

Abstract
While there is significant information concerning the consequences of cerebral ischemia on neuronal function, relatively little is known about functional responses of astrocytes, the predominant glial-cell type in the central nervous system (CNS). In this study, we asked whether focal ischemia would impact astrocytic Ca2+ signaling, a characteristic form of excitability in this cell type. In vivo Ca2+ imaging of cortical astrocytes was performed using two-photon (2-P) microscopy during the acute phase of photothrombosis-induced ischemia initiated by green light illumination of circulating Rose Bengal. Although whisker evoked potentials were reduced by over 90% within minutes of photothrombosis, astrocytes in the ischemic core remained structurally intact for a few hours. In vivo Ca2+ imaging showed that an increase in transient Ca2+ signals in astrocytes within 20 min of ischemia. These Ca2+ signals were synchronized and propagated as waves amongst the glial network. Pharmacological manipulations demonstrated that these Ca2+ signals were dependent on activation of metabotropic glutamate receptor 5 (mGluR5) and metabotropic γ-aminobutyric acid receptor (GABABR) but not by P2 purinergic receptor or A1 adenosine receptor. Selective inhibition of Ca2+ in astrocytes with BAPTA significantly reduced the infarct volume, demonstrating that the enhanced astrocytic Ca2+ signal contributes to neuronal damage presumably through Ca2+-dependent release of glial glutamate. Since astrocytes offer multiple functions in close communication with neurons and vasculature, the ischemia-induced increase in astrocytic Ca2+ signaling may represent an initial attempt for these cells to communicate with neurons or provide feed back regulation to the vasculature.
Keywords: two-photon imaging, blood flow, stroke, mGluR5, GABAB receptor, penumbra
Introduction
Cerebral ischemia is a major neurological disorder in which blood supply in the brain is reduced by cerebral thrombosis and embolism. Severe ischemia causes acute neuronal death within minutes in the ischemic core, whereas secondary neuronal death occurs in the penumbra within hours (Stapf and Mohr, 2002; Huang and McNamara, 2004). In experimental models of focal cerebral ischemia, several mechanisms including glutamate and Ca2+ toxicity, oxidative stress, inflammation, mitochondrial dysfunction, and acidosis are known to contribute to neuronal death (Barber and Demchuk, 2003; Xiong et al., 2004; Mattiasson et al., 2003). Astrocytes are the predominant glial-cell type in the CNS but little is known about functional impact of ischemia on astrocytes. Astrocytes make close contacts with neurons as well as with blood vessels by extending end-feet to ensheath the vessel walls in the brain (Bushong et al., 2002; Panickar and Norenberg, 2005). It is technically difficult to perform time lapse imaging studies to examine the response of astrocytes to ischemic insult in vivo using the conventional middle cerebral artery occlusion (MCAo) model (Karpiak et al., 1989). Focal ischemia induced by photothrombosis provides an alternative model for performing such in vivo time-lapse studies (Watson et al., 1985; Zhang et al., 2005). This model has been used to study structural and functional changes in neurons in the cortex using multi-photon microscopy (Enright and Zhang S.Murphy TH., 2007; Zhang et al., 2005). These studies demonstrated time-dependent damage of dendrites and neuronal death.
Astrocytes not only play supportive roles in maintaining structural integrity in brain, they also play an active role in supporting neuronal function. Astrocytes express a variety of receptors some of which are able to induce the release of chemical transmitters for interaction and communication with neurons and synapse (for reviews see (Haydon, 2001; Volterra and Meldolesi, 2005)). Functional studies of astrocytes under different neuronal injury and disease conditions will provide new insights regarding neuron-glia interactions. In this study, we used 2-P microscopy to study Ca2+ signaling in astrocytes in the ischemic core as well as in penumbra in the cortex during acute phase of cerebral ischemia induced by photothrombosis. Our results show that astrocytes retain their structural integrity for hours following photothrombosis and exhibit receptor-mediated Ca2+ oscillations in the ischemic region. We further provided evidence for the involvement of mGluR5 and GABAB receptors in mediating the increase in astrocytic Ca2+ signals. In addition, selective buffering of astrocytic Ca2+ resulted in protection against ischemia-induced brain damage. Taken together, our results demonstrate the important role of astrocytic Ca2+ in contribution to neuronal damage under ischemic condition.
Materials and Methods
Animals
Male FVB/NJ mice 5–7 weeks of age were purchased from The Jackson Laboratory (Bar Harbor, MA). All procedures were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the University of Pennsylvania and University of Missouri Institutional Animal Care and Use Committee.
Craniotomy surgery
Mice were anesthetized with an intraperitoneal (i.p.) injection of urethane (1.5–2.0 mg/g body weight) dissolved in artificial cerebral spinal fluid (ACSF) (in mM): 120 NaCl, 10 Hepes, 3.1 KCl, 2 CaCl2, 1.3 MgCl2, and 10 glucose, pH 7.4. Once the animal reached a surgical level of anesthesia, it was placed on a warm heating pad to maintain body temperature at 37°C for surgery. A circular craniotomy (2.0 mm in diameter) was made using a high speed drill over the somatosensory cortex at the coordinate of −0.8 mm from bregma and 2.0 mm lateral to the midline. A custom-made metal frame was attached to the skull with cyanocrylate glue, and the dura was then carefully removed with fine forceps.
In vivo fluorescent dye loading
For loading of the Ca2+ indicator fluo-4 into astrocytes, fluo-4 AM was dissolved in pluronic acid (20% pluronic acid plus 80% DMSO) to obtain a 10 µg/µl stock solution. This stock solution (2.5 µl) was mixed with 40 µl ACSF and applied to the dura free cortical surface within the craniotomy for 1 hr. In order to confirm that fluo-4 was taken up by astrocytes, sulforhodamine 101 (SR101, 100 µl) was applied onto the cortical surface for 1–5 minutes to label astrocytes. Bright SR101 labeled astrocytes were observed after 1 hr with a 2-P microscope. In order to image blood flow of the vasculature, we administered 100–150 µl of a 20 mg/ml solution of rhodamine- or FITC-dextran (70 kDa) in ACSF via a tail vein injection. To reduce movement artifact during imaging, a glass coverslip was glued on the metal frame above the craniotomy, and the gap between glass coverslip and cortex was filled with pre-melted 2% agarose in ACSF.
Photothrombosis-induced focal cerebral ischemia
To induce photothrombosis, the photosensitive dye Rose Bengal (RB) was dissolved in ACSF and injected through the tail vein at a dose of 0.03 mg/g body weight. In order to activate the dye, an area in the middle of the craniotomy was focally illuminated with a green light (535 ± 25 nm) from a mercury lamp for 2 min through a 10×0.3NA objective. For Ca2+ imaging, we illuminated an area of 0.6 mm in diameter; and for histological study, a larger area was illuminated (see Fig. 2 and Fig. 6). For simplicity, the illuminated area was regarded as the ischemic core region, and the surrounding area the penumbra. Ca2+ imaging was performed in the ischemic core and penumbra which can be easily identified based on the vasculature structure during experiments.
Fig. 2. Photothrombosis causes neuronal death and astrocyte activation in the cortex.
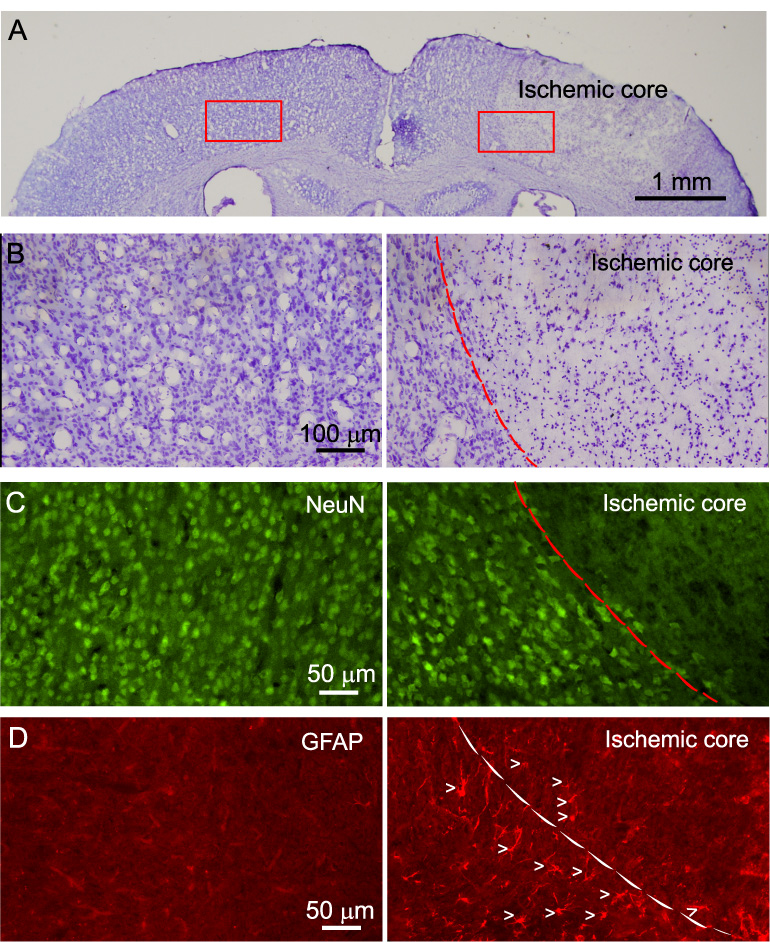
A) Nissl staining of brain sections showing cell damage in the ischemic region (right) 24 hr after photothrombosis. An area of 2 mm in diameter was illuminated in this experiment. B) High resolution micrographs of ischemic region (right) and corresponding region of contralateral side (left) from A. C) NeuN staining of a brain section showing neuronal death in the ischemic region (right) and healthy neurons in corresponding contralateral side (left). D) Enhanced GFAP expression in the transition zone between ischemic core and penumbra (right) after photothrombosis. The left panel shows low GFAP expression in the cortex in the contralateral side. Arrow heads indicate individual astrocytes. Dash line outlines the boundary between ischemic core and penumbral region.
Fig. 6. Effect of BAPTA on photothrombosis-induced infarct volume.
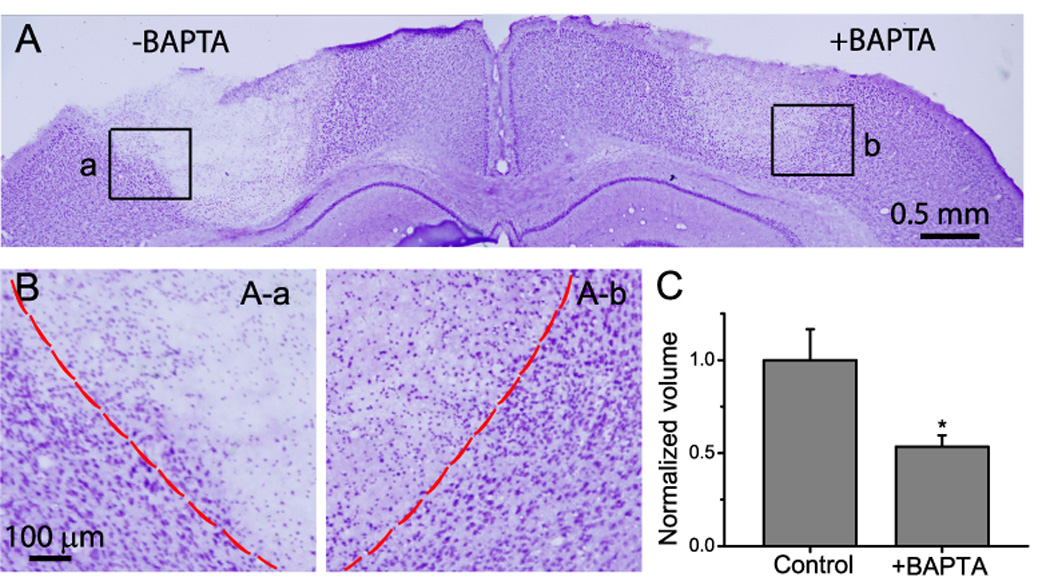
A) Nissl staining of brain section. Photothrombosis was induced on both sides of barrel cortex region at the same time. An area of 1 mm diameter was illuminated in this experiment. B) High resolution micrographs from the boxed areas in A showing distinct regions of ischemic cores and transition zones. C) Normalized infarct volume indicated neuronal protective role of BAPTA-AM. Data were obtained from N=6 mice. * p<0.05, t-test.
In vivo 2-P Ca2+ imaging in astrocytes
Mice were transferred to the stage of a 2-P microscope for in vivo imaging (Prairie Technologies Inc, WI). Images were obtained using an Ultima scan head attached to an Olympus BX51 microscope and a 60×/0.9 water immersion objective. A Chamelon Ti:Sapphire laser was used for 2-P excitation (820 nm). Time-lapse imaging was performed to track Ca2+ signals for a period of 5 min with acquisition rates of 1 image per second. For each animal, 15–25 astrocytes in 4–5 fields were imaged and 4–5 animals were used for each experimental condition. For drug administration, the glass coverslip and agarose were removed. A new glass coverslip was glued on the metal frame and the pre-melted 2% agarose containing the desired concentration of drug was applied to fill the gap between the cortex and glass coverslip. Imaging was performed within 15 min of drug administration. Throughout the period of the experiment (up to 3–4 hr), body temperature was maintained at 37°C using a heating pad (Fine Science Tool, CA). The mice were under a state of deep anesthesia with urethane as demonstrated by occasionally pinching the tail to check the response. It is reported that animals anesthetized with urethane maintain relative constant physiological parameters including heart beat and blood oxygen (Zhang et al., 2005; Murphy et al., 2008). In addition, dehydration was prevented by adding saline to the craniotomy.
Data analysis of Ca2+ signal
Data were analyzed using the Metamorph software (Universal Imaging, CA) to generate background-subtracted ΔF/Fo images, where Fo was resting fluorescence and ΔF was the change in florescence from the resting value. The magnitude of Ca2+ signal (ΔF/Fo.s) was defined as the area obtained by integrating ΔF/Fo over time (300 s) using the Origin software (OriginLab Corporation, MA). All-points histograms of ΔF/Fo were made with a bin of 0.05 units to study amplitude distribution of ΔF/Fo. Cross correlation of Ca2+ oscillations between cell pairs was computed using the data set of ΔF/Fo from two neighboring cells. Peak cross correlation coefficients within a lag time of 5 seconds were used to compare the synchrony of Ca2+ signaling between cells.
In vivo evoked potential (EP) recording
EPs were recorded using a glass electrode filled with ACSF using a low noise extracellular recording amplifier (Model 1800, A-M System, Inc., WA). Data were acquired with a Digidata 1320 interface and a pClamp 9 software (Axon instruments, CA).
Transcardial perfusion and histo- and immunocyto-chemistry studies
The procedure for transcardial perfusion has been described previously (Ding et al., 2007). Briefly, mice were anesthetized with halothane and transcardially perfused first with ice cold phosphate buffered saline (PBS) and then 4% paraformaldehyde in PBS (pH=7.4). After perfusion, the brain was post-fixed with 4% paraformaldehyde in PBS at 4°C for 30 min, and then transferred to 30% sucrose overnight to prevent ice crystal formation. Coronal sections of the brain (thickness 20 µm) were cut on a cryostat (Leica CM 1900), and were collected serially on pre-cleaned slides. NeuN staining for neurons was described previously. Briefly, brain sections were incubated with rabbit anti-NeuN (1:1000, Millipore) overnight, and subsequently incubated with FITC-conjugated goat anti-rabbit IgG (Millipore, CA) for 2 hr. Similar procedures were used for glial fibrillary acidic protein (GFAP) staining using rabbit polyclonal anti-GFAP (1:150, Sigma) and Rhodamine-conjugated donkey anti-rabbit IgG (Millipore). Nuclei were stained with DAPI (0.5 µg/ml in PBS) and mounted using anti-fade mounting medium. Stained sections were viewed with a Nikon epi-fluorescence microscope and analyzed using Metamorph (Universal Imaging Corp, CA). Nissl staining was performed by applying 0.2 % Cresyl violet for 2 min.
Infarct volume estimation
The infarct volume was determined by measuring areas showing loss of Nissl-staining in coronal brain sections (Vendrame et al., 2004). The areas of cerebral infarction were delineated and quantified using ImageJ (NIH). The total volume of ischemic tissue was calculated by multiplying the individual infarct area in the consecutive sections by the thickness of sections.
Statistical analysis
N values reported in in vivo studies represent the number of animals. In vivo Ca2+ imaging experiments were generally obtained from 15–25 astrocytes per animal and 4–5 animals per experimental condition. The data were averaged to obtain a single value per animal. Comparison of data from two different conditions was performed using unpaired t-test. Multiple comparisons of data were performed using one-way ANOVA test and differences between individual groups were determined using the Newman–Keuls post hoc comparison tests using the GraphPad Prism program version 4.0 (Graph Pad Software Inc., San Diego). * p<0.05. ** p<0.01.
Results
Photothrombosis-induced ischemia model
The cortical surface in the cranial window was exposed to green light to cause RB-induced cross-linking of platelets following RB injection. Blood flow rate was measured using linescan imaging on capillaries (Kleinfeld et al., 1998). Ten minutes after the induction of photothrombosis (RB+/Illumination+), blood flow rate in the capillary dropped from 398±30.9 µm/second to virtually 0 (9 capillaries from 5 animals) in the illuminated region (Fig. 1A). In addition, we recorded evoked potentials (EPs) in the ischemic core region stimulated by 100 ms whisker deflection by air puff before and after photothrombosis (Fig. 1B). The EP in the ischemic core region declined to 10% within 20 min of photothrombosis (N=3) indicating the loss of activity of neurons in the ischemic core region. Using SR101 to label astrocytes (Nimmerjahn et al., 2004), we found that they remained structurally intact for up to 3 hr following photothrombosis (Fig. 1C).
Fig. 1. Photo-activation of circulating Rose Bengal blocks local blood flow and reduces local evoked potential.
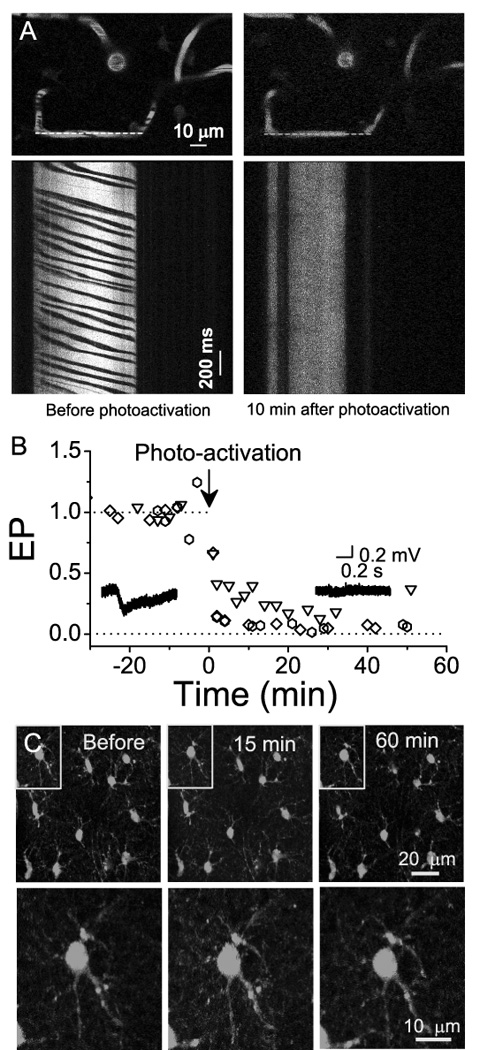
A) (Top) An image of vasculature labeled with rhodamine-dextran before (left) and 10 min after (right) photothrombosis. Linescan X-t images (lower panels) were performed at the regions identified by the dashed lines in the top panels. Right lower panel shows cessation of movement of blood cells (dark bands, lower left panel) following photothrombosis. B) Time course of evoked potential (EP) changes in the ischemic core stimulated by whisker deflection before and after photothrombosis. Data were recorded from 3 mice with different symbols representing each mouse. The insets in the figure show examples of averaged EPs before (left) and 40 min (right) after ischemia recorded in the core. C) Astrocytes were labeled using SR101 and maximum intensity projection images obtained from 20 Z-scan images (1 µm z-step interval) show the morphology of these glial cells before and following photothrombosis. Images in the bottom panel are enlargements of the boxed regions in the upper panels.
Histological examination was performed to confirm the irreversible tissue damage following photothrombosis. Twenty four hours after photothrombosis, mice were sacrificed and brain sections were subjected to Nissl staining and immunostaining for NeuN and GFAP, specific markers for neurons and astrocytes, respectively. Ipsilateral to the photothrombosis, Nissl staining (Fig. 2A and B) clearly showed a sharp transition zone between the normal and ischemic core region (dashed line in Fig. 2B). NeuN immunoreactivity demonstrated a loss of neurons in the ischemic core 24 hr after photothrombosis (Fig. 2C). We also observed the increased expression of GFAP in the penumbra (Fig. 2D), indicating activation of astrocytes after photothrombosis. These phenomena were observed in all mice subjected for histological examination (N=5).
Ischemia-induced increase in Ca2+ signaling in astrocytes
Incubation of fluo-4 AM on the cortical surface led to labeling of fluo-4 into cortical astrocytes that was confirmed with SR101 labeling (Fig 3A). When fluo-4 loaded ischemic core region was imaged, large repetitive and transient Ca2+ signals were observed in astrocytes within 20 min after photothrombosis (RB+/Illumination+, Fig. 3C–D). Ca2+ signals exhibited a high degree of synchrony among astrocytes in the same imaging field (Fig. 3D) as indicated by a large cross correlation coefficient between the adjacent astrocytes at zero lag time (Fig. 3E). Further quantification of data demonstrated enhanced Ca2+ signaling following photothrombosis (Fig. 3F). An all-points histogram of ΔF/Fo amplitude further demonstrated that photothrombosis not only increased the amplitude but also the frequency of Ca2+ signals (Fig. 3G). Ca2+ signals were sustained for the duration of the imaging period up to 3 hr (Fig. 3H).
Fig. 3. Astrocytes exhibit enhanced Ca2+ signals after photothrombosis.
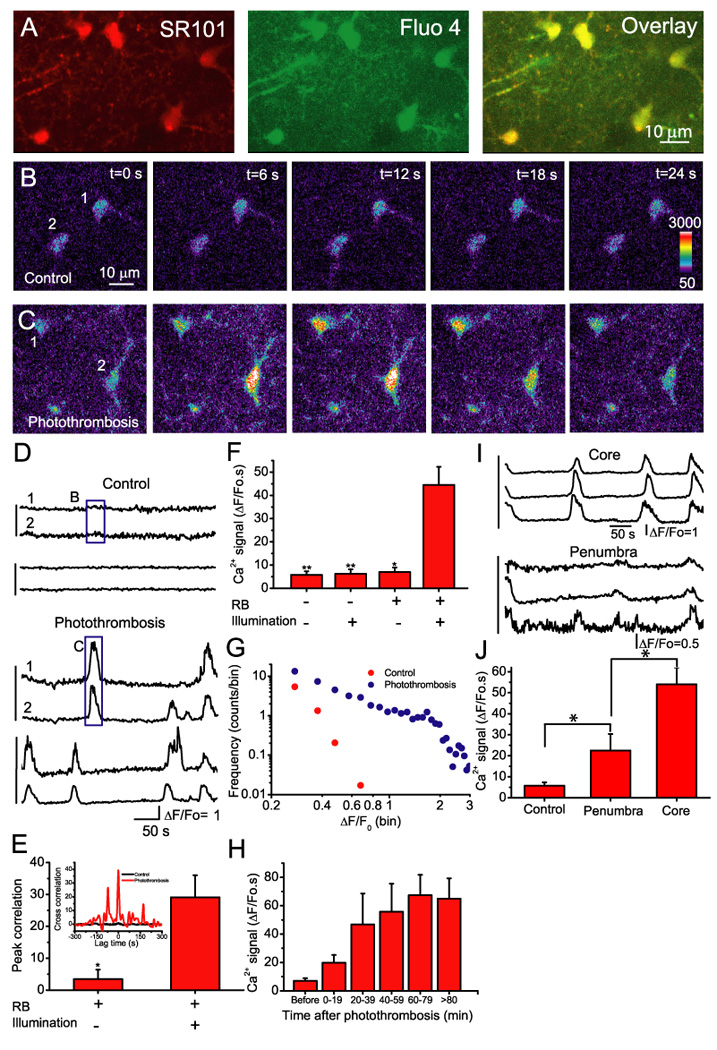
A). Cortical astrocytes double-labeled with fluo-4 and SR101 after photothrombosis. B–C) Representative images of astrocytes loaded with fluo-4 before (B) and after (C) induction of photothrombosis. D) Time course of somatic Ca2+ oscillations of astrocytes expressed as ΔF/Fo before (upper) and after (bottom) induction of photothrombosis. The vertical lines at the left indicate cell pairs that were recorded in the same image frame (also in (I)). The box regions correspond to the images in B and C as indicated. E) Cross correlation coefficients within a lag time of 5 s. The data were obtained from 5 pairs of astrocytes before photothrombosis and 9 pairs after photothrombosis from 4 mice. Inset: A representative cross correlation analysis of Ca2+ oscillations before (black) and after induction of focal ischemia (red). Statistical analysis was performed using t-test. *p <0.05. F) Summary of astrocytic Ca2+ signals expressed as an integral of ΔF/Fo traces over 300 s under different control conditions and following photothrombosis (RB+ and Illumination+). The data were collected 20–80 min after ischemia from N=4–9 mice. Statistical analyses between photothrombosis (RB+/illumination+) and each control condition (i.e. RB-/Illumination-, RB-/Illumination+ and RB+/Illumination-) were assessed using t-test. *p<0.05, **p<0.01. G) The frequency distributions of the amplitude of ΔF/Fo before and after photothrombosis (N=4). Data were binned in 0.05 unit of ΔF/Fo. H) Time course of somatic Ca2+ signal in astrocytes developed after photothrombosis. The data from each time interval of 20 min were averaged from 14–28 cells obtained from N=4–5 animals. The Ca2+ signal of the last bin was the average value from astrocytes imaged 80–150 min after photothrombosis. I) Time course of astrocytic Ca2+ signals in the ischemic core (upper) and penumbra (200 µm from the edge of core, lower). The data were recorded from the same mice. J) Summary of astrocytic Ca2+ signal in control condition (N=5), in ischemic core (N=9) and penumbra (N=4). Ca2+ signal of control condition (RB-/Illumination-) was the same as in F. *p<0.05, t-test.
The increased frequency of transient Ca2+ signals after photothrombosis was not caused by 2-P excitation, green light illumination or RB per se (Fig. 3F). First, we performed Ca2+ imaging without RB injection and illumination (RB-/Illumination-) of the cortex. Secondly, Ca2+ imaging was performed after RB injection but without green light illumination of the cortex (RB+/Illumination-). Finally, Ca2+ imaging was performed after illumination in the absence of RB injection (Illumination+/RB-). Stimulation of Ca2+ signals required the combination of RB and green light illumination (Fig. 3 F and H). These results are consistent with other reports indicating that astrocytic Ca2+ signaling events are rare in normal anesthetized mice (Wang et al., 2006; Takano et al., 2006; Ding et al., 2007).
To determine whether astrocytic Ca2+ signals are generated by the activation of G-protein-coupled receptors (GPCRs) we performed pharmacological evaluation of the role of mGluR5, GABABR and P2Y receptors in mediating astrocytic Ca2+ signals. Initially, specific agonists for these receptors were applied to the cortical surface to evaluate their ability to stimulate Ca2+ signals. As shown in Fig. 4A, Ca2+ signals could be induced by addition of 0.5 mM ATP and these signals were attenuated by the P2Y receptor antagonist suramin (0.2 mM). The mGluR5 selective agonist, 2-chloro-5-hydroxyphenylglycine (CHPG, 1 mM), induced astrocytic Ca2+ signals and was attenuated by the mGluR5 antagonist 2-Methyl-6-(phenylethynyl)-pyridine (MPEP, 30 µM) (Fig. 4C). The GABABR agonist, Baclofen (0.1 mM), also induced Ca2+ signals that were attenuated by its antagonist, CGP54626 (10 µM, Fig. 4B–C).
Fig. 4. Pharmacological manipulation of astrocytic Ca2+ signals in vivo.
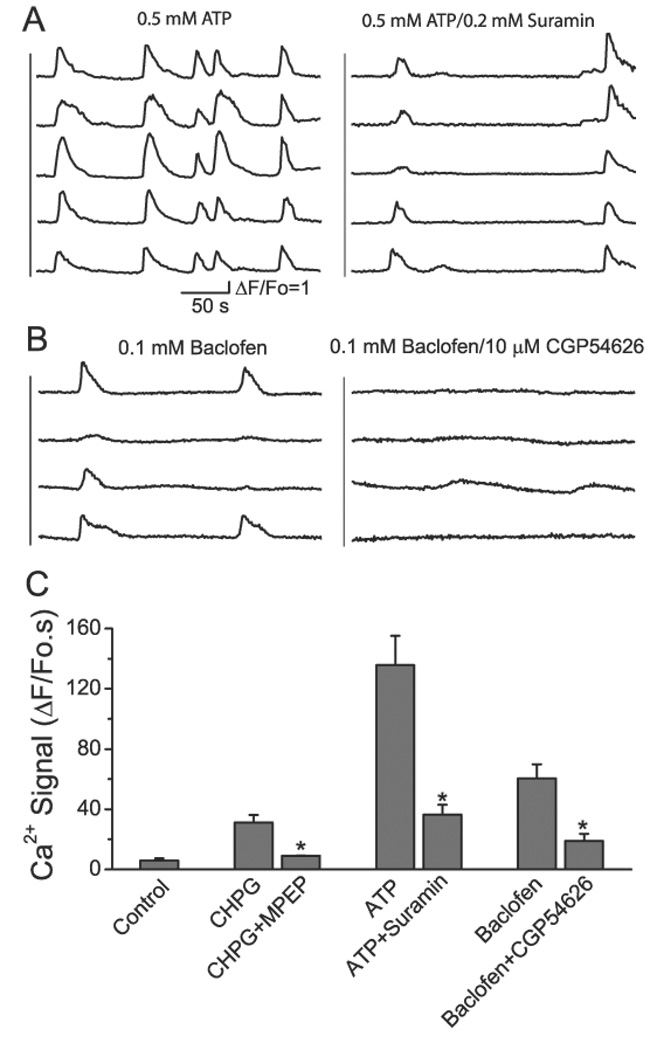
A) Astrocytic Ca2+ signals following administration of ATP (500 µM; left) and ATP together with Suramin (200 µM; right). B) Baclofen (left; 100 µM) alone and Baclofen together with CGP54626 (right; 10 µM). C) Summary of astrocytic Ca2+ signals induced by agonists in the presence and absence of their respective antagonists. Data are average values from N=4–6 mice in each condition. Statistical analyses (t-test) indicate significant attenuation of the Ca2+ signal by the respective antagonist compared to the signal in the presence of agonist alone. *p <0.05.
Using these receptor antagonists we determined the involvement of mGluR5, GABABR and P2Y receptors in mediating photothrombosis-induced Ca2+ signals. We first evaluated the involvement of mGluR5 and GABABR in stimulating Ca2+ signals because these transmitters are likely to be released by neurons and increase in the extracellular space following photothrombosis. After induction of Ca2+ signals following photothrombosis (30–40 min), agarose in the craniotomy was removed and mGluR5 antagonist MPEP (30 µM) was applied. MPEP significantly reduced Ca2+ signals by 62% (Fig. 5A and D; N = 4). Similarly cortical administration of the GABABR CGP54626 (10 µM) also significantly reduced Ca2+ signals by 57 % (Fig. 5B and D; N = 4). When MPEP and CGP54626 were administered together, the photothrombosis induced astrocytic Ca2+ signals was reduced by 74%. In these studies, sufficiently high but not toxic concentrations of antagonists were used to ensure full inhibition of these receptors.
Fig. 5. GABAB and mGluR5 receptors contribute to photothrombosis-induced astrocytic Ca2+ signaling.
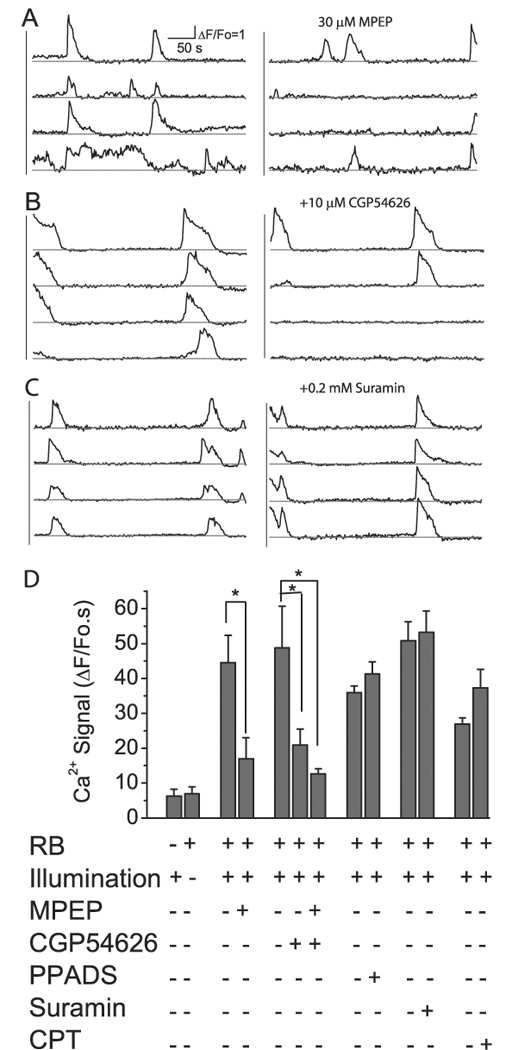
A–C) Astrocytic Ca2+ signals following photothrombosis before (left) and after (right) administration of different antagonists. D) Summary of the effects of antagonists on photothrombosis-induced astrocytic Ca2+ signals. Ca2+ signals in the absence of pharmaceutical reagents were collected 30–60 minutes following photothrombosis. Ca2+ signals in the presence of pharmaceutical reagents were collected 20–60 minutes after administration of these drugs. In each group of experiment, we recorded Ca2+ signal up to 40 min following photothrombosis to establish the base line Ca2+ signal before adding drug. The data were averaged from N=4–6 mice in each condition. Statistical analysis by ANOVA with post-hoc tests indicated significant difference comparing effects with and without appropriate antagonists. *p<0.05.
Although ATP-stimulated Ca2+ signals were effectively blocked by suramin, a non-specific inhibitor for P2Y receptors, suramin did not inhibit the photothrombosis-induced Ca2+ signal (Fig. 5C and D). Similarly, pyridoxal phosphate-6-azophenyl-2',4'-disulfonic acid (PPADS), a general P2 receptor antagonist, did not inhibit the Ca2+ signal (Fig. 5D). A possibility explanation for this result is that ATP released during ischemia is rapidly hydrolyzed by ectonucleotidases (Dunwiddie and Masino, 2001). However, the possibility that activation of adenosine receptor may contribute to ischemia-induced Ca2+ signals in astrocytes was eliminated by testing with 8-cyclopentyltheophylline (CPT), the A1 antagonist, which did not attenuate astrocytic Ca2+ signals after photothrombosis (Fig. 5D).
Clinical interventions aimed at restoring brain function have been targeted to inhibiting the evolvement of the penumbra. Here we asked whether astrocytes in the penumbra exhibit increased Ca2+ oscillations. Using vascular landmarks, we performed Ca2+ imaging in the penumbra (from the edge of ischemic core) in the time frame of 40 to 80 min after ischemia when Ca2+ signaling in the core region was fully developed. In comparison to control condition, the average Ca2+ signals in astrocytes around 200 µm from the core were significantly increased, but were much smaller than that in the ischemic core region (Fig. 3I–J). Our data are consistent with the idea that astrocytic Ca2+ waves are initiated in the core and subsequently propagate into the penumbra.
Brain protection by selective attenuation of astrocytic Ca2+ signal
Previously we have shown surface labeling with BAPTA AM can selectively inhibit the ATP-induced Ca2+ signals in astrocytes (Ding et al., 2007). Using this method, we determined whether the astrocytic Ca2+ signal contributes to neuronal death following ischemia. Photothrombosis was induced bilaterally in barrel cortices in the mouse brain and premelted agarose containing 200 µM BAPTA AM was subsequently applied onto one of the cranial windows to ensure continued delivery after photothrombosis. Premelted agarose without BAPTA AM was applied to the other cranial window to serve as a within-animal control. One day after ischemia, animals were perfused, sectioned and labeled with Cresyl violet. Infarct volumes were measured to assess the effect of BAPTA on brain damage. As shown in Fig. 6A and B, infarction is well demarcated either in the presence or in the absence of BAPTA. However, selective loading of the Ca2+ chelator BAPTA into astrocytes significantly reduced the infarct volume by 47% (Fig. 6C, N=6 mice).
Discussion
Studies of ischemia traditionally focus on blood flow, vasculature and neuronal function. However, recent advances in imaging and analysis of gene expression have revealed complex changes also occur in glial cells in response to the cerebral ischemic insult (Dirnag and Prillar, 2004). Since astrocytes are structurally and functionally interconnected with neurons and vasculature, an understanding of their physiological and metabolic changes at different stages of the ischemic insult is warranted. Photothrombosis ischemia model combined with 2-P microscopy provides a useful tool that can monitor cellular structure and physiological changes of astrocytes in vivo before and after induction of ischemia. In the present study, we first validated the photothrombosis-induced focal ischemic model by measuring blood flow rate, evoked potential and extent of tissue damage. Initiation of photothrombosis was associated with a loss of local blood flow and a decrease in evoked potentials. However, astrocytes were able to retain their structural integrity for hours. Histological study confirmed neuronal death and development of infarct 24 hr following photothrombosis. The results showed a distinct transition zone between the ischemic core and penumbra as revealed by Nissl and NeuN staining. Reactive astrocytes were observed around the penumbra as manifested by increased expression of GFAP. When 2-P microscopy was applied the core region to study Ca2+ signals in astrocytes, we found that photothrombosis led to an increase in frequency and amplitude of transient Ca2+ signals. These stimulated Ca2+ signals can not represent a response to sustained illumination because neither 2-P excitation nor green light illumination in the absence of Rose Bengal could stimulate astrocytic Ca2+ signals. Ca2+ signals were only stimulated upon cessation of local blood flow due to introduction of Rose Bengal and green light illumination. These Ca2+ signals were synchronized in the astrocytic network, a feature usually not observed in normal brain in anesthetized mice. The transient Ca2+ signals do not represent the response of dying astrocytes since these signals return to baseline and were repetitive. The ability for astrocytes to survive for a prolonged time following photothrombosis may be due to their glycogen store which can be used as energy source through glycolysis (Kasischke et al., 2004; Brown, 2004).
Since the basal level of Ca2+ in astrocytes was not changed within the recording time, these results suggest that astrocytes exhibit characteristic transient Ca2+ signals during the acute phase of photothrombosis. Using a similar photothrombosis model, Zhang et al (2005) showed the degeneration of neuronal dendrites within 30 min after ischemia. This time is concomitant with the increase in Ca2+ signaling in astrocytes. While the photothrombosis model has the advantage of studying physiological change of cells before and after ischemia, there are also limitations. For example, it is not possible to control reperfusion, thus tissue damage observed 24 hr later might be contributed by both ischemic and reperfusion injury.
There are two candidate mechanisms that could explain the increase in Ca2+ signals in astrocytes following photothrombosis: (1) loss of Ca2+ from mitochondria under anaerobic conditions, or (2) activation of GPCRs leading to the release of Ca2+ from intracellular stores. Although we did not formally test the role of mitochondria in this study, this mechanism is less likely given the repetitive nature of the Ca2+ signals. Instead, the ability for mGluR5 and GABABR antagonists to significantly attenuate astrocytic Ca2+ signals implies a role for receptor-mediated elevation of intracellular Ca2+ stores. On the other hand, PPADS and suramin, antagonists for P2X or P2Y receptors, as well as CPT, an A1 receptor antagonist, did not elicit an effect on the ischemia-induced Ca2+ signals in astrocytes. These results are in agreement with the notion that although ischemia induces rapid release of ATP, it is probably a rapid event that cannot be detected within the time frame of the experiment (Rossi et al., 2007).
We also observed increased astrocytic Ca2+ signal in the penumbra although it is smaller than that in the core region. These results suggest that synchronized Ca2+ signals amongst the astrocytic network might propagate beyond the ischemic core and trigger neuronal responses in the penumbra. Since enhanced astrocytic Ca2+ signals have been shown to induce the release of glutamate and D-serine which interact with NR2B subunit of NMDA receptors (Parpura and Haydon, 2000; Fellin et al., 2004; Haydon, 2001; Araque et al., 1998; Aguado et al., 2002; Angulo et al., 2004), it is possible that propagation of these glial Ca2+ waves could modulate neuronal function in the broader neuronal network. We have previously shown that in status epilepticus, enhanced astrocytic Ca2+ signals could activate neuronal NR2B subunit of N-methyl D-aspartate (NMDA) receptors, resulting in delayed neuronal death (Ding et al., 2007). Consequently, it is feasible that propagation of astrocytic Ca2+ signals in response to ischemia could contribute to delayed neuronal death known to occur in the penumbra. We tested this possibility by measuring the infarct volume following selective inhibition of astrocytic Ca2+ with BAPTA. Significant reduction of infarct volume was observed suggesting that the increased Ca2+ signals in astrocytes contribute to ischemic damage. Aside from glutamate release, Ca2+ elevation in astrocytes might alter other signaling pathways. For example, the activation of group I mGluR by DHPG increases the water permeability of aquaporin-4 channel through the phosphorylation by protein kinase G that might causes edema after ischemia (Gunnarson et al., 2008). In addition, given that Ca2+ signals in these cells can stimulate changes in vascular tone (Takano et al., 2006), elevation of astrocyte Ca2+ signals could also provide a mechanism for regulating local changes in blood flow.
In conclusion, our study using 2-P in vivo imaging demonstrated enhanced Ca2+ signals in astrocytes during the acute phase of ischemia induced by photothrombosis. These astrocytic Ca2+ signals were driven by the combined activation of mGluR5 and GABAB receptors. Selective buffering of astrocytic Ca2+ signals significantly reduce the infarct volume caused by photothrombosis raising the possibility that astrocytes contribute to neuronal death following ischemic injury.
Acknowledgements
The authors thank Dr. Grace Sun for critical reading of the manuscript and Dr. Agnes Simonyi for help in statistical analysis. Support for this project was provided by the American Heart Association to SD and grants from NINDS and NIMH to PGH.
References
- Aguado F, Espinosa-Parrilla JF, Carmona MA, Soriano E. Neuronal activity regulates correlated network properties of spontaneous calcium transients in astrocytes in situ. J Neurosci. 2002;22:9430–9444. doi: 10.1523/JNEUROSCI.22-21-09430.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angulo MC, Kozlov AS, Charpak S, Audinat E. Glutamate released from glial cells synchronizes neuronal activity in the hippocampus. J Neurosci. 2004;24:6920–6927. doi: 10.1523/JNEUROSCI.0473-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Sanzgiri RP, Parpura V, Haydon PG. Calcium elevation in astrocytes causes an NMDA receptor-dependent increase in the frequency of miniature synaptic currents in cultured hippocampal neurons. J Neurosci. 1998;18:6822–6829. doi: 10.1523/JNEUROSCI.18-17-06822.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber PA, Demchuk AMH. Biochemistry Ischemic Stroke. Advances in Neurology. 2003;92:151–164. [PubMed] [Google Scholar]
- Brown AM. Brain glycogen re-awakened. Journal of Neurochemistry. 2004;89:537–552. doi: 10.1111/j.1471-4159.2004.02421.x. [DOI] [PubMed] [Google Scholar]
- Bushong EA, Martone ME, Jones YZ, Ellisman MH. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci. 2002;22:183–192. doi: 10.1523/JNEUROSCI.22-01-00183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding S, Fellin T, Zhu Y, Lee SY, Auberson YP, Meaney DF, Coulter DA, Carmignoto G, Haydon PG. Enhanced Astrocytic Ca2+ Signals Contribute to Neuronal Excitotoxicity after Status Epilepticus. J Neurosci. 2007;27:10674–10684. doi: 10.1523/JNEUROSCI.2001-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirnag U, Prillar J. Focal cerebral ischemia: the multifaceted role of glial cells. In: Kettenmann H, Ransom B, editors. Neuroglia. New York: Oxford-520; 2004. [Google Scholar]
- Dunwiddie TV, Masino SA. The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci. 2001;24:31–55. doi: 10.1146/annurev.neuro.24.1.31. [DOI] [PubMed] [Google Scholar]
- Enright LE, Zhang S, Murphy TH. Fine mapping of the spatial relationship between acute ischemia and dendritic structure indicates selective vulnerability of layer V neuron dendritic tufts within single neurons in vivo. Journal of Cerebral Blood Flow & Metabolism. 2007;11:1–16. doi: 10.1038/sj.jcbfm.9600428. [DOI] [PubMed] [Google Scholar]
- Fellin T, Pascual O, Gobbo S, Pozzan T, Haydon PG, Carmignoto G. Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron. 2004;43:729–743. doi: 10.1016/j.neuron.2004.08.011. [DOI] [PubMed] [Google Scholar]
- Gunnarson E, Axehult G, Baturina G, Zelenin S, Zelenina M, Aperia A, Gunnarson E, Axehult G, Baturina G, Zelenin S, Zelenina M, Aperia A. Identification of a molecular target for glutamate regulation of astrocyte water permeability. GLIA. 2008;56:587–596. doi: 10.1002/glia.20627. [DOI] [PubMed] [Google Scholar]
- Haydon PG. GLIA: listening and talking to the synapse. Nature Reviews Neuroscience. 2001;2:185–193. doi: 10.1038/35058528. [DOI] [PubMed] [Google Scholar]
- Huang Y, McNamara JO. Ischemic stroke: “acidotoxicity” is a perpetrator. Cell. 2004;118:665–666. doi: 10.1016/j.cell.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Karpiak ST, Wakade CG, Karpiak SE, Tagliavia A, Wakade CG. Animal models for the study of drugs in ischemic stroke. Annu Rev. Pharmacol. Toxicol. 1989;29:403–429. doi: 10.1146/annurev.pa.29.040189.002155. [DOI] [PubMed] [Google Scholar]
- Kasischke KA, Vishwasrao HD, Fisher PJ, Zipfel WR, Webb WW. Neural Activity Triggers Neuronal Oxidative Metabolism Followed by Astrocytic Glycolysis. Science. 2004;305:99–103. doi: 10.1126/science.1096485. [DOI] [PubMed] [Google Scholar]
- Kleinfeld D, Mitra PP, Helmchen F, Denk W. Fluctuations and stimulus-induced changes in blood flow observed in individual capillaries in layers 2 through 4 of rat neocortex. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:15741–15746. doi: 10.1073/pnas.95.26.15741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattiasson G, Shamloo M, Gido G, Mathi K, Tomasevic G, Yi S, Warden CH, Castilho RF, Melcher T, Gonzalez-Zulueta M, Nikolich K, Wieloch T, Mattiasson G, Shamloo M, Gido G, Mathi K, Tomasevic G, Yi S, Warden CH, Castilho RF, Melcher T, Gonzalez-Zulueta M, Nikolich K, Wieloch T. Uncoupling protein-2 prevents neuronal death and diminishes brain dysfunction after stroke and brain trauma. Nature Medicine. 2003;9:1062–1068. doi: 10.1038/nm903. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Li P, Betts K, Liu R. Two-Photon Imaging of Stroke Onset In Vivo Reveals That NMDA-Receptor Independent Ischemic Depolarization Is the Major Cause of Rapid Reversible Damage to Dendrites and Spines. J Neurosci. 2008;28:1756–1772. doi: 10.1523/JNEUROSCI.5128-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Kerr JN, Helmchen F. Sulforhodamine 101 as a specific marker of astroglia in the neocortex in vivo. Nature Methods. 2004;1:31–37. doi: 10.1038/nmeth706. [DOI] [PubMed] [Google Scholar]
- Panickar KS, Norenberg MD. Astrocytes in cerebral ischemic injury: morphological and general considerations. GLIA. 2005;50:287–298. doi: 10.1002/glia.20181. [DOI] [PubMed] [Google Scholar]
- Parpura V, Haydon PG. Physiological astrocytic calcium levels stimulate glutamate release to modulate adjacent neurons. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:8629–8634. doi: 10.1073/pnas.97.15.8629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi DJ, Brady JD, Mohr C. Astrocyte metabolism and signaling during brain ischemia. Nat Neurosci. 2007;10:1377–1386. doi: 10.1038/nn2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapf C, Mohr JP. Ischemic stroke therapy. Annual Review of Medicine. 2002;53:453–475. doi: 10.1146/annurev.med.53.082901.104106. [DOI] [PubMed] [Google Scholar]
- Takano T, Tian GF, Peng W, Lou N, Libionka W, Han X, Nedergaard M. Astrocyte-mediated control of cerebral blood flow. Nat Neurosci. 2006;9:260–267. doi: 10.1038/nn1623. [DOI] [PubMed] [Google Scholar]
- Vendrame M, Cassady J, Newcomb J, Butler T, Pennypacker KR, Zigova T, Davis Sanberg C, Sanberg PR, Willing AE. Infusion of Human Umbilical Cord Blood Cells in a Rat Model of Stroke Dose-Dependently Rescues Behavioral Deficits and Reduces Infarct Volume. Stroke. 2004;35:2390–2395. doi: 10.1161/01.STR.0000141681.06735.9b. [DOI] [PubMed] [Google Scholar]
- Volterra A, Meldolesi J. Astrocytes, from brain glue to communication elements: the revolution continues. Nature Reviews Neuroscience. 2005;6:626–640. doi: 10.1038/nrn1722. [DOI] [PubMed] [Google Scholar]
- Wang X, Lou N, Xu Q, Tian GF, Peng WG, Han X, Kang J, Takano T, Nedergaard M, Wang X, Lou N, Xu Q, Tian GF, Peng WG, Han X, Kang J, Takano T, Nedergaard M. Astrocytic Ca2+ signaling evoked by sensory stimulation in vivo. Nature Neuroscience. 2006;9:816–823. doi: 10.1038/nn1703. [DOI] [PubMed] [Google Scholar]
- Watson BD, Dietrich WD, Busto R, Wachtel MS, Ginsberg MD. Induction of reproducible brain infarction by photochemically initiated thrombosis. Annals of Neurology. 1985;17:497–504. doi: 10.1002/ana.410170513. [DOI] [PubMed] [Google Scholar]
- Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J, Wei WL, MacDonald JF, Wemmie JA, Price MP, Welsh MJ, Simon RP. Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell. 2004;118:687–698. doi: 10.1016/j.cell.2004.08.026. [DOI] [PubMed] [Google Scholar]
- Zhang S, Boyd J, Delaney K, Murphy TH. Rapid Reversible Changes in Dendritic Spine Structure In Vivo Gated by the Degree of Ischemia. J Neurosci. 2005;25:5333–5338. doi: 10.1523/JNEUROSCI.1085-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]