

Abstract
A high-fat, high-calorie diet is associated with obesity and type 2 diabetes. However, the relative contribution of metabolic defects to the development of hyperglycaemia and type 2 diabetes is controversial. Accumulation of excess fat in muscle and adipose tissue in insulin resistance and type 2 diabetes may be linked with defective mitochondrial oxidative phosphorylation. The aim of the current study was to investigate acute effects of short-term fat overfeeding on glucose and insulin metabolism in young men. We studied the effects of 5 days’ high-fat (60% energy) overfeeding (+50%) versus a control diet on hepatic and peripheral insulin action by a hyperinsulinaemic euglycaemic clamp, muscle mitochondrial function by 31P magnetic resonance spectroscopy, and gene expression by qrt-PCR and microarray in 26 young men. Hepatic glucose production and fasting glucose levels increased significantly in response to overfeeding. However, peripheral insulin action, muscle mitochondrial function, and general and specific oxidative phosphorylation gene expression were unaffected by high-fat feeding. Insulin secretion increased appropriately to compensate for hepatic, and not for peripheral, insulin resistance. High-fat feeding increased fasting levels of plasma adiponectin, leptin and gastric inhibitory peptide (GIP). High-fat overfeeding increases fasting glucose levels due to increased hepatic glucose production. The increased insulin secretion may compensate for hepatic insulin resistance possibly mediated by elevated GIP secretion. Increased insulin secretion precedes the development of peripheral insulin resistance, mitochondrial dysfunction and obesity in response to overfeeding, suggesting a role for insulin per se as well GIP, in the development of peripheral insulin resistance and obesity.
High-fat and high-calorie diets along with a sedentary lifestyle have made type 2 diabetes a worldwide epidemic (Zimmet et al. 2001). Although many metabolic active organs may be involved, the pathophysiology of type 2 diabetes is characterized by three major defects, namely peripheral (muscle) insulin resistance, elevated hepatic glucose production and impaired insulin secretion (Defronzo, 2004). However, the relative contribution of peripheral and hepatic insulin resistance versus defective insulin secretion on development of hyperglycaemia is controversial, and may depend on the dominant underlying aetiology of this multi-factorial disease.
Altered lipid metabolism plays an important role in the pathogenesis of insulin resistance (Roden et al. 1996). Accumulation of excess fat as intramyocellular lipid (IMCL) in muscle tissue has been reported to be associated with reduced insulin sensitivity (Perseghin et al. 1999; Krssak et al. 1999), and it has been shown that mitochondrial function and expression of genes involved in oxidative phosphorylation including their key co-transcriptional factor peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) are commonly decreased in elderly and obese insulin resistant and type 2 diabetic subjects (Petersen et al. 2004, 2005; Patti et al. 2003). Dysregulation of IMCL metabolism in insulin resistance and type 2 diabetes may be linked with defective oxidative phosphorylation (Petersen et al. 2004; Befroy et al. 2007), and both short- and long-term fat exposure may play a key role in the development of impaired mitochondrial oxidative phosphorylation (Sparks et al. 2005; Brehm et al. 2006).
Previous studies have used intravenous lipid infusions to induce supraphysiological high levels of plasma FFA as a model to study the metabolic effects of high fat exposure, and thereby to mimic the state of overt type 2 diabetes commonly characterized by elevated FFA levels (Belfort et al. 2005; Bachmann et al. 2001). Other studies have used varying duration of either different types of overfeeding (Faeh et al. 2005; Clore et al. 1995; Cornier et al. 2006), or diets containing high amounts of fat (Bachmann et al. 2001; Westerbacka et al. 2005; Bisschop et al. 2001). However, these studies have mainly included rodents, obese human subjects and/or human subjects with a family history of diabetes with relatively small numbers (Westerbacka et al. 2005; Bachmann et al. 2001; Chanseaume et al. 2007; Chanseaume et al. 2006). To our knowledge, no study of short-term physiological ‘overfeeding’ has been conducted using a diet high in both fat and calories to study simultaneously the effects on multiple metabolic mechanisms relevant to the pathophysiology of type 2 diabetes in healthy human subjects without known predisposition to type 2 diabetes.
The aim of the present study was to examine the short-term effects of a Westernized diet, i.e. fat overfeeding, on insulin sensitivity in skeletal muscle and liver, and on β-cell function in a relatively large group of young, lean men without a family history of type 2 diabetes. Furthermore, we wanted to investigate the effect of such a diet on in vivo mitochondrial function, as well as on both global gene expression and on expression of PGC-1α and key oxidative phosphorylation genes, previously shown to be downregulated in type 2 diabetic patients, in skeletal muscle tissue (Patti et al. 2003; Mootha et al. 2003).
Methods
Subjects
Twenty-six young male volunteers were recruited from the Danish National Birth Registry. All men were born in 1979–1980 in Copenhagen. Subjects with a family history of diabetes (in 2 generations), body mass index (BMI) greater than 30 kg m−2, and with a high physical activity level (>10 h of exercise per week) were excluded from participation. None of the participants had known illness or took medications known to affect the study outcome. All subjects signed an informed consent before study participation. The protocol was approved by the ethical committee of Copenhagen county, and was in accordance with the Declaration of Helsinki.
Diet
The study was designed as a randomized cross-over study, where subjects were studied on two separate occasions 6–8 weeks apart (Fig. 1). The participants’ individual energy requirement was calculated by a WHO equation for men under the age of 30 years (FAO/WHO/UNU, 2007). The energy requirement was multiplied by a factor of 1.5 for physical activity level corresponding to a low physical activity level during the examination days.
Figure 1. Overview of study activities.

The study was designed as a cross-over study where the subjects received the diet in a randomized order. *Data not included.
All subjects had to start standardization of physical activity, alcohol consumption and diet 5 days prior the examinations (Fig. 1). Subjects kept a record of their standardization, so it could be repeated prior to the second period. Furthermore, subjects had to be weight stable (i.e. be on an isocaloric diet) and were asked to remain weight stable between the two examination periods. The high-fat intervention (‘challenge’) diet was delivered to participants for 5 days, starting 3 days prior to the first examination day. Besides the standardization period, the control experiment was optimized even further, by providing individualized meals the 3 days before the first examination.
The intervention diet was a Westernized diet high in fat and calories (HFHC diet) containing 50% extra energy where 60% of the energy came from fat, 32.5% from carbohydrate, and 7.5% from protein. The control diet was designed to reflect the participants’ habitual weight-maintaining diet with 35% of the energy coming from fat, 50% from carbohydrate, and 15% from protein. The fat proportion of the diet consisted of 1/3 monounsatured, 1/3 polyunsaturated and 1/3 saturated fatty acids. The average caloric content of the control diet was 11.8 ± 1.0 MJ versus 17.7 ± 1.4 MJ (mean ±s.d.) on the overfeeding diet.
The subjects were provided with five servings of food per day: breakfast (25%), snack (10%), lunch (25%), afternoon snack (10%) and dinner (30%). The meals did not vary from day to day, and were designed to be as common and regular as possible. Participants were expected to eat all of the provided food, but if there were for some reason any leftovers, the food was collected, weighed and subtracted from the total energy intake. The participants received the diet in a randomised order.
DEXA scanning
Body composition was assessed on the first examination day by DEXA (Lunar Radiation, Madison, WI, USA), followed by a 24 h respiratory chamber stay (data not included).
31Phosphorus magnetic resonance spectroscopy (31P-MRS)
On day 2, 31P-MRS was performed on two different muscle groups, the forearm flexor muscles and the tibialis anterior muscle. An Otsuka Electronics VivoSpec© spectrometer was used, interfaced to a 2.9 Tesla magnet (Magnex Scientific, Yarnton, UK) with a 26 cm bore diameter. Data acquisition and calculations were performed as described by Brøns et al. (2008).
Hyperinsulinaemic euglycaemic clamp
The examination was initiated at 07.00 h after an overnight fast and was carried out on day 3 as previously described in detail (Brøns et al. 2008). A primed-continuous infusion of [3-3H]glucose (bolus 10.9 μCi, 0.109 μCi min−1) was initiated at 0 h, and the examination was initiated by a 2 h basal period followed by an intravenous glucose tolerance test (IVGTT) to determine β-cell function. A glucose (20%) bolus of 0.3 g (kg body weight)−1 was infused i.v. over 1 min. Blood samples for glucose, insulin, and C-peptide were collected at −10, −4, −2, 0, 2, 4, 6, 8, 10, 15, 20 and 30 min.
Following the IVGTT a primed-continuous insulin infusion was initiated and fixed at 80 mU m−2 min−1 throughout the 180 min clamp. Steady-state was defined as the last 30 min of the basal and insulin clamp period, when tracer equilibrium was anticipated. Variable infusion of glucose (180 g l−1) enriched with [3-3H]glucose (110 μCi per 500 ml) was used to maintain euglycaemia (5 mmol l−1) during insulin infusion. During the clamp period, blood samples for measuring [3-3H]glucose and [3-3H]water were drawn at 15, 30, 45, 60, 90, 100, 110, 120, 135, 150, 160, 170 and 180 min. Steady-state was defined as the last 30 min of the basal and the insulin clamp period, when tracer equilibrium was anticipated. Oxygen consumption 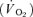 and carbon dioxide production
and carbon dioxide production 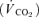 were measured during steady-state using indirect calorimetry with a computerized flow-through canopy gas analyser system (Deltatrac; Datex, Helsinki, Finland) as previously described (Vaag et al. 1995). Both steady-state periods were followed by excision of biopsies from m. vastus lateralis. Analytical procedures and calculations of glucose turnover and insulin secretion rates are described in the online Supplemental Material.
were measured during steady-state using indirect calorimetry with a computerized flow-through canopy gas analyser system (Deltatrac; Datex, Helsinki, Finland) as previously described (Vaag et al. 1995). Both steady-state periods were followed by excision of biopsies from m. vastus lateralis. Analytical procedures and calculations of glucose turnover and insulin secretion rates are described in the online Supplemental Material.
Quantitative real time-PCR
Extraction of total RNA from the muscle biopsies was performed with TRI reagent (Sigma-Aldrich). cDNA was synthesized using the QuantiTect® Reverse Transcription kit (Qiagen, Valencia, CA, USA). RT-PCR was performed using the ABI 7900 Sequence Detection System (Applied Biosystems, Foster City, CA, USA). Assays from Applied Biosystems were used: NDUFB6 (Hs00159583_m1), UQCRB (Hs00559884_m1), COX7A1 (Hs00156989_m1), ATP5O (Hs00426889_m1) and PGC-1α (Hs00173304_m1). All samples were run in duplicate, data were calculated using the standard curve method and normalized to the mRNA level of cyclophilin A (4326316E, Applied Biosystems).
Microarray analysis
Whole genome and pathway microarray analysis was performed in a subset of the subjects on RNA extracted from insulin-stimulated skeletal muscle tissue before and after the HFHC diet (Supplemental Material).
Statistics
Statistical analysis was performed with SAS Statistical Analysis Package (SAS Institute, Cary, NC, USA, v. 8.2). Student's paired t-test was used to detect statistical differences between the control and the HFHC diet. Correlations between MRS data and M-value were calculated using Spearman's or Pearson's (normally distributed data) correlation coefficient. A P-value of <0.05 was considered statistically significant. Data are presented as mean values ±s.d. or s.e.m.
Results
Clinical characteristics and plasma lipids
There were no differences in completeness of consumption of meals/snacks between the control and the HFHC diet. Additionally, there were no differences in body composition measured by DEXA scanning (data not shown). As shown in Table 1, 5 days of a HFHC diet had no statistical significant effect on weight, body composition, blood pressure, or total cholesterol levels. The HFHC diet resulted in a decrease in fasting plasma FFA, triglycerides, LDL cholesterol, VLDL cholesterol and in an increase in HDL cholesterol concentrations.
Table 1.
Clinical characteristics of study participants
| Control diet | HFHC diet | P-value | |
|---|---|---|---|
| Weight (kg) | 78.3 ± 9.1 | 78.6 ± 9.7 | 0.38 |
| BMI (kg m−2) | 23.4 ± 2.4 | 23.3 ± 2.5 | 0.91 |
| W/H-ratio | 0.88 ± 0.05 | 0.89 ± 0.05 | 0.25 |
| Systolic BP (mm Hg) | 137 ± 13 | 133 ± 14 | 0.14 |
| Diastolic BP (mm Hg) | 72 ± 7 | 73 ± 10 | 0.63 |
| ASAT (U l−1) | 22.73 ± 6.55 | 27.00 ± 7.13 | 0.02* |
| Fasting FFA (μmol l−1) | 334 ± 136 | 205 ± 82 | <0.01* |
| Insulin stimulated FFA (μmol l−1) (Clamp) | 9.3 ± 4.4 | 12.4 ± 6.4 | 0.01* |
| Fasting triglycerides (mmol l−1) | 0.92 ± 0.34 | 0.73 ± 0.35 | 0.02* |
| Fasting total cholesterol (mmol l−1) | 4.36 ± 0.83 | 4.18 ± 0.82 | 0.10 |
| Fasting LDL cholesterol (mmol l−1) | 2.56 ± 0.75 | 2.28 ± 0.78 | 0.01* |
| Fasting VLDL cholesterol (mmol l−1) | 0.42 ± 0.16 | 0.33 ± 0.16 | 0.02* |
| Fasting HDL cholesterol (mmol l−1) | 1.38 ± 0.22 | 1.56 ± 0.25 | <0.01* |
Data are means ±s.d.n= 26.
Significant differences between control and HFHC diet.
Insulin action and glucose metabolism
In the basal state prior to the euglycaemic hyperinsulinaemic clamp, the HFHC diet resulted in significant elevations of fasting glucose and C-peptide concentrations, as well as a borderline increase in insulin levels (Table 2). In addition, the liver enzyme aspartate aminotransferase (ASAT) was significantly increased after the HFHC diet (Table 1).
Table 2.
Basal and insulin stimulated metabolite concentrations and metabolic data from the hyperinsulinaemic euglycaemic clamp
| Control diet | HFHC diet | P-value | ||
|---|---|---|---|---|
| Blood glucose (mmol l−1) | Basal | 4.59 ± 0.46 | 5.05 ± 0.40 | <0.001* |
| Ins. stim. | 5.11 ± 0.31 | 5.17 ± 0.30 | 0.44 | |
| Serum insulin (pmol l−1) | Basal | 31.4 ± 14.2 | 43.4 ± 29.2 | 0.07 |
| Ins. stim. | 870 ± 232 | 867 ± 181 | 0.97 | |
| Serum C-peptide (pmol l−1) | Basal | 408 ± 146 | 529 ± 260 | 0.05* |
| Ins. stim. | 406 ± 279 | 453 ± 195 | 0.26 | |
| GOX (mg kg−1 FFM min−1) | Basal | 2.41 ± 0.83 | 2.53 ± 0.76 | 0.60 |
| Ins. stim. | 5.18 ± 0.80 | 5.03 ± 0.90 | 0.59 | |
| FOX (mg kg−1 FFM min−1) | Basal | 0.98 ± 0.39 | 0.98 ± 0.34 | 0.89 |
| Ins. stim. | 0.02 ± 0.25 | 0.16 ± 0.30 | 0.08 | |
| NOGM (mg kg−1 FFM min−1) | Ins. stim. | 7.54 ± 2.82 | 7.18 ± 2.73 | 0.61 |
| Rd (mg kg−1 FFM min−1) | Basal | 2.29 ± 0.57 | 2.97 ± 1.12 | 0.01* |
| Ins. stim. | 12.23 ± 2.38 | 11.78 ± 2.50 | 0.41 | |
| GF (mg kg−1 FFM min−1) | Basal | 1.64 ± 1.96 | 2.16 ± 4.52 | 0.60 |
| Ins. stim. | 4.62 ± 2.17 | 3.47 ± 2.55 | 0.05* | |
| Non-ox.GF (mg kg−1 FFM min−1) | Ins. stim. | −0.07 ± 2.47 | −0.95 ± 2.68 | 0.15 |
| EGS (mg kg−1 FFM min−1) | Basal | 0.87 ± 1.83 | 1.98 ± 4.14 | 0.23 |
| Ins. stim. | 7.60 ± 2.77 | 8.38 ± 3.10 | 0.35 |
Data are means ±s.d.*Significant differences between control and HFHC diet.
After overfeeding, a 26% increase in fasting hepatic glucose production (HGP) as well as an almost twofold increase in the hepatic insulin resistance index was observed (68.7 ± 6.7 vs. 113.7 ± 12.1 mg (kg FFM)−1 min−1 pmol−1 l−1) (Fig. 2A and B). Insulin-suppressed plasma FFA levels were increased during the clamp in response to overfeeding. There was no effect of overfeeding on insulin-mediated glucose uptake as determined by whole-body Rd (Table 2) or the clamp M-value (13.79 ± 0.45 vs. 13.29 ± 0.65 mg (kg FFM)−1 min−1) (Fig. 2C). However, insulin stimulated glycolytic flux (GF) was reduced by 25% by the HFHC diet. The HGP was fully suppressed during insulin stimulation and the non-oxidative glucose metabolism (NOGM), non-oxidative GF (Non-ox. GF) as well as the endogenous glucose storage (EGS) remained unchanged by the intervention (Table 2).
Figure 2. Basal hepatic glucose production and insulin resistance index and insulin stimulated glucose disposal.

A, hepatic glucose production (HGP) in the basal state. B, calculated hepatic insulin resistance index in the basal state. C, insulin stimulated glucose disposal, i.e. the clamp M-value. Control diet (grey) and HFHC diet (black). Data are means ±s.e.m. *P≤ 0.05, **P≤ 0.005.
Insulin secretion
The intravenous glucose tolerance test (IVGTT) of β-cell function showed that the AUC from 0–30 min for glucose was unaffected by the HFHC diet whereas there was a significant increased in both AUCinsulin and AUCC-peptide (P= 0.0004 and P= 0.01 respectively) (Fig. 3). Furthermore, the calculated first phase insulin response (FPIR) during the first 10 min of the IVGTT, was increased by overfeeding (Table 3).
Figure 3. AUC (0–30 min) for blood glucose, serum insulin and C-peptide during the IVGTT.

Filled squares, HFHC diet; filled triangles, control studies. Data are means ±s.d., *P≤ 0.05.
Table 3.
Results of the IVGTT before and after the HFHC diet
| Control diet | HFHC diet | P-value | |
|---|---|---|---|
| First phase insulin response (FPIR) | 1894 ± 1431 | 2604 ± 1793 | <0.001* |
| DI of insulin secretion (peripheral ins. action) | 0.29 ± 0.19 | 0.35 ± 0.19 | 0.04* |
| DI of insulin secretion (hepatic ins. action) | 0.38 ± 0.64 | 0.25 ± 0.21 | 0.36 |
Data are means ±s.d. The first phase insulin response (0–10 min) is used as a measure of insulin secretion. The disposition indices (DI) of insulin secretion are calculated to correct insulin secretion for the degree of in vivo peripheral and hepatic insulin action respectively. n= 26.
Significant differences between control and HFHC diet.
The disposition index (DI), based on in vivo insulin secretion and action, can be calculated because of an approximately hyperbolic relationship between the two measures (Bergman et al. 2002). When insulin secretion was expressed in relation to hepatic insulin resistance, the calculated DI was unaffected by overfeeding indication appropriate compensation. In contrast, when expressed in relation to peripheral insulin action, the DI increased paradoxically by overfeeding, indicating inappropriate over compensation (Table 3).
Gut hormones and adipokines
Fasting plasma levels of adiponectin, leptin, gastric inhibitory peptide (GIP) and pancreatic polypeptide (PP) were all increased in response to the HFHC diet (Table 4). Peptide YY (PYY) was borderline significantly increased by the HFHC diet. There were no changes in fasting plasma levels of amylin, ghrelin and glucagon-like peptide-1 (GLP-1).
Table 4.
Fasting plasma levels of gut hormones and adipokines
| Control diet | HFHC diet | P-value | |
|---|---|---|---|
| Adiponectin (μg ml−1) | 7.61 ± 3.91 | 8.63 ± 4.30 | <0.01* |
| Amylin (pmol l−1) | 353 ± 224 | 363 ± 223 | 0.56 |
| Ghrelin (pg ml−1) | 14.32 ± 6.27 | 15.2 ± 8.86 | 0.55 |
| Leptin (pg ml−1) | 3863 ± 2350 | 4657 ± 2678 | <0.001* |
| GIP (pg ml−1) | 53.99 ± 20.43 | 94.16 ± 62.71 | <0.01* |
| GLP-1 (pg ml−1) | 64.44 ± 30.69 | 69.85 ± 35.22 | 0.22 |
| PP (pg ml−1) | 51.24 ± 19.7 | 68.33 ± 36.12 | 0.05* |
| PYY (pg ml−1) | 103.7 ± 25.7 | 111.2 ± 31.4 | 0.07 |
Data are means ±s.d.
Significant differences between control and HFHC diet.
31P-MRS
The recovery rates of phosphocreatine (PCr) as well as of inorganic phosphate (Pi) after exercise representing well established measures of in vivo mitochondrial function and capacity (Prompers et al. 2006) was unaffected by HFHC diet (Table 5) in both the forearm flexor muscles (primarily type II fibres) and the tibialis anterior of the leg (primarily type I fibres). Besides a small unexplained increase in resting pH in the forearm flexor muscles in response to overfeeding, no other measures of mitochondrial function as determined by 31P-MRS during rest and after energy-depleting exercise were affected by the HFHC diet, in the two different types of muscles.
Table 5.
Results of 31P-MRS in the forearm flexor muscles (n= 19) and tibialis anterior of the leg (n= 16)
| Arm |
Leg |
|||||
|---|---|---|---|---|---|---|
| Control Diet | HFHC diet | P-value | Control Diet | HFHC diet | P-value | |
| Rest | ||||||
| PCr (mm) | 21.9 ± 2.2 | 21.4 ± 2.2 | 0.33 | 20.6 ± 3.4 | 20.7 ± 2.2 | 0.90 |
| PCr/Pi | 8.31 ± 1.97 | 7.80 ± 1.37 | 0.32 | 6.14 ± 1.99 | 6.15 ± 2.57 | 0.99 |
| pH | 7.01 ± 0.02 | 7.02 ± 0.02 | 0.04* | 6.98 ± 0.04 | 6.99 ± 0.03 | 0.37 |
| Recovery | ||||||
| PCr (mm) | 9.15 ± 3.25 | 9.67 ± 3.96 | 0.48 | 7.28 ± 4.74 | 5.23 ± 3.86 | 0.15 |
| PCr/Pi | 1.59 ± 1.02 | 1.69 ± 1.79 | 0.80 | 10.9 ± 31.2 | 31.1 ± 70.2 | 0.32 |
| pH | 6.32 ± 0.32 | 6.35 ± 0.27 | 0.68 | 6.74 ± 0.19 | 6.68 ± 0.22 | 0.31 |
| Vmax (mm s−1) | 0.34 ± 0.08 | 0.30 ± 0.09 | 0.17 | 0.45 ± 0.17 | 0.43 ± 0.15 | 0.58 |
| PCr t1/2 (s) | 68.0 ± 25.1 | 69.6 ± 24.9 | 0.74 | 30.4 ± 13.5 | 33.5 ± 11.6 | 0.48 |
| Pi t1/2 (s) | 45.3 ± 18.0 | 47.8 ± 20.5 | 0.61 | 21.4 ± 5.9 | 25.7 ± 10.1 | 0.12 |
Data are means ±s.d.
Significant differences between control and HFHC diet.
Qrt-PCR of PGC-1α and oxidative phosphorylation gene expressions
There were no changes in PGC-1α or oxidative phosphorylation gene expression levels in response to the HFHC diet during basal (Fig. 4A) and insulin stimulated conditions (Fig. 4B) in skeletal muscle tissue.
Figure 4. Oxidative phosphorylation and PGC-1α gene expression.

Expression of key oxidative phosphorylation genes as well as PGC-1α gene expression in response to control diet (grey) and HFHC diet (black) for the basal state (A) and during insulin stimulation (B). Data are means ±s.d.
Microarray analysis
There were no statistically significant effects of the HFHC diet on any single gene or gene pathways (Supplemental Material Table 1) after correcting for multiple comparisons.
Discussion
Our study demonstrated that 5 days’ intake of a high-fat diet with 50% extra calories resulted in a 26% increase of fasting hepatic glucose production as determined by [3-3H]glucose, in spite of a borderline increased fasting insulin level. As a consequence of the increased HGP, fasting glucose levels increased by as much as 0.46 mmol l−1 after overfeeding in these healthy young men. Although within the normal range, elevated fasting glucose does in itself constitute an independent risk factor for developing type 2 diabetes among young men (Park et al. 2006). We are unaware of any previous studies having shown such a significant increase in fasting glucose levels in young healthy subjects, without any a priori increased risk of developing type 2 diabetes in response to short-term overfeeding. Indeed, this finding alone underscores the significant deleterious effects of fat overfeeding per se in healthy humans even during short-term exposures equivalent to commonly occurring feast periods in most societies. When calculating the hepatic insulin resistance index (Abdul-Ghani et al. 2007), we expanded the finding of an elevated HGP after overfeeding to be due to hepatic insulin resistance. Overfeeding resulted in a significant increase of the liver enzyme ASAT, an indication of a stressed liver accumulating fat. Indeed, hepatic steatosis in human subjects is increasingly recognized as a key feature in the metabolic syndrome associated with development of hepatic insulin resistance (Kotronen & Yki-Jarvinen, 2008).
Fasting plasma FFA, TG, LDL and VLDL concentrations were decreased, whereas HDL cholesterol was increased, after high-fat overfeeding, possibly caused by the fatty acid composition of the diet. Indeed, decreased FFA levels was previously reported in some short-term overfeeding studies (Clore et al. 1995; Cornier et al. 2006), and is most likely due to a suppression of lipolysis in adipose tissue in the fasting state mediated by the increased insulin level. The low plasma FFA could also be due to an increased uptake into the muscle tissue, possibly resulting in FFA being stored within the cell as IMCL (Bachmann et al. 2001; Schrauwen-Hinderling et al. 2005). Lowered lipoprotein levels could be a result of an increased uptake into the adipose tissue due to stimulation of lipoprotein lipase by the increased fasting insulin levels during overfeeding. Accordingly, this finding could reflect an increased clearance and/or increased capacity for lipid storage. During the hyperinsulinaemic euglycaemic clamp, the very high insulin concentration failed to fully suppress the level of FFAs, possibly due to development of insulin resistance of the adipose tissue in response to fat overfeeding. However, in the fasting state, the degree of insulin resistance in the adipose tissue was not sufficient to prevent a reduction of fasting plasma FFA levels by insulin after overfeeding. Plasma FFAs play a major role in the (acute) control of whole-body lipid oxidation, and acute experimental manipulations reducing or increasing plasma FFA levels almost instantaneously either reduce or increase whole-body lipid oxidation rates (Vaag et al. 1991; Randle, 1998). However, in contrast to less physiological fat exposure experiments using intravenous lipid infusions, 5 days of high-fat overfeeding diet did not cause an increase in the lipid oxidation rate measured in the fasting state.
Despite the observed hepatic insulin resistance, whole-body insulin-stimulated glucose disposal was not decreased after 5 days of fat overfeeding. Short-term fructose (Faeh et al. 2005) and carbohydrate (Cornier et al. 2006) overfeeding did not have an effect on whole-body insulin sensitivity in lean healthy subjects either. To our knowledge, the finding of hepatic insulin resistance without the presence of muscle insulin resistance in response to only 5 days of high-fat overfeeding has not been shown in human subjects before. However, our data are supported by a study by Bisschop et al. showing that 11 days of a high-fat intake reduces the ability of insulin to suppress endogenous glucose production without affecting the insulin-stimulated glucose uptake in healthy men (Bisschop et al. 2001). Studies of animals have shown that short-term high-fat feeding causes hepatic insulin resistance preceding the subsequent more long-term development of peripheral insulin resistance (Kraegen, 1991; Kim et al. 2003).
The insulin-stimulated glycolytic flux was 25% decreased by the HFHC diet despite of normal whole-body insulin sensitivity. This finding is consistent with a previous report of decreased insulin-stimulated glycolysis preceding overt peripheral insulin resistance during intravenous lipid infusion in rats (Kim et al. 1996).
Mitochondrial dysfunction with impaired ATP synthesis due to reduced oxidative phosphorylation capacity may represent a primary pathogenic defect involved in the development of muscle insulin resistance in type 2 diabetes (Befroy et al. 2007) and aging (Petersen et al. 2003). Interestingly, high-fat and/or high-calorie diets has been shown to promote alterations in mitochondrial oxidative phosphorylation activity, suggesting that nutrition, both qualitatively and quantitatively could alter mitochondrial function (Chanseaume et al. 2006; Brehm et al. 2006; Sparks et al. 2005). In the current study, in vivo mitochondrial function measured by 31P-MRS before and after energy depleting exercise in two different muscle groups was found not to be affected by the HFHC diet. Furthermore, no differences in basal or insulin stimulated PGC-1α and oxidative phosphorylation gene expression were observed in response to the HFHC diet. Therefore, our data do not support the current hypothesis that short-term high-fat diet reduces the expression of nuclear genes encoding mitochondrial proteins and transcription factors involved in mitochondrial biogenesis in healthy young men (Sparks et al. 2005).
To examine further the extent to which short-term high-fat feeding influence gene expression and metabolism in skeletal muscle tissue, we performed whole genome microarray analyses. After correcting for multiple comparisons we were unable to detect significant up- or downregulations after the HFHC diet of either single genes or whole pathways potentially involved in glucose metabolism including receptor signalling, NFκB, PPARα/RXRα, PPAR, PTEN, PI3K/AKT and actin cytoskeleton signalling, as well as FXR/RXR activation, glycero phospholipid metabolism, glycolysis/gluconeogenesis, fatty acid metabolism or oxidative phosphorylation (Supplemental Material). All together, the array analyses are consistent with the qrt-PCR measurements of oxidative phosphorylation gene expressions, mitochondrial function as well as clamp insulin action showing no significant effect of high-fat over feeding in skeletal muscles. This supports the overall conclusion of hepatic – and not muscle insulin resistance – being the primary lesion responsible for the significant elevation of fasting blood glucose in healthy subjects in response to the HFHC diet. However, this may not hold true after longer periods of high-fat overfeeding.
Adipokines have diverse effects upon glucose and lipid metabolism, and play a crucial role in the development of insulin resistance and type 2 diabetes. Leptin is a satiety hormone and may increase peripheral insulin sensitivity as well as hepatic glucose production. Adiponectin increases both hepatic and peripheral insulin sensitivity (Fasshauer & Paschke, 2003). Thus, the significant increase of plasma leptin in response to the HFHC diet may contribute to the elevation of hepatic glucose production and the increase of both leptin and adiponectin may explain the lack of the otherwise expected development of overt peripheral insulin resistance. These data show that increased fasting levels of the satiety hormones PP and to some extent of PYY (Wren & Bloom, 2007) may act in concert with increased leptin levels to increase satiety after 5-days of high-fat feeding.
Acute intravenous lipid exposure and FFA elevation increases insulin secretion whereas chronic elevation of FFA may cause β-cell dysfunction as a consequence of lipotoxicity (Belfort et al. 2005; Kashyap et al. 2003). The first-phase insulin response (FPIR) to an intravenous glucose bolus is a sensitive measure of β-cell function (Cobelli et al. 2007). We found that the FPIR during the IVGTT was significantly elevated in response to high-fat overfeeding. This finding is in contrast to previous studies of intravenous lipid infusions in rodents (Kim et al. 1995; Reimer & Ahren, 2002). However, these studies have not looked at the effect of overfeeding, and we are unaware of studies that have examined the effect of short-term high-fat overfeeding on β-cell function in healthy human subjects. Nevertheless, because elevated plasma FFA levels per se may exert a deleterious effect on insulin synthesis and secretion (Cerf, 2007), our finding of elevated FPIR could be a result of a lower plasma FFA levels during overfeeding. This is to some extent supported by the finding of increased first and second phase insulin secretion after 48 h of suppression of lipolysis in people at risk of developing type 2 diabetes (Cusi et al. 2007).
Although the conventional view is that increased insulin secretion is a secondary and compensatory response to insulin resistance (Bergman et al. 2002), data suggest that the inverse scenario with increased insulin secretion preceding and possibly causing insulin resistance may characterize some metabolic states (Le Stunff & Bougnères, 1994). Interestingly, partly due to insufficient knowledge about the molecular feedback mechanisms and signals between β-cell and insulin action mediating the inverse relationship between the two, it has not even been debated the extent to which hepatic or peripheral insulin action should be used in the calculation of the disposition index, an issue clearly relevant to our data showing discordant results of a HFHC diet on hepatic versus peripheral insulin action. We therefore calculated the insulin secretion disposition index in two ways, including either hepatic or peripheral insulin action. When including hepatic insulin action, the disposition index was unaltered by high-fat feeding, indicating appropriate compensation of the β-cell. However, when including peripheral insulin action in the calculation, we found a disproportionately elevated insulin secretion in relation to peripheral insulin action. We interpret the data to indicate that the increased insulin secretion could possibly precede the development of peripheral insulin resistance in young men, thereby suggesting that hyperinsulinaemia may be causally related to the development of peripheral insulin resistance after longer periods of high-fat feeding. We are not able to determine the extent to which increased insulin secretion may even precede the development of hepatic insulin resistance.
GLP-1 and GIP are the main incretin hormones signalling from the gut to enhance pancreatic insulin secretion after food ingestion. The novel finding of a significant elevation of the fasting GIP level after the HFHC diet may explain or contribute to the elevation of insulin secretion. We believe that this represent some of the first evidence as to which mechanism that mediates and communicates the signal to the pancreas to increase insulin secretion during development of – and/or to compensate for – insulin resistance in response to overfeeding. In fact, the increase in plasma GIP levels in response to overfeeding may play a key role in the development of obesity in response to HFHC diets as indicated from the recent elegant knock-out experiments of the GIP receptor (Miyawaki et al. 2002). Finally, the increase of fasting blood glucose of approximately 0.5 mmol l−1 may also by itself contribute to the elevation of insulin secretion.
The selection of the composition of the control diet was made to ensure that the dietary intake was close to the habitual dietary intake of the subjects to achieve macronutrient balance. The composition of overfeeding diet was chosen to provide a high-energy density and a low satiating effect i.e. high-fat, low-protein. However, it is not possible with this study design to completely differentiate between the effects caused by high-fat and/or the hypercaloric intake per se. Additionally, the effect of short-term high-fat overfeeding may differ in subjects with a predisposition to type 2 diabetes.
In conclusion, short-term high-fat feeding increases fasting glucose as a consequence of increased hepatic glucose production. Increased insulin secretion, compensating for hepatic insulin resistance, may partly be mediated by elevated GIP secretion. We found that increased insulin secretion precedes the development of peripheral insulin resistance, mitochondrial dysfunction and obesity in response to overfeeding, suggesting a role for insulin as well GIP in the development of peripheral insulin resistance and obesity. Finally, adiponectin and leptin are likely to play significant roles in the short-term differential effects on hepatic versus peripheral insulin action during high-fat feeding.
Acknowledgments
Marianne Modest, Lars Sander Koch, Ib Terkelsen and Duncan Talbot provided technical support and assistance with the experiments. We are especially appreciative of all the young men who participated in this study. This study was funded by The Danish Diabetes Association, The European Foundation for the Study of Diabetes (EFSD), the EU 6th Framework EXGENESIS grant, The Danish Strategic Research Council and the Aase and Ejnar Danielsen Foundation. C.B. was granted a PhD scholarship from the Faculty of Life Sciences at the University of Copenhagen.
Glossary
Abbreviations
- AUC
area under the curve
- ASAT
aspartate aminotransferase
- DI
disposition index
- EGS
endogenous glucose storage
- FFM
fat free mass
- FOX
fat oxidation
- FPIR
first phase insulin response
- GF
glycolytic flux
- GIP
gastric inhibitory polypeptide
- GLP-1
glucagon-like peptide-1
- GOX
glucose oxidation
- IMCL
intramyocellular lipid
- IVGTT
intravenous glucose tolerance test
- M-value
insulin stimulated glucose disposal rate
- NOGM
non-oxidative glucose metabolism
- Non-ox. GF
non-oxidative glycolytic flux
- PCr
phosphocreatine
- Pi
inorganic phosphor
- 31P-MRS
31phosphorus magnetic resonance spectroscopy
- PP
pancreatic polypeptide
- PYY
peptide YY
- W/H-ratio
waist/hip ratio
Author contributions
C.B., A.A., A.V. designed the experiments. C.B., C.B.J., H.S., J.S.A., N.J.H., A.W., C.M.L. performed experiments. C.B., A.V., wrote the manuscript. S.J., E.N. performed gene expressions. All authors reviewed the manuscript.
Supplemental material
Online supplemental material for this paper can be accessed at:
http://jp.physoc.org/cgi/content/full/jphysiol.2009.169078/DC1
References
- Abdul-Ghani MA, Matsuda M, Balas B, DeFronzo RA. Muscle and liver insulin resistance indexes derived from the oral glucose tolerance test. Diabetes Care. 2007;30:89–94. doi: 10.2337/dc06-1519. [DOI] [PubMed] [Google Scholar]
- Bachmann OP, Dahl DB, Brechtel K, Machann J, Haap M, Maier T, Loviscach M, Stumvoll M, Claussen CD, Schick F, Haring HU, Jacob S. Effects of intravenous and dietary lipid challenge on intramyocellular lipid content and the relation with insulin sensitivity in humans. Diabetes. 2001;50:2579–2584. doi: 10.2337/diabetes.50.11.2579. [DOI] [PubMed] [Google Scholar]
- Befroy DE, Petersen KF, Dufour S, Mason GF, de Graaf RA, Rothman DL, Shulman GI. Impaired mitochondrial substrate oxidation in muscle of insulin-resistant offspring of type 2 diabetic patients. Diabetes. 2007;56:1376–1381. doi: 10.2337/db06-0783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belfort R, Mandarino L, Kashyap S, Wirfel K, Pratipanawatr T, Berria R, DeFronzo RA, Cusi K. Dose-response effect of elevated plasma free fatty acid on insulin signaling. Diabetes. 2005;54:1640–1648. doi: 10.2337/diabetes.54.6.1640. [DOI] [PubMed] [Google Scholar]
- Bergman RN, Finegood DT, Kahn SE. The evolution of β-cell dysfunction and insulin resistance in type 2 diabetes. Eur J Clin Invest. 2002;32(Suppl 3):35–45. doi: 10.1046/j.1365-2362.32.s3.5.x. [DOI] [PubMed] [Google Scholar]
- Bisschop PH, de Metz J, Ackermans MT, Endert E, Pijl H, Kuipers F, Meijer AJ, Sauerwein HP, Romijn JA. Dietary fat content alters insulin-mediated glucose metabolism in healthy men. Am J Clin Nutr. 2001;73:554–559. doi: 10.1093/ajcn/73.3.554. [DOI] [PubMed] [Google Scholar]
- Brehm A, Krssak M, Schmid AI, Nowotny P, Waldhausl W, Roden M. Increased lipid availability impairs insulin-stimulated ATP synthesis in human skeletal muscle. Diabetes. 2006;55:136–140. [PubMed] [Google Scholar]
- Brøns C, Jensen CB, Storgaard H, Alibegovic A, Jacobsen S, Nilsson E, Astrup A, Quistorff B, Vaag A. Mitochondrial function in skeletal muscle is normal and unrelated to insulin action in young men born with low birth weight. J Clin Endocrinol Metab. 2008;93:3885–3892. doi: 10.1210/jc.2008-0630. [DOI] [PubMed] [Google Scholar]
- Cerf ME. High fat diet modulation of glucose sensing in the β-cell. Med Sci Monit. 2007;13:RA12–RA17. [PubMed] [Google Scholar]
- Chanseaume E, Malpuech-Brugere C, Patrac V, Bielicki G, Rousset P, Couturier K, Salles J, Renou JP, Boirie Y, Morio B. Diets high in sugar, fat, and energy induce muscle type-specific adaptations in mitochondrial functions in rats. J Nutr. 2006;136:2194–2200. doi: 10.1093/jn/136.8.2194. [DOI] [PubMed] [Google Scholar]
- Chanseaume E, Tardy AL, Salles J, Giraudet C, Rousset P, Tissandier A, Boirie Y, Morio B. Chronological approach of diet-induced alterations in muscle mitochondrial functions in rats. Obesity Res. 2007;15:50–59. doi: 10.1038/oby.2007.511. [DOI] [PubMed] [Google Scholar]
- Clore JN, Helm ST, Blackard WG. Loss of hepatic autoregulation after carbohydrate overfeeding in normal man. J Clin Invest. 1995;96:1967–1972. doi: 10.1172/JCI118243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobelli C, Toffolo GM, Man CD, Campioni M, Denti P, Caumo A, Butler P, Rizza R. Assessment of β-cell function in humans, simultaneously with insulin sensitivity and hepatic extraction, from intravenous and oral glucose tests. Am J Physiol Endocrinol Metab. 2007;293:E1–15. doi: 10.1152/ajpendo.00421.2006. [DOI] [PubMed] [Google Scholar]
- Cornier MA, Bergman BC, Bessesen DH. The effects of short-term overfeeding on insulin action in lean and reduced-obese individuals. Metab Clin Exp. 2006;55:1207–1214. doi: 10.1016/j.metabol.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Cusi K, Kashyap S, Gastaldelli A, Bajaj M, Cersosimo E. Effects on insulin secretion and insulin action of a 48-h reduction of plasma free fatty acids with acipimox in nondiabetic subjects genetically predisposed to type 2 diabetes. Am J Physiol Endocrinol Metab. 2007;292:E1775–E1781. doi: 10.1152/ajpendo.00624.2006. [DOI] [PubMed] [Google Scholar]
- Defronzo RA. Dysfunctional fat cells, lipotoxicity and type 2 diabetes. Int J Clin Pract. 2004;58:9–21. doi: 10.1111/j.1368-504x.2004.00389.x. [DOI] [PubMed] [Google Scholar]
- Faeh D, Minehira K, Schwarz JM, Periasamy R, Park S, Tappy L. Effect of fructose overfeeding and fish oil administration on hepatic de novo lipogenesis and insulin sensitivity in healthy men. Diabetes. 2005;54:1907–1913. doi: 10.2337/diabetes.54.7.1907. [DOI] [PubMed] [Google Scholar]
- FAO/WHO/UNU. FAO Food and Nutrition Technical Report Series 1. WHO, Geneva. 2007. Human energy requirements. Report of a Joint FAO/WHO/UNU Expert Consultation, Rome, 17–24 October 2001. [Google Scholar]
- Fasshauer M, Paschke R. Regulation of adipocytokines and insulin resistance. Diabetologia. 2003;46:1594–1603. doi: 10.1007/s00125-003-1228-z. [DOI] [PubMed] [Google Scholar]
- Kashyap S, Belfort R, Gastaldelli A, Pratipanawatr T, Berria R, Pratipanawatr W, Bajaj M, Mandarino L, DeFronzo R, Cusi K. A sustained increase in plasma free fatty acids impairs insulin secretion in nondiabetic subjects genetically predisposed to develop type 2 diabetes. Diabetes. 2003;52:2461–2474. doi: 10.2337/diabetes.52.10.2461. [DOI] [PubMed] [Google Scholar]
- Kim JK, Wi JK, Youn JH. Plasma free fatty acids decrease insulin-stimulated skeletal muscle glucose uptake by suppressing glycolysis in conscious rats. Diabetes. 1996;45:446–453. doi: 10.2337/diab.45.4.446. [DOI] [PubMed] [Google Scholar]
- Kim SP, Ellmerer M, Van Citters GW, Bergman RN. Primacy of hepatic insulin resistance in the development of the metabolic syndrome induced by an isocaloric moderate-fat diet in the dog. Diabetes. 2003;52:2453–2460. doi: 10.2337/diabetes.52.10.2453. [DOI] [PubMed] [Google Scholar]
- Kim YB, Iwashita S, Tamura T, Tokuyama K, Suzuki M. Effect of high-fat diet on the gene expression of pancreatic GLUT2 and glucokinase in rats. Biochem Biophys Res Commun. 1995;208:1092–1098. doi: 10.1006/bbrc.1995.1446. [DOI] [PubMed] [Google Scholar]
- Kotronen A, Yki-Jarvinen H. Fatty liver: a novel component of the metabolic syndrome. Arterioscler Thromb Vasc Biol. 2008;28:27–38. doi: 10.1161/ATVBAHA.107.147538. [DOI] [PubMed] [Google Scholar]
- Kraegen EW. Development of muscle insulin resistance after liver insulin resistance in high-fat-fed rats. Diabetes. 1991;40:1397–1403. doi: 10.2337/diab.40.11.1397. [DOI] [PubMed] [Google Scholar]
- Krssak M, Falk Petersen K, Dresner A, DiPietro L, Vogel SM, Rothman DL, Shulman GI, Roden M. Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: a 1H NMR spectroscopy study. Diabetologia. 1999;42:113–116. doi: 10.1007/s001250051123. [DOI] [PubMed] [Google Scholar]
- Le Stunff, Bougnères P. Early changes in postprandial insulin secretion, not in insulin sensitivity, characterize juvenile obesity. Diabetes. 1994;43:696–702. doi: 10.2337/diab.43.5.696. [DOI] [PubMed] [Google Scholar]
- Miyawaki K, Yamada Y, Ban N, Ihara Y, Tsukiyama K, Zhou H, Fujimoto S, Oku A, Tsuda K, Toyokuni S, Hiai H, Mizunoya W, Fushiki T, Holst JJ, Makino M, Tashita A, Kobara Y, Tsubamoto Y, Jinnouchi T, Jomori T, Seino Y. Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat Med. 2002;8:738–742. doi: 10.1038/nm727. [DOI] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Park YW, Chang Y, Sung KC, Ryu S, Sung E, Kim WS. The sequential changes in the fasting plasma glucose levels within normoglycemic range predict type 2 diabetes in healthy, young men. Diabetes Res Clin Pract. 2006;73:329–335. doi: 10.1016/j.diabres.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R, Landaker EJ, Goldfine AB, Mun E, DeFronzo R, Finlayson J, Kahn CR, Mandarino LJ. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc Natl Acad Sci U S A. 2003;100:8466–8471. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perseghin G, Scifo P, De Cobelli F, Pagliato E, Battezzati A, Arcelloni C, Vanzulli A, Testolin G, Pozza G, Del Maschio A, Luzi L. Intramyocellular triglyceride content is a determinant of in vivo insulin resistance in humans: a 1H-13C nuclear magnetic resonance spectroscopy assessment in offspring of type 2 diabetic parents. Diabetes. 1999;48:1600–1606. doi: 10.2337/diabetes.48.8.1600. [DOI] [PubMed] [Google Scholar]
- Petersen KF, Dufour S, Shulman GI. Decreased insulin-stimulated ATP synthesis and phosphate transport in muscle of insulin-resistant offspring of type 2 diabetic parents. PLoS Med. 2005;2:e233. doi: 10.1371/journal.pmed.0020233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, DiPietro L, Cline GW, Shulman GI. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300:1140–1142. doi: 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350:664–671. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prompers JJ, Jeneson JA, Drost MR, Oomens CC, Strijkers GJ, Nicolay K. Dynamic MRS and MRI of skeletal muscle function and biomechanics. NMR Biomed. 2006;19:927–953. doi: 10.1002/nbm.1095. [DOI] [PubMed] [Google Scholar]
- Randle PJ. Regulatory interactions between lipids and carbohydrates: the glucose fatty acid cycle after 35 years. Diabetes Metab Rev. 1998;14:263–283. doi: 10.1002/(sici)1099-0895(199812)14:4<263::aid-dmr233>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Reimer MK, Ahren B. Altered β-cell distribution of pdx-1 and GLUT-2 after a short-term challenge with a high-fat diet in C57BL/6J mice. Diabetes. 2002;51:S138–S143. doi: 10.2337/diabetes.51.2007.s138. [DOI] [PubMed] [Google Scholar]
- Roden M, Price TB, Perseghin G, Petersen KF, Rothman DL, Cline GW, Shulman GI. Mechanism of free fatty acid-induced insulin resistance in humans. J Clin Invest. 1996;97:2859–2865. doi: 10.1172/JCI118742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrauwen-Hinderling VB, Kooi ME, Hesselink MK, Moonen-Kornips E, Schaart G, Mustard KJ, Hardie DG, Saris WH, Nicolay K, Schrauwen P. Intramyocellular lipid content and molecular adaptations in response to a 1-week high-fat diet. Obes Res. 2005;13:2088–2094. doi: 10.1038/oby.2005.259. [DOI] [PubMed] [Google Scholar]
- Sparks LM, Xie H, Koza RA, Mynatt R, Hulver MW, Bray GA, Smith SR. A high-fat diet coordinately downregulates genes required for mitochondrial oxidative phosphorylation in skeletal muscle. Diabetes. 2005;54:1926–1933. doi: 10.2337/diabetes.54.7.1926. [DOI] [PubMed] [Google Scholar]
- Vaag A, Alford F, Henriksen FL, Christopher M, Beck-Nielsen H. Multiple defects of both hepatic and peripheral intracellular glucose processing contribute to the hyperglycaemia of NIDDM. Diabetologia. 1995;38:326–336. doi: 10.1007/BF00400638. [DOI] [PubMed] [Google Scholar]
- Vaag A, Skøtt P, Damsbo P, Gall MA, Richter EA, Beck-Nielsen H. Effect of the antilipolytic nicotinic acid analogue acipimox on whole-body and skeletal muscle glucose metabolism in patients with non-insulin-dependent diabetes mellitus. J Clin Invest. 1991;88:1282–1290. doi: 10.1172/JCI115432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerbacka J, Lammi K, Hakkinen AM, Rissanen A, Salminen I, Aro A, Yki-Jarvinen H. Dietary fat content modifies liver fat in overweight nondiabetic subjects. J Clin Endocrinol Metab. 2005;90:2804–2809. doi: 10.1210/jc.2004-1983. [DOI] [PubMed] [Google Scholar]
- Wren AM, Bloom SR. Gut hormones and appetite control. Gastroenterology. 2007;132:2116–2130. doi: 10.1053/j.gastro.2007.03.048. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.