

Abstract
Background and purpose:
Cardioprotection against ischaemia by anaesthetic-induced preconditioning (APC) is well established. However, the mechanism underlying Ca2+ overload attenuation by APC is unknown. The effects of APC by isoflurane on the cardiac L-type Ca channel were investigated.
Experimental approach:
In a model of in vivo APC, Wistar rats were exposed to isoflurane (1.4%), delivered via a vaporizer in an enclosure, prior to thoracotomy. The Dahl S rats were similarly preconditioned to determine strain-dependent effects. Whole-cell patch clamp using cardiac ventricular myocytes was used to determine the L-type Ca2+ current (ICa,L) characteristics and calmodulin (CaM) levels were determined by Western blot analysis. Cytosolic Ca2+ levels were monitored using fluo-4-AM. Action potential (AP) simulations examined the effects of APC.
Key results:
In Wistar rats, APC significantly accelerated ICa,L inactivation kinetics. This was abolished when external Ca2+ was replaced with Ba2+, suggesting that Ca2+-dependent inactivation of ICa,L was modulated by APC. Expression levels of CaM, a determinant of ICa,L inactivation, were not affected. Attenuation of cytosolic Ca2+ accumulation following oxidative stress was observed in the APC group. Simulations showed that the accelerated inactivation of ICa,L resulted in a shortening of the AP duration. The Dahl S rat strain was resistant to APC and changes in ICa,L inactivation were not observed in cardiomyocytes prepared from these rats.
Conclusions and implications:
APC triggered persistent changes in the inactivation of cardiac L-type Ca channels. This can potentially lead to a reduction in Ca2+ influx and attenuation of Ca2+ overload during ischaemia/reperfusion.
Keywords: anaesthetic-induced preconditioning, cardioprotection, L-type calcium channel, volatile anaesthetics, isoflurane
Introduction
Ischemic preconditioning (IPC) confers cardioprotective effects whereby short, non-lethal ischemic episodes result in a significant reduction in infarct size following a subsequent prolonged ischaemia (Murry et al., 1986). Since IPC is invoked by a short, non-fatal ischemic period, its potential clinical application may be limited due to the difficulty in precisely timing the preconditioning ischemic stimulus. However, several pharmacological agents including volatile anaesthetics are known to mimic the cardioprotective effects of IPC (Cason et al., 1997; Kersten et al., 1997a). The efficacy of anaesthetic-induced preconditioning (APC) is similar to that of IPC, and mechanisms underlying its cardioprotective effects have been intensely investigated. The key components of the intracellular signal transduction pathways include G-proteins, protein kinase C (PKC)-δ and ε isoforms, protein tyrosine kinase, mitogen-activated protein kinase, reactive oxygen species, sarcolemmal and mitochondrial ATP-sensitive potassium channels (sarcKATP and mitoKATP channels respectively) (Kersten et al., 1997b; Tanaka et al., 2002; Zaugg et al., 2002; Ludwig et al., 2004; da Silva et al., 2004; Marinovic et al., 2006). Recent evidence suggests that the signalling pathways converge at the mitochondria and preserve their function, resulting in the cardioprotective effects afforded by preconditioning (Krolikowski et al., 2005; Riess et al., 2005; Ljubkovic et al., 2007). Nevertheless, key questions still remain unanswered on how the converging signalling cascades affect mitochondrial function and ultimately lead to cardioprotection.
In addition, whether there is a genetic component to APC has not been clearly established. Previous studies have reported species-dependent differences in response to myocardial ischaemia (Galinanes and Hearse, 1990). Furthermore, differential responses to resistance to myocardial ischaemia have been shown in different rat strains (Baker et al., 2000). Thus, a genetic component may also underlie the efficacy of cardioprotection by APC.
A physiologically beneficial outcome of APC that confers cardioprotection appears to be the attenuation of Ca2+ overload following ischaemia/reperfusion (Hoka et al., 1987; An et al., 2001; Varadarajan et al., 2002). However, the mechanism underlying this attenuation is unknown. A major source of myocardial Ca2+ entry is via the L-type Ca channel. Consequently, inhibition of the L-type Ca channel can result in reduced Ca2+ entry, and thus lead to diminished Ca2+ overload. On the other hand, Ca2+ entry via the L-type Ca channel may also contribute to preconditioning. A small increase in intracellular Ca2+ during IPC or Ca2+ preconditioning has been reported to trigger PKC-dependent pathways that contributed to the induction of cardioprotection (Miyawaki and Ashraf, 1997). This was abolished by blockade of Ca2+ influx via the reverse mode of the Na+/Ca2+ exchanger and the L-type Ca channel. Furthermore, atrial trabeculae extracted from patients taking L-type Ca channel blockers were resistant to cardioprotection by IPC (Cain et al., 1999). Though an increase in cytosolic Ca2+ by IPC would appear to counteract its cardioprotective effect, a rise in cytosolic Ca2+ during an initial episode of brief ischaemia can become attenuated in subsequent episodes of ischaemia (Smith et al., 1996). Thus, a transient ischaemic event can lead to a rapid adaptation of Ca2+ homeostasis during subsequent episodes.
Based on these previous reports, the L-type Ca channel may potentially play diverse but pivotal functional roles in protecting the myocardium during preconditioning. Ca2+ influx may contribute to the underlying cardioprotective signalling cascade, while its attenuation may contribute to a reduction in reperfusion injury. The consequence of a preconditioning stimulus on this channel has not been established. Furthermore, whether APC affects the molecular modulators of Ca channel function has not been investigated. For example, the effect of APC on CaM is unknown even though a major aspect of Ca2+-dependent inactivation is the involvement of CaM, through its association with the channel (Dick et al., 2008).
The purpose of this study was to investigate the effect of preconditioning on the cardiac L-type Ca channel current, ICa,L. We hypothesized that preconditioning triggers persistent changes in the biophysical profile of the L-type Ca channel and a physiologically relevant model of in vivo APC with isoflurane was utilized to test this hypothesis. Our results showed that APC triggered a persistent change in the inactivation profile of the L-type Ca channel, with a specific acceleration of Ca2+-dependent inactivation.
Methods
In vivo anaesthetic-induced preconditioning
This study was approved by the Institutional Animal Care and Use Committee of the Medical College of Wisconsin. Adult male Wistar and Dahl S rats (Harlan, Indianapolis, IN) were used for this study. Two strains of rats were chosen to determine whether the efficacy of APC was dependent on strain, which would be indicative of a genetic component. The rats were divided into two groups, non-APC (control) and APC (in vivo preconditioning). For in vivo APC, rats were exposed to 1.4% (1.0 minimum alveolar concentration) isoflurane (Baxter, Deerfield, IL) delivered via a vaporizer in an enclosure for 30 min followed by a 30 min recovery period prior to thoracotomy (Stadnicka et al., 2006). In the non-APC group, rats were not exposed to isoflurane prior to thoracotomy. This was a physiologically relevant model of preconditioning by APC. Since the rats were exposed to isoflurane in an enclosure, the animals were subjected to all the triggering events induced by the volatile anaesthetic and not only to those confined to the heart. This included any changes in the systemic input and contributions from the vasculature and the endothelium. This contrasts with the method of preconditioning using an isolated heart on a Langendorff apparatus where the preconditioning stimulus is applied at the level of the organ, in the absence of systemic input.
Isolation of cardiac ventricular myocytes
Cardiac ventricular myocytes were enzymatically isolated from adult male Wistar and Dahl S rats weighing 180–280 g as previously described (Aizawa et al., 2004). Briefly, rats were anaesthetized with thiobutabarbital (Inactin, 200 mg·kg−1, i.p.) and heparinized with 1000 U heparin (Baxter). Under anaesthesia, the heart was quickly removed and mounted on a Langendorff apparatus followed by retrograde perfusion through the aorta with a solution containing Joklik medium (Sigma-Aldrich, St. Louis, MO) and heparin at pH 7.23. After allowing for the blood to clear from the heart (for approximately 1–2 min), the perfusate was replaced with an enzyme solution containing Joklik medium, collagenase (Type II; Invitrogen, Carlsberg, CA), protease (Type XIV) and bovine serum albumin (Serologicals Proteins, Kankakee, IL) at pH 7.23. The solutions were gassed with 95% O2 and 5% CO2, and the temperature was maintained at 37°C. Following 15–20 min of perfusion with the enzyme solution, the ventricles were cut from the heart, minced and incubated in a shaking bath with the enzyme solution for 5–8 min. The resulting myocyte suspension was then filtered, centrifuged and stored in a modified Tyrode solution at room temperature. The myocytes were used for experiments within 10 h following the isolation procedure.
Solutions
The isolated cardiomyocytes were stored in a modified Tyrode solution that contained (in mmol·L−1): 132 NaCl, 4.8 KCl, 1 CaCl2, 1.2 MgCl2, 10 HEPES, 5 dextrose, and pH was adjusted to 7.4 with NaOH. For the recording of whole-cell ICa,L, the pipette solution contained (in mmol·L−1): 110 CsCl (substitute for potassium), 1 CaCl2, 1 MgCl2, 11 EGTA, 10 HEPES, 5 Mg-ATP, at pH 7.3 adjusted with CsOH. To isolate for ICa,L, the bath (external) solution contained (in mmol·L−1): 132 N-methyl-D-glucamine (substitute for sodium), 4.8 CsCl, 2 CaCl2, 2 MgCl2, 10 HEPES, 5 dextrose and 5 mmol·L−1 of 4-aminopyridine (transient outward potassium current antagonist), at pH 7.4 adjusted with HCl. For the investigation of the voltage-dependent inactivation and recovery from slow inactivation, CaCl2 was replaced with BaCl2, so Ba2+ was the charge carrier to remove Ca2+-dependent inactivation. All chemicals used in this study were obtained from Sigma-Aldrich (St. Louis, MO), unless otherwise noted.
Oxidative stress
A cell survival study was conducted to validate the in vivo APC model by comparing the tolerance against oxidative stress between the non-APC and APC myocytes (Marinovic et al., 2006). The cell suspension was transferred to a chamber on an inverted microscope (Eclipse TE2000-U; Nikon, Tokyo, Japan) and the myocytes were allowed to settle for 10 min, after which they eventually attached to the glass coverslip. They were then exposed to 0.4% Trypan blue solution for 2 min followed by washout with glucose-free Tyrode solution. Cells that were rod-shaped and not stained were considered to be viable and this number was counted. Following a 5 min perfusion with the glucose-free Tyrode solution, the myocytes were subjected to oxidative stress by perfusion with 200 µmol·L−1 H2O2 and 100 µmol·L−1 FeSO4 for 17 min. Subsequently, the myocytes were reperfused with glucose-free Tyrode solution for another 20 min. The myocytes were exposed to Trypan blue again and the living myocytes were counted to determine the percentage of cell death. For each group, 3–4 rats were utilized, and 1–3 experiments were conducted from myocytes isolated from each animal. The percentage of cell death was calculated from at least 200 myocytes for each experiment. For the survival study, the n number denotes the number of experiments that were conducted.
Electrophysiology
The whole-cell configuration of the patch clamp technique was used to record ICa,L. Patch pipettes were pulled from borosilicate glass capillary tubes (Garner Glass, Claremont, CA) using a horizontal micropipette puller (P-97; Sutter Instrument, Novato, CA), and heat polished using a microforge (MF-830; Narishige, Tokyo, Japan). The pipette resistances were in the range of 2–5 MΩ. ICa,L was monitored using a patch clamp amplifier (Axopatch 200B; Molecular Devices, Sunnyvale, CA) interfaced with a digitizer (Digidata 1322A; Molecular Devices) to a computer. Data acquisition and analysis were conducted using pClamp 9.2 (Molecular Devices), and additional analyses were performed using Origin 7 (OriginLab, Northampton, MA). All the experiments were performed under room temperature. For the patch clamp studies, 38 rats were utilized (19 each for the non-APC and APC groups). The n number denotes the number of myocytes from which the current recordings were obtained.
L-type Ca2+ current measurement
ICa,L was elicited by 400 ms duration test pulses from a holding potential of −80 to +50 mV in 10 mV increments at a rate of 0.33 Hz. A conditioning pulse (to −50 mV; 50 ms) was used to inactivate any residual cardiac sodium current before each test pulse. ICa,L was normalized to cell capacitance and the resultant current density was plotted against the test potentials to yield the current-voltage relationship.
Steady-state activation curve
The conductance was calculated with the following equation, G = I/(Vm − Vrev), where I is the current amplitude, Vm is the test potential and Vrev is the reversal potential. The corresponding steady-state activation curves were obtained by normalizing the conductance to the peak conductance. The normalized conductances (G/Gmax) were then plotted against the test potentials and fitted with the Boltzmann function,
where V1/2 is the voltage at half-maximal conductance and k is the slope factor.
Steady-state inactivation curve
The steady-state inactivation curves were obtained using a standard protocol, in which 500 ms conditioning pulses from a holding potential of −80 mV to +20 mV in 10 mV increments were followed by a 600 ms test pulse to 0 mV. Current amplitude was normalized to the peak current (I/Imax), and plotted against each conditioning potential. The steady-state inactivation curves were also fitted with the Boltzmann function,
to obtain V1/2, the voltage at which half the channels are available for opening, and k, the slope factor.
Recovery from fast and slow inactivation
A standard two-pulse protocol was used to monitor the recovery of ICa,L from fast inactivation. A 500 ms conditioning pulse to 0 mV was followed by a 600 ms test pulse to 0 mV in varying intervals from 0 to 550 ms in 50 ms increments. The holding potential was set at −80 mV. Current amplitude elicited by the test pulses were normalized to the peak current elicited by the conditioning pulse. The normalized currents were plotted against each time interval, and fitted with a single exponential function that yielded the time constant for recovery from fast inactivation.
To investigate the recovery from slow inactivation, Ba2+ was used as the charge carrier. The Ba2+ current, IBa, was elicited using a two-pulse protocol, in which a 5 s conditioning pulse to 0 mV was followed by a 50 ms test pulse to 0 mV in varying intervals from 1 to 15 s in 1 s increments from a holding potential of −80 mV. Current amplitude obtained during the test pulse was normalized to the peak current obtained during the conditioning pulse and plotted against the varying time intervals. The resultant plot was then fitted with a double exponential function to obtain the fast and slow time constants (τfast and τslow respectively) for the recovery from slow inactivation.
Ca2+ current inactivation kinetics and transmembrane charge transfer
ICa,L inactivation kinetics were quantified by fitting the current traces with a double exponential function of the form
where τfast and τslow are the fast and slow time constants, respectively, Afast and Aslow are the respective amplitudes of the time constants, and C is the final amplitude following the inactivation. The fraction of channels that gated either fast or slow was quantified by the respective amplitudes with the following equations:
In experiments where the voltage-dependent inactivation of ICa,L was investigated, Ba2+ substituted for Ca2+ as the charge carrier in the external solution to remove Ca2+-dependent inactivation and the Ba2+ current (IBa) was recorded.
To correlate the changes in Ca2+ influx to the changes in ICa,L inactivation kinetics, the transmembrane charge transfer was determined. This was calculated by integrating the area of ICa,L traces after the capacitive transients to the time point of 100 ms. This temporal duration was chosen to mimic the time course of a rat action potential at room temperature (Linz and Meyer, 2000).
Western blotting
Western blot analysis was carried out to determine the cytosolic levels of calmodulin (CaM). Ventricles were excised from both non-APC and APC hearts, and homogenized on ice in a solution that contained (in mmol·L−1): 20 Tris-HCl, 150 NaCl, 1 EDTA, 1 EGTA, 1% Triton X-100, and cocktails of protease inhibitors (complete mini; Roche, Indianapolis, IN) and phosphatase inhibitors (cocktail set II; Calbiochem, San Diego, CA), pH 7.4. The homogenate was centrifuged for 10 min at 10 000× g (4°C), and the resulting supernatant was centrifuged for 1 h at 100 000× g (4°C) to separate the cytosolic and membrane fractions. The total protein concentration was determined by a modified Lowry method (Bio-Rad, Hercules, CA). Equal amounts of proteins (100 µg) were separated on a 15% polyacrylamide gel and transferred to the nitrocellulose membrane (Bio-Rad). The membrane was blocked with 5% fat-free milk and incubated overnight at 4°C with a rabbit monoclonal CaM antibody (Epitomics, Burlingame, CA) at 1:1000 dilution. After being washed, the membrane was incubated for 1 h at room temperature with a horseradish peroxidase-conjugated anti-rabbit immunoglobulin G (Bio-Rad) at 1:10 000 dilution. Chemiluminescence was detected after incubation with Luminol/Enhancer solution (Pierce, Rockford, IL) using radiographic film. To verify equal protein loading, the membrane was re-probed with a mouse monoclonal anti-β-actin antibody at 1:1000 dilution followed by incubation with a horseradish peroxidase-conjugated anti-mouse immunoglobulin G (Bio-Rad) at 1:5000 dilution and developed on film. CaM/β-actin ratio was quantified using the ImageJ software (National Institutes of Health, Bethesda, MD).
Cytosolic Ca2+ measurement
A Ca2+-sensitive indicator, fluo-4-AM (2 µmol·L−1; Invitrogen, Carlsbad, CA) that preferentially loaded into the cytosol was used to assess cytosolic Ca2+. Images were obtained with a confocal microscope (Eclipse TE2000-U) equipped with a 40×/1.3 oil-immersion objective. Excitation at 488 nm was achieved with an argon laser. After 20 min of dye loading, followed by a 5 min dye washout, cells were exposed to an oxidative stress protocol identical to that used in the cell survival study described above. Images were taken every 5 min. Data were analysed with the Metamorph 6.1 software (Universal Imaging, West Chester, PA).
Simulation of the action potential
Simulations of the cardiac AP were conducted using Cellular Open Resource (COR), a modelling environment developed by the Oxford Cardiac Electrophysiology Group in the Department of Physiology at the University of Oxford (Great Britain) (Garny et al., 2003). Specifically, simulations were run using the Fox-McHarg-Gilmour (FMG) model on COR (Fox et al., 2002). This model was chosen for its ease of use in determining the effect of changes in the L-type Ca channel's Ca2+-dependent inactivation on the AP profile. To simulate an AP with the time course that mimicked that of a rat cardiac AP at room temperature, the FMG model was modified by adjusting the amplitudes of the following potassium (K) currents: Ito (transient outward K current), IKr (rapidly activating delayed rectifier K current), IKp (plateau K current) and IK1 (inward rectifier K current). Contributions from IKs (slowly activating delayed rectifier K current) were eliminated. In addition, since the original FMG model included an enhanced Ca2+-dependent inactivation of the L-type Ca channel, modifications were made to incorporate the ‘basal’ rate of Ca2+-dependent inactivation as described by the phase-2 Luo-Rudy model (Luo and Rudy, 1994). Specifically, the Ca2+-dependent inactivation gate at steady state was defined by
where [Ca2+]i is the intracellular Ca2+ concentration, and KmfCa is the half-saturation constant. For the initial conditions, KmfCa was set at 0.60 µmol·L−1 (Luo and Rudy, 1994).
Statistical analysis
Data are shown as mean ± SEM. One-way ANOVA followed by Scheffe's test was used to compare the data among the three groups in the cell survival study. The unpaired Student's t-test was used for comparisons between the non-APC and APC groups in the patch clamp studies and Ca2+ measurements. Statistical analyses were performed using the Origin 7 software and P < 0.05 was considered significantly different.
Results
Validation of the in vivo APC model: protection against oxidative stress
To validate the effectiveness of the in vivo APC model, a cell survival study was conducted to determine the efficacy of in vivo APC in inducing tolerance against oxidative stress injury on isolated myocytes obtained from the Wistar rats. Myocytes from the non-APC and APC groups were subjected to oxidative stress as described in Methods. As a time control, myocytes were perfused for 42 min with glucose-free Tyrode solution. As shown in Figure 1, in the time control group, the per cent cell death was low after this perfusion. In the non-APC group, oxidative stress resulted in a markedly increased proportion of dead cells and exposure to the APC protocol significantly attenuated this high percentage of cell death in myocytes following oxidative stress. These results using the Wistar rats confirmed that in vivo APC was very effective in decreasing cell death following oxidative stress. This cardioprotection persisted past the cell isolation procedure.
Figure 1.
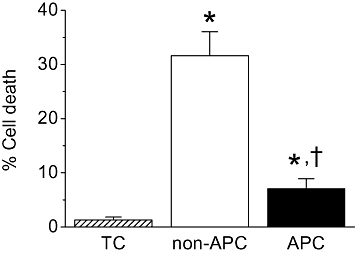
Effects of in vivo APC on cardiomyocyte survival after oxidative stress. Myocytes were isolated from Wistar rat hearts. In the non-APC group, oxidative stress significantly increased the percentage of cell death compared with the time control (TC) group. This effect of oxidative stress was significantly attenuated by in vivo APC. *P < 0.05 vs. TC, †P < 0.05 vs. non-APC. n = 7–10 per group.
Effects of in vivo APC on ICa,L steady-state activation, inactivation and recovery from inactivation
To characterize the effects of APC on ICa,L, whole-cell current recordings were made from isolated myocytes obtained from Wistar rats that had been previously preconditioned by isoflurane via the in vivo APC protocol. The ICa,L current-voltage relationship, the steady-state activation and inactivation curves, and the recovery from fast and slow inactivation were determined. Figure 2A depicts representative ICa,L traces recorded from ventricular myocytes isolated from a control (non-APC) rat heart. The current-voltage relationships obtained from the non-APC and APC groups suggested that the voltage dependence of activation was unaltered by APC (Figure 2B). The peak ICa,L densities were also not significantly different between the two groups with −7.6 ± 0.4 pA·pF–1 in the non-APC and −8.8 ± 0.5 pA·pF–1 in the APC groups (n = 21–22 per group). The absence of a significant shift in the voltage dependence of activation was confirmed by constructing the steady-state activation curve (Figure 2C, Table 1). In addition, the steady-state inactivation curves obtained from the non-APC and APC groups were not significantly different (Figure 2D, Table 1). Moreover, recovery from fast inactivation of ICa,L was not affected by in vivo APC (Figure 2E, Table 1). As ICa,L also exhibits slow inactivation, the effect of APC on recovery from slow inactivation was determined. As was the case for recovery from fast inactivation, APC had no significant effect on recovery from slow inactivation (Figure 2F, Table 1).
Figure 2.
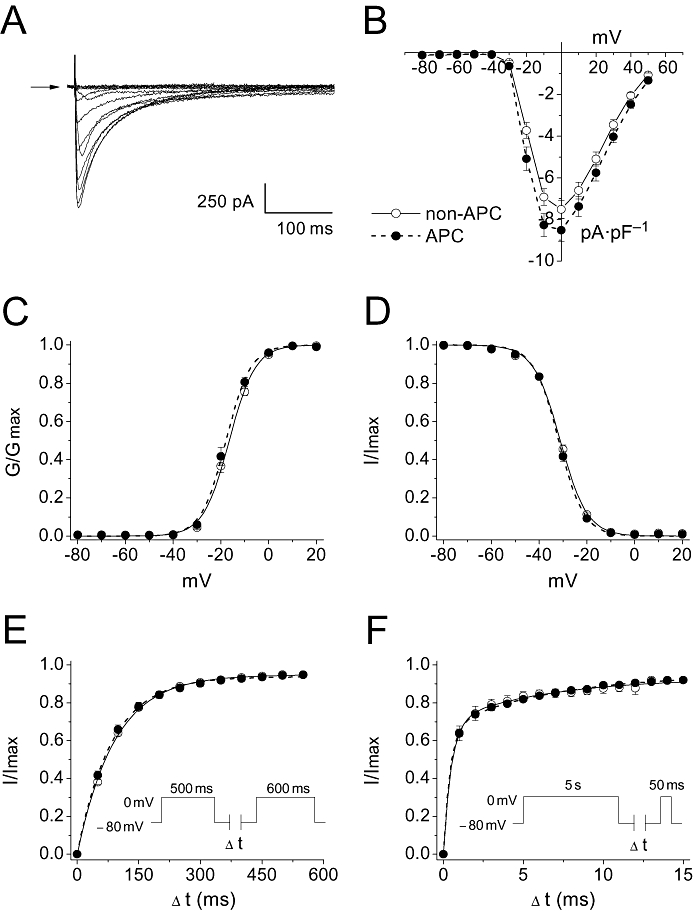
Effects of in vivo APC on the biophysical profile of ICa,L obtained in myocytes from non-APC and APC hearts. (A) Representative whole-cell ICa,L recorded from an isolated cardiomyocyte obtained from a non-APC heart. Arrow indicates zero current level. (B) Current-voltage relationship. The current-voltage relationship for the non-APC and APC groups are shown. No significant differences were observed (n = 21–22 per group). (C) Steady-state activation curves. Data were fitted with the Boltzmann function. No significant shifts in the steady-state activation curves were observed between the two groups (n = 21–22 per group). (D) Steady-state inactivation curves. Similarly to (C), data were fitted with the Boltzmann function. No significant shifts in the steady-state inactivation curves were observed between the two groups (n = 22 per group). (E) Recovery from fast inactivation. The inset depicts the standard two-pulse protocol that was utilized as described in Methods (n = 19 per group). (F) Recovery from slow inactivation. The inset depicts the two pulse protocol that was utilized as described in Methods (n = 4 per group). There were no significant differences in the recoveries from fast and slow inactivations between the non-APC and APC groups.
Table 1.
The effects of in vivo APC on the steady-state activation and inactivation curves, and recovery from fast and slow inactivation of ICa,L
| non-APC | APC | N per group | |
|---|---|---|---|
| Steady-state activation | |||
| V1/2 (mV) | −16.4 ± 0.6 | −17.8 ± 0.9 | 21–22 |
| k | 4.9 ± 0.2 | 4.6 ± 0.2 | |
| Steady-state inactivation | |||
| V1/2 (mV) | −31.0 ± 0.5 | −31.7 ± 0.5 | 22 |
| k | 5.1 ± 0.1 | 4.8 ± 0.1 | |
| Recovery from fast inactivation | |||
| τ (ms) | 91.8 ± 3.4 | 86.7 ± 4.0 | 19 |
| Recovery from slow inactivation | |||
| τfast (s) | 0.47 ± 0.03 | 0.42 ± 0.04 | 4 |
| τslow (s) | 6.4 ± 1.8 | 6.5 ± 1.1 | |
Data are presented as mean ± SEM.
Effects of in vivo APC on ICa,L inactivation kinetics and transmembrane charge transfer
Though in vivo APC did not result in any significant persistent changes in the steady-state activation and inactivation curves, and in the recovery from fast and slow inactivation, it did significantly accelerate ICa,L inactivation kinetics, as demonstrated in Figure 3A. Specifically, τfast in the voltage range of −20 to +10 mV, and τslow in the voltage range from −20 to +40 mV were significantly decreased compared with the non-APC group (n = 21–22 per group; Figure 3B). Additional analysis of ICa,L inactivation, fitted with the double exponential function (see Methods), revealed that APC induced a greater fraction of channels to favour fast inactivation as determined by the relative fraction of channels that gated fast (Figure 3c). This was accompanied by a corresponding decrease in the relative fraction of channels that gated slow (data not shown). These results were not affected by the time interval between cell isolation and the electrophysiological recordings, and persisted during the 10 h period the myocytes were used.
Figure 3.
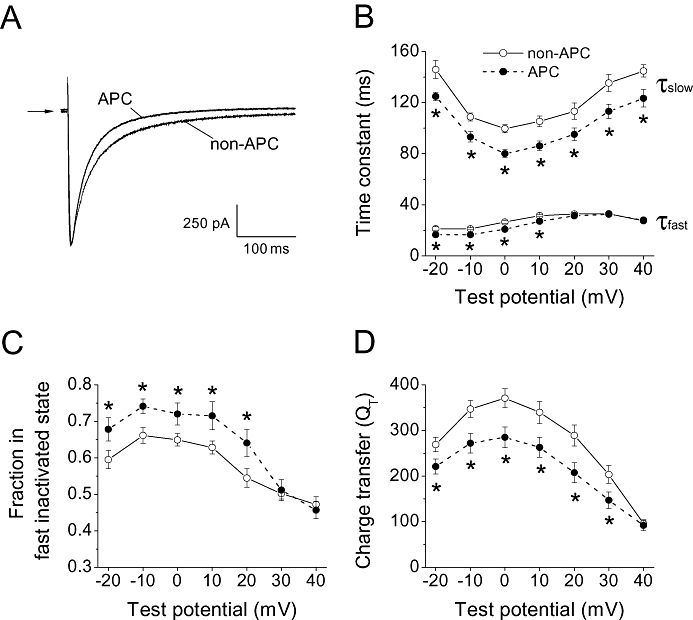
Effects of in vivo APC on voltage- and Ca2+-dependent inactivation of the cardiac L-type Ca channel. (A) Sample whole-cell ICa,L traces recorded from myocytes in the non-APC and APC groups monitored at 0 mV from a holding potential of −80 mV are shown. The current traces were superimposed. Arrow indicates zero current level. (B) The fast and slow time constants of the inactivation kinetics of ICa,L in myocytes from the non-APC and APC groups are plotted against test potentials. Compared with the non-APC group, APC significantly decreased τfast in the range from −20 to +10 mV and τslow in the range from −20 to +40 mV. (C) The fraction of channels that ‘fast inactivated’ was determined from the double exponential fit to the inactivating ICa,L traces. In the APC group, the fraction in the fast inactivated state was significantly increased compared to the non-APC group. (D) Changes in ICa,L inactivation was correlated to changes in the total influx of charge by integrating the area of ICa,L traces during a 100 ms period. The charge transfer was significantly decreased by APC in the range from −20 to +30 mV. *P < 0.05 vs. non-APC. n = 21–22 per group.
In order to quantify the change in ionic influx due to the acceleration of ICa,L inactivation, the transmembrane charge transfer was determined. To correlate Ca2+ influx during a rat ventricular AP, the charge transfer was calculated during 100 ms. As summarized in Figure 3D, a significant decrease in charge transfer resulted from the APC-triggered acceleration of ICa,L inactivation.
As the inactivation of ICa,L consists of voltage- and Ca2+-dependent components, experiments were conducted to determine which of the components, if not both, were affected by APC. The result above showed an increase in the fraction of channels in the fast inactivated state, which suggested that the effect of APC was likely to be on the Ca2+-dependent inactivation of the channel. Consequently, experiments were conducted to test this hypothesis. Ba2+ was utilized as the charge carrier to remove Ca2+-dependent inactivation. Under this condition, the inactivation of ICa,L was due to the voltage-dependent mechanism. As shown in Figure 4A, IBa exhibited markedly slower inactivation kinetics as expected. In contrast to its effect on ICa,L, APC did not affect IBa inactivation kinetics (Figure 4B). Under our recording conditions, inactivation of IBa was best fitted by a double exponential function (Findlay, 2002). This indicated that APC accelerated the Ca2+-dependent inactivation of ICa,L, but had no effect on voltage-dependent inactivation.
Figure 4.
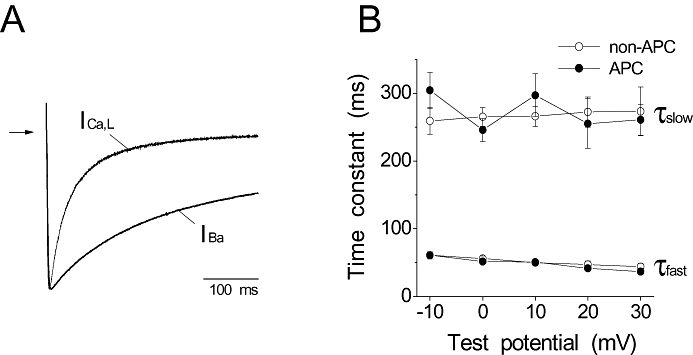
Effects of in vivo APC on the voltage-dependent inactivation of the cardiac L-type Ca channel. (A) Representative traces of ICa,L and IBa recorded at 0 mV from a holding potential of −80 mV using Ca2+ and Ba2+ as charge carriers, respectively, are shown. The currents were scaled and superimposed. Arrow indicates zero current level. Ba2+ as the charge carrier removed Ca2+-dependent inactivation of the L-type Ca channel. (B) The fast and slow time constants of the inactivation kinetics of IBa in the non-APC and APC groups are plotted against test potentials. No significant differences were observed in τfast and τslow between the non-APC and APC groups (n = 10 per group).
Cytosolic CaM levels
A major aspect of Ca2+-dependent inactivation is the involvement of CaM which associates with the channel. To investigate the mechanism of APC-induced changes in ICa,L inactivation kinetics, changes in the expression of cytosolic CaM were determined by Western blot analysis. CaM expression levels were compared between the non-APC and APC groups (n = 6 hearts per group). As shown in Figure 5, the cytosolic CaM expression levels, normalized to β-actin, were unchanged by APC. This suggested that APC did not induce a change in the expression levels of CaM.
Figure 5.
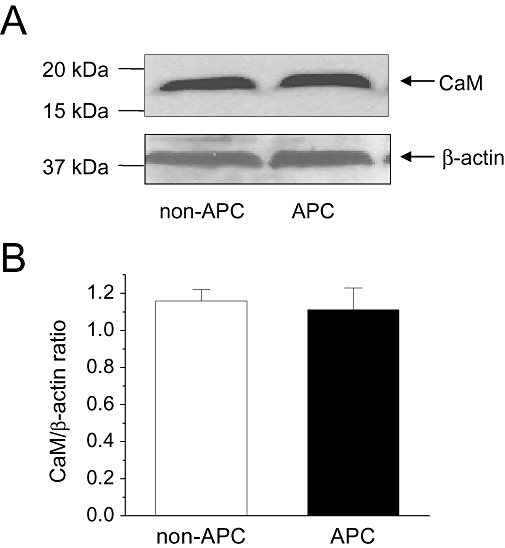
Cytosolic calmodulin (CaM) levels. Effects of in vivo APC on cytosolic CaM levels were determined by Western blot. (A) Representative results from one heart each from the non-APC and APC groups are shown. β-actin was used as the loading control. (B) Summary of the CaM/β-actin ratio in the non-APC and APC groups are shown. No significant differences were observed between two groups (n = 6 hearts per group).
Attenuation of cytosolic Ca2+ accumulation
Based on our results, in vivo APC significantly improved cell survival and also triggered the acceleration of ICa,L inactivation kinetics that could lead to diminished Ca2+ influx. However, the question of whether APC did indeed result in the attenuation of cytosolic Ca2+ accumulation in our model still remained. This was addressed by monitoring cytosolic Ca2+ with confocal microscopy. Using an identical oxidative stress protocol as in the cell survival study described above, cytosolic Ca2+ was monitored by loading isolated myocytes with the fluo-4-AM fluorescent indicator and monitored over time. Rats in the in vivo APC group were exposed to isoflurane prior to thoracotomy as described in Methods. As shown in Figure 6, cytosolic Ca2+ accumulation was observed in both the non-APC and APC groups following oxidative stress. However, in myocytes isolated from the APC group, this accumulation of Ca2+ was significantly attenuated as compared with that of the non-APC group. These results demonstrated that myocytes from the APC group were better protected against stress-induced Ca2+ accumulation.
Figure 6.
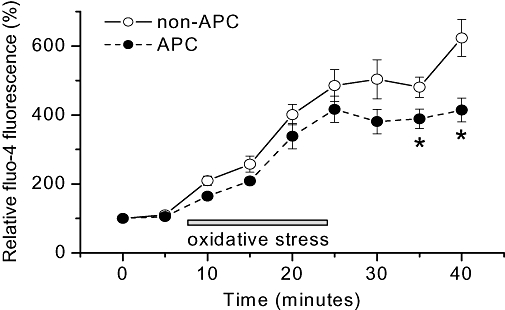
Cytosolic Ca2+ accumulation under stress conditions. Isolated cardiomyocytes were loaded with the cytosolic Ca2+ indicator, fluo-4-AM, and exposed to oxidative stress, followed by perfusion with glucose-free Tyrode solution. Fluo-4 fluorescence was monitored over time. Myocytes from both the non-APC and APC groups showed increases in cytosolic Ca2+ following stress as indicated by the increases in fluo-4 fluorescence. However, the increase in fluo-4 fluorescence was significantly reduced in the APC-treated myocytes. *P < 0.05 vs. non-APC. n = 10–12 per group.
Action potential simulation
To characterize the effects of accelerated ICa,L inactivation kinetics on the AP, simulations were conducted. Simulations were run using a modified FMG model of the cardiac AP as described in Methods. The resultant simulation is depicted in Figure 7A. As expected, the ICa,L activation and inactivation kinetics during an AP with dynamic changes in the membrane potential differed from those observed during a rectangular test pulse. In the modified FMG model, based on our experimental results, the descending phase of the ICa,L inactivation was accelerated by 23% in the APC-simulated ICa,L compared with the control-simulated ICa,L. This change in ICa,L inactivation kinetics resulted in a limited shortening of the APD, as shown in Figure 7B. In the control-simulated AP, the APD50 and APD90 were 83.0 and 98.1 ms, respectively, while in the APC-simulated AP, they were 77.8 and 92.9 ms.
Figure 7.
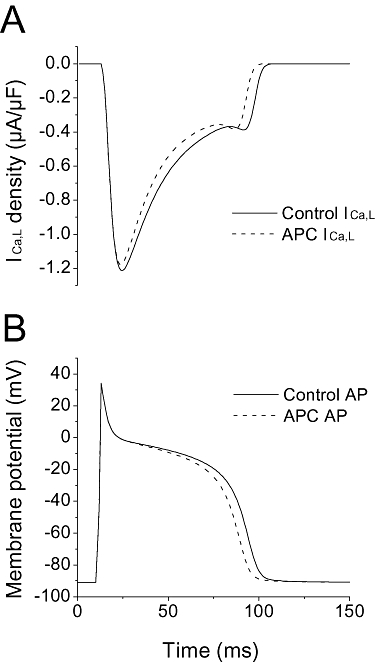
Simulation of the action potential. (A) Using a modified FMG model as described in Methods, inactivation kinetics were accelerated by 23% in the simulated APC ICa,L compared with the Control ICa,L. (B) Simulated changes in the AP (control vs. APC) are shown as the corresponding consequence of the accelerated ICa,L inactivation. The APD50 and APD90 were 83.0 and 98.1 ms, respectively, in the control-simulated AP, and 77.8 and 92.9 ms in the APC-simulated AP.
Although changes in CaM levels did not appear to account for the accelerated inactivation kinetics based on our Western blot analysis, AP simulations were run to test whether changes in Ca2+ sensitivity of the L-type Ca channel can lead to the observed changes in inactivation. To accommodate this change in Ca2+ sensitivity, the half-saturation constant for Ca2+ in the modified FMG model was adjusted (see Methods). We found that arbitrarily decreasing the half-saturation constant by approximately 28%, from 0.60 to 0.43 µmol·L−1, accelerated the inactivation of ICa,L by only 5% (Figure 8).
Figure 8.
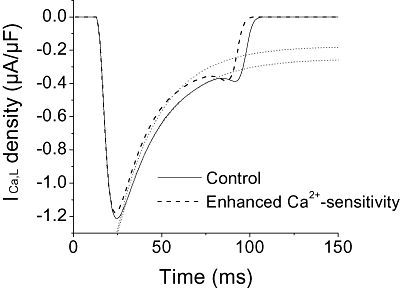
Effect of Ca2+ sensitivity on ICa,L inactivation. The effect of altering the sensitivity of L-type Ca channels to intracellular Ca2+ on ICa,L inactivation was simulated using the modified FMG model. Ca2+ sensitivity was enhanced by 28% (dashed lines) relative to the control. The exponential fits are depicted by the dotted lines. This enhancement of Ca2+ sensitivity induced only 5% change in the acceleration of the ICa,L inactivation.
Rat strain-dependent effects
Previous studies have reported on rat strain-dependent differences in resistance to myocardial ischaemia, indicative of a genetic component to cardioprotection (Baker et al., 2000). However, rat strain-dependent differences for APC have not been established. We conducted survival studies on cardiomyocytes from the Dahl S strain of rats and found that they showed resistance to the cardioprotective effects of in vivo APC (n = 5–9 per group; Figure 9a). This contrasted the effects of APC on the Wistar rats (Figure 1). Subsequently, to confirm that the observed changes in ICa,L inactivation were correlated with APC, we profiled the ICa,L inactivation in myocytes isolated from the Dahl S rats. As shown in Figure 9 (panels B and C), APC failed to alter ICa,L inactivation kinetics in myocytes obtained from the Dahl S rats (n = 9 per group). Thus, in a rat strain that is resistant to APC, changes in ICa,L inactivation were not evident.
Figure 9.
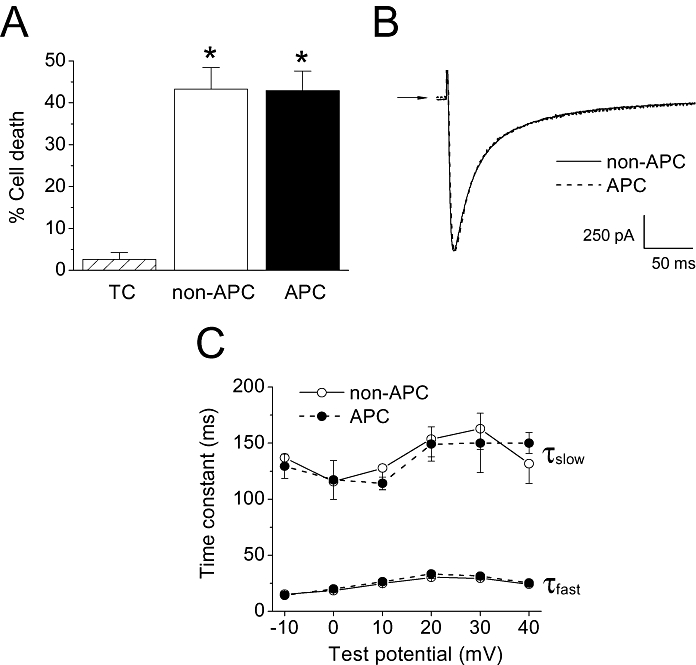
Effects of in vivo APC on the Dahl S strain of rats. (A) Cell survival study. Oxidative stress significantly increased the percentage of cell death in the non-APC group compared with the time control (TC) group. In the APC group, cell survival following oxidative stress was not significantly different from the APC group, indicating resistance to cardioprotection in the Dahl S rats. *P < 0.05 vs. TC. n = 5–9 per group. (B) Representative whole-cell ICa,L traces from Dahl S myocytes in the non-APC and APC groups. Currents were recorded at a test potential of 0 mV from a holding potential of −80 mV. The traces shown were superimposed. Arrow indicates zero current level. (C) ICa,L inactivation kinetics. The fast and slow time constants of current inactivation in myocytes from the non-APC and APC groups showed no significant differences in the Dahl S rats (n = 9 per group).
Discussion and conclusions
In the present study, we found that in vivo APC by isoflurane accelerated ICa,L inactivation kinetics, while all other measured biophysical characteristics of the L-type Ca channel remained unchanged. Although the acute inhibitory effects of volatile anaesthetics on the cardiac L-type Ca channel have been previously reported (Bosnjak et al., 1991; Pancrazio, 1996; Camara et al., 2001; Suzuki et al., 2002), this is the first report on an APC-triggered persistent change in a biophysical property of the channel. The voltage-dependent inactivation of the channel was not affected by in vivo APC, suggesting that the induced change in inactivation kinetics was due to the effects of APC on Ca2+-dependent inactivation. In addition, these observed changes in ICa,L inactivation in the Wistar rats were not evident in the Dahl S rats that were resistant to APC. Thus, the changes in ICa,L inactivation observed in the Wistar rats are a consequence of APC.
The mechanism underlying this observation is unknown. Based on the biophysical analysis, APC induced the L-type Ca channels to favour the fast inactivated state. Key components of Ca2+-dependent inactivation of the L-type Ca channel are modulated by CaM, whereby binding of Ca2+ to CaM that is bound to the carboxy-terminal tail of the α-subunit of the channel is a likely necessary step (Peterson et al., 1999; Van Petegem et al., 2005; Zhou et al., 2005). Using a general approach, we hypothesized that APC triggered changes in the CaM levels that can be linked to changes in the L-type Ca channel inactivation. Our results showed that APC did not induce changes in cytosolic CaM levels. Consequently, the accelerated inactivation of ICa,L was not due to changes in the expression levels of CaM. However, this does not exclude potential changes in the functional role of CaM in the inactivation of ICa,L. For example, changes in the Ca2+ binding to the C-lobe of CaM, purported to be a required step for Ca2+-dependent inactivation (Peterson et al., 1999; Alseikhan et al., 2002), can potentially be modified by APC. Additionally, since binding of apoCaM to the IQ motif of the L-type Ca channel is also a critical component of inactivation (Tang et al., 2003), changes in this interaction could also influence Ca2+-dependent inactivation.
Although the net physiological consequence of the APC-triggered change in ICa,L inactivation was not established, it can potentially contribute to reduced Ca2+ influx and to changes in cardiac electrophysiology. Regarding the former, since the L-type Ca channel is a major route of Ca2+ influx into the cytosol, the acceleration of ICa,L inactivation kinetics can potentially contribute to a decrease in Ca2+ overload, and subsequently reduce mitochondrial Ca2+ overload, and prevent or delay apoptosis. Our results showed a significant decrease in the total charge transfer as a result of the accelerated ICa,L inactivation by APC. Studies have indeed demonstrated that APC significantly reduced cytosolic Ca2+ concentration, relative to non-preconditioned hearts following global ischaemia (An et al., 2001; Varadarajan et al., 2002), and attenuated cytochrome c release from mitochondria (Qian et al., 2005). These effects can be correlated with a reduction in infarct size and with improved functional recovery following ischaemia/reperfusion.
In addition to the L-type Ca channel, contributions from other sources of Ca2+, for example, the roles of the Na+/Ca2+ exchanger and the ryanodine receptors on the sarcoplasmic reticulum, and the buffering capacity of the mitochondria, must also be considered in determining the precise mechanism(s) underlying Ca2+ overload attenuation, induced by APC. Our results showed that cytosolic Ca2+ accumulation was decreased following oxidative stress in myocytes obtained from preconditioned hearts. Although this demonstrated the functional efficacy of our APC model, the contribution from the L-type Ca channel in attenuating Ca2+ overload was uncertain as the Ca2+ measurements were conducted under in vitro conditions in non-contracting myocytes. Consequently, other mechanisms are likely to be involved. On the other hand, in a beating heart, cytosolic Ca2+ accumulation following stress might be greater due to influx of Ca2+ via the L-type Ca channel, which is a major source of Ca2+ entry into the cytosol. Thus, one can speculate that the acceleration of ICa,L inactivation kinetics triggered by APC would be likely to contribute to decreased Ca2+ accumulation in contracting myocytes. Overall, our results suggested that the preconditioned myocytes were better able to handle Ca2+ accumulation following stress.
With regard to cardiac electrophysiology, though the cardioprotective effect of APC is well established, in terms of reduction in infarct size, its effect against arrhythmias has not been firmly documented. For example, studies using the isolated heart model of APC showed that sevoflurane can delay the onset of ventricular fibrillation or have no effect on this type of arrhythmia (Novalija et al., 1999; Kevin et al., 2003). Based on our simulation studies, the accelerated ICa,L inactivation would result in the shortening of the cardiac APD. It is well established that APC enhances the opening of the sarcKATP channel and this effect can also lead to the shortening of the APD. Consequently, the acceleration of ICa,L inactivation could potentiate this effect, resulting in a further shortening of the APD and triggering re-entrant arrhythmias. Hence, a critical balance between the sarcKATP channel opening and acceleration of ICa,L inactivation may underlie some of the confusion regarding the effects of APC on cardiac rhythm. The overall effect on cardiac electrophysiology, thus, will depend on the cumulative effects the APC-triggered signalling cascade would have on the various sarcolemmal ion channels. Furthermore, it may also depend on the timing of the opening of the sarcKATP channel. Currently, the magnitude and duration of the activation of the sarcKATP channel during the various phases of preconditioning (stimulus, washout, ischaemia and reperfusion) are unknown, although a recent study suggested that the sarcKATP channels were more likely to open during the ischaemia/reperfusion period and contribute to cardioprotection (Marinovic et al., 2006; Stadnicka et al., 2006).
One intriguing but remarkable aspect of our findings is that only one biophysical characteristic of the L-type Ca channel, the rate of inactivation, was affected by APC. All other measured parameters remained unchanged. The direct, acute application of volatile anaesthetics is known to inhibit ICa,L amplitude and thus to depress cardiac contractility (Hanley et al., 2004). However, this inhibition is absent in the persistent effect triggered by isoflurane preconditioning. This change in the inactivation kinetics may allow for the fine tuning of Ca2+ entry without affecting the contractile ability of the cardiac myocytes. Furthermore, a recent study proposed that accelerating ICa,L inactivation while preserving peak ICa,L may be an effective way to prevent arrhythmias (Mahajan et al., 2008).
To date, studies systematically exploring the effect of APC on the cardiac sarcolemmal voltage-gated ion channels have been limited, despite the strong probability that the signalling mechanism triggered by APC can modulate the function of these proteins. In pursuit of the mechanism underlying APC, studies have focused on the intracellular signalling cascade and on mitochondrial function (Bienengraeber et al., 2005). With respect to the role of ion channels in APC, those not gated by voltage, specifically the sarcKATP and mitoKATP channels, have been intensely investigated. The roles of other ion channels, particularly those on the mitochondria such as the voltage-dependent anion and the calcium activated potassium channels and the adenine nucleotide translocase, have yet to be established.
In conclusion, the results of this study showed that in vivo APC triggered the acceleration of ICa,L inactivation kinetics in cardiomyocytes. Voltage-dependent inactivation of ICa,L was not affected, suggesting the involvement of Ca2+-dependent inactivation. Although a direct physiological consequence of this change in inactivation has yet to be determined, this alteration in the L-type Ca channel may contribute to the preservation of cardiomyocytes against Ca2+ overload and subsequent cell death.
Acknowledgments
This work was supported by grant No. PO1 GM066730 (Project III to WMK; Program Director ZJB) from the National Institutes of Health, Bethesda, MD, USA. The authors thank Dr Martin W. Bienengraeber (Assistant Professor, Departments of Anesthesiology and Pharmacology and Toxicology, Medical College of Wisconsin, Milwaukee, WI, USA), and Dr Garrett J. Gross (Professor, Department of Pharmacology and Toxicology, Medical College of Wisconsin, Milwaukee, WI, USA) for valuable discussions.
Glossary
Abbreviations:
- AP
action potential
- APC
anaesthetic-induced preconditioning
- APD
action potential duration
- CaM
calmodulin
- ICa,L
L-type calcium current
- IPC
ischemic preconditioning
- τ
time constant
Conflicts of interest
None.
References
- Aizawa K, Turner LA, Weihrauch D, Bosnjak ZJ, Kwok WM. Protein kinase C-epsilon primes the cardiac sarcolemmal adenosine triphosphate-sensitive potassium channel to modulation by isoflurane. Anesthesiology. 2004;101:381–389. doi: 10.1097/00000542-200408000-00019. [DOI] [PubMed] [Google Scholar]
- Alseikhan BA, DeMaria CD, Colecraft HM, Yue DT. Engineered calmodulins reveal the unexpected eminence of Ca2+ channel inactivation in controlling heart excitation. Proc Natl Acad Sci USA. 2002;99:17185–17190. doi: 10.1073/pnas.262372999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An J, Varadarajan SG, Novalija E, Stowe DF. Ischemic and anesthetic preconditioning reduces cytosolic [Ca2+] and improves Ca(2+) responses in intact hearts. Am J Physiol Heart Circ Physiol. 2001;281:H1508–H1523. doi: 10.1152/ajpheart.2001.281.4.H1508. [DOI] [PubMed] [Google Scholar]
- Baker JE, Konorev EA, Gross GJ, Chilian WM, Jacob HJ. Resistance to myocardial ischemia in five rat strains: is there a genetic component of cardioprotection? Am J Physiol Heart Circ Physiol. 2000;278:H1395–H1400. doi: 10.1152/ajpheart.2000.278.4.H1395. [DOI] [PubMed] [Google Scholar]
- Bienengraeber MW, Weihrauch D, Kersten JR, Pagel PS, Warltier DC. Cardioprotection by volatile anesthetics. Vascul Pharmacol. 2005;42:243–252. doi: 10.1016/j.vph.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Bosnjak ZJ, Supan FD, Rusch NJ. The effects of halothane, enflurane, and isoflurane on calcium current in isolated canine ventricular cells. Anesthesiology. 1991;74:340–345. doi: 10.1097/00000542-199102000-00022. [DOI] [PubMed] [Google Scholar]
- Cain BS, Meldrum DR, Cleveland JC, Jr, Meng X, Banerjee A, Harken AH. Clinical L-type Ca(2+) channel blockade prevents ischemic preconditioning of human myocardium. J Mol Cell Cardiol. 1999;31:2191–2197. doi: 10.1006/jmcc.1999.1039. [DOI] [PubMed] [Google Scholar]
- Camara AK, Begic Z, Kwok WM, Bosnjak ZJ. Differential modulation of the cardiac L- and T-type calcium channel currents by isoflurane. Anesthesiology. 2001;95:515–524. doi: 10.1097/00000542-200108000-00038. [DOI] [PubMed] [Google Scholar]
- Cason BA, Gamperl AK, Slocum RE, Hickey RF. Anesthetic-induced preconditioning: previous administration of isoflurane decreases myocardial infarct size in rabbits. Anesthesiology. 1997;87:1182–1190. doi: 10.1097/00000542-199711000-00023. [DOI] [PubMed] [Google Scholar]
- Dick IE, Tadross MR, Liang H, Tay LH, Yang W, Yue DT. A modular switch for spatial Ca2+ selectivity in the calmodulin regulation of CaV channels. Nature. 2008;451:830–834. doi: 10.1038/nature06529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findlay I. Voltage-dependent inactivation of L-type Ca2+ currents in guinea-pig ventricular myocytes. J Physiol. 2002;545:389–397. doi: 10.1113/jphysiol.2002.029637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox JJ, McHarg JL, Gilmour RF., Jr Ionic mechanism of electrical alternans. Am J Physiol Heart Circ Physiol. 2002;282:H516–H530. doi: 10.1152/ajpheart.00612.2001. [DOI] [PubMed] [Google Scholar]
- Galinanes M, Hearse DJ. Species differences in susceptibility to ischemic injury and responsiveness to myocardial protection. Cardioscience. 1990;2:127–143. [PubMed] [Google Scholar]
- Garny A, Kohl P, Noble D. Cellular Open Resource (COR): a public CellML based environment for modelling biological function. Int J Bifurcat Chaos. 2003;13:3579–3590. [Google Scholar]
- Hanley PJ, ter Keurs HE, Cannell MB. Excitation-contraction coupling in the heart and the negative inotropic action of volatile anesthetics. Anesthesiology. 2004;101:999–1014. doi: 10.1097/00000542-200410000-00027. [DOI] [PubMed] [Google Scholar]
- Hoka S, Bosnjak ZJ, Kampine JP. Halothane inhibits calcium accumulation following myocardial ischemia and calcium paradox in guinea pig hearts. Anesthesiology. 1987;67:197–202. doi: 10.1097/00000542-198708000-00008. [DOI] [PubMed] [Google Scholar]
- Kersten JR, Schmeling TJ, Pagel PS, Gross GJ, Warltier DC. Isoflurane mimics ischemic preconditioning via activation of K(ATP) channels: reduction of myocardial infarct size with an acute memory phase. Anesthesiology. 1997a;87:361–370. doi: 10.1097/00000542-199708000-00024. [DOI] [PubMed] [Google Scholar]
- Kersten JR, Orth KG, Pagel PS, Mei DA, Gross GJ, Warltier DC. Role of adenosine in isoflurane-induced cardioprotection. Anesthesiology. 1997b;86:1128–1139. doi: 10.1097/00000542-199705000-00017. [DOI] [PubMed] [Google Scholar]
- Kevin LG, Katz P, Camara AK, Novalija E, Riess ML, Stowe DF. Anesthetic preconditioning: effects on latency to ischemic injury in isolated hearts. Anesthesiology. 2003;99:385–391. doi: 10.1097/00000542-200308000-00020. [DOI] [PubMed] [Google Scholar]
- Krolikowski JG, Bienengraeber M, Weihrauch D, Warltier DC, Kersten JR, Pagel PS. Inhibition of mitochondrial permeability transition enhances isoflurane-induced cardioprotection during early reperfusion: the role of mitochondrial KATP channels. Anesth Analg. 2005;101:1590–1596. doi: 10.1213/01.ANE.0000181288.13549.28. [DOI] [PubMed] [Google Scholar]
- Linz KW, Meyer R. Profile and kinetics of L-type calcium current during the cardiac ventricular action potential compared in guinea-pigs, rats and rabbits. Pflugers Arch. 2000;439:588–599. doi: 10.1007/s004249900212. [DOI] [PubMed] [Google Scholar]
- Ljubkovic M, Mio Y, Marinovic J, Stadnicka A, Warltier DC, Bosnjak ZJ, et al. Isoflurane preconditioning uncouples mitochondria and protects against hypoxia-reoxygenation. Am J Physiol Cell Physiol. 2007;292:C1583–C1590. doi: 10.1152/ajpcell.00221.2006. [DOI] [PubMed] [Google Scholar]
- Ludwig LM, Weihrauch D, Kersten JR, Pagel PS, Warltier DC. Protein kinase C translocation and Src protein tyrosine kinase activation mediate isoflurane-induced preconditioning in vivo: potential downstream targets of mitochondrial adenosine triphosphate-sensitive potassium channels and reactive oxygen species. Anesthesiology. 2004;100:532–539. doi: 10.1097/00000542-200403000-00011. [DOI] [PubMed] [Google Scholar]
- Luo CH, Rudy Y. A dynamic model of the cardiac ventricular action potential. I. Simulations of ionic currents and concentration changes. Circ Res. 1994;74:1071–1096. doi: 10.1161/01.res.74.6.1071. [DOI] [PubMed] [Google Scholar]
- Mahajan A, Sato D, Shiferaw Y, Baher A, Xie LH, Peralta R, et al. Modifying L-type calcium current kinetics: consequences for cardiac excitation and arrhythmia dynamics. Biophys J. 2008;94:411–423. doi: 10.1529/biophysj.106.98590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinovic J, Bosnjak ZJ, Stadnicka A. Distinct roles for sarcolemmal and mitochondrial adenosine triphosphate-sensitive potassium channels in isoflurane-induced protection against oxidative stress. Anesthesiology. 2006;105:98–104. doi: 10.1097/00000542-200607000-00018. [DOI] [PubMed] [Google Scholar]
- Miyawaki H, Ashraf M. Ca2+ as a mediator of ischemic preconditioning. Circ Res. 1997;80:790–799. doi: 10.1161/01.res.80.6.790. [DOI] [PubMed] [Google Scholar]
- Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- Novalija E, Fujita S, Kampine JP, Stowe DF. Sevoflurane mimics ischemic preconditioning effects on coronary flow and nitric oxide release in isolated hearts. Anesthesiology. 1999;91:701–712. doi: 10.1097/00000542-199909000-00023. [DOI] [PubMed] [Google Scholar]
- Pancrazio JJ. Halothane and isoflurane preferentially depress a slowly inactivating component of Ca2+ channel current in guinea-pig myocytes. J Physiol. 1996;494:91–103. doi: 10.1113/jphysiol.1996.sp021478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson BZ, DeMaria CD, Adelman JP, Yue DT. Calmodulin is the Ca2+ sensor for Ca2+ -dependent inactivation of L-type calcium channels. Neuron. 1999;22:549–558. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- Qian LP, Zhu SS, Cao JL, Zeng YM. Isoflurane preconditioning protects against ischemia-reperfusion injury partly by attenuating cytochrome c release from subsarcolemmal mitochondria in isolated rat hearts. Acta Pharmacol Sin. 2005;26:813–820. doi: 10.1111/j.1745-7254.2005.00117.x. [DOI] [PubMed] [Google Scholar]
- Riess ML, Kevin LG, McCormick J, Jiang MT, Rhodes SS, Stowe DF. Anesthetic preconditioning: the role of free radicals in sevoflurane-induced attenuation of mitochondrial electron transport in Guinea pig isolated hearts. Anesth Analg. 2005;100:46–53. doi: 10.1213/01.ANE.0000139346.76784.72. [DOI] [PubMed] [Google Scholar]
- da Silva R, Grampp T, Pasch T, Schaub MC, Zaugg M. Differential activation of mitogen-activated protein kinases in ischemic and anesthetic preconditioning. Anesthesiology. 2004;100:59–69. doi: 10.1097/00000542-200401000-00013. [DOI] [PubMed] [Google Scholar]
- Smith GB, Stefenelli T, Wu ST, Wikman-Coffelt J, Parmley WW, Zaugg CE. Rapid adaptation of myocardial calcium homeostasis to short episodes of ischemia in isolated rat hearts. Am Heart J. 1996;131:1106–1112. doi: 10.1016/s0002-8703(96)90084-8. [DOI] [PubMed] [Google Scholar]
- Stadnicka A, Marinovic J, Bienengraeber M, Bosnjak ZJ. Impact of in vivo preconditioning by isoflurane on adenosine triphosphate-sensitive potassium channels in the rat heart: lasting modulation of nucleotide sensitivity during early memory period. Anesthesiology. 2006;104:503–510. doi: 10.1097/00000542-200603000-00018. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Aizawa K, Gassmayr S, Bosnjak ZJ, Kwok WM. Biphasic effects of isoflurane on the cardiac action potential: an ionic basis for anesthetic-induced changes in cardiac electrophysiology. Anesthesiology. 2002;97:1209–1217. doi: 10.1097/00000542-200211000-00026. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Weihrauch D, Kehl F, Ludwig LM, LaDisa JF, Jr, Kersten JR, et al. Mechanism of preconditioning by isoflurane in rabbits: a direct role for reactive oxygen species. Anesthesiology. 2002;97:1485–1490. doi: 10.1097/00000542-200212000-00021. [DOI] [PubMed] [Google Scholar]
- Tang W, Halling DB, Black DJ, Pate P, Zhang JZ, Pedersen S, et al. Apocalmodulin and Ca2+ calmodulin-binding sites on the CaV1.2 channel. Biophys J. 2003;85:1538–1547. doi: 10.1016/s0006-3495(03)74586-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Petegem F, Chatelain FC, Minor DL., Jr Insights into voltage-gated calcium channel regulation from the structure of the CaV1.2 IQ domain-Ca2+/calmodulin complex. Nat Struct Mol Biol. 2005;12:1108–1115. doi: 10.1038/nsmb1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadarajan SG, An J, Novalija E, Stowe DF. Sevoflurane before or after ischemia improves contractile and metabolic function while reducing myoplasmic Ca(2+) loading in intact hearts. Anesthesiology. 2002;96:125–133. doi: 10.1097/00000542-200201000-00025. [DOI] [PubMed] [Google Scholar]
- Zaugg M, Lucchinetti E, Spahn DR, Pasch T, Schaub MC. Volatile anesthetics mimic cardiac preconditioning by priming the activation of mitochondrial K(ATP) channels via multiple signaling pathways. Anesthesiology. 2002;97:4–14. doi: 10.1097/00000542-200207000-00003. [DOI] [PubMed] [Google Scholar]
- Zhou H, Yu K, McCoy KL, Lee A. Molecular mechanism for divergent regulation of Cav1.2 Ca2+ channels by calmodulin and Ca2+-binding protein-1. J Biol Chem. 2005;280:29612–29619. doi: 10.1074/jbc.M504167200. [DOI] [PubMed] [Google Scholar]