

Abstract
Background
Clinical trials suggest that anesthetic-induced preconditioning (APC) produces cardioprotection in humans, but the mechanisms of APC and significance of aging for APC in humans are not well understood. Here, the impact of age on the role of two major effectors of APC, mitochondria and sarcolemmal adenosine triphosphate-sensitive potassium (sarcKATP) channels, in preconditioning of the human atrial myocardium were investigated.
Methods
Right atrial appendages were obtained from adult patients undergoing cardiac surgery and assigned to mid-aged (MA) and old-aged (OA) groups. APC was induced by isoflurane in isolated myocardium and isolated cardiomyocytes. Mitochondrial oxygen consumption measurements, myocyte survival testing, and patch clamp techniques were used to investigate mitochondrial respiratory function and sarcKATP channel activity.
Results
After in vitro APC with isoflurane, the respiratory function of isolated mitochondria was better preserved after hypoxia-reoxygenation stress in MA than in OA. In isolated intact myocytes, APC significantly decreased oxidative stress-induced cell death in MA but not in OA, and isoflurane protection from cell death was attenuated by the sarcKATP channel inhibitor HMR-1098. Further, the properties of single sarcKATP channels were similar in MA and OA, and isoflurane sensitivity of pinacidil-activated whole cell KATP current was no different between MA and OA myocytes.
Conclusion
Anesthetic-induced preconditioning with isoflurane decreases stress-induced cell death and preserves mitochondrial respiratory function to a greater degree in MA than in OA myocytes; however, sarcKATP channel activity is not differentially affected by isoflurane. Therefore, effectiveness of APC in humans may decrease with advancing age partly because of altered mitochondrial function of myocardial cells.
PHARMACOLOGIC treatment with volatile anesthetics protects the myocardium from extensive injury by delaying cell death and limiting infarction during a subsequent ischemic injury.1 Thus far, the mechanisms of anesthetic-induced preconditioning (APC) have been studied primarily in animal models. However, there is evidence that APC has beneficial effects also in humans in vivo, i.e., during surgery.2-5 In vitro studies demonstrated that isoflurane may protect the contractile function of human right atrial trabeculae during anoxic stress by activating adenosine triphosphate-sensitive potassium (KATP) channels,6 and desflurane was shown to protect human myocardium from simulated ischemia-reperfusion via the mitochondrial but not the sarcolemmal KATP (sarcKATP) channels.7 Beneficial effects of APC were demonstrated in isolated cardiac myocytes as well.8 Although efficacy of ischemic preconditioning (IPC) in humans seems to be influenced by patient age,9,10 and effectiveness of APC decreases with advancing age in other species,11,12 the relation between age and protective effects of APC in humans has not been elucidated. Recently, we demonstrated that bioenergetics of cardiac mitochondria preconditioned with isoflurane are better preserved after hypoxic stress than bioenergetics of nonpreconditioned mitochondria.13 Further, in an isolated rat myocyte model, we showed that sarcKATP channel is an effector of protection afforded by isoflurane.14 Because the mechanisms of APC in humans are not fully understood, in the current study we investigated whether mitochondrial protection and opening of the sarcKATP channel contribute to APC in the human myocardium and whether advancing age has impact on those effectors of APC.
Materials and Methods
Patient Population
After approval by the institutional review board of the Medical College of Wisconsin, Milwaukee, Wisconsin, specimens of right atrial appendages were obtained from 89 adult patients of either sex (75% male, 25% female) who underwent cardiac surgery (coronary artery bypass graft surgery and, in a few cases, valve repair). No consent form was required for collecting discarded tissue. Two age groups were of interest in the current study: mid-aged (MA; 54 ± 7 yr; range, 33-60 yr; n = 38) and old-aged (OA; 74 ± 6 yr; range, 61-85 yr; n = 51). Clinically, patients of both groups exhibited an array of similar diseases, including coronary artery disease with or without previous myocardial infarction, hypertension, hyperlipidemia, atrial fibrillation, and diabetes mellitus (table 1). All patients were undergoing treatment with various medications, but the drug therapies and extracardiac conditions were undisclosed to us.
Table 1.
Clinical Characteristics of the Patients Studied
| Mid-aged | Old-aged | |
|---|---|---|
| Total number of patients | 38 | 51 |
| Mean age (range), yr | 54 ± 7 (33-60) | 74 ± 6 (61-85) |
| Sex, male/female | 33/5 | 32/16 |
| Underlying diseases | ||
| CAD ± MI | 35 (92%) | 48 (94%) |
| Hypertension | 23 (61%) | 26 (51%) |
| Atrial fibrillation | 7 (18%) | 9 (18%) |
| Hyperlipidemia | 13 (34%) | 15 (29%) |
| Diabetes mellitus | 5 (13%) | 8 (16%) |
| Tobacco use | 11 (29%) | 16 (31%) |
Values are number of patients or mean ± SD.
CAD = coronary artery disease; MI = myocardial infarction.
Preparation of Isolated Mitochondria
Mitochondria were isolated by a similar to previously reported procedure.13 Fat and connective tissue were removed from specimens of human right atrial appendages, and the myocardial tissue was minced into 1.0-mm pieces. Minced tissue was divided into two groups. In the first group, the control group, myocardium was immersed in normal mitochondrial isolation buffer. In the second group, the treated group, myocardium was exposed to the mitochondrial isolation buffer containing 1 minimum alveolar concentration (MAC) of isoflurane (0.5 mM) for 15 min to induce preconditioning. The isolation buffer was composed of 50 mM sucrose, 200 mM mannitol, 5 mM KH2PO4, 1 mM EGTA, 5 mM MOPS, and 0.1% bovine serum albumin, at pH 7.3. The myocardial tissue was homogenized twice for 5 s with T25 disperser (IKA-Werke, Staufen, Germany), and the homogenate was centrifuged at 800g. The supernatant was saved, and the pellet was rehomogenized with Potter-Elvehjem grinder (Weaton, Millville, NJ) in the bovine serum albumin-free isolation buffer containing 4.8 U/ml protease VIII (Sigma-Aldrich, St. Louis, MO). The supernatant and rehomogenized pellet were centrifuged at 6,000g, and the resulting pellets were purified in isolation buffer by homogenization with Potter-Elvehjem grinder and differential centrifugation at 800 and 6,000g. The mitochondrial pellet was resuspended in isolation buffer without EGTA, stored on ice, and used for experiments within 4 h. Protein concentration was determined by the modified Lowry assay using a detergent compatible protein assay kit (Bio-Rad, Hercules, CA). Isolation procedure was performed at +4°C. Consistency of mitochondrial isolation was routinely confirmed by measurement of citrate synthase activity (3.07 ± 0.17 U/mg protein).
Measurement of Mitochondrial Oxygen Consumption
Mitochondrial oxygen consumption was measured with an oxygen electrode (Hansatech Instruments, Norfolk, England, United Kingdom) at 30°C in the mitochondrial respiration buffer containing mitochondria at the final concentration of 1 mg protein/ml. The mitochondrial respiration buffer was composed of 130 mM KCl, 5 mM KH2PO4, 20mM MOPS, 2.5 mM EGTA, 1 μM Na4P2O7, and 0.1% bovine serum albumin, at pH 7.4. State 2 respiration was initiated with 5 mM pyruvate and 5 mM malate as substrates. The adenosine diphosphate (ADP)-stimulated oxygen consumption (state 3 respiration) was measured in the presence of 250 μM ADP, and the ADP-independent oxygen consumption (state 4 respiration) was monitored after ADP consumption. Hypoxic stress (partial pressure of oxygen approximately 0 mmHg for 15 min) was induced by bubbling the mitochondrial suspension in the chamber with nitrogen gas. After hypoxia, the chamber containing mitochondrial suspension was opened to room air to initiate a 10-min reoxygenation period. Oxygen consumption in the chamber was measured and the respiratory control ratio (RCR), defined as the ratio of oxygen consumption in state 3/state 4 respiration rate, was calculated to evaluate the degree of mitochondrial damage before and after exposure to 15 min of hypoxia and 10 min of reoxygenation.
Isolation of Cardiac Myocytes
Human atrial myocytes were isolated by modified procedure of Crumb et al.15 Briefly, right atrial specimens were placed in cold oxygenated cardioplegia solution (solution A) composed of 50 mM KH2PO4, 8 mM MgSO4, 10 mM NaHCO3,5 mM adenosine, 25 mM taurine, 140 mM glucose, and 100 mM mannitol, at pH 7.4 (0°-4°C) for 30-min transfer to the laboratory. The epicardial surface, fat, and fibrotic tissue were removed, and the myocardial tissue was minced into 1-mm pieces and placed in oxygenated ultralow Ca2+ wash solution (solution B) composed of 137 mM NaCl, 5 mM KH2PO4, 1 mM MgSO4, 10 mM taurine, 10 mM glucose, 5 mM HEPES, and 0.1 mM EGTA, at pH 7.4 (20°-23°C). Tissue was washed twice for 10 min in solution B and then incubated in 5 ml enzyme solution (solution C) containing 137 mM NaCl, 5 mM KH2PO4, 1 mM MgSO4, 10 mM taurine, 10 mM glucose, 5 mM HEPES, 0.1% bovine serum albumin, 1 mg/ml collagenase V (Sigma-Aldrich), and 0.5 mg/ml protease XXIV (Sigma-Aldrich) for 45 min at 37°C. Afterward, the tissue was incubated for 1 h at 37°C in solution C supplemented with 0.1 mM CaCl2 and 1 mg/ml collagenase V. The solution containing released cells was collected after 30 and 60 min of incubation and was centrifuged for 2 min at 1,000g, and the cell pellet was resuspended in the storage solution (solution D) composed of 25 mM KCl, 10 mM KH2PO4, 25 mM taurine, 0.5 mM EGTA, 22 mM glucose, and 55 mM glutamic acid, at pH 7.3 (20°-23°C). The procedure yielded approximately 20-40% of viable calcium-tolerant cells that were used for experiments within 4-5 h.
Myocyte Survival
Isolated myocytes were settled in a chamber on the stage of an inverted microscope (Olympus U-TB190; Tokyo, Japan) and superfused with glucose-free Tyrode buffer composed of 132 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, and 10 mM HEPES, at pH 7.4. Each experiment was started with a count of approximately 100-150 viable myocytes. Viability criteria were as follows: rod cell shape, distinct and regular cross-striations, and preserved membrane integrity. After the initial 2-min cell count, myocytes were superfused for 10 min with glucose-free Tyrode buffer and then subjected to 30 min of acute oxidative stress by H2O2 (Sigma-Aldrich). The concentration of H2O2 in superfusing buffer was increased in a cumulative manner from 0.001 to 10 mM in 5-min increments. Immediately after oxidative stress, viable myocytes were counted again, and the percentage of cell death was calculated. To induce APC, the myocytes were exposed for 5 min to 0.5 mM isoflurane and 5 min of anesthetic washout before the oxidative stress. Contribution of the sarcKATP channel to isoflurane-induced APC was investigated using the specific channel inhibitor HMR-1098 (a gift from Aventis Pharma, Frankfurt, Germany). HMR-1098 was present in the superfusing buffer throughout the experimental protocol. Experimental groups were as follows:
Time control (TC): 42 min of myocyte superfusion with Tyrode solution.
Oxidative stress (STRESS): 12 min of Tyrode superfusion followed by 30 min of H2O2.
APC with isoflurane (ISO + STRESS): 2 min of Tyrode superfusion, 5 min of isoflurane (0.5 mM), 5 min of isoflurane washout, 30 min of H2O2.
KATP channel blockade (+HMR): Protocol 3 was repeated in the continued presence of HMR-1098 (50 μM).
Tested in a separate experimental group, HMR-1098 alone had no significant effect on cell death (not shown).
Electrophysiology
The conventional single-electrode voltage clamp technique in whole cell and excised inside-out patch configurations was used to study the sarcKATP channel as reported before.16 For whole cell recordings, the intracellular/pipette solution contained 60 mM K-glutamate, 50 mM KCl, 10 mM HEPES, 1 mM CaCl2,1 mM MgCl2,2 mM EGTA, and 0.5 mM adenosine triphosphate dipotassium, at pH 7.2. The extracellular/bath solution contained 132 mM N-methyl-D-glucamine, 5 mM KCl, 1 mM CaCl2, 2 mM MgCl2, and 10 mM HEPES, at pH 7.4. The bath solution contained 200 nM nisoldipine (Miles-Pentex, West Haven, CT) to block L-type Ca2+ channels. The intracellular/bath solution for single channel recordings in inside-out configuration was composed of 145 mM KCl, 0.5 mM MgCl2, 1.0 mM EGTA, 10 mM HEPES, and 100 μM adenosine triphosphate dipotassium salt, at pH 7.2. The pipette solution facing the extracellular side of patches was composed of 145 mM KCl, 0.5 mM MgCl2, 0.5 mM CaCl2, and 10 mM HEPES, at pH 7.4.
Patch pipettes were pulled from borosilicate glass tubing (Garner Glass, Claremont, CA) using a horizontal PC-84 puller (Sutter, Novato, CA). Pipette tips were heat polished with microforge MF-83 (Narishige, Tokyo, Japan). Tip resistances of pipettes were 1.5-2.5 MΩ for whole cell recordings and 7-12 MΩ for single channel recordings. All recordings were made at room temperature using an EPC-7 patch clamp amplifier (List, Darmstadt-Eberstadt, Germany) with Digidata 1322A interface (Axon Instruments, Foster City, CA). Voltage protocols were generated with pClamp9 software (Axon Instruments). Whole cell sarcKATP channel current (IKATP) was monitored over time by applying a 200-ms test pulse to 0 mV from a holding potential of -40 mV every 15 s. Current traces were low-pass filtered at 3 kHz and digitized at 1 kHz. Current amplitude was measured at the end of the test pulse. Currents were normalized to cell capacitance, and current density (pA/pF) was plotted against time.
Single KATP channel activity was monitored continuously at membrane potential +40 mV using an EPC-7 amplifier, and 60-s recordings were made. The current signal was low-pass filtered at 500 Hz through an eight-pole Bessel filter and sampled at a rate of 1 kHz. The KATP channel was identified by unitary conductance, inhibition by intracellular adenosine triphosphate (1-2 mM), and blockade by glibenclamide (1 μM). In single channel analysis, half-amplitude threshold crossing was adopted for detecting the open state. The all-points amplitude histograms were constructed from 60-s recordings, and the channel open-state probability was determined from the ratios of the area under the peaks in the amplitude histograms fitted by a multigaussian distribution and was expressed as the cumulative open-state probability.16 Single channel data were analyzed using pClamp 9 software and Origin 7 software (OriginLab, Northampton, MA).
For in vitro treatment of isolated myocardium and isolated myocytes, liquid isoflurane (Baxter Healthcare Corp., Deerfield, IL) was dispersed in the appropriate buffer by sonication. The anesthetic-containing buffer was delivered to the incubation or recording chamber from glass syringe reservoirs by a gravity-fed perfusion system. The average concentration of isoflurane in all experiments was 0.51 mM, equivalent to 1.2 vol% or 1 MAC at 22°C. Concentrations of isoflurane in the buffer sampled from experimental chamber were determined by a gas chromatography method using a Shimadzu GC 8A chromatograph (Shimadzu, Kyoto, Japan).
Statistical Analysis
Data are reported as mean ± SD, and n refers to the number of experiments and reflects the number of patients. The statistical analyses of the data were performed using mixed analysis of variance models to compare treatment groups with random sample effect to adjust for correlation due to repeated measurements on the sample. The experiment-wise type I error rate was controlled using a Tukey-Kramer P value adjustment for the post hoc comparisons. The analyses were performed using SAS 9.1.3 (SAS Institute, Cary, NC) software. In mitochondria experiments, data were analyzed by a three-way (age by treatment by time point) analysis of variance model with random sample effect that was fitted to the state 3, state 4, and RCR values. In myocyte survival experiments, a two-way analysis of variance model with random sample effect was fitted to the arcsine-transformed cell death proportions. In whole cell patch clamp experiments, differences in sarcKATP channel current density were evaluated by a two-way analysis of variance model with random sample effect that was fitted to log-transformed current density. A value of P < 0.05 was considered statistically significant.
Results
Isoflurane Preserves Mitochondrial Oxygen Consumption in Human Atrial Myocardium
Oxygen consumption of isolated mitochondria was measured before (i.e., baseline) and after hypoxia-reoxygenation stress in MA and OA groups. Figures 1A and B present typical chart recordings obtained for oxygen consumption of mitochondria isolated from atrial myocardium of the MA (n = 8) and OA (n = 8) groups. In the MA group, the ADP-stimulated oxygen consumption (state 3 respiration) and ADP-independent oxygen consumption (state 4 respiration) of isoflurane treated mitochondria (fig. 1A, lower trace) were preserved better compared with control (fig. 1A, upper trace). In the OA group, state 3 respiration was decelerated and state 4 respiration was accelerated in both control and isoflurane-treated mitochondria after hypoxic stress (fig. 1B). Distinct respiration states, indicating coupled mitochondria, were well maintained after isoflurane preconditioning only in the MA group. Table 2 summarizes changes in RCR before and after hypoxic stress to mitochondria. In MA mitochondria (n = 8), RCR was significantly decreased after exposure to hypoxia (4.1 ± 0.6 and 1.9 ± 0.6 before and after hypoxia, respectively). In the isoflurane-preconditioned MA mitochondria, RCR was altered and tended to be lower, but the differences were not significant (4.0 ± 0.7 and 3.4 ± 0.8 before and after hypoxia, respectively). On the other hand, in mitochondria of the OA group (n = 8), RCR measured both under control conditions and after isoflurane preconditioning was significantly decreased, and the values were 4.4 ± 0.9 and 1.8 ± 0.6 (before and after hypoxia, respectively) in control mitochondria and 4.2 ± 0.8 and 1.9 ± 0.7 (before and after hypoxia, respectively) in isoflurane-preconditioned mitochondria. Because a higher RCR implicates better preserved mitochondrial oxygen consumption and integrity, those results suggest a preconditioning effect of isoflurane on mitochondrial function that is apparent only in the MA group, not in the OA group.
Fig. 1.

Typical chart recordings of mitochondrial respiration at baseline and after hypoxic stress in the mitochondria obtained from mid-aged (MA; A) and old-aged (OA; B) human atrial myocardium. Open circles = control, without anesthetic preconditioning (without APC); closed circles = isoflurane treated, with APC. Oxygen consumption was initiated by addition of pyruvate-malate, accelerated by addition of adenosine diphosphate (ADP; state 3 respiration), and decelerated after all ADP was consumed (state 4 respiration). After hypoxic stress, state 3 and state 4 respiration was preserved in mitochondria obtained from isoflurane-preconditioned myocardium of MA but not OA.
Table 2.
Effect of Isoflurane Preconditioning on Mitochondrial Respiration
| State 3, O2 nmol · mg protein-1 · min-1 |
State 4, O2 nmol · mg protein-1 · min-1 |
RCR |
||||
|---|---|---|---|---|---|---|
| CON | ISO | CON | ISO | CON | ISO | |
| Mid-aged | ||||||
| Baseline | 34.8 ± 11.1 | 39.6 ± 13.0 | 8.4 ± 2.0 | 9.7 ± 1.9 | 4.1 ± 0.6 | 4.0 ± 0.7 |
| After hypoxia | 23.8 ± 10.1* | 32.7 ± 11.0 | 13.1 ± 5.6 | 10.2 ± 4.0 | 1.9 ± 0.6* | 3.4 ± 0.8 |
| Old-aged | ||||||
| Baseline | 31.1 ± 13.2 | 29.3 ± 15.4 | 7.4 ± 3.4 | 6.9 ± 3.5 | 4.4 ± 0.9 | 4.2 ± 0.8 |
| After hypoxia | 18.9 ± 11.6* | 15.0 ± 7.5*† | 10.4 ± 7.1 | 8.7 ± 6.0 | 1.8 ± 0.6* | 1.9 ± 0.7*† |
Values are mean ± SD.
P < 0.05:
baseline vs. after hypoxia
after hypoxia in mid-aged vs. after hypoxia in old-aged.
CON = control (without anesthetic-induced preconditioning); ISO = 0.5 mM isoflurane; RCR = respiratory control ratio.
Isoflurane Protects Isolated Human Atrial Myocytes from Stress via sarcKATP Channel
To investigate whether preservation of mitochondrial respiration by isoflurane is associated with improved stress tolerance of isolated myocytes, we examined the effects APC by isoflurane on protection of human atrial myocytes from acute oxidative stress-induced cell death and tested whether sarcKATP channel contributes to this protection. Figure 2A shows that in the MA group (n = 6), 42 min of myocyte superfusion with Tyrode solution (TC) resulted in 15 ± 2% cell death. After 30 min of exposure to increasing concentrations of H2O2, cell death was significantly increased to 75 ± 5% (STRESS), but after 5 min of treatment of myocytes with isoflurane (0.5 mM) and 5 min of anesthetic washout, the H2O2-induced cell death was significantly attenuated to 21 ± 7% (ISO + STRESS). In the continued presence of HMR-1098 (50 μM), the protective effect was partly reversed and cell death increased to 46 ± 5% (+HMR). In general, responses of MA myocytes to stress and isoflurane were dynamic and uniform. By contrast, responses from OA myocytes (n = 6) were more blunted, and variability among patients was greater than in the MA group, as shown in figure 2B. The cell death of 21 ± 15% in TC significantly increased after STRESS to 52 ± 17%. This value was significantly lower than in MA myocytes, suggesting differential sensitivity to exogenous H2O2 in the OA and MA groups. After pretreatment with isoflurane (ISO + STRESS), the H2O2-induced cell death decreased to 33 ± 13%, but the difference was not statistically significant. In the presence of HMR-1098 (+HMR), the cell death was 47 ± 15%. These findings suggested age-related difference in sensitivity of isolated atrial myocytes to acute oxidative stress and protection by isoflurane, with greater sensitivity and protection in MA and less protection in OA myocytes. Further, we demonstrated that the sarcKATP channel contributes to APC-induced protection in human cardiomyocytes.
Fig. 2.
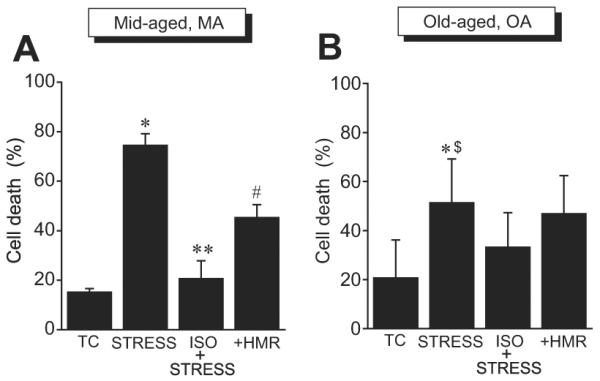
Isoflurane protects human atrial myocytes from stress-induced cell death via sarcolemmal adenosine triphosphate-sensitive potassium channel. Mid-aged (MA; A) and old-aged (OA; B) myocyte death was measured in time control (TC), after 30 min of oxidative stress with H2O2 (STRESS), after 5 min of cell treatment with isoflurane and 5 min of anesthetic washout before stress (ISO + STRESS), and after repeating the ISO + STRESS protocol in the continued presence of HMR-1098, the sarcolemmal adenosine triphosphate-sensitive potassium channel blocker (+HMR). Responses to stress and protection by isoflurane were greater in MA than in OA myocytes. Values are mean ± SD. P < 0.05: * STRESS versus respective TC in MA and OA; ** ISO + STRESS versus STRESS in MA; # +HMR versus all other groups in MA; $ STRESS in OA versus MA.
Characteristics of sarcKATP Channel in Human Atrial Myocytes
In inside-out patches from human right atrial myocytes, we tested for single sarcKATP channel activity. Screening of 99 patches from the MA group and 125 patches from the OA group showed that in both groups, the channel opening was infrequent and was found on average in 1 per 5 patches. However, the percentage of active patches was similar in MA (18%) and in OA (16%). These observations corroborated previous reports of relatively low activity of KATP channel in the human myocardium.17-20 Electrophysiologic characteristics of the channel, the amplitude of unitary current, and unitary conductance were typical for the cardiac KATP channel and were similar in MA and OA. Figure 3 shows example traces of unitary KATP current recorded at membrane potentials of +40 and -40 mV from human atrial myocyte and single channel current-voltage relation (n = 5). Calculated from linear regression fits, the single channel conductances of the inward and outward currents were 70 ± 1.3 and 53 ± 2.2 pS, respectively. Whole cell recordings of human atrial KATP channel current (fig. 4) demonstrated that basal density of current activated by the channel opener pinacidil was significantly higher in MA (7.3 ± 2 pA/pF, n = 4) than in OA (4.7 ± 0.4 pA/pF, n = 3). However, myocyte pretreatment with isoflurane produced a similar 1.6-fold increase in the pinacidil-activated current in both groups, and current density was 12.0 ± 2 pA/pF (n = 4) and 7.8 ± 1 (n = 4) in MA and OA, respectively.
Fig. 3.
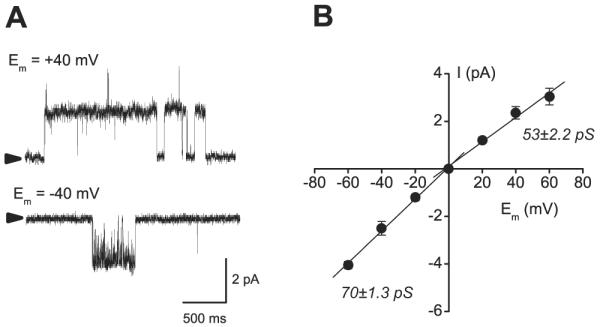
Single channel recordings of sarcolemmal adenosine triphosphate-sensitive potassium channel from human atrial myocytes. (A) Example traces of unitary current through sarcolemmal adenosine triphosphate-sensitive potassium channel recorded from inside-out patch of a mid-aged myocyte at +40 and -40 mV membrane potential (Em). The unitary current amplitude is characteristic for the cardiac sarcKATP channel. (B) Current-voltage relation for single human sarcolemmal adenosine triphosphate-sensitive potassium channel. Linear regression fits to data distribution revealed that the single channel conductance (γ) calculated from the slope values was 70 ± 1.3 pS for the inward current at negative membrane potentials and 53 ± 2.2 pS for the outward current at positive membrane potentials.
Fig. 4.

Treatment with isoflurane enhances sensitivity of human atrial sarcolemmal adenosine triphosphate-sensitive potassium channel to pinacidil in mid-aged (MA) and old-aged (OA). Whole cell recordings of sarcolemmal adenosine triphosphate-sensitive potassium channel current were made from MA and OA myocytes. Current was monitored over time by stepping every 15 s to 0 mV from a holding potential of -40 mV for 200 ms. Pinacidil-activated current was measured in MA and OA myocytes in control (A and B) and after cell exposure to isoflurane and washout (C and D). With or without isoflurane treatment, current activation by pinacidil was greater in MA than in OA. BASE = baseline; GLIB = 1 μM glibenclamide; ISO = 0.5 mM isoflurane; PIN = 30 μM pinacidil. (E) Summary data from experiments shown above. Values are mean ± SD. P < 0.05: * ISO/PIN versus PIN in MA and OA; # PIN in OA versus PIN in MA and ISO/PIN in OA versus ISO/PIN in MA.
Discussion
The current study shows that APC by isoflurane can be induced in isolated human myocardial cells and isolated mitochondria. We demonstrated that isoflurane treatment preserves the mitochondrial respiratory function during stress in MA but not in OA, thus uncovering a potential mechanism by which APC is modulated by aging. Better protection from stress-induced cell death in MA compared with OA was confirmed in a functional study on isolated intact myocytes, showing that the effectiveness of APC is greater in MA. Also, the study showed that whereas the KATP current density seems to decline with advancing age, the sarcKATP channel still could be activated by isoflurane in OA.
Mitochondria are involved in the process of aging, and various mitochondrial functions are impaired in the aging heart. However, some studies suggest that the electron transport system or oxidative phosphorylation is not affected by aging.21,22 The enzyme activities of complexes I, II, III, and IV of the mitochondrial respiratory chain were not decreased with age in the human left ventricular myocardium,21 and the mitochondrial respiratory function in aged human skeletal muscle was not altered compared with young skeletal muscle.22 Our results from experiments in human cardiac mitochondria isolated from control, nonpreconditioned myocardium showed that RCRs measured before the hypoxic stress in MA and OA were not significantly different from each other. It should be considered, however, that because of the limited amount of available atrial tissue, in our mitochondrial isolation we were unable to distinguish between two previously described mitochondrial subpopulations, subsarcolemmal and intrafibrillar mitochondria.23 There is strong evidence that aging decreases oxidative phosphorylation and increases oxidative stress to a greater extent in intrafibrillar compared with subsarcolemmal mitochondria.24 Therefore, by using a mixture of both mitochondrial subpopulations, we might have overlooked possible differences between MA and OA intrafibrillar mitochondria. Ischemic stress on mitochondria, on the other hand, damaged both mitochondrial subpopulation equally.25
Ischemic injury seems to be greater in aged hearts than in younger adult hearts.26,27 The recovery of developed tension after hypoxic stress is reduced in aged human right atrial trabeculae,26 and the postischemic function of Langendorff-perfused hearts is significantly deteriorated in aged rabbits.27 Other studies have reported that intracellular mechanisms leading to cell survival are impaired in the aged heart,28-30 and aging enhances activation of mitochondrial permeability transition pore, an important player in cell death.31 However, our results obtained from isolated mitochondria of control nonpre-conditioned human myocardium show that attenuation of RCR after hypoxia-reoxygenation stress in OA mitochondria was not significantly different from that in MA mitochondria. Therefore, responsiveness of mitochondrial respiratory function to ischemia does not seem to contribute to a greater ischemic damage in the aged heart. Further, intact myocyte response to stress also was different between MA and OA in that stress-induced cell death and protection by isoflurane were greater in MA, but both appeared lower (less stress-induced death and less isoflurane protection) in OA. There is no simple explanation for these findings, but one of the causes might be related to difference in reactive oxygen species (ROS) production and ROS handling between MA and OA individuals. It is well documented that production of endogenous ROS and ROS-induced damage both increase with advancing age32 and could impair stress responses of aged myocytes. MA individuals with lower levels of endogenous ROS might be more sensitive to exogenous ROS and also to protective effects of APC. Higher levels of endogenous ROS in OA individuals may increase organ damage due to increased cell death but may also desensitize cells to exogenous ROS. In addition, a large population of more damaged OA myocytes could be lost during enzymatic cell isolation, which might yield a smaller population of healthy cells that are more resistant to stress, thus artificially decreasing the stress-induced cell death in OA.
Cardiac mechanisms of APC have been investigated primarily in the animal models, but effectiveness of APC has been tested in clinical settings. Although large clinical trials have not been conducted, several smaller studies demonstrated beneficial effects of APC during surgery.2-5 Because animal studies demonstrated multiple mechanisms of APC, it is likely that the mechanisms of APC in humans are complex and multifactorial as well. Recent animal studies suggest that mitochondria play an important role in APC33,34; however, in humans, the role of mitochondria in APC has not been determined. Furthermore, the majority of patients undergoing cardiac surgery are of advanced age. Although it is known that APC is less effective in aging animals,11,12 the relation between advancing age and APC has not been defined in humans. Our study is the first to investigate the interrelation between APC, the function of isolated human cardiac mitochondria, and aging. Our experiments on isoflurane-preconditioned mitochondria demonstrated that RCR is well preserved after hypoxia-reoxygenation stress in MA but not in OA. Taken together, these findings suggest that mitochondria play an important role in APC in humans and that APC is attenuated in the aged heart through mitochondrial dysfunction. There seems to be a correlation between aging and reduced effectiveness of IPC as well. Fenton et al.35 demonstrated that phosphorylation of the signal transduction proteins, which play a role in the IPC-associated enhanced cell survival, was decreased in aged rats. Decreasing levels of connexin 43, the protein that builds cardiac gap junctions and also is localized in the mitochondria, contribute to the loss of IPC in aging mice.36 Those IPC mechanisms could be applicable to APC, because the mechanisms of APC and IPC are partly shared.37
Although isolated mitochondria experiments showed that aging-related dysfunction of mitochondrial respiration may prevent APC, isoflurane could still protect isolated intact aged human cardiomyocytes from stress-induced cell death. Indeed, pretreatment of MA myocytes with isoflurane improved cell survival during acute oxidative stress, whereas this effect was much less pronounced in OA myocytes, suggesting that aging-related changes in mitochondrial function could affect viability of MA and OA myocytes. The involvement of another potential effector of APC, the sarcKATP channel,14 in human cardiomyocyte protection was tested using HMR-1098, the sarcKATP channel blocker. HMR-1098 attenuated the protection by isoflurane, suggesting that in adult human atrial myocytes, protection from stress is mediated partly via sarcKATP channel. The properties of single sarcKATP channels from adult human right atrial myocytes, amplitude of unitary KATP current, slope conductance at negative membrane potentials, and weak inward rectification at positive membrane potentials were similar to those reported previously for human atrial myocytes by others,17,18 and no apparent differences were found between MA and OA. Although whole cell recordings showed that basal sensitivity to pinacidil, the channel opener, was higher in MA myocytes, the isoflurane sensitivity was similar in MA and OA. Taken together, differences in isoflurane protection of adult human myocytes can be accounted for by aging-related modifications of mitochondrial function, but not modulation of anesthetic sensitivity of the KATP channel. This lack of correlation between aging and KATP channel-mediated protection is not surprising because in our study, we were dealing with a population of adults only. It is possible that comparisons between more contrasting groups (pediatric/young vs. adults) would show distinct profiles of channel expression and anesthetic protection, because the expression and activity of sarcKATP channels undergo age-dependent changes in many species, including humans.38 In our study, the density of sarcKATP channels in adult human atrial myocytes was low compared with other species.14,17 Besides age and species differences, low KATP channel density could be a consequence of underlying disease, in particular atrial fibrillation, which was present in 18% of both MA and OA groups. There is evidence that atrial fibrillation may lead to electrical remodeling of myocardial cells39 by down-regulating certain cardiac plasma membrane ion channels, including the sarcKATP channel.18,40
There are several limitations to our study. First, atrial specimens were collected from patients who exhibited an array of cardiac diseases (disclosed) and noncardiac diseases (not disclosed, except hyperlipidemia and diabetes mellitus). Many of those diseases could impact the study outcome.41 In addition, all patients were undergoing long- or short-term drug therapies. There is evidence that medications such as β-adrenergic blockers42 or oral hypoglycemics43 may attenuate the efficacy of APC and therefore could influence study results. Although the majority of drugs are likely to be removed during isolation of cardiomyocytes or mitochondria, possible cellular effects of the long-term medications must be considered. Second, for obvious ethical reasons and restrictions, we were not able to conduct parallel control studies using atrial samples from healthy, nondiseased human hearts that would serve as baseline for studies on tissue from diseased hearts. Third, age was not the only variable between MA and OA patients. Another important variable was sex, and in our study there were more females in the OA group (33%) than in the MA group (13%). Estrogens are known to protect the heart by regulating gene expression, enhancing the transcription of SUR2A gene and increasing messenger RNA for SUR2A and the number of sarcKATP channels. These changes correlate with better protection of the female heart compared with the male heart.38 Aging seems to be associated with a greater decrease in number of sarcKATP channels in female than in male heart.38 Therefore, except for aging, sex-related factors could affect responses to APC, and indeed, it has been reported that delayed APC with isoflurane does not produce additional cardioprotection in adult female rabbits.44 Fourth, our study was conducted on myocytes and mitochondria isolated from the human right atrial myocardium. Despite obvious morphologic and functional differences between atrial and ventricular myocardium, both are susceptible to ischemia-reperfusion, and both are sensitive to ischemic preconditioning and anesthetic preconditioning. However, ischemia may differentially affect atrial and ventricular myocardium, e.g., infarction develops in ventricular myocardium and the left atrial myocardium,45 but not the right atrial myocardium. Therefore, we are aware that our findings from right atrial myocardium may not necessary reflect the underpinnings of mechanisms of APC in the ventricular myocardium.
In conclusion, in vitro preconditioning with isoflurane preserves mitochondrial respiration and enhances opening of the sarcKATP channel in human right atrial cardiomyocytes, thus protecting adult human mitochondria and cardiomyocytes from stress. Protection of mitochondrial respiration but not sarcKATP channel function declines with advancing age, suggesting a mechanism by which aging may modulate APC.
Acknowledgments
The authors thank Aniko Szabo, Ph.D. (Associate Professor of Biostatistics, Department of Population Health, Medical College of Wisconsin, Milwaukee, Wisconsin), for statistical consulting. Also, the authors thank Chiaki Kwok, M.S. (Research Technologist, Department of Anesthesiology, Medical College of Wisconsin), for assistance in isolation of mitochondria, and Mary Ziebell (Research Technologist, Department of Anesthesiology, Medical College of Wisconsin) for isoflurane measurements.
Supported in part by grant Nos. HL034708 and PO1GM066730 (to Dr. Bosnjak) from the National Institutes of Health, Bethesda, Maryland.
References
- 1.Kersten JR, Schmeling TJ, Pagel PS, Gross GJ, Warltier DC. Isoflurane mimics ischemic preconditioning via activation of KATP channels: Reduction of myocardial infarct size with an acute memory phase. ANESTHESIOLOGY. 1997;87:361–70. doi: 10.1097/00000542-199708000-00024. [DOI] [PubMed] [Google Scholar]
- 2.De Hert SG, Van der Linden PJ, Cromheecke S, Meeus R, Nelis A, Van Reeth V, ten Broecke PW, De Blier IG, Stockman BA, Rodrigus IE. Cardioprotective properties of sevoflurane in patients undergoing coronary surgery with cardiopulmonary bypass are related to the modalities of its administration. ANESTHESIOLOGY. 2004;101:299–310. doi: 10.1097/00000542-200408000-00009. [DOI] [PubMed] [Google Scholar]
- 3.Garcia C, Julier K, Bestmann L, Zollinger A, von Segesser LK, Pasch T, Spahn DR, Zaugg M. Preconditioning with sevoflurane decreases PECAM-1 expression and improves one-year cardiovascular outcome in coronary artery bypass graft surgery. Br J Anaesth. 2005;94:159–65. doi: 10.1093/bja/aei026. [DOI] [PubMed] [Google Scholar]
- 4.Lee MC, Chen CH, Kuo MC, Kang PL, Lo A, Liu K. Isoflurane preconditioning-induced cardio-protection in patients undergoing coronary artery bypass grafting. Eur J Anaesth. 2006;23:841–7. doi: 10.1017/S0265021506000354. [DOI] [PubMed] [Google Scholar]
- 5.Julier K, da Silva R, Garcia C, Bestmann L, Frascarolo P, Zollinger A, Chassot P-G, Schmid ER, Turina MI, von Segesser LK, Pasch T, Spahn DR, Zaugg M. Preconditioning by sevoflurane decreases biochemical markers for myocardial and renal dysfunction in coronary artery bypass graft surgery: A double-blinded placebo-controlled, multicenter study. ANESTHESIOLOGY. 2003;98:1315–27. doi: 10.1097/00000542-200306000-00004. [DOI] [PubMed] [Google Scholar]
- 6.Roscoe AK, Christensen JD, Lynch C., III Isoflurane, but not halothane, induces protection of human myocardium via adenosine A1 receptors and adenosine triphosphate-sensitive potassium channels. ANESTHESIOLOGY. 2000;92:1692–701. doi: 10.1097/00000542-200006000-00029. [DOI] [PubMed] [Google Scholar]
- 7.Hanouz JL, Yvon A, Massetti M, Lepage O, Babatasi G, Khayat A, Bricard H, Gerard JL. Mechanisms of desflurane-induced preconditioning in isolated human right atria in vitro. ANESTHESIOLOGY. 2002;97:33–41. doi: 10.1097/00000542-200207000-00006. [DOI] [PubMed] [Google Scholar]
- 8.Zaugg M, Lucchinetti E, Spahn DR, Pasch T, Schaub MC. Volatile anesthetics mimic cardiac preconditioning by priming the activation of mitochondrial KATP channels via multiple signaling pathways. ANESTHESIOLOGY. 2002;97:4–14. doi: 10.1097/00000542-200207000-00003. [DOI] [PubMed] [Google Scholar]
- 9.Lee TM, Su SF, Chou TF, Lee YT, Tsai CH. Loss of preconditioning by attenuated activation of myocardial ATP-sensitive potassium channels in elderly patients undergoing coronary angioplasty. Circulation. 2002;105:334–40. doi: 10.1161/hc0302.102572. [DOI] [PubMed] [Google Scholar]
- 10.Bartling B, Friedrich I, Silber RE, Simm A. Ischemic preconditioning is not cardioprotective in senescent human myocardium. Ann Thorac Surg. 2003;76:105–11. doi: 10.1016/s0003-4975(03)00186-3. [DOI] [PubMed] [Google Scholar]
- 11.Sniecinski R, Liu H. Reduced efficacy of volatile anesthetic preconditioning with advanced age in isolated rat myocardium. ANESTHESIOLOGY. 2004;100:589–97. doi: 10.1097/00000542-200403000-00019. [DOI] [PubMed] [Google Scholar]
- 12.Riess ML, Camara AK, Rhodes SS, McCormick J, Jiang MT, Stowe DF. Increasing heart size and age attenuate anesthetic preconditioning in guinea pig isolated hearts. Anesth Analg. 2005;101:1572–6. doi: 10.1213/01.ANE.0000181834.39483.0B. [DOI] [PubMed] [Google Scholar]
- 13.Ljubkovic M, Mio Y, Marinovic J, Stadnicka A, Warltier DC, Bosnjak ZJ, Bienengraeber M. Isoflurane preconditioning uncouples mitochondria and protects against hypoxia-reoxygenation. Am J Physiol Cell Physiol. 2007;292:C1583–90. doi: 10.1152/ajpcell.00221.2006. [DOI] [PubMed] [Google Scholar]
- 14.Marinovic J, Bosnjak ZJ, Stadnicka A. Distinct roles for sarcolemmal and mitochondrial adenosine triphosphate-sensitive potassium channels in isoflurane-induced protection against oxidative stress. ANESTHESIOLOGY. 2006;105:98–104. doi: 10.1097/00000542-200607000-00018. [DOI] [PubMed] [Google Scholar]
- 15.Crumb WJ, Jr, Pigott JD, Clarkson CW. Comparison of Ito in young and adult human atrial myocytes: Evidence for developmental changes. Am J Physiol. 1995;268:H1335–42. doi: 10.1152/ajpheart.1995.268.3.H1335. [DOI] [PubMed] [Google Scholar]
- 16.Stadnicka A, Marinovic J, Bienengraeber M, Bosnjak ZJ. Impact of in vivo preconditioning by isoflurane on adenosine triphosphate-sensitive potassium channels in the rat heart: Lasting modulation of nucleotide sensitivity during early memory period. ANESTHESIOLOGY. 2006;104:503–10. doi: 10.1097/00000542-200603000-00018. [DOI] [PubMed] [Google Scholar]
- 17.Heidbuchel H, Vereecke J, Carmeliet E. Three different potassium channels in human atrium: Contribution to the basal potassium conductance. Circ Res. 1990;66:1277–86. doi: 10.1161/01.res.66.5.1277. [DOI] [PubMed] [Google Scholar]
- 18.Koumi SI, Martin RL, Sato R. Alterations in ATP-sensitive potassium channel sensitivity to ATP in failing human hearts. Am J Physiol. 1997;272:H1656–65. doi: 10.1152/ajpheart.1997.272.4.H1656. [DOI] [PubMed] [Google Scholar]
- 19.Zunkler BJ, Henning B, Ott T, Hildebrandt AG, Fleck E. Effects of tolbutamide on ATP-sensitive K+ channels from human right atrial cardiac myocytes. Pharmacol Toxicol. 1997;80:69–75. doi: 10.1111/j.1600-0773.1997.tb00286.x. [DOI] [PubMed] [Google Scholar]
- 20.Verkerk AO, Veldkamp MW, van Ginneken ACG, Bouman LN. Biphasic response of action potential duration to metabolic inhibition in rabbit and human ventricular myocytes: Role of transient outward current and ATP-regulated potassium current. J Mol Cell Cardiol. 1996;28:2443–56. doi: 10.1006/jmcc.1996.0237. [DOI] [PubMed] [Google Scholar]
- 21.Miro O, Casademont J, Casals E, Perea M, Urbano-Marquez A, Rustin P, Cardellach F. Aging is associated with increased lipid peroxidation in human hearts, but not with mitochondrial respiratory chain enzyme defects. Cardiovasc Res. 2000;47:624–31. doi: 10.1016/s0008-6363(00)00122-x. [DOI] [PubMed] [Google Scholar]
- 22.Hutter E, Skovbro M, Lener B, Prats C, Rabol R, Dela F, Jansen-Durr P. Oxidative stress and mitochondrial impairment can be separated from lipofuscin accumulation in aged human skeletal muscle. Aging Cell. 2007;6:245–56. doi: 10.1111/j.1474-9726.2007.00282.x. [DOI] [PubMed] [Google Scholar]
- 23.Matlib MA, Rebman D, Ashraf M, Rouslin W, Schwartz A. Differential activities of putative subsarcolemmal and interfibrillar mitochondria from cardiac muscle. J Mol Cell Cardiol. 1981;13:163–70. doi: 10.1016/0022-2828(81)90213-3. [DOI] [PubMed] [Google Scholar]
- 24.Judge S, Jang YM, Smith A, Hagen T, Leeuwenburgh C. Age-associated increases in oxidative stress and antioxidant enzyme activities in cardiac interfibrillar mitochondria: Implications for the mitochondrial theory of aging. FASEB J. 2005;19:419–21. doi: 10.1096/fj.04-2622fje. [DOI] [PubMed] [Google Scholar]
- 25.Lesnefsky EJ, Gudz TI, Migita CT, Ikeda-Saito M, Hassan MO, Turkaly PJ, Hoppel CL. Ischemic injury to mitochondrial electron transport in the aging heart: Damage to the iron-sulfur protein subunit of electron transport complex III. Arch Biochem Biophys. 2001;385:117–28. doi: 10.1006/abbi.2000.2066. [DOI] [PubMed] [Google Scholar]
- 26.Mariani J, Ou R, Bailey M, Rowland M, Nagley P, Rosenfeldt F, Pepe S. Tolerance to ischemia and hypoxia is reduced in aged human myocardium. J Thorac Cardiovasc Surg. 2000;120:660–7. doi: 10.1067/mtc.2000.106528. [DOI] [PubMed] [Google Scholar]
- 27.McCully JD, Toyoda Y, Wakiyama H, Rousou AJ, Parker RA, Levitsky S. Age and gender-related differences in ischemia/reperfusion injury and cardioprotection: Effects of diazoxide. Ann Thorac Surg. 2006;82:117–23. doi: 10.1016/j.athoracsur.2006.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taylor RP, Starnes JW. Age, cell signaling and cardioprotection. Acta Physiol Scand. 2003;178:107–16. doi: 10.1046/j.1365-201X.2003.01132.x. [DOI] [PubMed] [Google Scholar]
- 29.Liu P, Xu B, Cavalieri TA, Hock CE. Attenuation of antioxidative capacity enhances reperfusion injury in aged rat myocardium after MI/R. Am J Physiol Heart Circ Physiol. 2004;287:H2719–27. doi: 10.1152/ajpheart.00317.2004. [DOI] [PubMed] [Google Scholar]
- 30.Wittnich C, Su J, Boscarino C, Belanger M. Age-related differences in myocardial hydrogen ion buffering during ischemia. Mol Cell Biochem. 2006;285:61–7. doi: 10.1007/s11010-005-9055-9. [DOI] [PubMed] [Google Scholar]
- 31.Mather M, Rottenberg H. Aging enhances the activation of the permeability transition pore in mitochondria. Biochem Biophys Res Commun. 2000;273:603–8. doi: 10.1006/bbrc.2000.2994. [DOI] [PubMed] [Google Scholar]
- 32.Juhaszova M, Rabuel C, Zorov DB, Lakatta EG, Sollott SJ. Protection in the aged heart: Preventing the heart-break of old age? Cardiovasc Res. 2005;66:233–44. doi: 10.1016/j.cardiores.2004.12.020. [DOI] [PubMed] [Google Scholar]
- 33.Bienengraeber MW, Weihrauch D, Kersten JR, Pagel PS, Warltier DC. Cardioprotection by volatile anesthetics. Vascul Pharmacol. 2005;42:243–52. doi: 10.1016/j.vph.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 34.Stadnicka A, Marinovic J, Ljubkovic M, Bienengraeber MW, Bosnjak ZJ. Volatile anesthetic-induced cardiac preconditioning. J Anesth. 2007;21:212–9. doi: 10.1007/s00540-006-0486-6. [DOI] [PubMed] [Google Scholar]
- 35.Fenton RA, Dickson EW, Dobson JG., Jr Inhibition of phosphatase activity enhances preconditioning and limits cell death in the ischemic/reperfused aged rat heart. Life Sci. 2005;77:3375–88. doi: 10.1016/j.lfs.2005.05.047. [DOI] [PubMed] [Google Scholar]
- 36.Boengler K, Konietzka I, Buechert A, Heinen Y, Garcia-Dorado D, Heusch G, Schulz R. Loss of ischemic preconditioning's cardioprotection in aged mouse hearts is associated with reduced gap junctional and mitochondrial levels of connexin 43. Am J Physiol Heart Circ Physiol. 2007;292:H1764–9. doi: 10.1152/ajpheart.01071.2006. [DOI] [PubMed] [Google Scholar]
- 37.Zaugg M, Schaub MC. Signaling and cellular mechanisms in cardiac protection by ischemic and pharmacological preconditioning. J Muscle Res Cell Motil. 2003;24:219–49. doi: 10.1023/a:1026021430091. [DOI] [PubMed] [Google Scholar]
- 38.Jovanovic S, Jovanovic A. Sarcolemmal KATP channel in ageing. Ageing Res Rev. 2004;3:199–214. doi: 10.1016/j.arr.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 39.Dobrev D, Ravens U. Remodeling of cardiomyocyte ion channels in human atrial fibrillation. Basic Res Cardiol. 2003;98:137–48. doi: 10.1007/s00395-003-0409-8. [DOI] [PubMed] [Google Scholar]
- 40.Balana B, Dobrev D, Wettwer E, Christ T, Knaut M, Ravens U. Decreased ATP-sensitive K+ current density during chronic human atrial fibrillation. J Mol Cell Cardiol. 2003;35:1399–405. doi: 10.1016/s0022-2828(03)00246-3. [DOI] [PubMed] [Google Scholar]
- 41.Kehl F, Krolikowski JG, Mraovic B, Pagel PS, Warltier DC, Kersten JR. Hyperglycemia prevents isoflurane-induced preconditioning against myocardial infarction. ANESTHESIOLOGY. 2002;96:183–8. doi: 10.1097/00000542-200201000-00032. [DOI] [PubMed] [Google Scholar]
- 42.Lange M, Smul TM, Blomeyer CA, Redel A, Klotz KN, Roewer N, Kehl F. Role of the β1-adrenergic pathway in anesthetic and ischemic preconditioning against myocardial infarction in the rabbit heart in vivo. ANESTHESIOLOGY. 2006;105:503–10. doi: 10.1097/00000542-200609000-00014. [DOI] [PubMed] [Google Scholar]
- 43.Miura H, Wachtel RE, Loberiza FR, Saito T, Miura M, Nicolosi AC, Gutter-man DD. Diabetes mellitus impairs vasodilation to hypoxia in human coronary arterioles: Reduced activity of ATP-sensitive potassium channels. Circ Res. 2003;92:151–8. doi: 10.1161/01.res.0000052671.53256.49. [DOI] [PubMed] [Google Scholar]
- 44.Wang C, Chiari PC, Weihrauch D, Krolikowski JG, Warltier DC, Kersten JR, Pratt PF, Jr, Pagel PS. Gender-specificity of delayed preconditioning by isoflurane in rabbits: Potential role of endothelial nitric oxide synthase. Anesth Analg. 2006;103:274–80. doi: 10.1213/01.ANE.0000230389.76351.0C. [DOI] [PubMed] [Google Scholar]
- 45.Medrano GA, de Micheli A, Osornio Vargas A. Experimental left atrial infarct. Arch Inst Cardiol Mex. 1986;56:283–8. [PubMed] [Google Scholar]