

Abstract
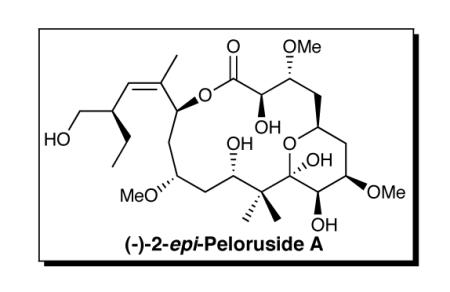
A convergent synthesis of (-)-2-epi-Peloruside A has been achieved. Highlights include implementation of multicomponent Type I Anion Relay Chemistry (ARC) to unite 2-TBS-1,3-dithiane with two epoxides to construct the eastern hemisphere, a late-stage dithiane union to secure the complete, fully functionalized carbon backbone, and Yamaguchi macrolactonization, which led to (-)-2-epi-peloruside A via an unexpected epimerization at C(2).
In 2000 Northcote and co-workers reported the isolation and relative stereochemistry of (+)-peloruside A (1),1 an architecturally complex marine metabolite produced by the sponge Mycale (Carmia). Although a microtubule-stabilizing agent with potency similar to Taxol,2 recent studies reveal that (+)-peloruside A competes competitively for the laulimalide binding site, at a newly discovered microtubule site.3
Our interest in (+)-peloruside A (1) emanated from the synthetic challenge, in conjunction with the opportunity to showcase the synthetic utility of dithiane linchpin tactics, in particular the use of the three-component union of trialkylsilyl dithianes with diverse electrophiles, a synthetic tactic we now recognize as Type I Anion Relay Chemistry (ARC).4
Structurally (+)-peloruside A (1) is comprised of 10-stereogenic centers, a Z-trisubstituted olefin, and a six-membered hemi-ketal ring, inscribed in a 16-membered macrolactone. Not surprisingly, the structural complexity, interesting biological activity, and scarcity, has led to considerable interest from both the chemical5 and biological communities.6
In 2003, De Brabander and co-workers7 achieved an elegant total synthesis of unnatural (−)-peloruside A, thus permitting assignment of the absolute configuration. Shortly thereafter (2005), the Taylor group8 reported the first total synthesis of natural (+)-peloruside A, followed in 2008 by a second total synthesis from the Ghosh laboratory.9 We report here completion of the total synthesis of (−)-2-epi-peluroside A (28, Scheme 5), the result of a surprising, late stage epimerization (vide infra) that procluded access to (+)-peloruside A (1).
Scheme 5.
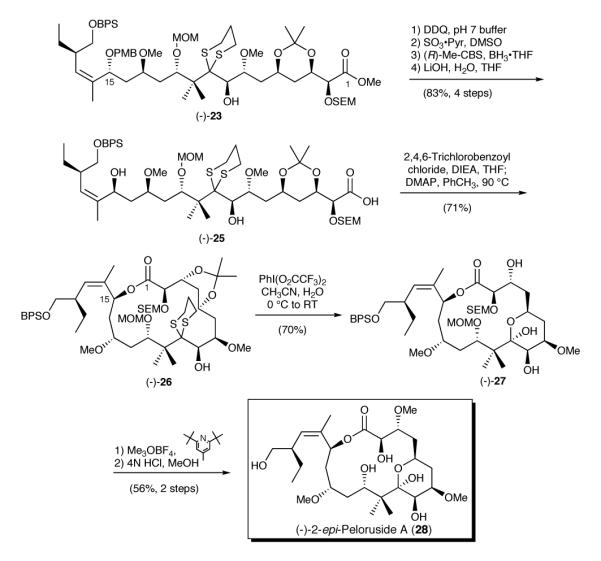
Synthesis of (-)-2-epi-Peloruside A
Shortly after the report by Northcote and co-workers,1 we initiated a synthetic venture directed toward the total synthesis of (+)-peloruside A (1).10 Our endgame strategy called for formation of the inscribed tetrahydropyran ring after macrocyclization (Scheme 1). Central to this scenario was a flexible route that would permit either acid or alcohol activation to achieve macrolactonization. Taken together, (+)-peloruside A (1) was envisioned to arise from macrolide 2 upon removal of the dithiane and isopropylidene protecting groups. To construct the macrolactone precursor, we would employ union of a dithiane 3 with aldehyde 4, followed by appropriate functional group adjustments.
Scheme 1.
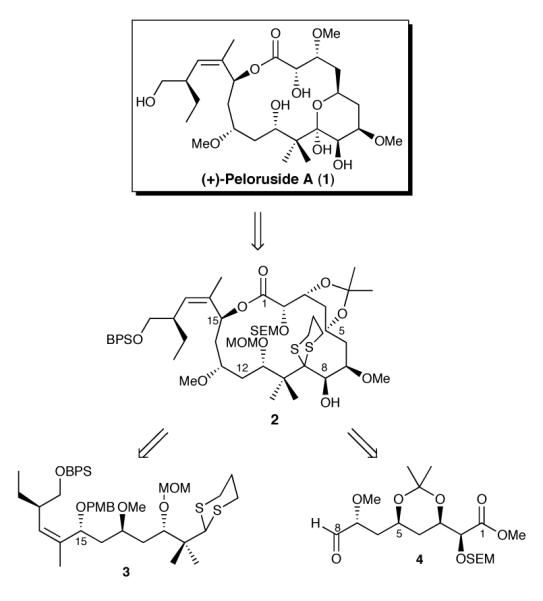
(+)-Peloruside A Retrosynthesis
Construction of dithiane (−)-3 began with known homoallylic alcohol (+)-5 (Scheme 2),11 which was protected as the BPS-ether. Ozonolysis furnished aldehyde (+)-6. Installation of the trisubstituted Z-olefin was next achieved via a Still-Gennari modification of the Horner-Wadsworth-Emmons olefination12 to yield ester (-)-8 in 89% yield as a single diastereomer. Next, enal (-)-9, available by a two-step reduction/oxidation sequence, was subjected to a Brown asymmetric allylation13 to generate alcohol (-)-10 in a highly diastereoselective fashion (>20:1).14,15 Protection of the resulting alcohol as the PMB ether, followed by selective dihydroxylation16 of the terminal olefin and oxidative cleavage furnished (-)-11, the requisite aldehyde for the proposed Mukaiyama aldol.17 Toward this end, reaction of (-)-11 with the silyl-enol ether derived from ketone 1218 led to β-hydroxy ketone (-)-13 with >20:1 diastereoselectivity at C(13).14 Ketone (-)-13 was then subjected to a SmI2 promoted Evans-Tishchenko reduction19 to generate (-)-14, possessing the correct stereochemistry at C(11).14
Scheme 2.
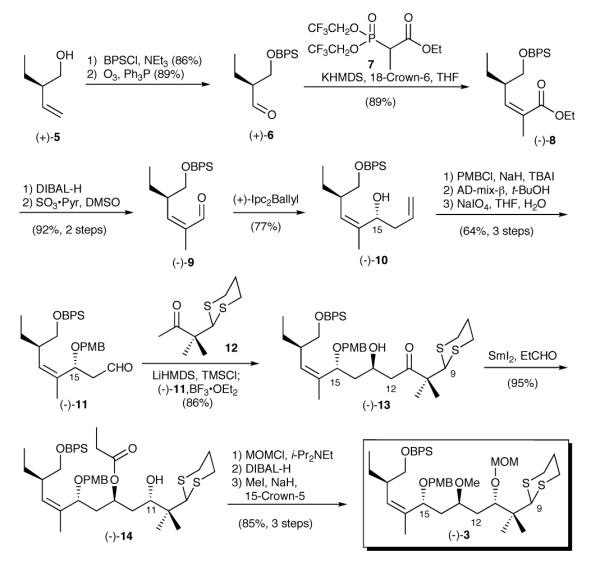
Synthesis of Dithiane (-)-3
Completion of dithiane (-)-3 entailed formation of the MOM-ether, reductive removal of the ethyl ester with DIBAL-H, and generation of the methyl ether. The overall sequence to (-)-3, the dithiane coupling partner, proved highly efficient, proceeding with a longest linear sequence of fourteen steps and in 21.4% overall yield from (+)-5.
Construction of aldehyde (+)-4 was designed specifically to demonstrate the utility of our multicomponent Type I ARC protocol, employing epoxide (+)-16, readily prepared from known epoxide (-)-1520 and epoxide (+)-1821 (Scheme 3). Toward this end, addition of the lithium anion of TBS-1,3-dithiane (17) to epoxide (+)-16, followed by a solvent controlled Brook rearrangement (HMPA) and addition of epoxide (+)-18 furnished alcohol (+)-1914 in 65% yield. Methyl ether formation, followed by removal of both the TBS and 1,3-dithiane moieties led to ketone (+)-20. We next called upon a hydroxyl directed 1,3-syn reduction,22 followed in turn by acetonide formation23 and removal of the benzyl ether via hydrogenolysis to generate alcohol (+)-21. Completion of (+)-4, the aldehyde coupling partner, was achieved in five steps. First, alcohol (+)-21 was converted via a three-step sequence to the corresponding methyl ester, and then subjected to oxidative removal of the PMB moiety to provide (-)-22. Parikh-Doering oxidation24 of the resultant terminal hydroxyl then furnished aldehyde (+)-4 in 87 % yield. The synthesis of (+)-4 also proved efficient, proceeding with a longest linear sequence of thirteen steps and in 12.9% overall yield from (-)-15.
Scheme 3.
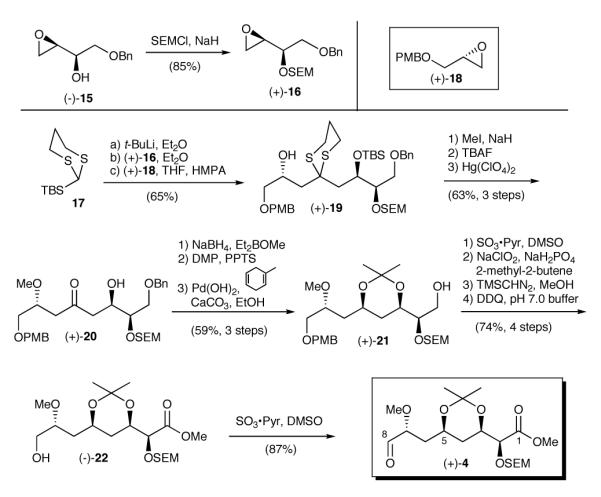
Synthesis of Aldehyde (+)-4
With advanced coupling fragments (-)-3 and (+)-4 in hand, we turned to their union (Scheme 4). Reaction of the lithium anion derived from dithiane (-)-3, with aldehyde (+)-4, in the presence of HMPA, led to alcohol (-)-23 as a mixture at C(8) favoring the desired isomer (ca. 9:1) presumably under Felkin-Anh control25. Importantly, union of (-)-3 and (+)-4 furnished the complete, stereochemically correct, carbon backbone of (+)-peloruside A. Formation of seco-acid (-)-24 was next readily achieved, in two steps, by removal of the PMB ether (DDQ) and saponification of the methyl ester (LiOH). Unfortunately, all attempts to achieve macrolactonization26 via the Mitsunobu protocol proved unsuccessful; only recovery of starting material or complete decomposition occurred.
Scheme 4.
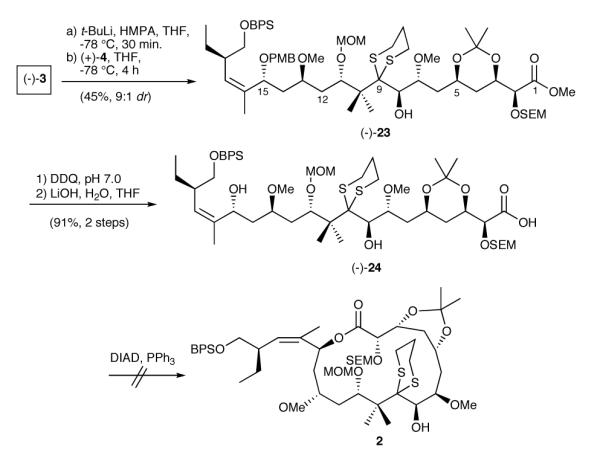
Efforts Toward (+)-Peloruside A
Undeterred, and with acid activation for macrolactonization as a backup, we inverted the C(15) hydroxyl (Scheme 5). The inversion required three steps: deprotection of the PMB-ether, oxidation of the derived secondary hydroxyl, and CBS reduction27 to provide the requisite C(15) stereogenicity. Saponification then furnished seco-acid (−)-25, setting the stage for macrolactonization. Here we encountered what proved to be an unexpected result. Execution of the Yamaguchi protocol28 involving acid activation and cyclization generated a macrolide in 71% yield, albeit with complete (!) epimerization at C(2) to furnish (−)-26, a result that went undetected until after global deprotection.
To understand after the fact (vide infra) this surprising result, we initiated a series of computational studies of (-)-26 and the corresponding desired C2-epimer. Initial conformational searches were preformed using Macromodel 7.2 software.29 The resulting low energy conformers were then clustered according to the macrocyclic ring torsions with the representative structures subjected to full geometry optimization at the B3LYP/6-31G(d,p) level of theory. The undesired, albeit observed, epimer (-)-26 was found to be more stable by 1.8 kcal/mol. Not surprisingly, the lowest energy conformations of the two compounds possess different macrocyclic ring conformations with the major torsional differences residing in the C1-C2 bond. As seen in Figure 1, the C2 hydrogen of (-)-26 adopts a favorable eclipsed conformation with the C1 carbonyl due to A(1,3) strain,30 while remainder of the macrocyclic ring does not show additional eclipsed interactions. The epimer, epi-26, on the other hand, takes up a bisected rather than an eclipsed conformation at C2, resulting in different C2-C3 and C5-C6 bond torsions around the protected 1,3-diol. In addition, the lowest energy macrocyclic ring conformer has one eclipsing interaction between the C7 methoxy and the C8 hydroxyl groups.
Figure 1.
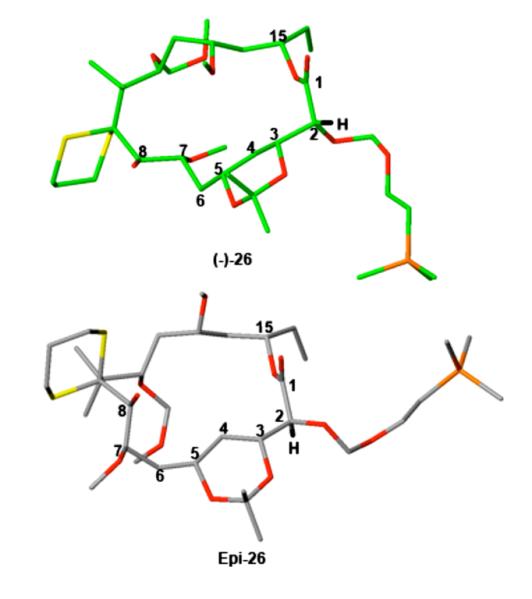
The observed (-)-26 epimer in green and the desired C2-epimer in grey, with the hydrogen at C2 shown in black. The C15 side chain has been omitted for better view.
Unaware at the time of the the C(2) epimerization, treatment of macrolide (-)-26 with the Stork reagent [PhI(O2CCF3)2]31 resulted in concomitant hydrolysis of the 1,3-dithiane, removal of the isopropylidene protecting group, and hemi-ketal formation to yield (-)-27.32 Selective methylation of the C(3) hydroxyl group with Meerwein’s reagent (Me3OBF4),33 followed by global deprotection employing 4N HCl in MeOH then delivered what was revealed by extensive 1-D and 2-D NMR analyses to be (-)-2-epi-peloruside A (28).
In summary the synthesis of 2-epi-peloruside A (28) has been achieved with a longest linear sequence of 25 steps and in 0.56% overall yield. Pleasingly, this synthetic venture demonstrates the utility of both dithiane linchpins and the multicomponent Type I ARC tactic.
Supplementary Material
Acknowledgment
Support was provided by National Institutes of Health (National Cancer Institute) through grant CA-19033, American Cancer Society for a Postdoctoral Fellowship (J.M.C.), partially funded by the National Fisheries Institute (PF-03-005-01-CDD), the Japanese Society for the Promotion of Science for a Postdoctoral fellowship (N.F.), and the University of Pennsylvania for a graduate fellowship (W.M.W.). Computational studies were supported by the National Science Foundation under Grant No. 0131132.
Footnotes
Supporting Information Available Spectroscopic and analytical data for compounds 6-28 and selected experimental and computational procedures. This material is available free of charge via the internet at http://pubs.acs.org.
References
- (1).West LM, Northcote PT, Battershill CN. J. Org. Chem. 2000;65:445. doi: 10.1021/jo991296y. [DOI] [PubMed] [Google Scholar]
- (2).Hood KA, Bäckström BT, West LM, Northcote PT, Berridge MV, Miller JH. Anticancer Drug Des. 2001;16:155. [PubMed] [Google Scholar]
- (3).Huzil JT, Chik JK, Slysz GW, Freedman H, Tuszynski J, Taylor RE, Sackett DL, Schriemer DC. J. Mol. Biol. 2008;378:1016. doi: 10.1016/j.jmb.2008.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Smith AB, III, Kim W—S, Wuest WM. Angew. Chem. Int. Ed. 2008;47:7082. doi: 10.1002/anie.200802301. [DOI] [PMC free article] [PubMed] [Google Scholar]; Smith AB, III, Wuest WM. Chem Comm. 2008 doi: 10.1039/b810394a. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Smalley MK, Hoye TR, Tennakoon M. Abstracts Amer. Chem. Soc. 2001 APR;221 [Google Scholar]; (b) Paterson I, Di Francesco ME, Kühn T. Org. Lett. 2003;5:599. doi: 10.1021/ol034035q. [DOI] [PubMed] [Google Scholar]; (c) Ghosh AK, Kim J-H. Tetrahedron Lett. 2003;44:3967. doi: 10.1016/S0040-4039(03)00744-5. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ghosh AK, Kim J-H. Tetrahedron Lett. 2003;44:7659. doi: 10.1016/j.tetlet.2003.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Taylor RE, Jin M. Org. Lett. 2003;5:4959. doi: 10.1021/ol0358814. [DOI] [PubMed] [Google Scholar]; (f) Vergin JB, Ryba TD. Abstracts Amer. Chem. Soc. 2003 MAR;225 [Google Scholar]; (g) Heady TN, Crimmins MT. Abstracts Amer. Chem. Soc. 2003 MAR;225 [Google Scholar]; (h) Liu B, Zhou W-S. Org. Lett. 2004;6:71. doi: 10.1021/ol036058a. [DOI] [PubMed] [Google Scholar]; (i) Engers DW, Bassindale MJ, Pagenkopf BL. Org. Lett. 2004;6:663. doi: 10.1021/ol036393z. [DOI] [PubMed] [Google Scholar]; (j) Gurjar MK, Pedduri Y, Ramana CV, Puranik VG, Gonnade RG. Tetrahedron Lett. 2004;45:387. [Google Scholar]; (k) Stocker BL, Teesdale-Spittle P, Hoberg JO. Eur. J. Org. Chem. 2004:330. [Google Scholar]; (l) Roulland E, Ermolenko MS. Org. Lett. 2005;7:2225. doi: 10.1021/ol050588k. [DOI] [PubMed] [Google Scholar]; (m) Owen RM, Roush WR. Org. Lett. 2005;7:3941. doi: 10.1021/ol0514303. [DOI] [PubMed] [Google Scholar]; (n) Chen Z—L, Zhou WS. Tetrahedron Lett. 2006;47:5289. [Google Scholar]
- (6).(a) Hamel E, Day BW, Miller JH, Jung MK, Northcote PT, Ghosh AK, Curran DP, Cushman M, Nicolaou KC, Paterson I, Sorensen EJ. Mol. Pharmacol. 2006;70:1555. doi: 10.1124/mol.106.027847. [DOI] [PubMed] [Google Scholar]; (b) Hood KA, West LM, Rouwe B, Northcote PT, Berridge MV, Wakefield SJ, Miller JH. Cancer Res. 2002;62:3356. [PubMed] [Google Scholar]; (c) Gaitanos TN, Buey RM, Díaz JF, Northcote PT, Spittle P. Teesdale, Andreu JM, Miller JH. Cancer Res. 2004;64:5063. doi: 10.1158/0008-5472.CAN-04-0771. [DOI] [PubMed] [Google Scholar]
- (7).Liao X, Wu Y, De Brabander JK. Angew. Chem., Int. Ed. 2003;42:1648. doi: 10.1002/anie.200351145. [DOI] [PubMed] [Google Scholar]
- (8).Jin M, Taylor RE. Org. Lett. 2005;7:1303. doi: 10.1021/ol050070g. [DOI] [PubMed] [Google Scholar]
- (9).Ghosh AK, Xu X, Kim J—H, Xu C—X. Org. Lett. 2008;10:1001. doi: 10.1021/ol703091b. [DOI] [PubMed] [Google Scholar]
- (10).Zheng J. Ph. D. Thesis. University of Pennsylvania; 2003. ; also see: Wuest WM. Ph. D. Thesis. University of Pennsylvania; 2008.
- (11).Xu Z, Johannes CW, Houri AF, La DS, Cogan DA, Hofilena GE, Hoveyda AH. J. Am. Chem. Soc. 1997;119:10302. [Google Scholar]
- (12).Still WC, Gennari C. Tetrahedron Lett. 1983;24:4405. [Google Scholar]
- (13).Racherla US, Brown HC. J. Org. Chem. 1991;56:401. [Google Scholar]
- (14).The absolute stereochemistry of alcohols (-)-10 C(15), (-)-13 C(13), (+)-19 C(7), were established by conversion to the corresponding Mosher’s esters and analyzed using the Kakisawa method. See supporting information and Ohtani I, Kusumi T, Kashman Y, Kakisawa H. J. Am. Chem. Soc. 1991;113:4092.
- (15).The stereochemistry of the C(15) hydroxyl is epimeric to that in the natural product, but is essential to set the C(13) hydroxyl stereochemistry in the Mukaiyama aldol reaction between (-)-11 and 12.
- (16).The sterically bulky ligand in AD-mix-β provides selectivity for the terminal olefin.
- (17).Mukaiyama T, Banno K, Narasaka K. J. Am. Chem. Soc. 1974;96:7503. [Google Scholar]
- (18).Ketone 12 was readily prepared in six steps from isobutyraldehyde.
- (19).Evans DA, Hoveyda AH. J. Am. Chem. Soc. 1990;112:6447. [Google Scholar]
- (20).Hungerbühler E, Seebach D. Helv. Chim. Acta. 1981;64:687. [Google Scholar]
- (21).Smith AB, III, Pitram SM, Boldi AM, Gaunt MJ, Sfouggatakis C, Moser WH. J. Am. Chem. Soc. 2003;125:14435. doi: 10.1021/ja0376238. [DOI] [PubMed] [Google Scholar]
- (22).Chen K-M, Hardtmann GE, Prasad K, Repic O, Shapiro MJ. Tetrahedron Lett. 1987;28:155. [Google Scholar]
- (23).(a) Rychnovsky SD, Skalitsky DJ. Tetrahedron Lett. 1990;31:945. [Google Scholar]; (b) Rychnovsky SD, Rogers B, Yang G. J. Org. Chem. 1993;58:3511. [Google Scholar]; (c) Evans DA, Rieger DL, Gage JR. Tetrahedron Lett. 1990;31:7099. [Google Scholar]
- (24).Parikh JR, Doering WE. J. Am. Chem. Soc. 1967;89:5505. [Google Scholar]
- (25).Chérest M, Felkin H, Prudent N. Tetrahedron Lett. 1968:2199.Anh NT, Eisenstein O. Nouv. J. Chem. 1977;1:61.Anh NT. Top. Curr. Chem. 1980;88:145.For assignment of the C(8) hydroxyl stereochemistry see reference 32.
- (26).Hungerbühler E, Seebach D. Helv. Chim. Acta. 1981;64:687. [Google Scholar]
- (27).Corey EJ, Helal CJ. Angew. Chem., Int. Ed. 1998;37:1986. doi: 10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- (28).Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. Bull Chem. Soc. Jpn. 1979;52:1989. [Google Scholar]
- (29).MacroModel, Schrödinger, L.L.C. 1500 S.W. First Avenue, Suite 1180, Portland, OR 97201-5815.
- (30).(a) Hoffman R. Chem. Rev. 1989;89:1841. [Google Scholar]; (b) Allinger NL. J. Am. Chem. Soc. 1969;91:337. [Google Scholar]
- (31).Stork G, Zhao K. Tetrahedron Lett. 1989;30:287. [Google Scholar]
- (32).Bis-MOM ether formation of (-)-27 permitted confirmation of the relative stereochemistry of C(5,7,8 and 9) by NOESY experiments. See supporting information.
- (33).Schaper UA. Synthesis. 1981:794. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.