

Abstract
Autographa californica multiple nucleopolyhedrovirus (AcMNPV) encodes two proteins that possess properties typical of single-stranded DNA-binding proteins (SSBs), late expression factor-3 (LEF-3), and a protein referred to as DNA-binding protein (DBP). Whereas LEF-3 is a multi-functional protein essential for viral DNA replication, transporting helicase into the nucleus, and forms a stable complex with the baculovirus alkaline nuclease, the role for DBP in baculovirus replication remains unclear. Therefore, to better understand the functional role of DBP in viral replication, a DBP knockout virus was generated from an AcMNPV bacmid and analyzed. The results of a growth curve analysis indicated that the dbp knockout construct was unable to produce budded virus indicating that dbp is essential. The lack of DBP does not cause a general shutdown of the expression of viral genes, as was revealed by accumulation of early (LEF-3), late (VP39), and very late (P10) proteins in cells transfected with the dbp knockout construct. To investigate the role of DBP in DNA replication, a real-time PCR-based assay was employed and showed that, although viral DNA synthesis occurred in cells transfected with the dbp knockout, the levels were less than that of the control virus suggesting that DBP is required for normal levels of DNA synthesis or for stability of nascent viral DNA. In addition, analysis of the viral DNA replicated by the dbp knockout by using pulsed-field gel electrophoresis failed to detect the presence of genome-length DNA. Furthermore, analysis of DBP from infected cells indicated that similar to LEF-3, DBP was tightly bound to viral chromatin. Assessment of the cellular localization of DBP relative to replicated viral DNA by immunoelectron microscopy indicated that, at 24 hours post-infection, DBP co-localized with replicated DNA at distinct electron-dense regions within the nucleus. Finally, immunoelectron microscopic analysis of cells transfected with the dbp knockout revealed that DBP is required for the production of normal-appearing nucleocapsids and for the generation of the virogenic stroma.
Introduction
The Baculoviridae consists of a diverse family of rod shaped viruses that contain circular covalently closed dsDNA genomes that range in size from 80 to 180 kbp. Baculovirus infections are restricted to invertebrates and the most well studied example is the Autographa californica multiple nucleopolyhedrovirus (AcMNPV) which is pathogenic for several species of Lepidoptera. Upon entry of AcMNPV into a susceptible host, replication occurs in the nucleus to generate two virus types. Budded virus (BV) are produced from nucleocapsids that become enveloped during egress through the plasma membrane that has been modified by the viral fusion protein GP64. In contrast, occlusion derived virions (ODV) are produced from nucleocapsids that remain in the nucleus where they are enveloped prior to becoming occluded within a crystalline matrix comprised of polyhedrin. Whereas BV is associated with systemic infection, ODV mediates lateral transmission between insects when released into the environment upon death of the host.
It has been determined through the use of a transient replication assay that six baculovirus gene products are required for viral DNA replication (Kool et al., 1994; Todd et al., 1995). These include an activator of transcription (ie-1), a helicase (p143), a DNA polymerase (dnapol), late expression factor-1 (lef-1) that functions as a primase, lef-2 that serves as a primase accessory factor, and lef-3, a single-stranded DNA-binding protein. More recently, the lef-11 gene product was shown to be required for replication of bacmid DNA in tissue culture (Lin and Blissard, 2002).
Previously, a search for additional DNA-interacting proteins from BmN cells infected with Bombyx mori nucleopolydedrovirus (BmNPV) identified a 37-kDa protein designated as DNA-binding protein, or DBP (Mikhailov et al., 1998). Interestingly, subsequent biochemical analysis of DBP after purification to near homogeneity indicated that it possessed properties of a bone-fide single-stranded DNA-binding protein (SSB). These properties include a much higher affinity for ssDNA over dsDNA, the ability to protect bound DNA substrates from exonuclease digestion, and the ability to unwind duplex DNA substrates in a dose-dependant manner without ATP (Mikhailov et al., 1998). Proteolytic digestion of the purified protein followed by mass spectroscopy analysis indicated that DBP was encoded by ORF16 of BmNPV, and homologs of this gene have been identified in all baculovirus genomes sequenced, except for one infectious for Diptera, that also lacks an identifiable homolog of lef-3 (Okano et al., 2006). In addition, analysis of the temporal expression pattern of DBP in BmNPV-infected cells indicated that it is expressed as an early gene, initially detectable by 4 hours post-infection (h.p.i.) and peaking at 14 h.p.i. and is not a virion structural protein (Okano et al., 1999). Using confocal microscopy to characterize the localization of DBP in infected cells, it was shown that at early times, DBP colocalizes with viral DNA, LEF-3, and IE-1, however, by 14 h.p.i., DBP became more diffuse and did not appear to colocalize with IE-1 or LEF-3 (Okano et al., 1999). The location of DBP at the viral DNA replication sites was confirmed for AcMNPV, a close relative of BmNPV (Mainz et al., 2002). A putative homologous region (hr) replication origin and two replication factors, IE-1 and helicase, were found to promote localization of both baculovirus SSBs, LEF-3 and DBP, to subnuclear foci (Nagamine et al, 2006). These observations suggest that DBP may be closely associated with replicating DNA during the early stages of infection, or could possibly function in DNA processing during the later stages of infection.
The observation that baculoviruses encode two single-stranded DNA-binding proteins raises interesting questions regarding their roles in DNA replication and processing. In this report we describe the construction of an AcMNPV bacmid lacking the dbp gene (AcMNPV ORF25) and the results of a series of experiments designed to gain insight into the function of DBP during infection.
Results
Analysis of budded virus production and gene expression
To determine whether dbp was required for budded virus production, a growth curve analysis was performed to monitor the amount of budded virus (BV) produced from cells transfected with the dbp knockout (dbp-KO), dbp repaired (dbp-rep), and control (AcGUS) bacmids. For these analyses, cell supernatants were collected every 24 hours up to 120 hours post-transfection (h.p.t.) and titered by end-point dilution assays. As shown in Fig. 1, no infectious BV was detected in supernatants collected from cells transfected with the dbp-KO bacmid at any time-point post-transfection. In contrast, beginning at 24 h.p.t. and continuing to 120 h.p.t., the budded virus titer from cells transfected with the dbp repair virus was similar to the level of BV produced from cells transfected with the infectious control bacmid, indicating that re-inserting the dbp ORF at the polyhedrin locus of the original dbp knockout was sufficient to restore DBP expression (Fig. 2A) and repair the non-infectious phenotype (Fig. 1) and confirmed that the lack of BV production in the null mutant is directly due to deletion of the dbp gene.
Fig. 1.
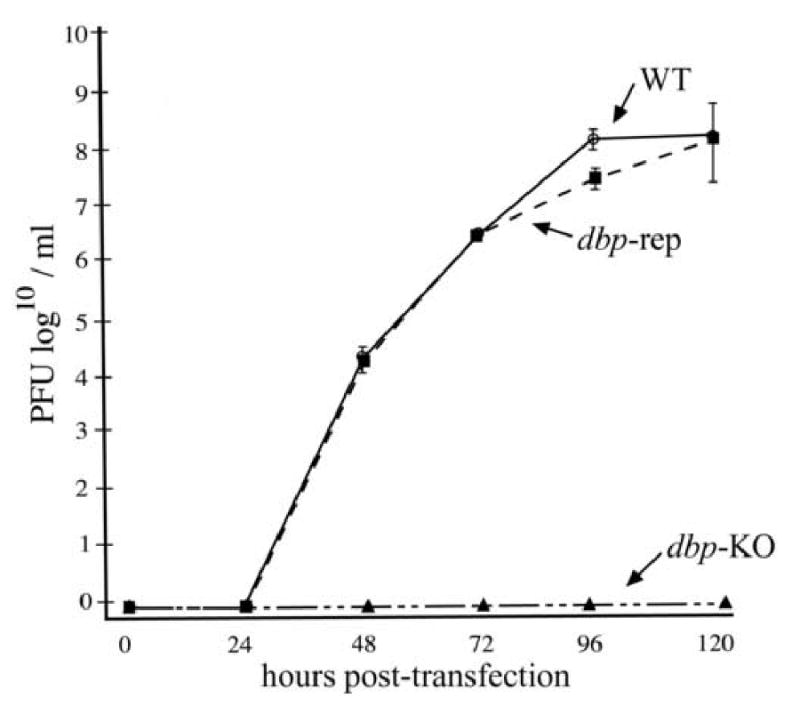
Analysis of budded virus production from bacmid transfected cells. At the indicated time-points, the supernatants from Sf-9 cells transfected with equimolar amounts of the dbp knockout bacmid (dbp-KO), the dbp repair bacmid (dbp-rep), or a control bacmid (AcGUS) were removed and the titers determined by TCID50 end-point dilution assays. The points indicate averages from transfections performed in triplicate and error bars represent standard deviations.
Fig. 2.
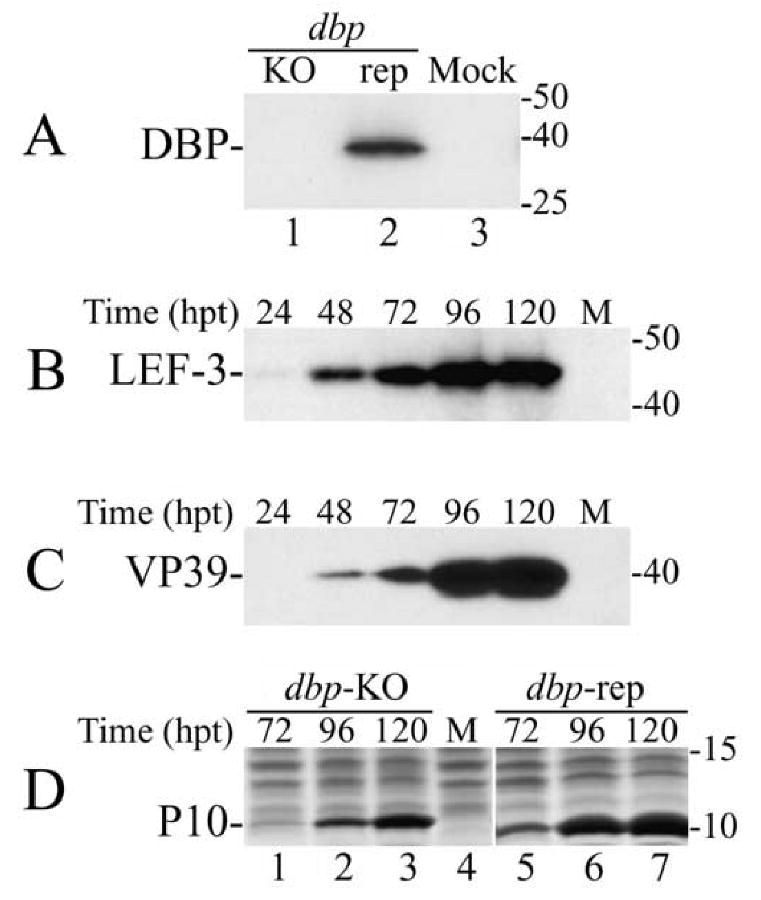
Western blot analysis of the synthesis of viral proteins in Sf-9 cells transfected with the dbp knockout (dbp-KO) bacmid. (A) The cells were transfected with the dbp-KO bacmid (lane 1), the repair (rep) bacmid (lane 2), or were mock transfected (lane 3) and harvested at 72 h.p.t. Portions from the cell extracts that corresponded to 0.5 × 104 cells were analyzed by SDS-15% PAGE. The blot was developed with polyclonal antibodies to DBP. (B-C) Time-course of the synthesis of LEF-3 and VP39 in Sf-9 cells transfected with the dbp-KO bacmid. The transfected cells were harvested at the times indicated above the respective lanes. Portions of cell extracts that correspond to 105 cells were subjected to SDS-12% PAGE. The blot was first developed with polyclonal antibodies to LEF-3 (B), and then with monoclonal antibody to VP39 (C). (D) Time-course of the synthesis of P10 in Sf-9 cells transfected with the dbp-KO bacmid (lanes 1-3) or with the repair bacmid (lanes 5-7). The transfected cells were harvested at times indicated above the respective lanes. Portions from the cell extracts that corresponded to 1 ×105 cells were subjected to SDS-15% PAGE. The gel was stained with Coomassie brilliant blue. Lanes “M” show the extract from mock transfected cells. Protein standards (in kDa) are shown on the right.
To rule out the possibility that the lack of BV production in cells transfected with the dbp-KO bacmid was due to a defect in viral gene expression, we investigated whether DBP was necessary for the expression of different classes of viral proteins. The accumulation of early (LEF-3), late (VP39), and very late (P10) proteins in cells transfected with the dbp-KO bacmid was monitored at different times post transfection (Fig. 2). The cell extracts were subjected to SDS-PAGE, and the amount of LEF-3 and VP39 was determined by using Western blotting and specific antibodies (panels B and C), whereas the abundant very late protein P10 was directly visualized by staining with Coomassie blue (panel D). The results of this analysis showed that all these proteins were synthesized in the cells lacking DBP (Fig. 2B, C, and D). The more robust expression of P10 by the dbp repair construct is likely due to the fact that it is an infectious virus. These data indicate that DBP is not essential for synthesis of viral products and confirms previous evidence that DBP is not a member of the LEF-family of viral factors that are required for synthesis of late (and very late) viral products.
Quantitative analysis of viral DNA replication
To evaluate whether the lack of budded virus production by the dbp knockout was due to the defect in DNA synthesis, a quantitative DNA replication assay was performed. Because the dbp knockout bacmid was unable to produce infectious virus as indicated by the growth curve analysis, replicated viral DNA from cells transfected with the dbp knockout was compared to DNA replicated by a bacmid lacking the gp64 gene that encodes an envelope fusion protein required for infectivity. Therefore, this construct serves as a non-infectious control (Vanarsdall, 2006). Additionally, the region used for PCR amplification contains four DpnI restriction sites and total DNA extracts were treated with DpnI prior to PCR amplification which serves to eliminate all input bacmid DNA (see Material and Methods). The results of this analysis showed that in cells transfected with the dbp knockout bacmid, although viral DNA synthesis occurs, the level of nascent DNA produced is reduced relative to the gp64 control knockout (Fig 3A). This reduction in DNA synthesis appeared to occur throughout the time-course. At the final time-point of 96 h.p.t., the level of DNA synthesized by the dbp knockout was equal to about 50% of the level of DNA synthesized by the gp64 knockout control virus (Fig. 3A).
Fig. 3.
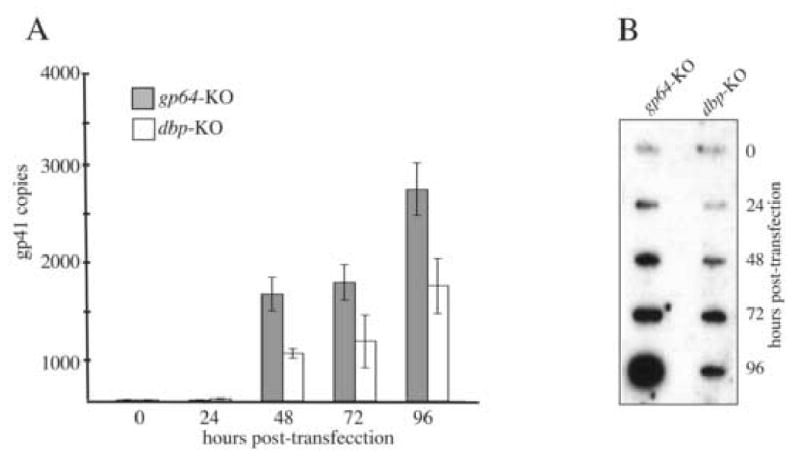
Analysis of viral DNA replication. (A) The bar graph represents the amount of nascent viral DNA synthesized in Sf-9 cells transfected with either the gp64 knockout virus serving as the non-infectious control or the dbp knockout virus. At the designated time-point, total DNA was isolated, treated with DpnI enzyme to digest input bacmid DNA, and analyzed by real-time PCR using a taqman probe and primer set that amplifies a 100 bp region of the AcMNPV gp41 open reading frame. The values displayed are the averages from transfections performed in triplicate with error bars indicating the standard deviations. (B) Slot-blot analysis of replicated viral DNA. At the indicated time-points, one well of a six-well plate of Sf-9 cells transfected with either the gp64 or dbp knockout virus was harvested at the indicated time points in 1 ml PBS and 100 μl of the cell suspension was removed, processed, and applied to nylon membrane using a slot-blot apparatus. The membrane was probed with 32P-labeled AcMNPV genomic DNA.
To confirm the results of the real-time PCR analysis, replicated viral DNA was analyzed by slot-blot hybridization. Although not as quantitative as real-time PCR, the results of the slot-blot analysis also showed that during a 96-hour time-course the signal produced with DNA extracts from dbp knockout transfected cells was substantially lower than the signal produced with DNA extracts from cells transfected with the gp64 knockout control (Fig. 3B). Therefore, although not absolutely required for DNA synthesis, DBP appears to be necessary for producing normal levels of viral DNA.
Characterization of replicated DNA by field-inversion gel electrophoresis
Baculovirus DBP has been shown to have a high affinity for single-stranded DNA and localizes to viral replication foci in nuclei. Based on these observations, DBP may be required for the production or maintenance of newly replicated viral genomes. Therefore, to determine what effect deleting dbp had on the electrophoretic properties of the nascent viral DNA, field-inversion gel electrophoresis was performed. For these analyses, whole cells transfected with either the dbp knockout or the gp64 knockout control virus were harvested at selected times and the DNA was purified in situ (see Materials and Methods), and separated under conditions that resolve DNA in the size range of 15 to 200 kbp. In addition, to determine whether full-length genomes are produced, prior to electrophoresis the DNA samples were digested in situ with the restriction enzyme Eco81I that cuts the genome at a single site. Under these conditions, host-cell DNA is digested to produce fragments in a range of about 20 to 70 kbp (Fig. 4A). However, newly replicated DNA isolated from cells transfected with the gp64 knockout control bacmid at 24 to 72 h.p.t. showed the presence of genome-length DNA of 140 kbp (see arrow) and DNA in the compression zone at >200 kbp (Fig. 4A and B). The compression zone contains DNA of variable size greater than ∼200 kbp that is not resolved by the FIGE program used for these analyses. Using a different program it was found to contain DNA of ∼200-700 kbp (Okano et al, 2007). A portion of viral DNA also remained trapped in the wells of the agarose gel and subgenome fragments <100 kbp are also evident. These results are consistent with previous reports characterizing baculovirus genomes and represent the normal pattern of viral DNA under these conditions (Okano et. al, 2007). In contrast, extracts from cells transfected with the dbp knockout produced a different DNA pattern. Although subgenome-size DNA fragments were evident and increasing amounts of viral DNA were present in the wells of the agarose gel from 24 to 72 h.p.t. at similar levels to that observed from the gp64 knockout control samples, no viral DNA was observed that migrated to the compression zone >200 kbp) or as genome-length DNA of 140 kbp from cells transfected with the dbp knockout (Fig. 4A and B). Therefore these results suggest that the DNA replicated by the dbp knockout is structurally distinct when compared to normal replicating virus.
Fig. 4.
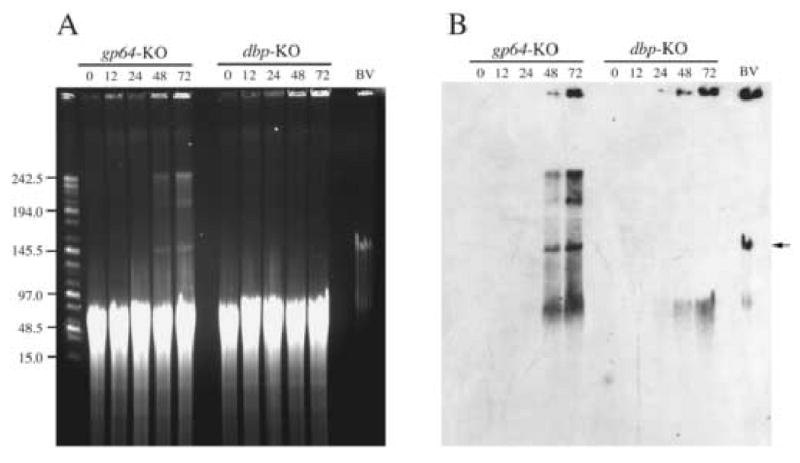
Field-inversion gel electrophoresis of replicated viral DNA. Total DNA was analyzed from Sf-9 cells transfected with either the gp64 knockout serving as the control (gp64-KO) or the dbp knockout (dbp-KO) as described in Material and Methods. Panel A represents the ethidium bromide stained agarose gel and panel B shows the results the Southern blot after hybridization with labeled AcMNPV bacmid DNA. The bacmid contructs that were used for transfection and the time samples were harvested post-transfection are indicated at the top of the panels. The size of the DNA marker in kilo-bases is shown on the left of panel A. “BV” represents viral DNA extracted and processed from extra-cellular budded virus. All samples were digested with Eco81I which cuts the genome at a single site prior to electrophoresis. The arrow indicates the position of full length genomic DNA.
Association of DBP with sub-nuclear structures
Previous reports (Okano et al., 1999; Mainz et al., 2002; Nagamine et al, 2006) indicated that DBP localizes to the nuclei of baculovirus infected cells. In order to identify possible nuclear substructures that recruit and interact with DBP, we fractionated nuclei from AcMNPV-infected cells using a standard protocol for isolation of the nuclear matrix. A similar fractionation scheme was employed previously to demonstrate that the viral protein pp31 associates with the nuclear matrix (Guarino et al., 1992). The fractionation procedure was carried out at early (10 h.p.i.) and late (20 h.p.i.) stages of virus replication and the extraction of DBP was compared to another viral DNA-binding protein, LEF-3 (Fig. 5A). The cells were disrupted by Dounce homogenization, and the nuclear and cytoplasmic fractions were separated by centrifugation. Most DBP and LEF-3 were present in the cytoplasmic fraction of infected cells harvested at 10 h.p.i. (Fig. 5A, lane 1). In contrast, the yield of DBP and LEF-3 was lower in the cytoplasmic than the nuclear fraction for cells harvested at 20 h.p.i. (Fig. 5A, compare lane 2 to lane 8). The appearance of nuclear proteins in the cytoplasmic fraction might be caused by the leakage of soluble proteins from nuclei that often occurs during cell fractionation in aqueous solutions. The higher proportion of DBP and LEF-3 with nuclear structures at 20 h.p.i. than at 10 h.p.i. suggests that the number of binding sites for DBP and LEF-3 in nuclei may have increased in the course of infection which might be caused by multiplication of the viral genome. Washing nuclei in a buffer containing Nonidet P-40 extracted additional DBP and LEF-3 (Fig. 5A, lanes 3 and 4). Treatment of nuclei with DNase and RNase released only a small amount of DBP, but no LEF-3 (Fig. 5A, lanes 5 and 6). Hence, the fragmentation of nucleic acids by these enzymes does not solubilize DBP or LEF-3 from nuclei. Further treatment with 2 M NaCl, which should release fragmented DNA and all chromatin-bound proteins, solubilized all of the residual DBP and LEF-3 associated with the nuclei (Fig. 5A, lanes 7 and 8). The nuclear matrix was obtained after an additional treatment with DNase and RNase (Fig. 5A, lanes 9 and 10) and resulted in final fractions that contained approximately 6 to 8% of total cellular protein including the cellular pool of the nuclear matrix marker protein, lamin, but only traces of DBP or LEF-3 (Fig. 5A, lanes 11 and 12). Therefore these results indicate that neither DBP nor LEF-3 are components of the nuclear matrix, and suggests that they presumably associate with chromatin. This conclusion is in agreement with the initial identification of DBP and LEF-3 as DNA-binding proteins.
Fig. 5.
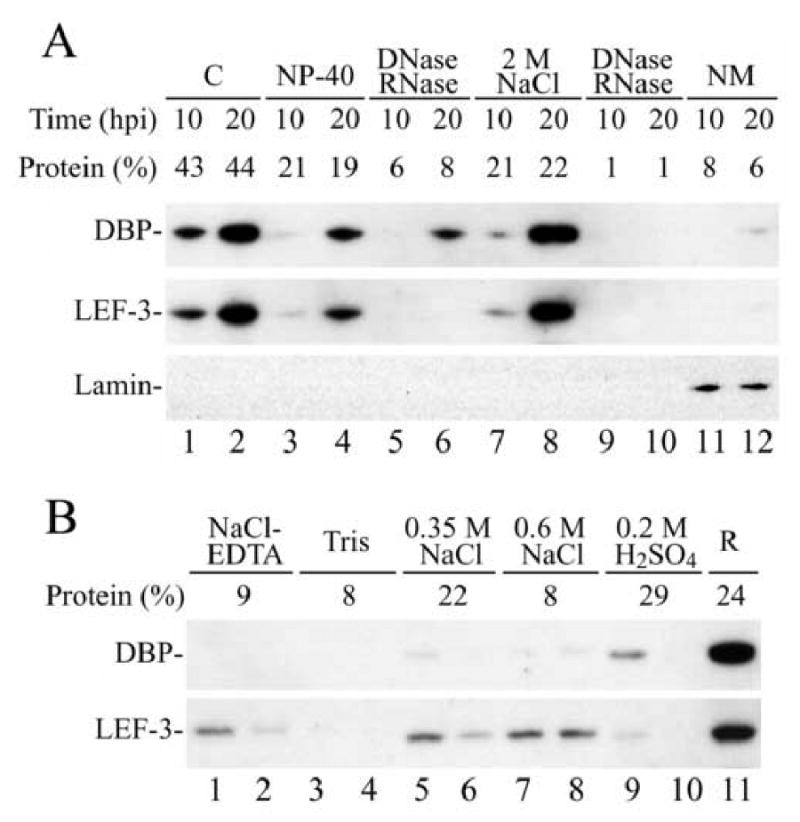
Western blot analysis of the association of DBP and LEF-3 with subnuclear structures. (A) AcMNPV-infected Sf-9 cells were harvested at 10 or 20 h.p.i. and homogenized in a hypotonic buffer (see Materials and Methods). Nuclei were separated from the cytoplasmic fraction “C” (lanes 1 and 2) by centrifugation and were sequentially washed with buffer containing 1% Nonidet P-40 (lanes 3 and 4), treated with DNase and RNase (lanes 5 and 6), extracted with buffer containing 2 M NaCl (lanes 7 and 8), treated again with DNase and RNase (lanes 9 and 10), and finally the nuclear matrix “NM” was collected (lanes 11 and 12). A portion from each fraction that corresponded to 0.9 × 105 cells was analyzed by SDS-11% PAGE. The blot was first developed with polyclonal antibodies to DBP, then with polyclonal antibodies to LEF-3, and finally with monoclonal antibody to Drosophila lamin. The amount of protein in each fraction was estimated by optical densitometry of the Coomassie stained gel and is shown as the percentage of total cellular protein above the respective lane. (B) Western blot analysis of the association of DBP and LEF-3 with chromatin structures. Nuclei were isolated from AcMNPV-infected Sf-9 cells harvested at 20 h.p.i. as described in the Materials and Methods and were sequentially extracted twice with buffers containing 75 mM NaCl, 25 mM EDTA (lanes 1 and 2), 10 mM Tris, pH 8.0 (lanes 3 and 4), 0.35 M NaCl (lanes 5 and 6), 0.6 M NaCl (lanes 7 and 8), and with 0.2 M H2SO4 (lanes 9 and 10). Lane R presents the nuclear residue. A portion from each fraction that corresponded to 1.2 × 105 cells was analyzed by SDS-11% PAGE. The blot was first developed with polyclonal antibodies to DBP, and then with polyclonal antibodies to LEF-3. Percentage of nuclear protein extracted by each solution is shown above the lanes.
Wilson and Miller (1986) suggested that nascent viral DNA undergoes packaging by host histones into nucleosomal fibrils that are similar to cellular chromatin. In order to identify chromatin components that interact with DBP or LEF-3, chromatin-associated proteins of nuclei from cells harvested at 20 h.p.i. were fractionated using different buffers following a standard protocol (Busch, 1984). Washing with each buffer was carried out twice and each pair of extracts was analyzed sequentially. At first, the nuclei were washed with a buffer containing 75 mM NaCl and 25 mM EDTA (Fig. 5B, lanes 1 and 2), and then with a buffer containing 10 mM Tris, pH 8.0 (Fig. 5B, lanes 3 and 4). This treatment released approximately 17% of total nuclear protein but no DBP and only a minor amount of LEF-3. The next washing step was performed with a buffer containing 0.35 M NaCl that should release the high mobility and low mobility groups (HMG and LMG) of non-histone chromatin proteins. This fraction contained only traces of DBP and a small portion of LEF-3 (Fig. 5B, lanes 5 and 6). Washing with 0.6 M NaCl yields extracts containing predominantly histone H1, but not core histones. This fraction contained traces of DBP and some LEF-3 (Fig. 5B, lanes 7 and 8). Treatment with 0.2 M H2SO4 yields core histones. This was directly confirmed by staining the gels after SDS-PAGE with Coomassie blue (data not shown). This fraction also contained minimal amounts of DBP and LEF-3 (Fig. 5B, lanes 9 and 10). The major portion of both DBP and LEF-3 was recovered in the residue fraction (R) that remained after all the treatments described above and therefore contains proteins tightly associated with DNA (Fig. 5B, lane 11). Since DBP and LEF-3 were retained under conditions when most of the non-histone and histone proteins were eluted indicates that these host proteins do not provide primary binding sites for DBP and LEF-3 in chromatin.
Electron microscopic analysis
To determine whether the deletion of dbp effects nucleocapsid structures, electron microscopic analysis was performed on thin sections generated from Sf-9 cells transfected with either the gp64 knockout control or the dbp knockout bacmid. Virus nucleocapsids were examined by staining thin sections with a monoclonal antibody specific for the major capsid protein, VP39. As expected, cells transfected with the gp64 control bacmid displayed features characteristic of baculovirus infection. These features include an enlarged nucleus, rearrangement of host chromatin, and the presence of an electron-dense virogenic stroma (Fig. 6A). In addition, abundant rod-shaped nucleocapsids stained with gold label were seen within the nucleus (Fig. 6A, inset). In contrast, cells that were transfected with the dbp knockout, although showed an enlarged nucleus and rearranged host chromatin, lacked an organized virogenic stroma (Fig. 6B). Furthermore, cells transfected with the dbp knockout did not contain the rod-shaped nucleocapsids seen in the control virus. Instead, clusters of gold label representing the major capsid protein VP39 were observed associating with amorphous electron-dense structures (Fig. 6C and D). Notably, these clusters of gold label were primarily localized to the periphery of the nucleus near the inner nuclear membrane (Fig. 6D). Therefore, these results demonstrate that a bacmid lacking DBP is unable to form a virogenic stroma and is defective in nucleocapsid production. Notably, the defect in nucleocapsid production is not predicted to be the result of a structural deficiency, as DBP is not a component of extracellular virus. This was confirmed by Western blot analysis on purified virions (data not shown) and is similar to the results previously reported for BmNPV (Okano et al, 1999).
Fig. 6.
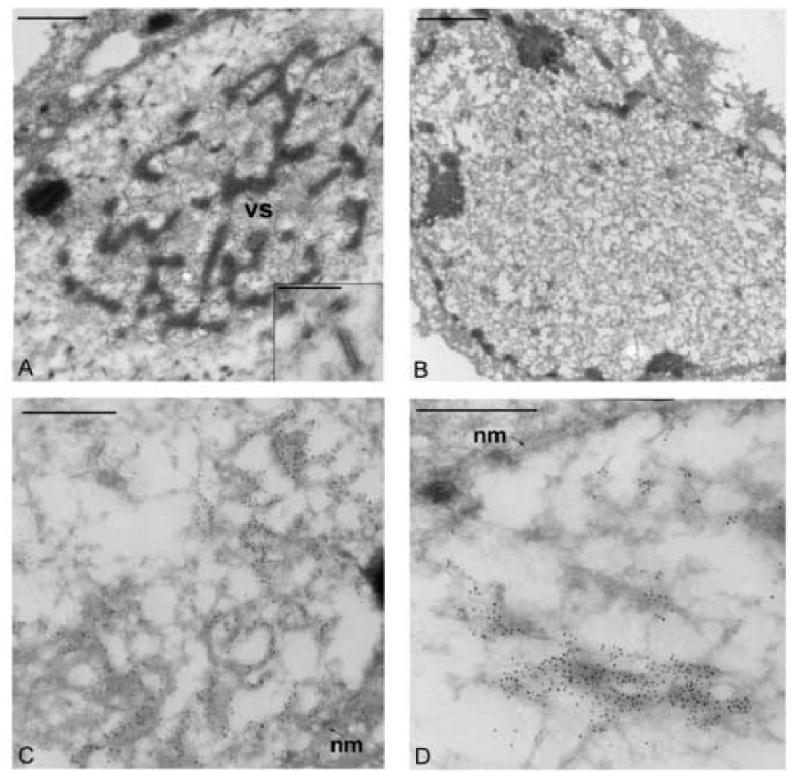
Electron microscopic analysis of thin sections from Sf-9 transfected cells stained with a VP39 antibody. (A) Image of a cell transfected with the gp64-KO control bacmid showing the electron dense virogenic stroma (VS) within the nucleus. The inset panel shows a higher magnification of nucleocapsids immuno-stained with the monoclonal antibody to the major capsid protein VP39. (B) Image of a cell transfected with the dbp-KO bacmid construct showing the lack of an organized virogenic stroma (C and D). Images of a portion of the nucleus from cells transfected with the dbp-KO bacmid construct. The gold label can be seen localized to regions near the nuclear membrane (nm). The bars in panels A and B represent 1 μm and the bars in panels C, D, and the inset represent 0.5 μm.
To investigate the localization of DBP relative to replicated viral DNA in infected cells, additional immunoelectron microscopy experiments were performed. For these experiments, BV derived from the replication competent AcGUS construct was used to infect cell monolayers that were cultured in medium supplemented with the DNA precursor BrdU. Because host cell DNA replication is halted by virus infection, the BrdU in the culture medium is selectively incorporated into newly synthesized viral DNA (Braunagel et al., 1998; Ikeda and Kobayashi, 1999). To detect both DBP and the newly synthesized viral DNA in infected cells, monolayers were harvested at 24 h.p.i., processed for electron microscopy, and the thin sections co-stained with a monoclonal antibody against BrdU and a polyclonal antibody against DBP followed by goat-anti mouse and goat anti-rabbit secondary antibodies conjugated to 20 nm and 10 nm gold particles, respectively. Microscopic analysis of these thin sections revealed numerous well-defined electron-dense regions that were evenly distributed throughout the nucleus (Fig. 7A). Interestingly, these electron-dense regions were specifically labeled with 20 nm as well as 10 nm gold particles (Fig 7B-D). Therefore these results indicate that at 24 h.p.i. both newly replicated viral DNA and DBP are localized to distinct domains within the nucleus.
Fig. 7.
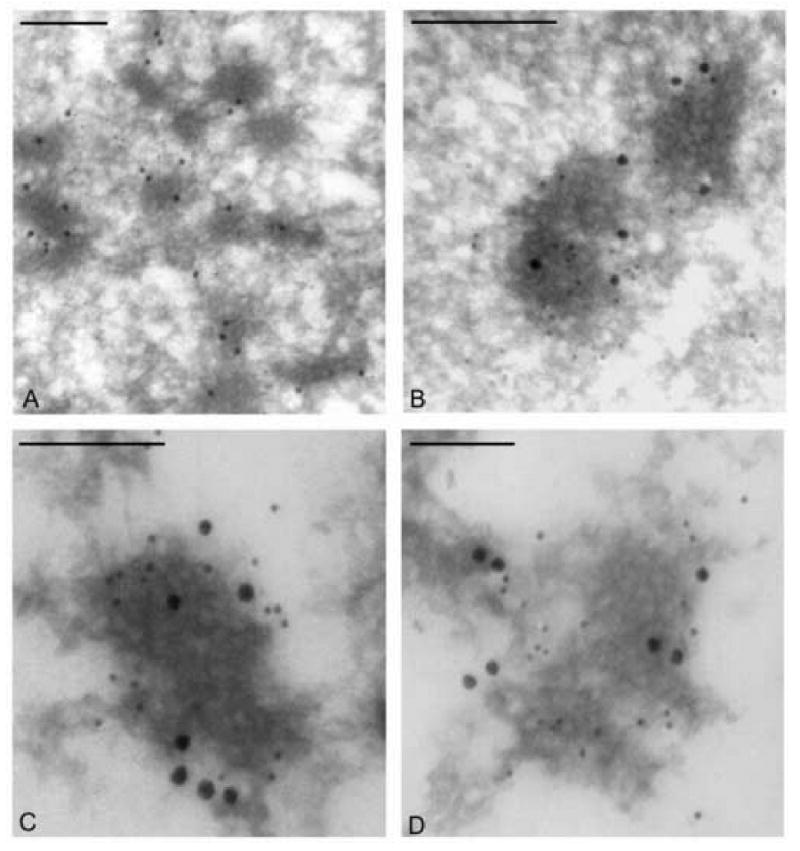
Electron microscopic analysis of Sf-9 cells infected with a DBP repair virus and treated with BrdU. (A-D) Images showing portions of the nucleus from Sf-9 cells at 24 h.p.i. Thin sections were co-stained with a monoclonal antibody to BrdU (20 nm gold) and a polyclonal antibody to DBP (10 nm gold). The bars represent 0.5 μm.
Discussion
Baculovirus DBP was first described after being purified from BmNPV infected cells and subsequently shown to possess properties of a bone fide ssDNA-binding protein, although its role in virus replication was unclear (Mikhailov et al., 1998). To determine whether DBP is an essential factor required for viral replication, we employed the AcMNPV bacmid system to produce a dbp knockout construct and characterized its ability to replicate in Sf-9 cells. The results of a growth curve analysis monitoring the amount of BV produced from cells transfected with the dbp knockout construct indicated a complete block in BV production (Fig. 1) thereby confirming that dbp is an essential gene. Another baculovirus single-stranded DNA-binding protein LEF-3, is also an essential gene and has been implicated in viral DNA replication based on the results of a transient DNA replication assay (Kool et al., 1994). However, DBP was not identified as being required for DNA replication in this assay. To investigate whether DBP was required to replicate viral DNA in the course of an infection, a real-time PCR assay was used to monitor viral DNA synthesis in bacmid-transfected cells. This analysis indicated that, although nascent viral DNA was synthesized in the absence of DBP, the level of newly replicated DNA was reduced to about 50% of that produced by the non-infectious gp64 knockout control virus (Fig. 3). These results confirmed that DBP is not essential for DNA synthesis and therefore is not likely to be involved in the viral replisome. However, the fact that the overall level of DNA synthesis was reduced in the absence of DBP does suggest that DBP may be a stimulatory factor or may indirectly affect genome replication. Although viral DNA was synthesized at about 50% the level of the controls in cells transfected with the dbp knockout bacmid, we did not detect production of budded virions. This suggests that the DNA synthesized in the absence of DBP might be abnormal and not suitable for packaging into viral particles. To investigate this possibility, the nature of the DNA replicated by the dbp knockout virus was analyzed by field-inversion gel electrophoresis after digesting the DNA samples with Eco81I, an enzyme that cuts the genome once. This allowed resolution of monomeric genomes regardless of whether they were fully maturated (e.g. circularized) or in the form of a replication intermediate such as a large concatemer. A similar strategy has been used previously to analyze viral DNA replicated in Sf-9 cells (Vanarsdall et al., 2006) and demonstrates that baculoviral replicative intermediates, synthesized by control viruses such as the gp64–KO virus, include genome-length DNA that migrates at ∼140 kbp, an intermediate species that migrates near the compression zone at >200 kbp, and DNA that remains in the wells of the agarose gel. In contrast, the DNA replicated by the dbp knockout did not appear to yield genome-length DNA or the intermediate species that migrates near the compression zone (Fig. 4). However, at 24, 48 and 72 h.p.t., increasing amounts of viral DNA was present in the wells of the gel and some fragmented DNA smaller than genome length was also detected. The failure to detect genome-length DNA in cells transfected with the dbp knockout indicated that, although the viral DNA synthesis is able to proceed, it is unable to produce the high molecular weight intermediates and full-size genomes.
DBP is an abundant protein in cells infected with BmNPV, but it was not found among structural proteins from budded or occlusion-derived virions by Western blot analysis (Okano et al., 1999). To investigate if nucleocapsid production was affected by a lack of DBP, immunoelectron microscopy was performed on sections of cells transfected with the dbp knockout bacmid and stained with a monoclonal antibody to the capsid protein, VP39 (Fig. 6). Interestingly, these experiments revealed regions within nuclei that stained for VP39, but lacked rod-shaped nucleocapsids. These results suggested that DBP may be directly required for nucleocapsid assembly or that nucleocapsids cannot be formed in the absence of mature full-size viral genomes. Another interesting observation was the lack of a well-organized virogenic stroma in cells transfected with the dbp knockout virus. Although the components that make up the virogenic stroma are not well defined, results from a previous report suggested that viral nucleic acids are present within these structures (Young et al., 1993). Therefore, both complete viral genomes and DBP may be required for nucleocapsid production and also might be essential constituents of the virogenic stroma.
Additional immunoelectron microscopic analysis showed colocalization of DBP with newly replicated DNA at 24 h.p.i. (Fig. 7). Interestingly, immuno-staining of both DBP and newly replicated DNA was limited to electron-dense regions within infected cell nuclei that are predicted to represent DNA replication foci (Okano et al, 1999; Mainz et al., 2002). These data confirmed that DBP is likely associated with or located in close proximity to nascent viral DNA.
Fractionation of nuclei and chromatin isolated from AcMNPV-infected cells showed that DBP and LEF-3 are not components of the nuclear matrix. Both proteins are tightly associated with chromatin, but cellular histones do not mediate the linkage. It was suggested earlier that host cell histones organize viral DNA into nucleosome fibrils that are similar to cellular chromatin (Wilson and Miller, 1986). Our data suggest that DBP and LEF-3 are not incorporated into the nucleosomal structure. Instead, due to their intrinsic DNA-binding activity, DBP and LEF-3 might directly interact with ssDNA generated by replication and recombination of the viral genome. DBP and LEF-3 also showed similar, but not identical patterns during fractionation. A larger portion of LEF-3 than DBP was released from the chromatin at NaCl concentrations of 0.35 M and 0.6 M (Fig. 5B, lanes 5-8). This result is in agreement with the earlier observation that LEF-3 elutes from ssDNA-cellulose at lower salt concentration than DBP (Mikhailov et al., 1998).
It remains uncertain what role DBP plays during genome replication. This and previous reports suggest that DBP may serve as an integral component of the virogenic stroma and may directly protect nascent viral DNA against attacks by nucleases and facilitate the assembly of viral particles. The fact that the mutant virus lacking DBP displays a reduced DNA replication phenotype and is unable to generate mature genomes implies also that DBP may stimulate replication or maintain the stability of the DNA replisome during synthesis. Alternatively, the reduced level of DNA replication may implicate DBP in a DNA processing mechanism such as recombination. A similar phenotype in which DNA replication is reduced has been described for bacteriophage T4 genomes harboring mutations in the recombination proteins that promote strand invasion of homologous DNA (Cunningham and Berger, 1977; Hashimoto and Yonesaki, 1991; Melamede and Wallace, 1977). Therefore if a similar mechanism were used during baculovirus replication and involves DBP, it may also entail several additional factors, some of which may interact directly with DBP. To this end, employing a proteomics approach to identify candidates that interact with DBP may provide significant insight into the role for DBP during replication.
We have now characterized a number of genes implicated in baculovirus DNA replication by the characterization of deletion mutants. Mutants deleted for DNA polymerase (Vanarsdall et al, 2005) were unable to initiate DNA replication. In contrast, deletions of VLF-1 (Vanarsdall et al, 2006), alkaline nuclease (Okano et al, 2007), and DBP, although defective in infectious virus and nucleocapsid production, were capable of replicating DNA. However, although the VLF-1 deletion appeared to synthesize normal genomic DNA, DNA produced by the alkaline nuclease knockout appeared to be fragmented, and the DNA generated by the DBP deletion mutant appeared to be synthesized at lower levels and we could not detect complete genome-size DNA. The correlation of defective nucleocapsid production with the inability to produce genome size DNA in the DBP and alkaline nuclease knockouts suggests that intact, correctly process DNA may be a prerequisite for nucleocapsid assembly. Deletion of VLF-1 produced distinct tube-like structures and when it was repaired with a VLF-1 Y335F point mutant, normal-appearing nucleocapsids were produced, but they were still non infectious. We interpreted these data to indicate the VLF-1 is required both for normal capsid assembly and for a final DNA processing event. The requirement for normal capsid assembly could reflect the involvement of VLF-1 as an essential capsid structural component. As described above, DBP does not appear to be a structural component, but its deletion does result in defective appearing nucleocapsids. Since it is not a virion structural protein this was attributed to a defect in DNA processing such that properly assembled nucleocapsids could not be produced.
Material and Methods
Cells and antibiotics
Spodoptera frugiperda (Sf-9) cells were cultured in Sf-900 II serum-free medium (Invitrogen) with added penicillin G (50 units/ml), streptomycin (50 units/ml,Whittaker Bioproducts), and fungizone (amphotericin, 375 ng/ml, Invitrogen) as previously described (Harwood et al., 1998).
Bacmid construction, purification, and transfection
The control bacmid used for these studies (gp64-KO) is a non-infectious bacmid construct lacking the gp64 ORF that encodes for the baculovirus major envelope fusion glycoprotein and has been described previously (Vanarsdall, 2006). A bacmid construct lacking the DNA-binding protein (dbp-KO) was generated with the λ Red homologous recombination system in Escherichia coli using a previously described protocol (Vanarsdall et al., 2004). Briefly, primers dbp-KO-1, (5′-ATTTATTGTTCAATAATAACAAATATTCCAGGCTTAAAAGCTAACGAATAGCGATTGTGTA GGCTGGAGCTGCT-3′) and DBP-KO-2, (5′-CCACAGCAGACAGCGCACGTCGGTAGCATGGCAACTAAACGCAAGATTGGCATATGAATA TCCTCCTTAGTTCC-3′) were used to generate a linear chloramphenicol acetyl transferase (CAT) marker gene including the bacterial promoter from plasmid pKD3 (Datsenko and Wanner, 2000) and includes 50 nucleotides of sequence homology to the 5′ and 3′ region of the dbp open reading frame (ORF25). This fragment was subsequently electroporated into E. coli DH10B cells containing bacmid bMON14272 (Invitrogen) and plasmid pKD46 encoding the λ Red recombination genes gam, beta, and exo (Datsenko and Wanner, 2000). Potential bacmid knockout clones were selected on LB plates containing chloramphenicol and isolated colonies were screened for insertion of the CAT gene at the dbp locus by PCR using primers Ac20970, (5′-CGCCCAGATACTGGTTTACG-3′), that anneals to a genomic region outside the recombination locus and primer CATR2 (5′-TTGTTACACCGTTTTCCATGAGC-3′) that is specific for the CAT gene. To confirm that the observed phenotype is due to deletion of the target gene and not from secondary mutations, a repair virus was constructed by re-inserting the dbp ORF under control of its native promoter at the polyhedrin locus by Tn7 mediated transposition. This was accomplished by PCR amplifying the dbp ORF and native promoter with primers DBP-1, (5-′ATGAATTCTGATAAAATAAAACGGGGGC-3′) and DBP-2 (5′-GCTCTAGATTATTGTTCAATAATAACAAATATTC-3′), cloning the PCR product into PCR2.1 TOPO vector (Invitrogen), removing the insert and ligating it into the NotI and HindIII sites of plasmid pfbie-GUS(Eco81I) as described previously (Vanarsdall, 2006).
Bacmid DNA was purified from 0.5 liter cultures using the Large-Construct or Maxi purification kits (Qiagen) according to the manufacturer's instructions and 2 μg of purified DNA was used for transfecting Sf-9 cells (0.9 × 106 cells/well) seeded in a six-well plate via a cationic liposome method (Campbell, 1995). Briefly, bacmid DNA was mixed with 200 μl of SF-900 II medium containing 10 μl of liposomes and incubated at 27°C for 30-45 minutes. After incubation, the DNA solution was increased to 1 ml with SF-900 II medium and overlaid onto freshly plated Sf-9 cells and transferred to 27°C and allowed to incubate for 4 h. After 4 h incubation, the transfection media was removed and the cells were replenished with 2 ml of fresh SF-900 II medium and returned to 27°C.
Virus growth curve
To determine the titers of virus supernatants, Sf-9 cells were transfected as described above and at the indicated time-points the supernatants were collected and the titers determined using a TCID50 end-point dilution assay that employed the GUS reporter gene as a marker for infection (O'Reilly et al., 1992). Cells seeded onto 96-well plates were incubated with transfected tissue culture supernatants for 7 days and then lysed with 100 μl of cell extraction buffer (50 mM sodium phosphate, 10 mM EDTA, 5 mM β-mercaptoethanol, 0.1% sodium n-lauroylsarcosine, 0.1% Triton X-100) containing 2mM 4-methylumbelliferyl β-D-glucuronide (4-MUG). After lysing the cells, the plates were incubated overnight at 37°C and then analyzed with UV light to determine wells that were positive for viral infection.
Sub-nuclear localization of DBP and LEF-3
Sf-9 cells (30 ml, ∼106 /ml) infected with AcMNPV at a MOI of 5 were collected at 10 and 20 h.p.i. The cells were washed in phosphate-buffered saline (PBS) and then in a hypotonic buffer A (20 mM Hepes-HCl (pH 7.5), 5 mM KCl, 1.5 mM MgCl2, 1 mM DTT) and a set of protease inhibitors (1 mM phenylmethylsulfonyl fluoride, 1 μM pepstatin, 5 μg/ml leupeptin, 5 μg/ml aprotinin, 2 μg/ml E64). After swelling for 10 min in the buffer A, the cells were disrupted in a Dounce homogenizer, and nuclei were precipitated by centrifugation for 10 min at 1,000 × g. The nuclei were washed for 10 min in the buffer A supplemented with 1% Nonidet P-40 and pelleted for 10 min at 2,000 × g. The detergent-washed nuclei were treated with DNase I (50 μg/ml) and RNase A (10 μg/ml) in buffer A for 1 h at room temperature and were pelleted for 10 min at 3,000 × g. The nuclease-treated nuclei were extracted for 20 min in a mixture containing equal volumes of buffer A and 4 M NaCl and were centrifuged for 10 min at 10,000 × g. The nuclear residue was treated again with nucleases: DNase I (20 μg/ml) and RNase A (10 μg/ml) in buffer A for 30 min at room temperature, and the nuclear matrix was precipitated by centrifugation for 10 min at 10,000 × g. The volume of each fraction was equal to 1.5 ml. Portions from the fractions obtained by fractionation were subjected to SDS-11% PAGE and Western blotting using polyclonal antibodies to DBP and LEF-3, and monoclonal antibody ADL101 to Drosophila lamin Dm0. The protein content of the fractions was estimated by scanning lanes in the gel stained by Coomassie brilliant blue.
Chromatin fractionation
Nuclei were purified from Sf-9 cells (∼106 /ml) infected with AcMNPV at a MOI of 5 at 20 h.p.i. The cells were washed in PBS and then incubated for 10 min on ice in 1.5 ml of buffer B (20 mM Tris-HCl (pH 8.0), 140 mM NaCl, 3 mM MgCl2, 1 mM DTT, 1% Nonidet P-40) and a set of protease inhibitors described above. The nuclei were centrifuged for 10 min at 1,000 × g, resuspended in 1.5 ml of buffer B, incubated for 10 min and centrifuged again. The purified nuclei were then subjected to chromatin fractionation as described previously with minor modifications (Busch, 1984). The nuclei were sequentially extracted with buffers containing: a) 20 mM Tris-HCl (pH 8.0), 75 mM NaCl, 25 mM EDTA, 1 mM DTT, 0.5 mM PMSF; b) 10 mM Tris-HCl (pH 8.0), 1 mM EDTA, 1 mM DTT, 0.5 mM PMSF; c), 20 mM Tris-HCl (pH 8.0), 0.35 M NaCl, 1 mM EDTA, 1 mM DTT, 0.5 mM PMSF; d) 20 mM Tris-HCl (pH 8.0), 0.6 M NaCl, 1 mM EDTA, 1 mM DTT, 0.5 mM PMSF; f) 0.2 M H2SO4. Extractions were performed twice. The volume of each fraction was equal to 1.5 ml (steps a to d) or 0.75 ml (f). Portions from the fractions obtained were subjected to SDS-12% PAGE and Western blotting using polyclonal antibodies to DBP and LEF-3. The protein content of the fractions was estimated by scanning lanes in the gel stained by Coomassie brilliant blue.
Quantitative DNA replication assay
To assess viral DNA replication, a quantitative real-time PCR (Q-PCR) assay was performed as previously described (Vanarsdall et al., 2005) with the following modifications. To prepare total DNA for analysis, Sf-9 cells transfected with bacmid DNA, harvested into 1 ml PBS, centrifuged at 5000 rpm for 3 min and the cell pellet placed at -80° C. DNA was extracted from cell pellets with 100 μl of cell lysis buffer (10 mM Tris-pH 8.0, 100 mM EDTA, 20 μg/ml RNAase A, 1% SDS) and incubated for 30 min at 37° C before adding 80 μg/ml of proteinase K and continuing incubation overnight at 65°C. DNA was extracted once with 100 μl of buffer-saturated phenol-chloroform and the aqueous layer was removed and diluted to a total volume of 300 μl with sterile water. Prior to PCR, 5μl of total DNA from each time-point was digested with 2U of DpnI restriction enzyme (Fermentas) overnight in 20 μl total reaction volume. This digested residual input bacmid DNA. The PCR reaction was performed with 2 μl of the digested DNA added to Platinum Taq PCR SuperMix UDG (Invitrogen) according to the manufacturer's instructions with primers (5′-CGTAGTGGTAGTAATCGCCGC-3′) and (5′-AGTCGAGTCGCGTCGCTTT-3′) used at 100nM final concentration and the dual-labeled probe (5′-FAM-AGTTCGAGGGCGACACCCTG-TAM-3′) at a final concentration of 100 nM. These primers anneal to genomic regions that span four DpnI restriction sites located in the gp41 open reading frame. Samples were analyzed on an ABI Prism 7500 sequence detection system under the following conditions: 50°C for 2 min, 95°C for 2 min, and 45 cycles of 95°C for 30 s, and 60°C for 30 s.
Slot-blot analysis
Sf-9 cells were transfected with bacmid DNA and at the designated time-point, cells from one well of a six-well plate were harvested in 1 ml of PBS and 100 μl of the cell suspension was removed and stored at −80° C. DNA was analyzed by boiling the cell samples at 100° for 10 min, quickly cooling on ice for 5 min, and then applying to Hybond N+ membranes (Amersham) using a hybri-slot manifold (BRL). The membrane was hybridized with [α 32P]dATP labeled bacmid DNA overnight at 55° C in 6 × SSC at ∼1 × 106 CPM/ml of buffer, washed twice with 2 × SSC at 65° C, and once with 0.2 × SCC at 70° C.
Field-inversion gel-electrophoresis
Sf-9 cells were transfected as described above and at selected time-points cells were harvested, washed once with PBS, and mixed with 0.8% low-melting agarose and poured into a plug mold. The agarose plugs were then treated with 10 mM Tris HCl (pH 8.0), 100mM EDTA, 1% N-lauroyl sarcosine, and 200 μg/ml proteinase K at 50°C overnight. After washing several times with 10 mM Tris-HCl (pH 8.0), samples were stored at 4°C until further use. To prepare DNA for electrophoresis, agarose pieces were digested with 30U of Eco81I and/or 10U of DpnI in 100 μl of reaction volume overnight. After digestion, the agarose pieces were inserted into the wells of a 1% slab gel made with 0.5 × TBE buffer (45 mM Tris-borate, pH 8.0, 1 mM EDTA). The DNA was separated by field inversion gel electrophoresis (FIGE) with a MJ Research PPI-200 programmable pulse inverter using program 4 (initial reverse time; 0.05 min, reverse increment; 0.01 min, initial forward time; 0.15 min, forward increment; 0.03 min, number of steps; 81, reverse increment; 0.001 min, forward increment; 0.003) and run at 8 V/cm for 17 hr at 4°C. MidRange PFG marker I (New England Biolabs) was used as DNA size markers. The DNA was transferred to a nylon membrane and hybridized with viral genomic DNA using the alkaline phosphate direct labeling system (Amersham) as described previously (Vanarsdall et al., 2004) or by 32P-labeled bacmid DNA.
Immunoelectron microscopy
To analyze nucleocapsid production, Sf-9 cells were transfected as stated above, harvested at 72 h.p.t., and prepared for immunoelectron microscopy as described previously (Russell and Rohrmann, 1990). A mouse monoclonal antibody to AcMNPV VP39 (a generous gift from Dr. Loy Volkman) was used as undiluted tissue culture supernatant. The goat anti-mouse IgG 10 nm gold secondary antibody was used at a dilution of 1:50. Images were obtained with a Phillips EM 300 electron microscope.
To investigate DBP localization with viral DNA, Sf-9 cells were infected with a parental AcGUS budded virus stock at an MOI of 5. At 12 h.p.i., the cell supernatant was removed and replaced with culture medium containing BrdU labeling reagent at a 1:100 dilution (Zymed Laboratories). Cell were harvested at 24 h.p.i. and processed as described above. Prior to antibody staining, grids were floated on 20 μl drops of 5 N HCl for 20 min and washed briefly with 10 mM Tris HCl (pH 7.4). Antigens were detected simultaneously with a primary antibody mixture containing a monoclonal antibody to BrdU (Oncogene) diluted 1:50 and a polyclonal antibody to baculovirus DBP (Okano et al., 1999) diluted 1:500. A secondary antibody mixture was prepared with a goat anti-mouse IgG conjugated to 20 nm gold and a goat anti-rabbit IgG conjugated to 10 nm gold. Both secondary antibodies were diluted to 1:50. Images were obtained with a Phillips EM 300 electron microscope.
Acknowledgments
This research was supported by National Institutes of Health Grant GM060404 (to G. F. R.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Braunagel SC, Parr R, Belyavskyi M, Summers MD. Autographa californica nucleopolyhedrovirus infection results in Sf9 cell cycle arrest at G2/M phase. Virology. 1998;244:195–211. doi: 10.1006/viro.1998.9097. [DOI] [PubMed] [Google Scholar]
- Busch H. Ubiquitination of proteins. Methods Enzymol. 1984;106:238–62. doi: 10.1016/0076-6879(84)06025-0. [DOI] [PubMed] [Google Scholar]
- Campbell MJ. Lipofection reagents prepared by a simple ethanol injection technique. Biotechniques. 1995;18:1027–32. [PubMed] [Google Scholar]
- Cunningham RP, Berger H. Mutations affecting genetic recombination in bacteriophage T4D. I. Pathway analysis. Virology. 1977;80:67–82. doi: 10.1016/0042-6822(77)90381-6. [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97:6640–5. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarino LA, Dong W, Xu B, Broussard DR, Davis RW, Jarvis DL. The baculovirus phosphoprotein pp31 is associated with the virogenic stroma. J Virol. 1992;66:7113–7120. doi: 10.1128/jvi.66.12.7113-7120.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harwood SH, Li L, Ho PS, Preston AK, Rohrmann GF. AcMNPV late expression factor-5 interacts with itself and contains a zinc ribbon domain that is required for maximal late transcription activity and is homologous to elongation factor TFIIS. Virology. 1998;250:118–134. doi: 10.1006/viro.1998.9334. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Yonesaki T. The characterization of a complex of three bacteriophage T4 recombination proteins, uvsX protein, uvsY protein, and gene 32 protein, on single-stranded DNA. J Biol Chem. 1991;266:4883–8. [PubMed] [Google Scholar]
- Ikeda M, Kobayashi M. Cell-cycle perturbation in Sf9 cells infected with Autographa californica nucleopolyhedrovirus. Virology. 1999;258(1):176–88. doi: 10.1006/viro.1999.9706. [DOI] [PubMed] [Google Scholar]
- Kool M, Ahrens C, Goldbach RW, Rohrmann GF, Vlak JM. Identification of genes involved in DNA replication of the Autographa californica baculovirus. Proc Natl Acad Sci USA. 1994;91:11212–11216. doi: 10.1073/pnas.91.23.11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin G, Blissard GW. Analysis of an Autographa californica nucleopolyhedrovirus lef-11 knockout: LEF-11 is essential for viral DNA replication. J Virol. 2002;76:2770–9. doi: 10.1128/JVI.76.6.2770-2779.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainz D, Quadt I, Knebel-Mörsdorf D. Nuclear IE2 structures are related to viral DNA replication sites during baculovirus infection. J Virol. 2002;76:5198–5207. doi: 10.1128/JVI.76.10.5198-5207.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melamede RJ, Wallace SS. Properties of the nonlethal recombinational repair x and y mutants of bacteriophage T4.II. DNA synthesis. J Virol. 1977;24:28–40. doi: 10.1128/jvi.24.1.28-40.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhailov VS, Mikhailova AL, Iwanaga M, Gomi S, Maeda S. Bombyx mori nucleopolyhedrovirus encodes a DNA -binding protein capable of destabilizing duplex DNA. J Virol. 1998;72:3107–3116. doi: 10.1128/jvi.72.4.3107-3116.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagamine T, Kawasaki Y, Matsumoto S. Induction of a subnuclear structure by the simultaneous expression of baculovirus proteins, IE1, LEF3, and P143 in the presence of hr. Virology. 2006;352:400–407. doi: 10.1016/j.virol.2006.04.034. [DOI] [PubMed] [Google Scholar]
- O'Reilly DR, Miller L, Luckow VA. Baculovirus Expression Vectors : A Laboratory Manual. xiii. W.H. Freeman; New York: 1992. p. 347. [Google Scholar]
- Okano K, Mikhailov VS, Maeda S. Colocalization of baculovirus IE-1 and two DNA-binding proteins, DBP and LEF-3, to viral replication factories. J Virol. 1999;73:110–119. doi: 10.1128/jvi.73.1.110-119.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano K, Vanarsdall AL, Rohrmann GF. A Baculovirus alkaline nuclease knock out construct produces fragmented DNA and aberrant capsids. Virology. 2007;359:46–54. doi: 10.1016/j.virol.2006.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano K, Vanarsdall AL, Mikhailov VS, Rohrmann GF. Conserved molecular systems of the Baculoviridae. Virology. 2006;344:77–87. doi: 10.1016/j.virol.2005.09.019. [DOI] [PubMed] [Google Scholar]
- Russell RL, Rohrmann GF. A baculovirus polyhedron envelope protein: immunogold localization in infected cells and mature polyhedra. Virology. 1990;174:177–84. doi: 10.1016/0042-6822(90)90066-z. [DOI] [PubMed] [Google Scholar]
- Todd JW, Passarelli AL, Miller LK. Eighteen baculovirus genes, including lef-11, p35, 39K, and p47, support late gene expression. J Virol. 1995;69:968–974. doi: 10.1128/jvi.69.2.968-974.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanarsdall AL, Okano K, Rohrmann GF. Characterization of a baculovirus with a deletion of vlf-1. Virology. 2004;326:191–201. doi: 10.1016/j.virol.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Vanarsdall AL, Okano K, Rohrmann GF. Characterization of the replication of a baculovirus mutant lacking the DNA polymerase gene. Virology. 2005;331:175–80. doi: 10.1016/j.virol.2004.10.024. [DOI] [PubMed] [Google Scholar]
- Vanarsdall AL, Okano K, Rohrmann GF. Characterization of the role of VLF-1 in baculovirus capsid structure and DNA processing. J Virol. 2006;80:1724–33. doi: 10.1128/JVI.80.4.1724-1733.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson ME, Miller LK. Changes in the nucleoprotein complexes of a baculovirus DNA during infection. Virology. 1986;151:315–328. doi: 10.1016/0042-6822(86)90052-8. [DOI] [PubMed] [Google Scholar]
- Young JC, MacKinnon EA, Faulkner P. The architecture of the virogenic stroma in isolated nuclei of Spodoptera frugiperda cells in vitro infected by Autographa californica nuclear polyhedrosis virus. J Struct Biol. 1993;110:141–153. [Google Scholar]