys. 1986;249:273–285. doi: 10.1016/0003-9861(86)90003-2. [DOI] [PubMed] [Google Scholar]
- 64.Gupta K, Selinsky BS, Kaub CJ, Katz AK, Loll PJ. J Mol Biol. 2004;335:503–518. doi: 10.1016/j.jmb.2003.10.073. [DOI] [PubMed] [Google Scholar]
- 65.Kurumbail RG, Stevens AM, Gierse JK, McDonald JJ, Stegeman RA, Pak JY, Gildehaus D, Miyashiro JM, Penning TD, Seibert K, Isakson PC, Stallings WC. Nature. 1996;384:644–648. doi: 10.1038/384644a0. [DOI] [PubMed] [Google Scholar]
- 66.van der Donk WA, Tsai A-L, Kulmacz RJ. Biochemistry. 2002;41:15451–15458. doi: 10.1021/bi026938h. [DOI] [PubMed] [Google Scholar]
- 67.Wei C, Kulmacz RJ, Tsai A-L. Biochemistry. 1995;34:8499–8512. doi: 10.1021/bi00026a034. [DOI] [PubMed] [Google Scholar]
- 68.Chen YN, Marnett LJ. FASEB J. 1989;3:2294–2297. doi: 10.1096/fasebj.3.11.2506093. [DOI] [PubMed] [Google Scholar]
- 69.Loll PJ, Picot D, Ekabo O, Garavito RM. Biochemistry. 1996;35:7330–7340. doi: 10.1021/bi952776w. [DOI] [PubMed] [Google Scholar]
- 70.Rowlinson SW, Kiefer JR, Prusakiewicz JJ, Pawlitz JL, Kozak KR, Kalgutkar AS, Stallings WC, Kurumbail RG, Marnett LJ. J Biol Chem. 2003;278:45763–45769. doi: 10.1074/jbc.M305481200. [DOI] [PubMed] [Google Scholar]
- 71.Loll PJ, Sharkey CT, O’Connor SJ, Dooley CM, O’Brien E, Devocelle M, Nolan KB, Selinsky BS, Fitzgerald DJ. Mol Pharmacol. 2001;60:1407–1413. doi: 10.1124/mol.60.6.1407. [DOI] [PubMed] [Google Scholar]
- 72.Selinsky BS, Gupta K, Sharkey CT, Loll PJ. Biochemistry. 2001;40:5172–5180. doi: 10.1021/bi010045s. [DOI] [PubMed] [Google Scholar]
- 73.Ormö M, Regnström K, Wang Z, Que LJ, Sahlin M, Sjöberg B-M. J Biol Chem. 1995;270:6570–6576. doi: 10.1074/jbc.270.12.6570. [DOI] [PubMed] [Google Scholar]