

Abstract
Three 9,10-di-O-(−)-camphanoyl–7,8,9,10-tetrahydro-benzo[h]chromen-2-one (7-carbon-DCK) analogs (3a–c) were synthesized and evaluated for inhibition of HIV-1 replication in H9 lymphocytes. All three new carbon bioisosteres of the anti-HIV lead DCK showed anti-HIV activity. Compound 3a had an EC50 value of 0.068 μM, which was comparable to that of DCK in the same assay. The preliminary results indicated that 7-carbon-DCK analogs merit attention as potential HIV-1 inhibitors for further development into clinical trials candidates.
Keywords: Anti-HIV activity, DCK, Bioisostere
3′,4′-Di-O-(−)-camphanoyl-(+)-cis-khellactone (DCK, 1) demonstrated extremely potent inhibitory activity against HIV-1 replication in H9 lymphocytic cells with an EC50 value of 2.56 × 10−4 μM and a therapeutic index (TI) of 1.37 × 105 in our prior research.1 In subsequent structural modification studies, numerous DCK derivatives were synthesized and at least 20 DCK analogs have shown promising inhibitory activity against HIV-1 replication in H9 lymphocytes.2 Among them, 3-methyl, 4-methyl, and 5-methyl substituted DCKs were much more potent than DCK and AZT in the same assay with EC50 and TI values ranging from 5.25 × 10−5 to 2.39 × 10−7 μM and 2.15 × 106 to 3.97 × 108, respectively.3 In addition, a preliminary mechanistic study showed that 3-hydroxymethyl-4-methyl DCK inhibits HIV reverse transcriptase (RT) via a different mechanism of action from those of current clinical anti-HIV/AIDS drugs.4 It was also found that DCK analogs are strongly synergistic with approved drugs such as AZT and act at a point in the virus life cycle immediately following the target for AZT and nevirapine.4 In our recent research on structural modification of 4-methyl DCK (2), the ring oxygen atom in the A or C ring of DCK was replaced by a sulfur atom, and these sulfur-containing analogs also exhibited potent inhibitory effects on HIV-1 replication in H9 lymphocytes.5,6 Moreover, gem-dimethyl substitution at the 8-position was found to be preferable to larger alkyl substituents or hydrogen atoms.7 In a continuing effort to identify the pharmacophores in this class of potent anti-HIV agents, we designed a new series of DCK analogs, namely 7-carbon-DCK derivatives (3a–c). In these compounds, a methylene group replaces the oxygen in the C ring of DCK. Thus, these analogs are bioisosteres of DCK, and the effect of the 7-oxygen atom on the anti-HIV activity of DCK-type compounds can be further explored. In addition, to help determine the possible impact of the 8,8-dimethyl groups, both unsubstituted (3a–b) and dimethylated (3c) analogs were prepared. Herein, we report the synthesis of compounds 3a–c and their preliminary anti-HIV bioassay results (Fig. 1).
Figure 1.
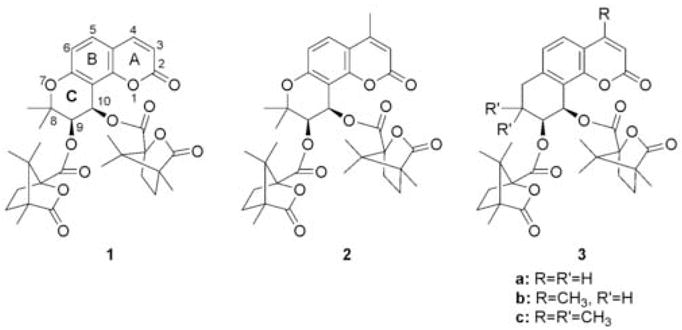
Structures of DCK (1), 4-methyl DCK (2) and 7-carbon-DCK analogs (3a–c).
The synthesis of 3a–b was accomplished by a 7-step sequence, as illustrated in Scheme 1. The key intermediates 7,8-dihydro-benzo[h]chromen-2-one (10a) and its 4-methyl analog (10b) were prepared according to the procedure reported in our prior work.8 Sharpless asymmetric dihydroxylation (AD) of 10a and 10b afforded dihydroxy derivatives 11a and 11b in moderate yield (45–49%).9 Finally, 7-carbon-DCK analogs 3a and 3b were obtained in 62% and 82% yields, respectively, by acylation of 11a and 11b with (S)-(−)-camphanic chloride in CH2Cl2 at room temperature with pyridine as acid scavenger.
Scheme 1.
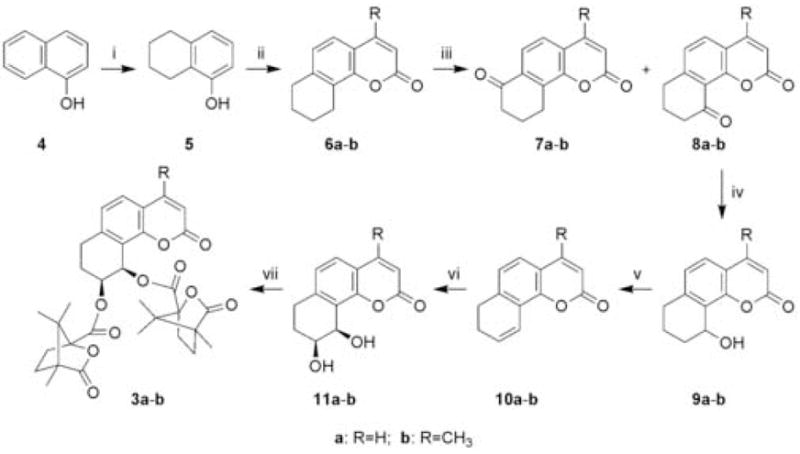
Synthesis of 3c: (i) t-BuOK/THF, reflux, 5 h, CH3I, 0.5 h; (ii) H2, Pd-C, CH3SO3H, HOAc, CH3COOC2H5, C2H5OH, rt, 36 h; (iii) BBr3/CH2Cl2, −78°C; (iv) CH3COCH2COOC2H5, POCl3, Benzene, reflux, 24 h, 66.0%; (v) CrO3, HOAc, rt, 30 h, (yield: 17=39.0%, 18=19.9%); (vi) NaBH4, CH3OH, 0.5 h, (yield: 70.3%); (vii) 2% H2SO4, 120–130°C, 5 h, (yield: 95.0%); (viii) AD-mix-α (K2OsO4·2H2O, K3Fe(CN)6, (DHQ)2PHAL, K2CO3), t-butanol/H2O 1:1, CH3SO2NH2, rt, 32 h, (yield: 75.5%); (ix) (S)-camphanic chloride, Et3N, DMAP, CH2Cl2, rt, 4 h, (yield: 81.2%).
As shown in Scheme 2, the preparation of 3c followed a slightly different synthetic route with 5-methoxy-1-tetralone (12) as starting material. Dimethylation of 12 with CH3I in the presence of t-BuOK afforded 2,2-dimethyl-5-methoxy-1-tetralone (13) in 91% yield.10 Reduction of dimethylated tetralone 13 with H2 catalyzed with 10% Pd-C gave 1,2,3,4-tetrahydro-5-methoxy-2,2-dimethylnaphthalene (14) quantitatively.11 Demethylation of 14 with BBr3 resulted in the formation of phenol derivative 15 in 98% yield.11 The remaining synthetic steps followed those detailed above for 3a–b from phenol 5. The target compound 3c was thus obtained in an overall yield of 5% via a six-step reaction sequence starting from 15.
Scheme 2.
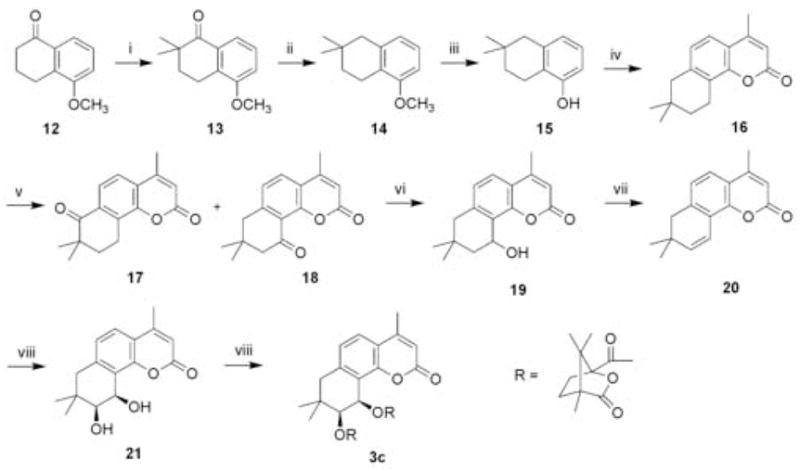
Synthesis of 3a–b: (i) Raney Ni-Al alloy, 1% aq. KOH/water, 90°C, 2 h; (ii) L-Malic acid, H2SO4, HOAc, 140°C, 6 h (R=H); CH3COCH2COOC2H5, POCl3, 100°C, 18 h (R=CH3); (iii) CrO3, HOAc, rt, 3 days; (iv) NaBH4, CH3OH, 1 h; (v) 2% H2SO4, reflux, 6–16 h; (vi) AD-mix-α (K2OsO4·2H2O, K3Fe(CN)6, (DHQ)2PHAL, K2CO3), t-butanol/H2O 1:1, rt, 2–3 days; (vii) (S)-camphanic chloride, pyridine, DMAP, CH2Cl2, rt, 24 h.
The anti-HIV activities of compounds 3a–c were evaluated in H9 lymphocytes, with AZT as the reference compound. The bioassay data are shown in Table 1 and indicated that all three compounds inhibited HIV replication and had reasonable therapeutic index (TI) values. Compounds 3a and 3b had significant EC50 values of 0.068 and 0.083 μM, respectively. Thus, the presence of the C-4 methyl in these 7-carbon DCK analogs did not lead to increased potency, in contrast to results with DCK and 4-methyl DCK. Although an absence of gem-dimethylation was detrimental in the 7-oxy DCK series,7 it was hard to make a definitive conclusion in the 7-carbon DCK series (comparison of 3c with 3b) due to solubility problems with 3c. However, the 7-carbon analog 3b was more potent and had a higher TI than the corresponding demethylated 7-oxy DCK derivative, 2′,2′-dihydro-4-methyl DCK (EC50 = 6.9 μM, TI > 6).7 Compound 3b was also more potent against HIV replication but was more cytotoxic than the analogous 7-thio analog (EC50 = 0.141 μM, TI = 1,110).6
Table 1.
Anti-HIV data of compounds 3a–c in acutely infected H9 lymphocytes
| Compound | IC50 (μM)a | EC50 (μM)b | TIc |
|---|---|---|---|
| 3a | 57.2 | 0.068 | 841 |
| 3b | 54.4 | 0.083 | 659 |
| 3cd | >39.4 | <0.39 | >100 |
| DCKe | >16.1 | 0.049 | >328 |
| 4-Me DCKe | >38.9 | 0.0059 | >6,600 |
| AZT | 500 | 0.0137 | 36,520 |
Concentration that inhibits uninfected H9 cell growth by 50%.
Concentration that inhibits viral replication by 50%.
TI = therapeutic index IC50/EC50.
More precise data could not be determined due to solubility problems.
Further structural modification and biological screening are in progress as these promising bioassay results demonstrate that 7-carbon-DCK analogs merit attention as potential HIV-1 inhibitors.
Supplementary Material
Acknowledgments
This research was supported by grants from the National Natural Science Foundation of China (No. 20272010 and 20672022) awarded to P. Xia and Y. Wang, respectively, a research grant (20020246069) for Ph.D. program from the National Education Administration of China awarded to P. Xia, and Grant AI-33066 from the National Institute of Allergies and Infectious Diseases awarded to K. H. Lee.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Huang L, Kashiwada Y, Cosentino LM, Fan S, Chen CH, McPhail AT, Fujioka T, Mihashi K, Lee KH. J Med Chem. 1994;37:3947. doi: 10.1021/jm00049a014. [DOI] [PubMed] [Google Scholar]
- 2.Yu D, Suzuki M, Xie L, Morris-Natschke SL, Lee KH. Med Res Rev. 2003;23:322. doi: 10.1002/med.10034. [DOI] [PubMed] [Google Scholar]
- 3.Xie L, Takeuchi Y, Cosentino LM, Lee KH. J Med Chem. 1999;42:2662. doi: 10.1021/jm9900624. [DOI] [PubMed] [Google Scholar]
- 4.Xie L, Yu D, Wild C, Allaway G, Turpin J, Smith PC, Lee KH. J Med Chem. 2004;47:756. doi: 10.1021/jm030416y. [DOI] [PubMed] [Google Scholar]
- 5.Xia P, Yin ZJ, Chen Y, Zhang Q, Zhang BN, Xia Y, Yang ZY, Kilgore N, Wild C, Morris-Natschke SL, Lee KH. Bioorg Med Chem Lett. 2004;14:3341. doi: 10.1016/j.bmcl.2004.03.051. [DOI] [PubMed] [Google Scholar]
- 6.Chen Y, Zhang Q, Zhang BN, Xia P, Xia Y, Yang ZY, Kilgore N, Wild C, Morris-Natschke SL, Lee KH. Bioorg Med Chem. 2004;12:6383. doi: 10.1016/j.bmc.2004.09.038. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Q, Chen Y, Xia P, Xia Y, Yang ZY, Yu DL, Morris-Natschke SL, Lee KH. Bioorg Med Chem Lett. 2004;14:5855. doi: 10.1016/j.bmcl.2004.09.032. [DOI] [PubMed] [Google Scholar]
- 8.Wang Y, Huang SX, Xia P. Synth Commun. 2005;35:3141. [Google Scholar]
- 9.Sharpless KB, Amberg W, Bennani YL, Crispino GA, Hartung J, Jeong KS, Kwong HL, Morikawa K, Wang ZM, Xu D, Zhang XL. J Org Chem. 1992;57:2768. [Google Scholar]
- 10.Gabriel Garcia J, Enas JD, Fronczek FR, VanBrocklin HF. J Org Chem. 1994;59:8299. [Google Scholar]
- 11.Bayston DJ, Fraser JL, Ashton MR, Baxter AD, Polywka MEC, Moses E. J Org Chem. 1998;63:3137. [Google Scholar]
- 12.Physical and spectral data for 3a–c: 9,10-Di-O-(−)-camphanoyl–7,8,9,10-tetrahydro-benzo[h]chromen-2-one (3a) mp 120–122 °C; 1H NMR (CDCl3, 300 MHz) δ0.94–1.13 (m, 18H, camphanoyl CH3), 1.61–2.56 (m, 10H, 8-H, camphanoyl CH2), 3.07–3.17 (m, 2H, 7-H), 5.31–5.40 (m, 1H, 9-H), 6.38 (d, J = 9.3 Hz, 1H, 3-H), 6.82 (d, J = 2.7 Hz, 1H, 10-H), 7.12 (d, J = 8.1 Hz, 1H, 6-H), 7.44 (d, J = 8.1 Hz, 1H, 5-H), 7.67 (d, J = 9.6 Hz, 1H, 4-H). ESI-MS m/z (%): 615.25 (M+Na+, 100). HR-MS: calcd for C33H36O10Na+ 615.2201, found 615.2191. 9,10-Di-O-(−)-camphanoyl–4-methyl–7,8,9,10-tetrahydro-benzo[h]chromen-2-one (3b) mp 159–161 °C; 1H NMR (CDCl3, 300 MHz) δ0.96–1.12 (m, 18H, camphanoyl CH3), 1.61–2.56 (m, 10H, 8-H, camphanoyl CH2), 2.42 (s, 3H, 4-CH3), 3.00–3.20 (m, 2H, 7-H), 5.31–5.38 (m, 1H, 9-H), 6.24 (s, 1H, 3-H), 6.83 (d, J = 2.7 Hz, 1H, 10-H), 7.12 (d, J = 8.1 Hz, 1H, 6-H), 7.55 (d, J = 8.1 Hz, 1H, 5-H). ESI-MS m/z (%): 606.30 (M +, 19). HR-MS: calcd for C34H38O10Na+ 629.2357, found 629.2367. 9,10-Di-O-(−)-camphanoyl–4,8,8-trimethyl–7,8,9,10-tetrahydro-benzo[h]chromen-2-one (3c) mp 148–150 °C; 1H NMR (CDCl3, 300 MHz) δ0.92–1.30 (m, 24H, camphanoyl CH3, 8-CH3), 1.61–1.76 (m, 2H, camphanoyl CH2), 1.86–1.98 (m, 2H, camphanoyl CH2), 2.11–2.28 (m, 2H, camphanoyl CH2), 2.45 (s, 3H, 4-CH3), 2.50–2.61 (m, 2H, camphanoyl CH2), 2.82–3.02 (m, 2H, 7-H), 5.35 (d, J = 5.1 Hz, 1H, 9-H), 6.25 (s, 1H, 3-H), 6.76 (d, J = 5.1 Hz, 1H, 10-H), 7.11 (d, J = 8.4 Hz, 1H, 6-H), 7.59 (d, J = 8.1 Hz, 1H, 5-H). ESI-MS m/z (%): 657.30 (M+Na+, 100). HR-MS: calcd for C36H42O10Na+ 657.2670, found 657.2690.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.