

Abstract
Autophagy is an intracellular bulk degradation system that plays a vital role in maintaining cellular homeostasis. This degradation process involves dynamic membrane rearrangements resulting in the formation of double-membraned autophagosomes. However, the driving force for generating curvature and deformation of isolation membranes remains a mystery. Bif-1, also known as SH3GLB1 or Endophilin B1, was originally discovered as a Bax-binding protein. Bif-1 contains an amino-terminal N-BAR (Bin-Amphiphysin-Rvs) domain and a carboxy-terminal SH3 (Src-homology 3) domain and displays membrane binding and bending activities. It has been shown that Beclin1 is involved in the nucleation of autophagosomal membranes through an unknown mechanism. Interestingly, Bif-1 forms a complex with Beclin1 through UVRAG and promotes the activation of the class III PI3 kinase, Vps34, in mammalian cells. In response to nutrient starvation, Bif-1 accumulates in punctate foci where it co-localizes with LC3, Atg5, and Atg9. Furthermore, Bif-1-positive, crescent-shaped small vesicles expand by recruiting and fusing with Atg9-positive small membranes to complete autophagosome formation. This review highlights the role of Bif-1 in the regulation of autophagy and discusses the potential involvement of Bif-1 in the biogenesis of membranes for the formation of autophagosomes.
Keywords: Bif-1/Endophilin B1, BAR domain, membrane curvature, SH3 domain, PI3KC3-Beclin1-UVRAG complex, autophagy
Introduction
Autophagy is an evolutionally conserved intracellular process for the bulk degradation of cytoplasmic components that is initiated in response to environmental changes(1, 2). This degradation process plays a critical role in the maintenance of cellular homeostasis by recycling nutrients under starvation conditions. In addition, autophagy prevents the accumulation of misfolded proteins and malfunctioning organelles such as mitochondria; failure to remove damaged organelles could result in the induction of cellular stresses such as reactive oxygen species(3, 4). Accumulating evidence indicates that autophagic degradation is involved in a wide variety of physiological and pathophysiological processes including development, aging, neurodegeneration, cancer, and infectious diseases(5–7). It is generally agreed that autophagy is a cytoprotective mechanism. However, many studies have also suggested that autophagy plays a pro-death role under certain circumstances, especially when apoptosis is inhibited(6, 8–10). It is not difficult to imagine that excessive induction of autophagy could result in the overconsumption of functional proteins and organelles, ultimately leading to cell death. However, it remains to be further evaluated whether autophagy observed in dying cells is a bona fide cell death program or simply a morphological feature of cell death with autophagic vacuoles(11). Nevertheless, there is great potential to manipulate autophagy for therapeutics.
In mammalian cells, autophagy is initiated when cytoplasmic constituents are sequestered into cup-shaped membrane structures known as phagophores or isolation membranes(2, 12, 13). The phagophores are extended and sealed to form double-membraned autophagosomes, which then fuse with endosomes or lysosomes to become single-membraned autolysosomes, resulting in the degradation of the sequestered components. To date, 31 genes that are involved in autophagy have been identified by genetic screens in yeast and are thus termed AuTophaGy-related (ATG) genes(14–16). Of these, 18 ATG genes have recently been named AP-ATG genes(16, 17), as they are indispensable for the formation of autophagosomes. While many of these genes have been characterized for their involvement in autophagy, the molecular machineries that regulate the generation of phagophores and the origin of autophagosomal membranes remain far from clear.
Bif-1, also known as Endophilin B1 or SH3GLB1, is a member of the endophilin protein family, and was originally discovered as a Bax-binding protein(18, 19). Interestingly, recent studies suggest that Bif-1 is involved in mitochondrial fission and COPI-vesicle formation(20, 21). Moreover, we have found that Bif-1 interacts with Beclin1 (the mammalian orthologue of yeast Atg6) through UVRAG (ultraviolet irradiation resistant-associated gene) to regulate the activation of the class III PI3 kinase, PI3KC3 (also known as Vps34), and the induction of autophagy in mammalian cells(10). Loss of Bif-1 significantly suppresses PI3KC3 activation and the formation of autophagosomes in both HeLa cells and mouse embryonic fibroblasts (MEFs)(10). In response to nutrient starvation, a portion of Bif-1 accumulates in foci in the cytoplasm. Interestingly, the starvation-induced Bif-1 foci colocalize with microtubule-associated protein light chain 3 (LC3), as well as Atg5(10). While LC3 is the most widely used marker for autophagosomes, Atg5 has been shown to be present on the phagophore throughout the elongation step of autophagosome formation, but is absent from completed autophagosomes(22–24). Therefore, these results suggest that Bif-1 is involved in the early stages of autophagosome formation and may play a role in the biogenesis and/or expansion of phagophores. Indeed, Bif-1 has been found to co-localize with Atg9-positive vesicles(25); formation and trafficking of these vesicles are essential for the biogenesis and expansion of autophagosomal membranes during the induction of autophagy(26–28). While recent studies have shed light on the role of Bif-1 in autophagy, the precise molecular mechanism by which Bif-1 regulates the formation of autophagosomes remains to be examined. In this review, we summarize the current knowledge of Bif-1-mediated autophagy and endocytic trafficking and discuss the potential role of Bif-1 in the biogenesis and/or expansion of autophagosomal membranes.
The endophilin protein family
Endophilins are an evolutionally conserved family of proteins that are involved in a variety of intracellular membrane dynamics (Fig. 1). Based on sequence similarity, this family of proteins can be categorized into two groups: the Endophilin A and Endophilin B subfamilies(29). In mammalian systems, three Endophilin A proteins have been characterized, Endophilin A1-3, and two Endophilin B proteins have been identified, Bif-1/SH3GLB1/Endophilin B1 and SH3GLB2/Endophilin B2(29, 30). While Endophilin A1 is well known to be involved in the formation of endocytic vesicles at the plasma membrane(29), Bif-1 is localized in the cytosol and regulates the membrane dynamics of organelles such as the Golgi complex and mitochondria, as well as autophagosomes(10, 20, 21, 25). All endophilins contain an amino-terminal N-BAR (Bin-Amphiphysin-Rvs) domain and a carboxy-terminal SH3 (Src-homology 3) domain. While the SH3 domain plays a key role in forming complexes with proteins containing a proline-rich region (PRR), the N-BAR domain is composed of three anti-parallel helixes and is required for binding to lipid bilayers and inducing membrane curvature(10, 30–38). In addition, the helixes of the N-BAR domain form a coiled-coil structure which allows endophilins to dimerize with each other. Recent structural analyses of endophilin N-BAR domains revealed three major functional regions that are required for binding to and tubulating liposomes in vitro: an N-terminal amphipathic helix (H0), an amphipathic helix inserted in helix 1 (H1I) and a crescent-shaped main body that is formed by dimerized BAR domains(34, 35). Whereas the H0 region plays a crucial role in binding to membranes, the H1I region is inserted into membranes and drives membrane curvature in cooperation with the dimerized BAR main body. Moreover, Endophilin A1 mutants lacking the H1I domain were only found as monomers in vitro(35), suggesting that this region is also important for the dimerization of endophilins. In addition, the H1I region seems to be responsible for the ability of endophilins to target to specific sites within the cell, as substitution of this region of Bif-1 for the Endophilin A1 H1I domain alters Bif-1 localization from the cytosol to the plasma membrane(34).
Figure 1. The domain structure of the endophilin protein family.

(a) Schematic representation of human Endophilin A and B proteins. All endophilins are composed of an N-terminal N-BAR domain and a C-terminal SH3 domain. The helix 0 (H0) and helix 1 insertion (H1I) regions located in the N-BAR domain are well-conserved between the Endophilin A and B families. (b) Structure-based alignment of Bif-1/Endophilin B1/SH3GLB1 among species. The N-BAR and SH3 domains, as well as the H0 region are evolutionally conserved. Notably, protein structure prediction analysis revealed that the H1I region is missing in the S. pombe yeast homologue of Bif-1; however, this region is conserved among all multicellular organisms that we investigated. The GenBank accession numbers of endophilins shown in (a) and (b) are: human Endophilin A1/SH3GL2, NP_003017; human Endophilin A2/SH3GL1, NP_003016; human Endophilin A3/SH3GL3, Q99963; human Bif-1/Endophilin B1/SH3GLB1, NP_057093; human Endophilin B2/SH3GLB2, NP_064530; mouse Bif-1, NP_062337; Xenopus Bif-1, NP_001016072; zebrafish Bif-1, NP_001017617; Ciona Bif-1, XP_002125621; Drosophila Bif-1, NP_725873; C. elegans Bif-1, NP_741756; S. pombe Bif-1, NP_595695.
Bif-1 and autophagosome biogenesis
The process of autophagy involves multiple steps, including initiation, cargo packaging, vesicle nucleation, vesicle expansion and completion, retrieval, docking and fusion, and lysosomal degradation of the vesicles and their contents(2, 12, 13). Vesicle nucleation is an early step in autophagosome formation that generates double-membraned structures. It has been well demonstrated that the Beclin1-PI3KC3 complex is required for this step(6); however, the machinery that supplies the driving force for membrane curvature is essentially unknown. Interestingly, Bif-1 has been shown to bind to Beclin1, through UVRAG, and regulate the activation of PI3KC3 and the formation of autophagosomes(10, 25). Moreover, Bif-1 has intrinsic membrane curvature-inducing activity(32), indicating that this protein might play a role in the biogenesis of isolation membranes. To date, many candidates have been proposed for the source of autophagosomal membranes including membranes from the ER, Golgi, mitochondria, endosomes, and de novo assembled membranes(26, 27, 39). Atg9 is one of the best characterized molecules for investigating the biogenesis of autophagosomes, as this protein is the only member of the AP-ATG proteins that has been shown to be anchored to membranes(40–42). In the yeast system, Atg9 cycles between perivacuolar sites, known as phagophore assembly sites (PAS), and peripheral sites including mitochondria(26, 39). In contrast, the mammalian orthologue of yeast Atg9, mAtg9, has been shown to translocate from the trans-Golgi network (TGN) to peripheral sites, including late endosomes, during starvation. However, mAtg9 has not been shown to localize to mitochondria(26, 27). Loss of Atg9, or inhibition of Atg9 trafficking, renders both yeast and mammalian cells unable to form autophagosomes(26, 27, 39), suggesting that Atg9-containing vesicles are a source of membranes for the biogenesis and/or expansion of autophagosomes. The activation of PI3KC3 appears to play a key role in the translocation of mAtg9 from the TGN during starvation, as inhibition of PI3KC3 activation by treatment with LY294002 suppresses this event(28). Reportedly, Bif-1, in addition to mAtg9, Beclin1 and PI3KC3, can be localized to the Golgi complex under normal culture conditions(21, 28, 32, 43). During the induction of autophagy, Bif-1 concentrates in punctate structures in the cytosol where it co-localizes with mAtg9(25), implying that Bif-1, together with the UVRAG-Beclin1-PI3KC3 complex, may regulate the formation of Atg9-vesicles at the Golgi complex for the biogenesis of autophagosomal membranes (Fig. 2). Consistently, we have recently used time-lapse microscopy to show that small Bif-1-positive vesicles fuse with Atg9-positive small membrane compartments in response to nutrient starvation(25). Moreover, this membrane fusion results in cup-shaped membrane structures that expand by recruiting new Atg9-positive membranes. These results suggest that Bif-1 may also promote the expansion of isolation membranes. Reportedly, the expansion of autophagosomal membranes is mediated through membrane-tethering and hemifusion by Atg8, the yeast homologue of mammalian LC3(44). Interestingly, it has also been shown that the generation of membranes with high curvature by endophilin N-BAR domains is important for the fusion of liposomes(35). Thus, it is tempting to investigate whether Bif-1 plays a role in the regulation of Atg8/LC3-mediated membrane fusion for the biogenesis of autophagosomes.
Figure 2. Schematic model of the role of Bif-1 in autophagy.
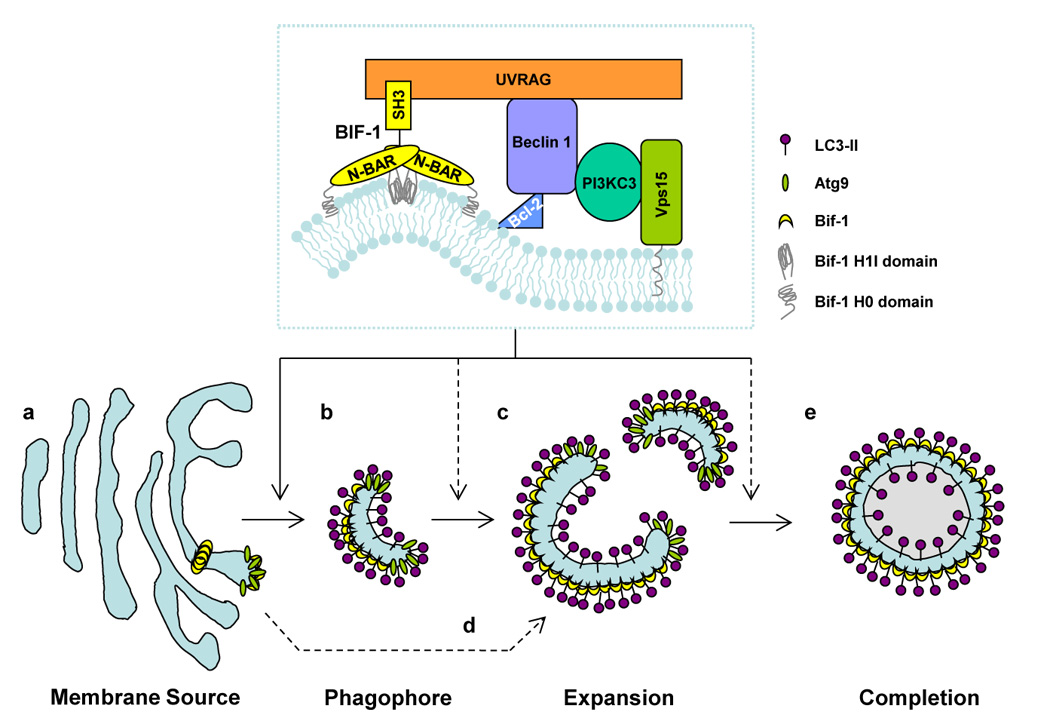
Bif-1 collaborates with PI3KC3 in regulating the biogenesis, expansion and completion of autophagosomes. In response to autophagic signals, Bif-1 tubulates the TGN to generate Atg9-Bif-1-containing vesicles (a). The resultant vesicles serve as the source of phagophores/isolation membranes (b), which are then nucleated and expanded by fusion with other Atg9-Bif-1-containing vesicles (c). Alternatively, Atg9-Bif-1-positive vesicles are delivered to phagophores/isolation membranes, which are de novo synthesized or supplied from unknown donors, as source membranes for the expansion and completion of autophagosomes (d). The membrane is eventually sealed to become a double-membraned autophagosome (e). Atg9 is then released from the vacuole, while Bif-1 remains attached to the autophagosome.
It has been reported that Bif-1 is indispensable for the formation of COPI-vesicles in certain types of cells, such as MEFs and adult mouse lung fibroblasts, but is dispensable for COPI-vesicle formation in CHO cells and mouse L cells(21). As loss of Bif-1 suppresses the formation of autophagosomes in MEFs(10), COPI-vesicles could be another candidate for the source of autophagosomal membranes. However, this is unlikely, as our preliminary results indicate that knockdown of Bif-1 in mouse L cells in which BARS (Brefeldin-A ADP-ribosylated substrate), but not Bif-1, plays a key role in COPI-vesicle formation(21), still results in the suppression of autophagosome formation (Y. Takahashi and H.G. Wang, unpublished data). Recently, it has been reported that PI3P-enriched Ω-like shaped membrane structures are generated in the proximity of the ER. These so-called omegasomes co-localize with markers of autophagosomes during starvation and thus are proposed to be an origin of autophagosomal membranes(45). As the formation of omegasomes depends on Beclin1, as well as PI3KC3(45), it is of interest to determine whether Bif-1 may also play a role in the omegasome formation. Clearly, further studies are required to define the source of autophagosomal membranes.
The role of mammalian PI3KC3 complexes and Bif-1 in the induction of autophagy
The activation of PI3KC3 plays an essential role in the induction of autophagy, as the inhibition of PI3KC3 kinase activity, by treatment with wortamannin or 3-methyladenine, suppresses autophagosome formation(46–48). In addition, PI3KC3 is also involved in vesicle transport, including endocytic trafficking(48). It has been shown that PI3KC3/Vps34 can form two distinct complexes in yeast; complex I (Vps15-Vps34-Atg6-Atg14) and complex II (Vps15-Vps34-Atg6-Vps38), which are specifically involved in autophagy and vacuolar protein sorting, respectively(47) (Fig. 3a, b). In mammalian cells, PI3KC3 has also been shown to interact with Atg6/Beclin1(43, 49, 50), as well as Vps15(48, 51, 52). Moreover, recent studies have identified putative mammalian counterparts of yeast Atg14 and Vps38; mAtg14/Barkor and UVRAG, respectively(53–55), suggesting that the yeast PI3KC3/Vps34 complexes may be conserved in mammalian systems (Figure 3c, d). Indeed, it has recently been shown that UVRAG, together with Beclin1, promotes the activation of PI3KC3 in a Vps15-dependent manner(51). To support the notion that the mAtg14/Barkor complex (Vps15-PI3KC3-Beclin1-mAtg14/Barkor) is a mammalian counterpart of the yeast Vps34 complex I, it was shown that mAtg14/Barkor contains a coiled-coil domain necessary for binding to Beclin1, co-localizes with LC3 and Atg5 during starvation, and is required for the formation of autophagosomes(54, 55). However, Beclin1 can functionally replace Atg6 in yeast for PI3KC3/Vps34-mediated autophagy, but not vacuolar protein sorting(49, 50). Consistently, knockdown of Beclin1 expression has little affect on endocytotic internalization and trafficking in U-251 human glioblastoma cells(56). Furthermore, UVRAG was originally found to interact with Beclin1 and enhance the interaction of Beclin1 with PI3KC3 to promote the activation of PI3KC3 and the formation of autophagosomes(53). In addition, although two additional Beclin1-interacting proteins, Ambra1 (activating molecule in Beclin1-regulated autophagy)(57) and VMP-1 (the vacuole membrane protein 1)(58) have been identified to be required for autophagosome formation in mammalian systems, their yeast orthologues have not been found. A recent study has also shown that UVRAG regulates the maturation of autophagosomes and the fusion of endosomes through its interaction with the class C Vps complex(59). Therefore, it seems that the mammalian PI3KC3 complex I (Vps15-PI3KC3-Beclin1-mAtg14/Barkor) and II (Vps15-PI3KC3-Beclin1-UVRAG) are not functionally equivalent to the yeast Vps34 complex I and II, which are distinct from each other by specifically localizing to PAS and endosomes, respectively(60) (Figure 3).
Figure 3. Schematic model of Bif-1 regulation of PI3KC3 and UVRAG complexes.

(a, b) The yeast Vps34 complexes. The yeast Vps34 complex I plays an essential role in autophagy at the vesicle nucleation step by recruiting ubiquitin-like Atg proteins to PAS (a), whereas the Vps34 complex II is required for the vacuolar protein sorting system (b). (c–e) The mammalian PI3KC3 complexes and UVRAG complexes. In mammalian systems, the counterparts of yeast Vps34 complex I and II have been identified as the PI3KC3 complex I (c) and II (d), respectively. However, evidence suggests that both PI3KC3 complex I and II are important for autophagy in mammalian systems. (d) Bif-1 has been shown to interact with the complex II and promote the activation of PI3KC3 and the formation of autophagosomes. Moreover, Bif-1 together with the PI3KC3 complex II might be involved in Atg9-containing vesicle formation/trafficking. (e) Bif-1 may regulate UVRAG binding to the class C Vps complex for autophagosome/endosome maturation.
Although recent studies have suggested that mAtg14/Barkor and UVRAG play a key role in the localization of the PI3KC3 complex to PAS/autophagosomes and/or the activation of PI3KC3(53–55, 61), precisely how these proteins work with Beclin1 to regulate the activation of PI3KC3 activity and the induction of autophagy are currently unknown. An interesting molecule that may help address this question is the N-BAR and SH3 domain-containing protein, Bif-1, which has been shown to interact with Beclin1 through UVRAG(10). UVRAG consists of an N-terminal PRR and a potential calcium-dependent phospholipid binding C2 domain (C2D), followed by a central coiled-coil domain (CCD). The CCD is required for binding to Beclin1, but is insufficient for UVRAG to promote autophagy, as deletion of the N-terminal region containing the PRR and C2D greatly reduces the formation of autophagosomes induced by ectopic expression of UVRAG(53). Intriguingly, Bif-1 interacts with UVRAG through its SH3 domain(10). Since the SH3 domain is capable of binding to proline-rich sequences (PxxP)(62), UVRAG might bind to the Bif-1 SH3 domain through its PRR to bridge Bif-1 and the Beclin1-PI3KC3 complex. Although it has been shown that both the SH3 and the N-BAR domains are required for Bif-1 to promote autophagy(10), the molecular role of the N-BAR domain in the formation of autophagosomes remains to be determined. Our preliminary results suggest that a Bif-1 mutant lacking the H0 domain, which is responsible for Bif-1 binding to membranes, cannot rescue autophagosome formation in Bif-1-deficient MEFs and HeLa cells (Y. Takahashi and H.G. Wang, unpublished data). Thus, it will be interesting to determine whether the ability of Bif-1 to drive membrane curvature is responsible for the formation of autophagosomes. This can be achieved using mutants of Bif-1, such as those lacking the H1I domain, which can bind to lipid bilayers, but cannot regulate membrane dynamics. Moreover, it will be important to determine if the ability of Bif-1 to bind to membranes and/or force membrane curvature plays a regulatory role in the activation of PI3KC3. It has been reported that the activation of PI3-kinase to produce PI3P is strongly dependent on the membrane curvature of lipid particles in vitro(63). Since Bif-1 has been shown to bind to lipid bilayers and transform liposomes into narrow tubules in vitro(32), it is tempting to speculate that Bif-1 could promote the activation of PI3KC3 not only by forming a complex with Beclin1 through UVRAG, but also by forcing membrane curvature through its N-BAR domain, thus inducing the formation of autophagosomes. To support this theory, it has been shown that restoration of the Bif-1 SH3 domain alone failed to rescue starvation-induced autophagosome formation in Bif-1-deficient cells, despite the fact that the SH3 domain of Bif-1 is sufficient for interaction with UVRAG and is indispensable for Bif-1-mediated PI3KC3 activation(10).
In contrast to previous studies(53, 59, 61), UVRAG was also reported to co-localize with Rab9, a protein known to localize on late endosomes, but not with LC3(54). In addition, knockdown of UVRAG had minimal effects on the formation of autophagosomes, supporting the notion that UVRAG is a mammalian orthologue of yeast Vps38(54). The discrepancy regarding the involvement of UVRAG in autophagosome formation may be due to differences in the experimental systems examined, such as transient versus stable transfection and/or a difference in cell types studied. Localization of UVRAG may be affected by culture conditions, such as cell confluency and stimulation by certain transfection systems. In addition, ectopic expression of GFP-UVRAG itself may cause the aggregation and formation of foci in an autophagy-independent manner, such as those that are observed when cells are transfected with GFP-LC3(64). Interestingly, it has been reported that many GFP foci were observed, even under nutrient-rich conditions, in NIH3T3 cells stably transfected with GFP-UVRAG(54), whereas results from our group did not show any punctate signals in HeLa cells transiently transfected with flag-UVRAG (Y. Takahashi and H.G. Wang, unpublished data). However, we did observe that flag-UVRAG signals accumulate in foci in the cytosol of HeLa cells during nutrient starvation (Y. Takahashi and H.G. Wang, unpublished data). In the future, it will be important to determine if these starvation-induced UVRAG foci co-localize with markers of isolation membranes and autophagosomes, as well as early and late endosomes. Notably, we also observed that a significant portion of HeLa cells undergo cell death after transient transfection of flag-UVRAG, but not flag-Beclin 1, flag-PI3KC3 or an empty vector. Since excess amounts of autophagy could lead to cell death(6, 8, 9), it is of interest to determine the mechanisms behind the cell death induced by ectopic expression of flag-UVRAG.
Bif-1 and endocytosis
It has been shown that PI3KC3 is involved not only in the regulation of autophagy, but also endocytosis, especially internal vesicle formation in late endosomes(48, 65). Knockdown of PI3KC3 expression in U-251 cells does not affect internalization of the epidermal growth factor receptor (EGFR), but reduces the rate of dephosphorylation and degradation of EGFR induced by EGF stimulation(48). Moreover, a recent study has shown that UVRAG interacts with the class C Vps complex, an essential component for tethering endosomal membranes(66, 67), and participates in the maturation of autophagosomes in a Beclin1-independent manner(59). Ectopic expression of UVRAG did not affect ligand stimulation-induced EGFR internalization, but promoted the endocytic trafficking and degradation of EGFR by accelerating endosomal fusion with lysosomes(59). As Bif-1 has been shown to interact with the UVRAG-Beclin1 complex to promote the activity of PI3KC3(10), it would be assumed that loss of Bif-1 should therefore result in a defect in endocytic vesicle trafficking and a reduction in EGFR degradation. Similar to PI3KC3(48), our preliminary data indicate that Bif-1 does not regulate the internalization step of the endocytic pathway. We found that knockdown of Bif-1 did not inhibit the internalization of EGF or the uptake of a fluid phase marker, horseradish peroxidase (C.L. Meyerkord, Y. Takahashi and H.G. Wang, unpublished data). Surprisingly, however, loss of Bif-1 did not suppress, but rather accelerated the co-localization of internalized EGF with LAMP-1, a marker for late endosomes/lysosomes (C.L. Meyerkord, Y. Takahashi and H.G. Wang, unpublished data). Moreover, the rate of EGF-stimulated degradation of total and phosphorylated EGFR was enhanced in Bif-1 knockdown HeLa cells as compared to control cells (C.L. Meyerkord, Y. Takahashi and H.G. Wang, unpublished data), indicating that endocytic vesicle trafficking and receptor degradation were promoted by loss of Bif-1 expression. Consistent with our findings, it has recently been shown that loss of Bif-1 accelerated the endocytic trafficking and degradation of NGF-Trk receptor tyrosine kinase A (TrkA) in PC12 cells(68). One possible explanation for the discrepancy between the effect of UVRAG and Bif-1 on the endocytic system could be that Bif-1 switches UVRAG to the PI3KC3 complex II from the class C Vps complex(Fig. 3e). As mentioned above, we have found that the SH3 domain of Bif-1 is required for its interaction with UVRAG (10). Interestingly, the C2D, along with the C-terminus of UVRAG, is required for its binding to class C Vps(59). As the C2D is located adjacent to the PRR, the binding of Bif-1 may alter the conformation of UVRAG and thus decrease the affinity of UVRAG for class C Vps. If this is the case, however, overexpression of Bif-1 may inhibit autophagosomal maturation. Further studies are warranted to determine the molecular mechanisms by which loss of Bif-1 accelerates the endocytic process and affects the crosstalk between autophagy and endocytic degradation.
Bif-1, Bax, Beclin1 and autophagy
As previously mentioned, Bif-1 was originally discovered by yeast two-hybrid screens aimed at identifying novel Bax-binding proteins(30, 31). Bax is a pro-apoptotic member of the Bcl-2 protein family which, once activated, promotes the release of apoptogenic factors, such as cytochrome c, from the mitochondria to initiate the caspase cascade(69–71). Loss of Bif-1 suppresses the activation of Bax and mitochondrial apoptosis induced by serum starvation, tunicamycin treatment, and detachment from the extracellular matrix(72, 73). In contrast, enhanced activation of caspase-3/7 and induction of apoptosis were observed in nutrient-deprived Bif-1-knockout cells(10). The mechanisms by which Bif-1 influences the decision of a cell to undergo apoptosis or to induce autophagy are currently unknown. Interestingly, the interaction of Bif-1 with Bax occurs prior to Bax activation and decreases after Bax conformational change(72). Moreover, Bcl-2, an anti-apoptotic protein, has been shown to suppress the induction of autophagy by binding to Beclin1(74). Therefore, Bif-1 might liberate Beclin1 from Bcl-2 by releasing Bax and promoting Bax binding to Bcl-2(75), thus allowing free Beclin1 to associate with Bif-1 to induce autophagy.
Notably, it has been shown that most mitochondrial Bax accumulates at scission sites on the surface of mitochondria, where it co-localizes with Drp1, a mediator of mitochondrial fission(76). Fragmentation of mitochondria plays a critical role in the induction of apoptosis, as this fission event occurs prior to cytochrome c release and caspase activation(77). Interestingly, heterogeneous expression of human Bax in yeast does not affect the induction of apoptosis, but promotes the fragmentation of the mitochondrial network and degradation of mitochondria through a mitochondria-selective autophagic degradation pathway known as mitophagy(78). Furthermore, overexpression of Fis1, a mitochondrial protein, promotes the fission of mitochondria and has been reported to trigger the induction of autophagy(79). Since Bif-1 has been shown to translocate to mitochondria(20, 72) where it interacts with and activates Bax during the induction of apoptosis(72), Bif-1, together with Bax, may also contribute to the regulation of mitophagy.
Conclusions and Perspectives
Evidence from recent studies has shown that Bif-1 plays a role in multiple intracellular processes that are involved in dynamic membrane rearrangements(10, 20, 25, 32). Due to its unique structure, Bif-1 is proposed to regulate these intracellular events by inducing membrane curvature. Remarkably, recent structural analyses of the endophilin N-BAR domain identified three functional regions, the H0, H1I, and crescent-shaped BAR dimer(34, 35). These studies provided valuable information for further characterization of the role of Bif-1 in the regulation of membrane dynamics. We are currently using various Bif-1 mutants lacking one or more of these domains to determine whether the function of Bif-1 in apoptosis, autophagy, mitochondrial fission and endocytic trafficking is dependent on its ability to bind to and/or bend membranes.
Another important question that remains to be answered is what regulates the ability of Bif-1 to generate membrane curvature. Post-translational modification(s) could be a mechanism by which this occurs, as Bif-1 has recently been shown to be phosphorylated by Src kinase on tyrosine 80 (Y80)(73). This site is located within the H1I region of the Bif-1 N-BAR domain, suggesting that the ability of Bif-1 to promote membrane curvature may be regulated by the status of phosphorylation at Y80. Consistently, mutation of amino acids within the H1I domain to negatively charged amino acids almost completely abrogated the liposome tubulation activity of Bif-1 in vitro(35). Interestingly, phosphorylation at Y80 was shown to negatively regulate the interaction of Bif-1 with Bax(73), indicating that membranes with high-curvature might be a key factor for the Bif-1-Bax interaction. Indeed, the interaction of Bif-1 with Bax was observed on mitochondria or liposomes, but not in the cytosol or solution(72, 80). It is of interest to investigate the effect of phosphorylation of Bif-1 on its ability to mediate autophagosome formation. In addition to post-translational regulation, the translocation of Bif-1 to certain target sites might be regulated through a positive-feedback loop, as the crescent shape of dimerized BAR domains is capable of sensing and binding to membranes with high-curvature(81). That is, Bif-1 could generate membrane curvature, which may cause accumulation of Bif-1 on target sites that would further enhance the curvature of these membranes.
Investigations into the precise role of Bif-1 in autophagy have just begun. Results from previous studies suggest a possible role for Bif-1 in the regulation of membrane biogenesis and the expansion of autophagosomal membranes by generating Atg9-containing vesicles as a source of membranes(10, 25, 32). Intracellular localization profiles indicate that Bif-1, together with the UVRAG-Beclin1-PI3KC3 complex, may regulate the formation of Atg9-containing vesicles at the TGN(25, 28, 43). Thus, the Golgi complex may be a donor of membranes for autophagosome formation and/or expansion. As mentioned above, the trafficking of Atg9-containg vesicles during starvation has been shown to be suppressed by the treatment with a PI3KC3 inhibitor(28). Thus, it is tempting to determine if the mAtg14/Barkor complex and/or the UVRAG complex, together with Bif-1, plays a key role in the regulation of Atg9-containing vesicles to supply a membrane source for autophagosome formation. Notably, however, loss of Bif-1 does not completely suppress the formation of autophagosomes(10). Moreover, Bif-1 knockout mice develop normally, which is different from AP-Atg knockout mice(10), which are embryonically lethal(82) or die at an early neonatal stage(83, 84). If Bif-1 is essential for the formation of autophagosomes, then why is the phenotype of Bif-1 deficiency so different from that of AP-Atg knockouts? The existence of functional homologue(s) of Bif-1 could be a potential answer to this question. Endophilin B2/SH3GLB2, another member of the Endophilin B family of proteins, is the best candidate to functionally compensate for Bif-1. To investigate this, we are currently generating mice lacking both of these Endophilin B family proteins. Further studies are anticipated to clarify the role of the Endophilin B family proteins in the biogenesis of autophagosomal membranes and the regulation of autophagy.
Acknowledgements
The work is supported by grants from the James & Esther King Biomedical Research Program to Y.T., National Institutes of Health, American Cancer Society, and Flight Attendant Medical Research Institute to H.G.W.
Abbreviations
- Bif-1
Bax-interacting factor 1
- BAR
Bin-Amphiphysin-Rvs
- SH3
Src-homology 3
- UVRAG
ultraviolet irradiation resistant-associated gene
- PI3KC3
phosphatidylinositol 3-kinase class 3
- Vps
vacuolar protein sorting
- Atg
autophagy-related
- AP-Atg
autophagosome-formation Atg
- LC3
microtubule-associated protein light chain 3
- COPI
coat protein complex I
- H0
helix 0
- H1I
helix 1 insert
- ER
endoplasmic reticulum
- PAS
phagophore assembly site
- TGN
trans-Golgi network
- PRR
proline rich region
- C2D
calcium-dependent phospholipid binding C2 domain
- CCD
central coiled-coil domain
- EGF
epidermal growth factor
References
- 1.Yoshimori T. Autophagy: a regulated bulk degradation process inside cells. Biochem Biophys Res Commun. 2004 Jan 9;313(2):453–458. doi: 10.1016/j.bbrc.2003.07.023. [DOI] [PubMed] [Google Scholar]
- 2.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004 Apr;6(4):463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 3.Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S, et al. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 2007 Jul 1;21(13):1621–1635. doi: 10.1101/gad.1565707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, et al. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007 Jun 1;21(11):1367–1381. doi: 10.1101/gad.1545107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008 Feb 28;451(7182):1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008 Jan 11;132(1):27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kundu M, Thompson CB. Autophagy: basic principles and relevance to disease. Annu Rev Pathol. 2008;3:427–455. doi: 10.1146/annurev.pathmechdis.2.010506.091842. [DOI] [PubMed] [Google Scholar]
- 8.Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004 Dec;6(12):1221–1228. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]
- 9.Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, et al. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004 Jun 4;304(5676):1500–1502. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]
- 10.Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol. 2007 Oct;9(10):1142–1151. doi: 10.1038/ncb1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008 Dec;9(12):1004–1010. doi: 10.1038/nrm2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoshimori T, Noda T. Toward unraveling membrane biogenesis in mammalian autophagy. Curr Opin Cell Biol. 2008 Aug;20(4):401–407. doi: 10.1016/j.ceb.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 13.Eskelinen EL. New insights into the mechanisms of macroautophagy in mammalian cells. Int Rev Cell Mol Biol. 2008;266:207–247. doi: 10.1016/S1937-6448(07)66005-5. [DOI] [PubMed] [Google Scholar]
- 14.Suzuki K, Ohsumi Y. Molecular machinery of autophagosome formation in yeast, Saccharomyces cerevisiae. FEBS Lett. 2007 May 22;581(11):2156–2161. doi: 10.1016/j.febslet.2007.01.096. [DOI] [PubMed] [Google Scholar]
- 15.Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007 Oct;9(10):1102–1109. doi: 10.1038/ncb1007-1102. [DOI] [PubMed] [Google Scholar]
- 16.Mizushima N. Autophagy: process and function. Genes Dev. 2007 Nov 15;21(22):2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 17.Kabeya Y, Kawamata T, Suzuki K, Ohsumi Y. Cis1/Atg31 is required for autophagosome formation in Saccharomyces cerevisiae. Biochem Biophys Res Commun. 2007 May 4;356(2):405–410. doi: 10.1016/j.bbrc.2007.02.150. [DOI] [PubMed] [Google Scholar]
- 18.Pierrat B, Simonen M, Cueto M, Mestan J, Ferrigno P, Heim J. SH3GLB, a new endophilin-related protein family featuring an SH3 domain. Genomics. 2001 Jan 15;71(2):222–234. doi: 10.1006/geno.2000.6378. [DOI] [PubMed] [Google Scholar]
- 19.Cuddeback SM, Yamaguchi H, Komatsu K, Miyashita T, Yamada M, Wu C, et al. Molecular cloning and characterization of Bif-1. A novel Src homology 3 domain-containing protein that associates with Bax. J Biol Chem. 2001 Jun 8;276(23):20559–20565. doi: 10.1074/jbc.M101527200. [DOI] [PubMed] [Google Scholar]
- 20.Karbowski M, Jeong SY, Youle RJ. Endophilin B1 is required for the maintenance of mitochondrial morphology. J Cell Biol. 2004 Sep 27;166(7):1027–1039. doi: 10.1083/jcb.200407046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang JS, Zhang L, Lee SY, Gad H, Luini A, Hsu VW. Key components of the fission machinery are interchangeable. Nat Cell Biol. 2006 Dec;8(12):1376–1382. doi: 10.1038/ncb1503. [DOI] [PubMed] [Google Scholar]
- 22.Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, et al. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol. 2001 Feb 19;152(4):657–668. doi: 10.1083/jcb.152.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J. 2000 Nov 1;19(21):5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008 Feb 16;4(2):151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takahashi Y, Meyerkord CL, Wang HG. BARgaining membranes for autophagosome formation: Regulation of autophagy and tumorigenesis by Bif-1/Endophilin B1. Autophagy. 2008 Jan 1;4(1):121–124. doi: 10.4161/auto.5265. [DOI] [PubMed] [Google Scholar]
- 26.Noda T, Kim J, Huang WP, Baba M, Tokunaga C, Ohsumi Y, et al. Apg9p/Cvt7p is an integral membrane protein required for transport vesicle formation in the Cvt and autophagy pathways. J Cell Biol. 2000 Feb 7;148(3):465–480. doi: 10.1083/jcb.148.3.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamada T, Carson AR, Caniggia I, Umebayashi K, Yoshimori T, Nakabayashi K, et al. Endothelial nitric-oxide synthase antisense (NOS3AS) gene encodes an autophagy-related protein (APG9-like2) highly expressed in trophoblast. J Biol Chem. 2005 May 6;280(18):18283–18290. doi: 10.1074/jbc.M413957200. [DOI] [PubMed] [Google Scholar]
- 28.Young AR, Chan EY, Hu XW, Kochl R, Crawshaw SG, High S, et al. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci. 2006 Sep 15;119(Pt 18):3888–3900. doi: 10.1242/jcs.03172. [DOI] [PubMed] [Google Scholar]
- 29.Huttner WB, Schmidt A. Lipids, lipid modification and lipid-protein interaction in membrane budding and fission--insights from the roles of endophilin A1 and synaptophysin in synaptic vesicle endocytosis. Curr Opin Neurobiol. 2000 Oct;10(5):543–551. doi: 10.1016/s0959-4388(00)00126-4. [DOI] [PubMed] [Google Scholar]
- 30.Pierrat B, Simonen M, Cueto M, Mestan J, Ferrigno P, Heim J. SH3GLB, a new endophilin-related protein family featuring an SH3 domain. Genomics. 2001;71(2):222–234. doi: 10.1006/geno.2000.6378. [DOI] [PubMed] [Google Scholar]
- 31.Cuddeback SM, Yamaguchi H, Komatsu K, Miyashita T, Yamada M, Wu C, et al. Molecular cloning and characterization of Bif-1: A novel SH3 domain- containing protein that associates with Bax. J Biol Chem. 2001;276(23):20559–20565. doi: 10.1074/jbc.M101527200. [DOI] [PubMed] [Google Scholar]
- 32.Farsad K, Ringstad N, Takei K, Floyd SR, Rose K, De Camilli P. Generation of high curvature membranes mediated by direct endophilin bilayer interactions. J Cell Biol. 2001 Oct 15;155(2):193–200. doi: 10.1083/jcb.200107075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Modregger J, Schmidt AA, Ritter B, Huttner WB, Plomann M. Characterization of Endophilin B1b, a brain-specific membrane-associated lysophosphatidic acid acyl transferase with properties distinct from endophilin A1. J Biol Chem. 2003 Feb 7;278(6):4160–4167. doi: 10.1074/jbc.M208568200. [DOI] [PubMed] [Google Scholar]
- 34.Masuda M, Takeda S, Sone M, Ohki T, Mori H, Kamioka Y, et al. Endophilin BAR domain drives membrane curvature by two newly identified structure-based mechanisms. Embo J. 2006 Jun 21;25(12):2889–2897. doi: 10.1038/sj.emboj.7601176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gallop JL, Jao CC, Kent HM, Butler PJ, Evans PR, Langen R, et al. Mechanism of endophilin N-BAR domain-mediated membrane curvature. Embo J. 2006 Jun 21;25(12):2898–2910. doi: 10.1038/sj.emboj.7601174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ringstad N, Nemoto Y, De Camilli P. The SH3p4/Sh3p8/SH3p13 protein family: binding partners for synaptojanin and dynamin via a Grb2-like Src homology 3 domain. Proc Natl Acad Sci U S A. 1997 Aug 5;94(16):8569–8574. doi: 10.1073/pnas.94.16.8569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Micheva KD, Ramjaun AR, Kay BK, McPherson PS. SH3 domain-dependent interactions of endophilin with amphiphysin. FEBS Lett. 1997 Sep 8;414(2):308–312. doi: 10.1016/s0014-5793(97)01016-8. [DOI] [PubMed] [Google Scholar]
- 38.Weissenhorn W. Crystal structure of the endophilin-A1 BAR domain. J Mol Biol. 2005 Aug 19;351(3):653–661. doi: 10.1016/j.jmb.2005.06.013. [DOI] [PubMed] [Google Scholar]
- 39.Reggiori F, Shintani T, Nair U, Klionsky DJ. Atg9 cycles between mitochondria and the pre-autophagosomal structure in yeasts. Autophagy. 2005 Jul;1(2):101–109. doi: 10.4161/auto.1.2.1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Webber JL, Young AR, Tooze SA. Atg9 trafficking in Mammalian cells. Autophagy. 2007 Jan–Feb;3(1):54–56. doi: 10.4161/auto.3419. [DOI] [PubMed] [Google Scholar]
- 41.He C, Klionsky DJ. Atg9 trafficking in autophagy-related pathways. Autophagy. 2007 May–Jun;3(3):271–274. doi: 10.4161/auto.3912. [DOI] [PubMed] [Google Scholar]
- 42.Mari M, Reggiori F. Atg9 trafficking in the yeast Saccharomyces cerevisiae. Autophagy. 2007 Mar–Apr;3(2):145–148. doi: 10.4161/auto.3608. [DOI] [PubMed] [Google Scholar]
- 43.Kihara A, Kabeya Y, Ohsumi Y, Yoshimori T. Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep. 2001 Apr;2(4):330–335. doi: 10.1093/embo-reports/kve061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nakatogawa H, Ichimura Y, Ohsumi Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell. 2007 Jul 13;130(1):165–178. doi: 10.1016/j.cell.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 45.Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008 Aug 25;182(4):685–701. doi: 10.1083/jcb.200803137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. Distinct classes of phosphatidylinositol 3'-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem. 2000 Jan 14;275(2):992–998. doi: 10.1074/jbc.275.2.992. [DOI] [PubMed] [Google Scholar]
- 47.Kihara A, Noda T, Ishihara N, Ohsumi Y. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J Cell Biol. 2001 Feb 5;152(3):519–530. doi: 10.1083/jcb.152.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Backer JM. The regulation and function of Class III PI3Ks: novel roles for Vps34. Biochem J. 2008 Feb 15;410(1):1–17. doi: 10.1042/BJ20071427. [DOI] [PubMed] [Google Scholar]
- 49.Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999 Dec 9;402(6762):672–676. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 50.Furuya N, Yu J, Byfield M, Pattingre S, Levine B. The evolutionarily conserved domain of Beclin 1 is required for Vps34 binding, autophagy and tumor suppressor function. Autophagy. 2005 Apr;1(1):46–52. doi: 10.4161/auto.1.1.1542. [DOI] [PubMed] [Google Scholar]
- 51.Yan Y, Flinn RJ, Wu H, Schnur RS, Backer JM. hVps15 but not calcium/calmodulin is required for the activity and regulation of hVps34 in mammalian cells. Biochem J. 2008. Oct 28, [DOI] [PMC free article] [PubMed]
- 52.Volinia S, Dhand R, Vanhaesebroeck B, MacDougall LK, Stein R, Zvelebil MJ, et al. A human phosphatidylinositol 3-kinase complex related to the yeast Vps34p-Vps15p protein sorting system. EMBO J. 1995 Jul 17;14(14):3339–3348. doi: 10.1002/j.1460-2075.1995.tb07340.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, et al. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol. 2006 Jul;8(7):688–699. doi: 10.1038/ncb1426. [DOI] [PubMed] [Google Scholar]
- 54.Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 Forms Two Distinct Phosphatidylinositol 3-Kinase Complexes with Mammalian Atg14 and UVRAG. Mol Biol Cell. 2008. Oct 8, [DOI] [PMC free article] [PubMed]
- 55.Sun Q, Fan W, Chen K, Ding X, Chen S, Zhong Q. Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proc Natl Acad Sci U S A. 2008 Dec 9;105(49):19211–19216. doi: 10.1073/pnas.0810452105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zeng X, Overmeyer JH, Maltese WA. Functional specificity of the mammalian Beclin-Vps34 PI 3-kinase complex in macroautophagy versus endocytosis and lysosomal enzyme trafficking. J Cell Sci. 2006 Jan 15;119(Pt 2):259–270. doi: 10.1242/jcs.02735. [DOI] [PubMed] [Google Scholar]
- 57.Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, et al. Ambra1 regulates autophagy and development of the nervous system. Nature. 2007 Jun 28;447(7148):1121–1125. doi: 10.1038/nature05925. [DOI] [PubMed] [Google Scholar]
- 58.Ropolo A, Grasso D, Pardo R, Sacchetti ML, Archange C, Lo Re A, et al. The pancreatitis-induced vacuole membrane protein 1 triggers autophagy in mammalian cells. J Biol Chem. 2007 Dec 21;282(51):37124–37133. doi: 10.1074/jbc.M706956200. [DOI] [PubMed] [Google Scholar]
- 59.Liang C, Lee JS, Inn KS, Gack MU, Li Q, Roberts EA, et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol. 2008 Jul;10(7):776–787. doi: 10.1038/ncb1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Obara K, Sekito T, Ohsumi Y. Assortment of phosphatidylinositol 3-kinase complexes--Atg14p directs association of complex I to the pre-autophagosomal structure in Saccharomyces cerevisiae. Mol Biol Cell. 2006 Apr;17(4):1527–1539. doi: 10.1091/mbc.E05-09-0841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liang C, Feng P, Ku B, Oh BH, Jung JU. UVRAG: a new player in autophagy and tumor cell growth. Autophagy. 2007 Jan–Feb;3(1):69–71. doi: 10.4161/auto.3437. [DOI] [PubMed] [Google Scholar]
- 62.Mayer BJ. SH3 domains: complexity in moderation. J Cell Sci. 2001 Apr;114(Pt 7):1253–1263. doi: 10.1242/jcs.114.7.1253. [DOI] [PubMed] [Google Scholar]
- 63.Hubner S, Couvillon AD, Kas JA, Bankaitis VA, Vegners R, Carpenter CL, et al. Enhancement of phosphoinositide 3-kinase (PI 3-kinase) activity by membrane curvature and inositol-phospholipid-binding peptides. Eur J Biochem. 1998 Dec 1;258(2):846–853. doi: 10.1046/j.1432-1327.1998.2580846.x. [DOI] [PubMed] [Google Scholar]
- 64.Kuma A, Matsui M, Mizushima N. LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: caution in the interpretation of LC3 localization. Autophagy. 2007 Jul–Aug;3(4):323–328. doi: 10.4161/auto.4012. [DOI] [PubMed] [Google Scholar]
- 65.Johnson EE, Overmeyer JH, Gunning WT, Maltese WA. Gene silencing reveals a specific function of hVps34 phosphatidylinositol 3-kinase in late versus early endosomes. J Cell Sci. 2006 Apr 1;119(Pt 7):1219–1232. doi: 10.1242/jcs.02833. [DOI] [PubMed] [Google Scholar]
- 66.Peterson MR, Emr SD. The class C Vps complex functions at multiple stages of the vacuolar transport pathway. Traffic. 2001 Jul;2(7):476–486. doi: 10.1034/j.1600-0854.2001.20705.x. [DOI] [PubMed] [Google Scholar]
- 67.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000 Dec 1;290(5497):1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wan J, Cheung AY, Fu WY, Wu C, Zhang M, Mobley WC, et al. Endophilin B1 as a novel regulator of nerve growth factor/TrkA trafficking and neurite outgrowth. J Neurosci. 2008 Sep 3;28(36):9002–9012. doi: 10.1523/JNEUROSCI.0767-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chipuk JE, Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008 Apr;18(4):157–164. doi: 10.1016/j.tcb.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Reed JC. Proapoptotic multidomain Bcl-2/Bax-family proteins: mechanisms, physiological roles, and therapeutic opportunities. Cell Death Differ. 2006 Aug;13(8):1378–1386. doi: 10.1038/sj.cdd.4401975. [DOI] [PubMed] [Google Scholar]
- 71.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008 Jan;9(1):47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 72.Takahashi Y, Karbowski M, Yamaguchi H, Kazi A, Wu J, Sebti SM, et al. Loss of Bif-1 suppresses Bax/Bak conformational change and mitochondrial apoptosis. Mol Cell Biol. 2005 Nov;25(21):9369–9382. doi: 10.1128/MCB.25.21.9369-9382.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yamaguchi H, Woods NT, Dorsey JF, Takahashi Y, Gjertsen NR, Yeatman T, et al. SRC directly phosphorylates Bif-1 and prevents its interaction with Bax and the initiation of anoikis. J Biol Chem. 2008 Jul 4;283(27):19112–19118. doi: 10.1074/jbc.M709882200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005 Sep 23;122(6):927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 75.Hsu YT, Youle RJ. Nonionic detergents induce dimerization among members of the Bcl-2 family. J Biol Chem. 1997;272(21):13829–13834. doi: 10.1074/jbc.272.21.13829. [DOI] [PubMed] [Google Scholar]
- 76.Karbowski M, Lee YJ, Gaume B, Jeong SY, Frank S, Nechushtan A, et al. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J Cell Biol. 2002 Dec 23;159(6):931–938. doi: 10.1083/jcb.200209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Suen DF, Norris KL, Youle RJ. Mitochondrial dynamics and apoptosis. Genes Dev. 2008 Jun 15;22(12):1577–1590. doi: 10.1101/gad.1658508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kissova I, Plamondon LT, Brisson L, Priault M, Renouf V, Schaeffer J, et al. Evaluation of the roles of apoptosis, autophagy, and mitophagy in the loss of plating efficiency induced by Bax expression in yeast. J Biol Chem. 2006 Nov 24;281(47):36187–36197. doi: 10.1074/jbc.M607444200. [DOI] [PubMed] [Google Scholar]
- 79.Gomes LC, Scorrano L. High levels of Fis1, a pro-fission mitochondrial protein, trigger autophagy. Biochim Biophys Acta. 2008 Jul–Aug;1777(7–8):860–866. doi: 10.1016/j.bbabio.2008.05.442. [DOI] [PubMed] [Google Scholar]
- 80.Etxebarria A, Terrones O, Yamaguchi H, Landajuela A, Landeta O, Antonsson B, et al. Endophilin B1/Bif-1 stimulates BAX activation independently from its capacity to produce large-scale membrane morphological rearrangements. J Biol Chem. 2008. Dec 11, [DOI] [PMC free article] [PubMed]
- 81.Peter BJ, Kent HM, Mills IG, Vallis Y, Butler PJ, Evans PR, et al. BAR domains as sensors of membrane curvature: the amphiphysin BAR structure. Science. 2004 Jan 23;303(5657):495–499. doi: 10.1126/science.1092586. [DOI] [PubMed] [Google Scholar]
- 82.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003 Dec 9;100(25):15077–15082. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004 Dec 23;432(7020):1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 84.Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005 May 9;169(3):425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]