

Abstract
Background and purpose:
The mechanisms responsible for phase 2 (infarct-related) ventricular arrhythmias remain unclear. We have investigated the role of α1 and β1 adrenoceptor activation and the interaction of this with infarct neutrophil accumulation, in anaesthetized rats.
Experimental approach:
Neutrophil-replete Sprague-Dawley rats (n = 8–9 per group) were anaesthetized and randomized to receive vehicle, prazosin (0.5 mg·kg−1 i.v.), atenolol (4 mg·kg−1 i.v.) or their combination prior to left main coronary artery occlusion. A further group was depleted of neutrophils and received both atenolol and prazosin. Coronary ligation in all groups was maintained for 240 min.
Key results:
Atenolol and prazosin treatment lowered heart rates and blood pressures respectively, but neither agent given alone affected the incidence of phase 2 ventricular tachycardia or fibrillation. However, co-administration of atenolol with prazosin reduced phase 2 ventricular premature beats (log10-transformed totals were 1.25 ± 0.26 vs. 2.43 ± 0.18 in controls; P < 0.05). Neutrophil depletion attenuated this antiarrhythmic effect (log10-transformed total ventricular premature beats were 1.66 ± 0.35; P > 0.05 vs. controls).
Conclusions and implications:
Phase 2 arrhythmias appear to depend in part on a complex interaction between catecholamines and neutrophils. A model of this interaction is proposed.
Keywords: neutrophil, catecholamines, myocardial infarction, phase-2 arrhythmias, ventricular fibrillation, anaesthesia, rat
Introduction
Ventricular fibrillation (VF) is the main cause of sudden cardiac death (Smith and Cain, 2006). VF and other ventricular arrhythmias can occur spontaneously in several contexts in which the underlying pathology, electrophysiology, pharmacology and consequent causative mechanisms are different (Clements-Jewery et al., 2005). These include the acute phase of myocardial ischaemia and the subacute phase of evolving myocardial infarction. Susceptibility to arrhythmias is manifested differently during these phases in animal models, with the arrhythmias occurring during acute ischaemia (up to 30 min after coronary obstruction) termed phase 1 arrhythmias and those occurring after 1.5 h of sustained ischaemia during the subacute phase of infarct evolution (Harris, 1950; Clark et al., 1980; Johnston et al., 1983) termed phase 2 arrhythmias (Curtis, 1998).
In contrast to phase 1 arrhythmias, the mechanisms underlying phase 2 arrhythmias are not well understood. On the basis of the observation that phase 2 arrhythmias occur in rats in vivo but do not occur in isolated buffer-perfused rat hearts subjected to coronary ligation (Ravingerova et al., 1995; Clements-Jewery et al., 2002a), two key factors absent from the isolated perfused heart, namely blood components (such as neutrophils) and cardiac adrenoceptor activation, have been proposed as potential mediators of phase 2 arrhythmias (Clements-Jewery et al., 2005). Possible roles of these factors are supported by what is already known regarding the pathophysiological processes accompanying the onset of myocardial ischaemia. For example, release of catecholamines within the infarcting tissue has been well-characterized (Schömig et al., 1984; Lameris et al., 2000). Moreover, the accumulation of neutrophils within the infarct occurs over a similar time-course to that of the appearance of phase 2 arrhythmias (Mullane et al., 1984; Sasaki et al., 1988).
However, neither catecholamines nor neutrophils have been proven to mediate phase 2 arrhythmias. Moreover, there is some evidence to suggest that catecholamines and neutrophils are insufficient on their own to mediate phase 2 arrhythmias (Clements-Jewery et al., 2002a). This includes the finding that catecholamine replenishment in isolated, buffer-perfused, rat hearts subjected to coronary ligation was unable to restore susceptibility to phase 2 arrhythmias to the level observed in vivo (Clements-Jewery et al., 2002a). Additionally, phase 2 VF and other arrhythmias were not abolished in an anaesthetized rat model in which neutrophils were depleted prior to coronary ligation by injection of an anti-rat antineutrophil antiserum (Clements-Jewery et al., 2007). Finally, while Botting et al. (1983) concluded that labetalol and propranolol had little influence on susceptibility to phase 2 arrhythmias, interpretation of the data in this study is complex owing to the excess mortality that occurred. Therefore, the best available evidence suggests catecholamines do not appear to be sufficient, and that neutrophils do not appear to be necessary, for phase 2 arrhythmogenesis, although this conclusion is not yet firm owing to the insufficiency of available data. It therefore remains a possibility that catecholamines and neutrophils do contribute to phase 2 arrhythmia susceptibility. In addition, in analogy with arrhythmia mediation in the phase 1 setting (Baker and Curtis, 2004), it is possible that these two ‘mediators’ interact, perhaps in a complex manner, to facilitate phase 2 arrhythmias. Presently this possibility is entirely untested.
In vitro studies, using isolated perfused hearts, with neutrophils are not feasible owing to unavoidable non-specific neutrophil activation in the perfusion circuit (Clements-Jewery et al., 2002b). Therefore, we chose to use an in vivo rat model to examine the role of catecholamines (and the role of their interaction with neutrophil accumulation within the infarct) in causing phase 2 arrhythmias. Phase 2 arrhythmias have been observed in anaesthetized (Clark et al., 1980) and conscious rats (Curtis et al., 1984; Opitz et al., 1995), but as anaesthetized animals are preferable to conscious animals for ethical reasons, in the present study we used an anaesthetized rat model. Specifically, α- and/or β-adrenoceptor antagonists were administered to neutrophil-replete and neutrophil-depleted anaesthetized rats subjected to permanent coronary ligation, and the effects on phase 2 arrhythmias were monitored.
Methods
All experiments were performed in accordance with the United Kingdom Home Office Guide on the Operation of the Animals (Scientific Procedures) Act 1986.
Procedure for neutrophil depletion
Male Sprague-Dawley rats (290–418 g; Bantin and Kingman, Hull, UK) were randomized to receive 2 mL·kg−1 i.p. rabbit anti-rat anti-neutrophil antiserum (abbreviated hereafter as antiserum) or normal rabbit serum as control (Accurate Chemicals, Westbury, NY, USA) at least 17 h before coronary occlusion. The administration of the antiserum in this way results in complete depletion of circulating neutrophils whereas administration of normal rabbit serum has no effect on circulating neutrophils (Clements-Jewery et al., 2007).
Surgical procedure for placement of the coronary ligature
Rats, pretreated with antiserum or normal rabbit serum as described above, were anaesthetized by injection of sodium pentobarbitone (60 mg·kg−1 i.p). Sufficient anaesthesia was determined by loss of pedal paw and eye-blink reflexes. Body temperature was monitored by a rectal temperature probe and maintained at 37 ± 1°C by means of a heated operating table. The left femoral vein (for administration of further anaesthetic; 0.05–0.1 mL of 60 mg·mL−1 pentobarbitone) and right femoral artery (for recording of blood pressure) were isolated by blunt dissection and cannulated (18 G and 22 G Abbocath-T catheters respectively). The right femoral vein was also cannulated for drug administration. The arterial cannula was attached to a polyethylene line flushed with heparinised saline (3000 U·L−1) that was connected to a pressure transducer (Becton Dickinson DTX™Plus). The venous cannula was attached to a similar line flushed with heparinised saline. To allow respiration using a pump (model 683; Harvard Apparatus, Edenbridge, UK), the trachea was also cannulated. To place the coronary ligature, the heart was exposed by intercostal incision followed by rib retraction. Following intercostal incision, the rat was immediately ventilated with room air by means of the pump at a rate of 52 strokes min−1 and stroke volume of 1–1.5 mL·100 g−1 rat weight to maintain arterial blood gases and blood pH in the normal range (Milmer and Clough, 1983). The heart was suspended in a pericardial cradle and the suture (Ethicon Prolene 5/0) sewn around the left main coronary artery beginning underneath the left atrial appendage. The ends of the coronary ligature were sewn through the flared end of a section of guide tubing to create a traction-type occluder, and the chest evacuated and sewn up such that the occluder protruded from the chest. Coronary occlusion was performed subsequently by pulling on the protruding ends of the suture to tighten the ligature.
Arrhythmia diagnosis and electrocardiogram (ECG) analysis
The ECG was recorded using standard limb electrodes in configuration lead II connected to a Powerlab™ system (ADInstruments, Chalgrove, UK) and was used to assess arrhythmias in accordance with the Lambeth Conventions (Walker et al., 1988). Ventricular premature beats (VPBs) were defined as premature QRS complexes occurring independently of a P wave, and hence mean values included individual VPBs, bigeminy and salvos (Walker et al., 1988). The number of VPBs occurring during a specified time period in each heart was log10-transformed to produce a Gaussian-distributed variable for calculation of group mean values (Johnston et al., 1983). QT intervals at the point of 90% repolarization (QT90), heart rate and PR intervals were also measured from the ECG, as previously described (Ridley et al., 1992). The ECG was recorded at a sampling rate of 1 kHz, allowing millisecond precision for measurement of ECG intervals. Blood pressure was also recorded using the Powerlab™ system.
Episodes of VF were terminated by manual cardioversion. This was successful in >90% of circumstances where VF occurred.
Blood analysis and methods for total and differential white blood cell counts
Blood samples (approximately 0.7 mL) were taken from the arterial line at 15 and 180 min following coronary ligation and analysed using a Stat Profile 9 Blood Gas Analyser (Nova Biomedical, Deeside, UK) to confirm that blood gas parameters and blood K+ values were within acceptable ranges. The sample taken after 180 min of ischaemia was also used for white blood cell total and differential counts. Removed blood samples were replaced with equivalent volumes of i.v. saline to maintain blood volume.
White blood cell counts were performed according to established methods (Pitchford et al., 2003). For total white blood cell counts, 10 µL of the blood sample taken at 180 min of ischaemia were added to 90 µL of haemolysis solution containing 0.1% methylene blue in 1% acetic acid, and mixed thoroughly. White blood cells were counted using an improved Neubauer haemocytometer and a 20× objective lens. For differential white blood cell counts, 10 µL of the same blood sample were smeared onto microscope slides and stained using the DiffQuik stain system (Gamidor Ltd., Didcot, UK). One hundred white blood cells were identified in total for each smear at 40× magnification. Together with the total white blood cell count, this was used to calculate the circulating numbers of each white blood cell type (mononuclear, neutrophils, eosinophils).
Assessment of cardiac myeloperoxidase content
Cardiac myeloperoxidase (MPO) content, an index of neutrophil accumulation, was assessed according to the method of Mullane et al. (1985). Samples from the involved and uninvolved zones of the excised heart at the end of the experiment were frozen in liquid nitrogen and stored at −80°C until use. Cardiac tissue samples were homogenized in 0.5% hexadecyltrimethylammonium bromide (HETAB) in 50 mmol·L−1 potassium phosphate buffer (pH 6.0), in the ratio of 1 mL HETAB per 100 mg tissue. The homogenate was then centrifuged at 13 000× g for 20 min at 4°C. Supernatant samples were frozen at −80°C until the time of assay where-upon thawed samples were kept on ice. To measure supernatant MPO activity, 50 µL of each sample was added to 50 µL of o-dianisidine dihydrochloride (0.025% in phosphate buffer with 0.5% HETAB) in a 96-well plate. The reaction was started by addition of 50 µL of 0.01% hydrogen peroxide and the increase in optical density at 510 nm was measured over 3 min. Duplicates were run for each sample, and the average value used to calculate the number of units of MPO present by interpolation with a standard curve established using dilutions of a commercially available preparation of human neutrophil MPO (Sigma Myeloperoxidase: 1 unit is defined as causing an increase in optical density of 1 min−1 at pH 7 at 25°C with guaiacol substrate). Protein in the supernatant was then determined according to the bicinchoninic acid method of Smith et al. (Smith et al., 1985) by interpolation with a standard curve established using dilutions of bovine serum albumin. Duplicates were run for each sample, and the average value used for calculation of protein content. MPO concentration was expressed as units of MPO (mg protein)−1.
Measurement of involved zone size and infarct size
At the end of the experiment, the heart was excised and briefly washed in heparinised saline. With the coronary ligature in place, the aorta was cannulated and 1–2 mL of blue disulphine dye flushed through the heart. Following another brief rinse in saline, stained (uninvolved) tissue was separated from pink (involved) tissue using a pair of scissors and weighed. The respective sections were then cut into strips and incubated in 1% triphenyl tetrazolium chloride (TTC) at 37°C for 20 min. The resulting tissue was placed in formal saline for 1–2 days, following which infarct size was calculated by separating brick red-stained (uninfarcted) tissue from unstained (infarcted) tissue using scissors, and weighing the respective sections. It was assumed that samples taken for MPO assay from the involved and uninvolved zones (which were therefore not incubated with TTC) were completely infarcted and completely uninfarcted respectively.
Experimental groups
Acute α- and/or β-adrenoceptor blockade (with prazosin and atenolol), with and without neutrophil depletion, were tested for effects on phase 1 and phase 2 arrhythmia susceptibility. Five experimental groups were used (n = 8–9 per group), four of which involved neutrophil-replete rats (i.e. pretreated with normal rabbit serum). These neutrophil-replete groups were: vehicle control (equivalent volume of saline/propylene glycol vehicle), acute atenolol treatment (4 mg·kg−1 bolus + continuous infusion at a rate of 0.24 mg·kg−1·h−1), acute prazosin treatment (0.5 mg·kg−1 bolus + continuous infusion at a rate of 0.03 mg·kg−1·h−1), and a group treated with a single solution containing atenolol and prazosin to achieve the same doses. A further group was used in which rats were pretreated with anti-neutrophil antiserum and received atenolol and prazosin. Bolus doses of drugs or drug vehicle were given 10 min prior to coronary ligation. Figure 1 shows the scheme of the experimental protocol.
Figure 1.
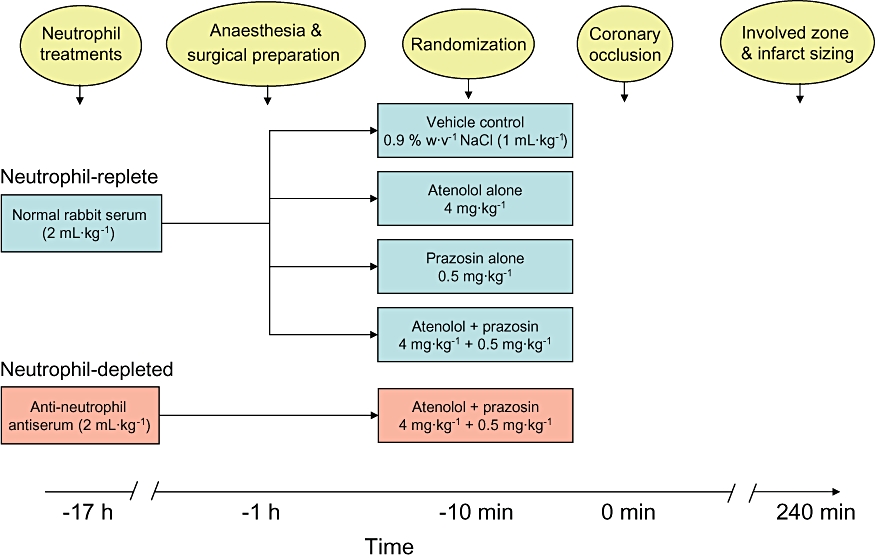
Diagram illustrating the experimental protocol (elaborated in Methods).
Rationale for drug doses
Bolus doses of atenolol (4 mg·kg−1) and prazosin (0.5 mg·kg−1) were chosen for their ability to cause an approximate 15-fold shift in the dose-response (tachycardic or pressor respectively) to bolus isoproterenol (initial dose 0.07 mL of 0.004 mg·mL−1) in the case of atenolol, or adrenaline (initial dose 0.07 mL of 0.02 mg·mL−1) in the case of prazosin, in three anaesthetized rats, not subjected to coronary ligation (data not shown). No evidence of PR prolongation was found in ECG recordings following administration of these doses of the antagonists (data not shown), indicating the absence of drug-induced Na+ channel blockade. The maintenance infusions were calculated to be 0.1% of the bolus dose per minute.
Drugs and solutions
All chemicals were obtained from Sigma-Aldrich (UK). For administration of atenolol and prazosin to the rats, drugs were made up such that the volume of solution delivered to the rat as the initial bolus was 1 mL·kg−1. Thus, atenolol was dissolved in 0.9% w·v−1 NaCl solution to yield a concentration of 4 mg·mL−1, and prazosin was dissolved in 0.9% w·v−1 NaCl solution containing 40% propylene glycol to yield a concentration of 0.5 mg·mL−1. Initial experiments were conducted with prazosin dissolved in ultrapure water, but this frequently caused sufficient haemolysis in the venous line to elevate blood K+ such that these rats had to be excluded from analysis. However, where blood K+ remained below the exclusion limit, these initial experiments were kept. Identity of the solutions was not known by the experimental operator, and the choice of experiment was made by reference to a randomization table.
Exclusion criteria
Rats were excluded from analysis if involved zone size was <30% [associated with a very low likelihood of VF during ischaemia (Johnston et al., 1983)], or if rats that did not have phase 2 VF also had blood K+ during the phase 2 period (90–240 min of ischaemia) of >5.0 mmol·L−1[blood K+ > 5.0 mmol·L−1 is highly antiarrhythmic (Clements-Jewery et al., 2005)]. They were also excluded if systolic blood pressure was <80 mmHg immediately prior to administration of drug and coronary occlusion, if ventricular tachycardia (VT) occurred in the 5 min period preceding occlusion, or if rats died early (from phase 1 VF unresponsive to manual cardioversion or from haemodynamic collapse independent of VF). The numbers of rats excluded using these criteria were 1, 4, 3, 0 and 9 respectively. Excluded animals were replaced to maintain group sizes of 8–9.
Statistics
Gaussian distributed variables (expressed as mean ± SEM) were subjected to analysis of variance followed by Dunnett's or Tukey's tests where appropriate. Binomially distributed variables were compared using Mainland's contingency tables (Mainland et al., 1956) as previously described (Curtis and Hearse, 1989). P < 0.05 was taken as indicative of a statistically significant difference between values. Arrhythmia incidences were expressed as the percentage of hearts in each group experiencing each arrhythmia during specified time intervals.
Results
Haemodynamic effects of acute treatment with atenolol and/or prazosin
β1-blockade was confirmed in atenolol treated groups by significant reductions in heart rate. Heart rate (Figure 2) was lower 1 min before coronary ligation (values at this time point were 407 ± 20 beats·min−1 in controls, and 345 ± 8, 334 ± 8 and 337 ± 10 beats·min−1 in neutrophil-replete atenolol-treated, neutrophil replete atenolol- and prazosin-treated and neutrophil-depleted atenolol- and prazosin-treated rats respectively; all P < 0.05 vs. controls) and this difference was maintained for the duration of the experiment (Figure 2).
Figure 2.

Heart rate during ischaemia in the different experimental groups. The arrow indicates the point at which drugs were administered. Coronary ligation occurred at 0 min. *Denotes P < 0.05 for neutrophil-replete atenolol treated group versus vehicle controls; #denotes P < 0.05 for neutrophil-replete atenolol and prazosin-treated group versus vehicle controls; §denotes P < 0.05 for neutrophil-depleted atenolol and prazosin-treated rats versus vehicle controls. Values for the atenolol-treated groups were significantly different (P < 0.05) from those in the control group at all time points after drug administration.
α1-blockade was confirmed by the acute reduction in blood pressure in response to prazosin versus the control group (Figure 3). Systolic blood pressure was reduced in all three prazosin-treated groups prior to ligation (1 min before ligation, values were 98 ± 5, 75 ± 2 and 93 ± 4 mmHg in neutrophil-replete prazosin-treated, neutrophil-replete atenolol- and prazosin-treated, and neutrophil-depleted atenolol- and prazosin-treated rats respectively; all P < 0.05 vs. 120 ± 10 mmHg in controls). However, the blood pressure-lowering effect of prazosin treatment was lost following coronary ligation, despite the continued infusion of prazosin throughout the duration of the experiment. In the neutrophil-replete prazosin-treated rats, the hypotensive effect of prazosin was lost immediately following coronary ligation, whereas in the neutrophil-replete and depleted rats treated with atenolol and prazosin, the hypotensive effect was lost more slowly (after 60 min and 30 min ischaemia respectively).
Figure 3.
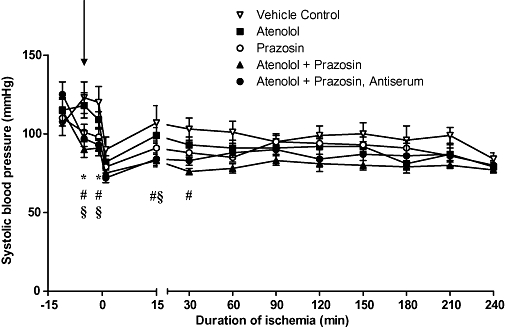
Systolic blood pressure in the different experimental groups. Compared with vehicle controls, prazosin treatment resulted in significantly lower systolic blood pressure prior to coronary ligation. Blood pressure in the prazosin-treated groups was lower as a trend for the duration of the experiment. Treatments were administered 5 min before coronary ligation, shown by the arrow. *Denotes P < 0.05 for neutrophil-replete prazosin-treated rats versus vehicle controls; #denotes P < 0.05 for neutrophil-replete atenolol + prazosin-treated rats versus vehicle controls; §denotes P < 0.05 for neutrophil-depleted atenolol + prazosin-treated rats versus vehicle controls.
Circulating white blood cells and cardiac MPO activity
As in our previously published study, antiserum pretreatment significantly depleted circulating neutrophils (Figure 4). Circulating mononuclear cells were also significantly reduced in antiserum-pretreated rats. There were no significant differences in numbers of circulating neutrophils or eosinophils between the four groups pretreated with normal rabbit serum. Surprisingly, circulating mononuclear cells were significantly reduced in neutrophil-replete atenolol-treated and prazosin-treated rats.
Figure 4.
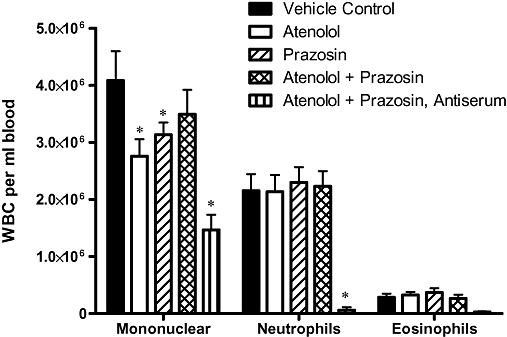
Circulating white blood cell types in the different experimental groups. Blood samples for white blood cell (WBC) counting were taken at 180 min after the onset of ischaemia. *Denotes P < 0.05 versus vehicle control. The apparent reduction of circulating eosinophils by antiserum treatment did not reach statistical significance.
As expected, antiserum pretreatment also significantly reduced cardiac MPO activity in involved zone samples taken at the end of the experiment (i.e. after 240 min continuous ischaemia) from 236 ± 69 mU·mg−1 protein in normal rabbit serum pretreated vehicle controls, to 9 ± 2 mU·mg−1 protein (P < 0.05) in the antiserum-pretreated rats. There were no significant differences in involved zone MPO activity between the four normal rabbit serum-pretreated groups (data not shown).
ECG variables
Atenolol and/or prazosin treatment had no effect on PR intervals measured from the ECG at any time point during the experiment (data not shown; values ranged from 40–44 ms in all groups; P = NS), or on the pattern of QT90 changes following coronary ligation (data not shown; representative values from vehicle controls 1 min before, and 1, 15 and 180 min after coronary ligation were 41 ± 2, 28 ± 3, 53 ± 1 and 57 ± 4 ms respectively).
Effects of atenolol/prazosin treatment on susceptibility to arrhythmias
The incidence of phase 2 VT (five out of nine rats) and VF (two out of nine) in vehicle controls were too low to allow detection of antiarrhythmic effects. Nevertheless, even as a trend, there was clearly no indication of any protection afforded by atenolol, prazosin or antiserum (Table 1). However, with the combination of atenolol and prazosin in neutrophil-replete rats, there were no episodes of phase 2 VF, the incidence of phase 2 VT was one out of eight and the number of VPBs was statistically significantly reduced (Figure 5). Surprisingly, these protective effects, including the significant reduction in VPB number, were partly lost when the combination of prazosin and atenolol was tested in antiserum treated rats (Figure 5, Table 1).
Table 1.
Incidences of ventricular tachycardia (VT) and ventricular fibrillation (VF) in the phase 2 period (90–240 min) in the different groups
| Group | VT incidence | VF incidence | |
|---|---|---|---|
| Neutrophil-replete | Vehicle control | 5 out of 9 | 2 out of 9 |
| Atenolol only | 6 out of 9 | 2 out of 9 | |
| Prazosin only | 6 out of 9 | 3 out of 9 | |
| Atenolol + prazosin | 1 out of 8 | 0 out of 8 | |
| Neutrophil-depleted | Atenolol + prazosin | 3 out of 9 | 2 out of 9 |
There were no significant differences between groups.
Figure 5.
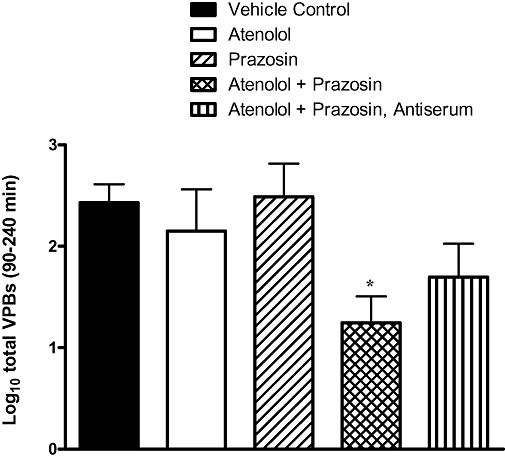
Total ventricular premature beats (VPBs) occurring in the different experimental groups in the phase 2 period (90–240 min of ischaemia). Values are log10-transformed. *Denotes P < 0.05 versus vehicle control group.
While not the main focus of the present study, the effect of the different treatments on the incidence of phase 1 arrhythmias was also examined. There was a similar pattern of effects to those on phase 2 arrhythmias. The incidence of phase 1 VF (three out of nine) in controls was too low to allow detection of antiarrhythmic effects. There were no significant differences between groups in phase 1 VT incidence (ranging from five out of nine to eight out of nine). However, in neutrophil-replete rats treated with the combination of atenolol and prazosin, the number of VPBs was significantly lower than in vehicle controls (log10-transformed totals were 1.33 ± 0.32 vs. 2.26 ± 0.12 in vehicle controls; P < 0.05). As with phase 2 VPBs, this effect of atenolol and prazosin was lost when neutrophils were additionally depleted by antiserum pretreatment (the log10-transformed total was 1.92 ± 0.14; P > 0.05 vs. vehicle controls).
Blood K+, involved zone and infarct size
There were no differences in blood K+ concentration in the different treatment groups compared with vehicle controls at either 15 or 180 min following coronary ligation. Involved zone size (area at risk; Table 2) was similar in the various groups (P = NS), and there was no effect of any treatment on infarct size expressed as a percentage of the area at risk (Figure 6; P = NS).
Table 2.
Area at risk (involved zone size) as a percentage of total ventricular weight in the different groups
| Group | Area at risk (% of total ventricular weight) | |
|---|---|---|
| Neutrophil-replete | Vehicle control | 43 ± 2 |
| Atenolol only | 45 ± 2 | |
| Prazosin only | 44 ± 2 | |
| Atenolol + prazosin | 43 ± 2 | |
| Neutrophil-depleted | Atenolol + prazosin | 43 ± 2 |
There were no significant differences between groups.
Figure 6.
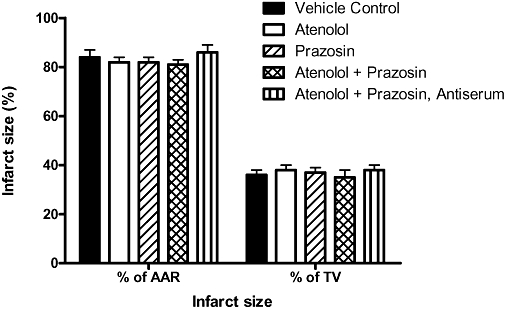
Infarct size in the different experimental groups expressed as a percentage of the area at risk (AAR) and of total ventricular weight (TV). Values were not statistically significant from vehicle controls in any of the groups.
Discussion
Relevance of the present experiments
It has been argued that for a drug to be capable of achieving benefit against sudden cardiac death, it must be capable of suppressing all types of life-threatening arrhythmias that may contribute to sudden cardiac death in the population (Clements-Jewery et al., 2005). This includes acute ischaemia-related (phase 1) ventricular arrhythmias and infarct evolution-related (phase 2) arrhythmias. The present studies were focused on the mechanisms and suppression of the latter.
Previous studies have indicated that neutrophil accumulation and cardiac adrenergic receptor stimulation by themselves are insufficient to cause phase 2 arrhythmias. For example, abolishing neutrophil accumulation in the evolving infarct by antineutrophil antiserum-induced neutropenia was found not to diminish the incidence of phase 2 VF, VT or VPBs (Clements-Jewery et al., 2007). Likewise, in vitro catecholamine replenishment to isolated buffer-perfused rat hearts did not restore susceptibility to phase 2 VF (Clements-Jewery et al., 2002a). On the other hand, these studies did not conclusively prove that neutrophils and catecholamines play no role in phase 2 arrhythmogenesis. In the case of catecholamines, our previous data came from in vitro studies. Additionally, no studies have addressed the possibility of an interaction between neutrophils and sympathetic stimulation in mediating phase 2 VF. The aims of the present study were therefore twofold: to investigate the role of sympathetic stimulation in mediating phase 2 VF in an in vivo model, and to investigate whether phase 2 VF might also result from an interaction between neutrophils and catecholamines in this model.
Efficacy of interventions
In the present experiments, the roles of neutrophil infarct accumulation and sympathetic stimulation in mediating phase 2 VF were probed respectively by pretreatment with antineutrophil antiserum and acute α1- and β1-adrenoceptor blockade with prazosin and/or atenolol, respectively, prior to coronary ligation. The effectiveness of these pretreatments was demonstrated by the occurrence of neutropenia in the case of the antiserum pretreatment, and by reductions in heart rate and/or blood pressure with acute atenolol and/or prazosin administration.
The reduction of blood pressure in the presence of prazosin alone was not sustained throughout the experiment, in spite of the continued infusion of prazosin. It is not clear why this was so, but a similar time course of effects was previously observed following bolus prazosin administration to conscious rats (Dynon et al., 1983). The effect cannot be explained by a reflex tachycardia, because of the absence of an increase in heart rate in the prazosin-treated group above vehicle controls. An increase in plasma volume due to intravenous saline infusion is also unlikely as the volume administered for this purpose was the same in all groups, although additional mechanisms mediated by α2- and β-adrenoceptors which would be unaffected by prazosin may have acted to increase plasma volume/blood pressure over the course of the experiment. The fact that blood pressure tended to be lower in the two groups additionally treated with atenolol gives some support to this assertion. Additionally, it is of interest that acute administration of 0.5 mg·kg−1 prazosin to rabbits in which reflex heart rate responses were abolished by autonomic blockade resulted in a short-lasting depression of blood pressure followed by a pressor response (Hamilton et al., 1985). However, the precise molecular nature of this pressor response, which matches the blood pressure effects seen in the present study, has not been established. What is known about the pharmacokinetics of prazosin suggests there was sufficient prazosin present in our experiments during the period when phase 2 arrhythmias occur (90–240 min). Thus, in man, the half-life of prazosin in the plasma is around 2.5 h (Jaillon, 1980), and in the rabbit, which may have a similar rate of prazosin elimination to the rat, the half-life of prazosin in the plasma following a single i.v. bolus dose of 0.1 mg·kg−1 (one fifth of that in the present experiments) was found to be around 2 h but with significant α1-blockade still present after 4 h (dose ratio for phenylephrine pressor response > 2) (Hamilton et al., 1985). Therefore, we infer that in our experiments there will have been significant α-blockade present during the phase 2 period (90–240 min).
Evaluation of the potential role of sympathetic stimulation on phase 2 arrhythmias
In the present study, neither the acute administration of atenolol nor prazosin alone to neutrophil-replete rats reduced the incidence of phase 2 arrhythmias. The results obtained with atenolol alone accord with previously published studies on the effect of β-blockers on phase 2 arrhythmias. Thus, with the exception of pindolol (Lepran et al., 1983), all other β-blockers investigated including propranolol (Botting et al., 1983) and oxprenolol (Campbell et al., 1984) have been found to be ineffective against phase 2 VF or other arrhythmias. Surprisingly, until now, the effect of α-adrenergic antagonists on phase 2 arrhythmias has not been independently assessed. The present results indicate that α-blockade alone does not reduce susceptibility to phase 2 arrhythmias.
However, when prazosin was administered with atenolol to neutrophil-replete rats, the occurrence of VPBs in the phase 2 period was significantly reduced, and the overall phase 2 arrhythmia susceptibility appeared to be lower in these rats. These effects on arrhythmias could not be accounted for by changes in blood K+ concentration, the size of the ischaemic substrate (area at risk or involved zone size) or infarct size, but were lost when rats were additionally depleted of neutrophils. Indeed, infarct size relative to the size of the area at risk was high in all groups, probably reflecting the long duration of ischemia and the lack of collateralization in the rat heart. In passing, we would note that the fact that no differences were found between neutrophil-replete and neutrophil-depleted groups in infarct size suggests that the presence or absence of neutrophils had no influence on the development of infarction following permanent occlusion.
These data imply that co-activation of α1- and β1-adrenoceptors by endogenous catecholamines may be proarrhythmic and neutrophil accumulation within the infarct territory may be antiarrhythmic in the phase 2 period. These data may be understood by proposing a model in which the balance between proarrhythmic and antiarrhythmic factors underlies susceptibility to phase 2 arrhythmias whereby when proarrhythmic factors outweigh antiarrhythmic factors, the probability of occurrence of arrhythmias is increased whereas, when antiarrhythmic factors outweigh proarrhythmic factors, the probability of occurrence of arrhythmias is diminished. Thus, when the proarrhythmic action of catecholamines is blocked by adrenergic antagonists, the antiarrhythmic effect of neutrophils predominates. In the absence of neutrophils (depletion by means of antineutrophil antiserum pretreatment), the balance is shifted again in favour of other unknown proarrhythmic factors, increasing arrhythmia susceptibility.
One intriguing possibility with regard to the mechanism of these observed effects is that the pro- and antiarrhythmic effects of catecholamines and neutrophils is the result of a direct interaction, that is, that catecholamines directly modulate neutrophil function by inhibiting neutrophil chemotaxis and/or the release of biochemical factors that are antiarrhythmic (Figure 7). In support of this possibility, previous studies on the effects of direct adrenergic stimulation on neutrophil function have revealed that β-adrenergic stimulation by catecholamines inhibits release of lysosomal enzymes, generation of superoxide and other free radicals, and neutrophil chemotaxis (Zurier et al., 1974; Nielson, 1987; Harvath et al., 1991). Neutrophils are also known to express β2 and α2-adrenoceptors on their cell surface (Elenkov et al., 2000). However, the present data suggest that the antiarrhythmic effect of adrenergic antagonism is not the result of any effect on neutrophil chemotaxis, as neutrophil accumulation within the infarct was not significantly different in the four neutrophil-replete groups.
Figure 7.
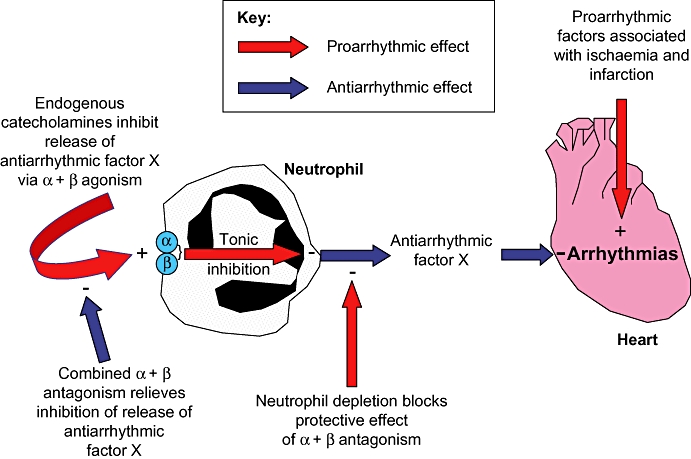
Proposed mechanism to explain the involvement of neutrophils in the antiarrhythmic effect of α- and β-adrenoceptor antagonism. See text for detailed elaboration.
The idea that neutrophils release as yet unidentified mediators which are protective against arrhythmias and this release is reduced by co-stimulation of adrenoceptors on the surface of neutrophils has several problems. One is that selective β1 and α1-adrenoceptor antagonists were used in the present study, while previous studies in the literature have in contrast revealed a β2 and α2-adrenoceptor-dependent regulation of neutrophils and other leukocytes (Elenkov et al., 2000). Another problem is that the use of antineutrophil antiserum in the present study reduced mononuclear leukocytes in addition to neutrophils, so the intervention was not completely selective. Therefore, any effects of the adrenoceptor antagonists used may also have involved actions on other white blood cells. Moreover, there is also no evidence from the present study that effects resulted exclusively from direct modulation of neutrophil function by the adrenoceptor antagonists used. Finally, the inference that catecholamines are proarrhythmic is also inconsistent with findings from perfused heart studies in which catecholamine repletion failed to restore the appearance of phase 2 arrhythmias (absent ordinarily in isolated perfused hearts) (Clements-Jewery et al., 2002a). This, however, may lend further credence to the idea that the proarrhythmic effects of catecholamines occurs through inhibition of release of neutrophil-derived antiarrhythmic factors, as these would be absent in isolated crystalloid perfused hearts (blood-free). Alternatively, the sympathetic drive that hearts ordinarily encounter in vivo during infarct evolution may not have been adequately restored in previous studies using the isolated heart model (Clements-Jewery et al., 2002a), meaning that the model was not fit for purpose. Ultimately, a conscious rat model of phase 2 arrhythmias may be necessary to resolve any model-dependent idiosyncrasies. Nonetheless, we contend that the present data warrant further investigation of the possible antiarrhythmic effect of neutrophils.
Effect of atenolol and prazosin on circulating white blood cells
Atenolol and prazosin independently reduced circulating numbers of mononuclear white blood cells (comprising lymphocytes and monocytes) in the present study. However, when atenolol and prazosin were administered together, the effect was lost. This seemingly surprising observation is in accordance with previous studies that have shown that lymphocytes are mobilized following acute catecholamine administration (Benschop et al., 1996). There is some evidence for the involvement of both β- and α-adrenoceptors in this response. For example, prazosin has been shown to decrease the number of spleen lymphocytes while increasing blood granulocytes (Maestroni et al., 1992), and β-adrenoceptor stimulation has been proposed to increase the rate of haematopoiesis (Byron, 1972). Modulation of adrenoceptors may therefore influence numbers of circulating lymphocytes directly by affecting production of lymphocytes or indirectly by modifying blood and lymph flow in regional circulations, and thereby delivery of lymphocytes into the circulation and to organs (Elenkov et al., 2000). Indeed, the present effects of atenolol and prazosin may be the complex result of inhibition of several independent mechanisms that operate simultaneously under normal circumstances – for example, slowing hematopoiesis but at the same time increasing lymphocyte delivery into the circulation by changing the regional distribution of blood flow. These possibilities are far to complex to interrogate in the context of a single study.
Limitations
Although we were able to probe phase 2 arrhythmia mechanisms via alterations in VPBs, the incidence of VF was too low in controls to permit detection of VF suppression at an acceptable level of statistical significance. To rectify this would require group sizes of 30. Given that the present study picked up significant changes in VPBs and a trend to effects on VT and VF, it would be difficult to justify such a large increase in group sizes. Nevertheless, we cannot extrapolate present findings, with confidence, specifically to regulation of VF susceptibility. It is important to emphasize that VF is the lethal arrhythmia associated with ischaemic heart disease. In the present study it is implied that VPBs represent surrogate biomarkers for lethal arrhythmias. In humans, there is no good evidence that this notion is valid; indeed, spontaneous VPB occurrence in the absence of acute regional ischaemia is known to not predict VF liability in humans (Kennedy et al., 1985) and drugs that suppress VPBs in patients with ischaemic heart disease can increase VF susceptibility (Pratt and Moye, 1995). Therefore, we do not suggest that the present findings may be extrapolated to the elucidation of lethal arrhythmias, without further validation of VPB susceptibility in acute ischaemia as a surrogate biomarker for VF.
The antiserum was not selective for neutrophils. We found this previously (Clements-Jewery et al., 2007). This means that any suppression of arrhythmias by the antiserum could be attributable, in part, to off-target effects. However, given that the antiserum did not itself suppress arrhythmias, this limitation does not have substantial consequence. Finally, the present study was performed in ‘healthy’ rats, whereas the effects of coronary occlusion are known to be influenced by the presence of independent disease-related risk factors (Ferdinandy et al., 2007).
Conclusion
Reduction of phase 2 arrhythmia susceptibility was only partly obtained in neutrophil-replete rats with α- and β-adrenoceptor blockade. The precise mechanism of action is unclear. As additional neutrophil depletion [which has no effect alone on phase 2 arrhythmias (Clements-Jewery et al., 2007)] attenuated these antiarrhythmic effects, neutrophils appear to have a protective effect on arrhythmia development in the phase 2 period. A conscious animal model of phase 2 arrhythmias may be required to better establish this mechanism.
Acknowledgments
We thank the British Heart Foundation for funding this project (PG03/054).
Glossary
Abbreviations:
- VF
ventricular fibrillation
- VPB
ventricular premature beat
- VT
ventricular tachycardia
Conflicts of interest
None.
References
- Baker KE, Curtis MJ. Left regional cardiac perfusion in vitro with platelet-activating factor, norepinephrine and K+ reveals that ischaemic arrhythmias are caused by independent effects of endogenous ‘mediators’ facilitated by interactions, and moderated by paradoxical antagonism. Br J Pharmacol. 2004;142(2):352–366. doi: 10.1038/sj.bjp.0705767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benschop RJ, Rodriguez-Feuerhahn M, Schedlowski M. Catecholamine-induced leukocytosis: early observations, current research, and future directions. Brain Behav Immun. 1996;10(2):77–91. doi: 10.1006/brbi.1996.0009. [DOI] [PubMed] [Google Scholar]
- Botting JH, Johnston KM, Macleod BA, Walker MJ. The effect of modification of sympathetic activity on responses to ligation of a coronary artery in the conscious rat. Br J Pharmacol. 1983;79(1):265–271. doi: 10.1111/j.1476-5381.1983.tb10520.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byron JW. Evidence for a ß-adrenergic receptor initiating DNA synthesis in haemopoietic stem cells. Exp Cell Res. 1972;71(1):228–232. doi: 10.1016/0014-4827(72)90283-2. [DOI] [PubMed] [Google Scholar]
- Campbell CA, Parratt JR, Kane KA, Bullock G. Effects of prolonged administration of oxprenolol on severity of ischaemic arrhythmias, enzyme leakage, infarct size, and intracellular cardiac muscle action potentials. J Cardiovasc Pharmacol. 1984;6(3):369–377. doi: 10.1097/00005344-198405000-00001. [DOI] [PubMed] [Google Scholar]
- Clark C, Foreman MI, Kane KA, McDonald FM, Parratt JR. Coronary artery ligation in anesthetized rats as a method for the production of experimental dysrhythmias and for the determination of infarct size. J Pharmacol Methods. 1980;3(4):357–368. doi: 10.1016/0160-5402(80)90077-7. [DOI] [PubMed] [Google Scholar]
- Clements-Jewery H, Hearse DJ, Curtis MJ. Independent contribution of catecholamines to arrhythmogenesis during evolving infarction in the isolated rat heart. Br J Pharmacol. 2002a;135(3):807–815. doi: 10.1038/sj.bjp.0704509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements-Jewery H, Hearse DJ, Curtis MJ. The isolated blood-perfused rat heart: an inappropriate model for the study of ischaemia- and infarction-related ventricular fibrillation. Br J Pharmacol. 2002b;137(7):1089–1099. doi: 10.1038/sj.bjp.0704977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements-Jewery H, Hearse DJ, Curtis MJ. Phase 2 ventricular arrhythmias in acute myocardial infarction: a neglected target for therapeutic antiarrhythmic drug development and for safety pharmacology evaluation. Br J Pharmacol. 2005;145(5):551–564. doi: 10.1038/sj.bjp.0706231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements-Jewery H, Hearse DJ, Curtis MJ. Neutrophil ablation with anti-serum does not protect against phase 2 ventricular arrhythmias in anaesthetised rats with myocardial infarction. Cardiovasc Res. 2007;73(4):761–769. doi: 10.1016/j.cardiores.2006.12.017. [DOI] [PubMed] [Google Scholar]
- Curtis MJ. Characterisation, utilisation and clinical relevance of isolated perfused heart models of ischaemia-induced ventricular fibrillation. Cardiovasc Res. 1998;39(1):194–215. doi: 10.1016/s0008-6363(98)00083-2. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Hearse DJ. Ischaemia-induced and reperfusion-induced arrhythmias differ in their sensitivity to potassium: implications for mechanisms of initiation and maintenance of ventricular fibrillation. J Mol Cell Cardiol. 1989;21(1):21–40. doi: 10.1016/0022-2828(89)91490-9. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, MacLeod BA, Walker MJ. Antiarrhythmic actions of verapamil against ischaemic arrhythmias in the rat. Br J Pharmacol. 1984;83(2):373–385. doi: 10.1111/j.1476-5381.1984.tb16497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dynon MK, Jarrott B, Louis WJ. Tissue distribution and hypotensive effect of prazosin in the conscious rat. J Cardiovasc Pharmacol. 1983;5(2):235–239. doi: 10.1097/00005344-198303000-00012. [DOI] [PubMed] [Google Scholar]
- Elenkov IJ, Wilder RL, Chrousos GP, Vizi ES. The sympathetic nerve – an integrative interface between two supersystems: the brain and the immune system. Pharmacol Rev. 2000;52(4):595–638. [PubMed] [Google Scholar]
- Ferdinandy P, Schulz R, Baxter GF. Interaction of cardiovascular risk factors with myocardial ischemia/reperfusion injury, preconditioning, and postconditioning. Pharmacol Rev. 2007;59(4):418–458. doi: 10.1124/pr.107.06002. [DOI] [PubMed] [Google Scholar]
- Hamilton CA, Reid JL, Vincent J. Pharmacokinetic and pharmacodynamic studies with two alpha-adrenoceptor antagonists, doxazosin and prazosin in the rabbit. Br J Pharmacol. 1985;86(1):79–87. doi: 10.1111/j.1476-5381.1985.tb09437.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris AS. Delayed development of ventricular ectopic rhythms following experimental coronary occlusion. Circulation. 1950;1:1318–1328. doi: 10.1161/01.cir.1.6.1318. [DOI] [PubMed] [Google Scholar]
- Harvath L, Robbins JD, Russell AA, Seamon KB. cAMP and human neutrophil chemotaxis. Elevation of cAMP differentially affects chemotactic responsiveness. J Immunol. 1991;146(1):224–232. [PubMed] [Google Scholar]
- Jaillon P. Clinical pharmacokinetics of prazosin. Clin Pharmacokinet. 1980;5(4):365–376. doi: 10.2165/00003088-198005040-00004. [DOI] [PubMed] [Google Scholar]
- Johnston KM, MacLeod BA, Walker MJ. Responses to ligation of a coronary artery in conscious rats and the actions of antiarrhythmics. Can J Physiol Pharmacol. 1983;61(11):1340–1353. doi: 10.1139/y83-193. [DOI] [PubMed] [Google Scholar]
- Kennedy HL, Whitlock JA, Sprague MK, Kennedy LJ, Buckingham TA, Goldberg RJ. Long-term follow-up of asymptomatic healthy subjects with frequent and complex ventricular ectopy. N Engl J Med. 1985;312(4):193–197. doi: 10.1056/NEJM198501243120401. [DOI] [PubMed] [Google Scholar]
- Lameris TW, de Zeeuw S, Alberts G, Boomsma F, Duncker DJ, Verdouw PD, et al. Time course and mechanism of myocardial catecholamine release during transient ischemia in vivo. Circulation. 2000;101(22):2645–2650. doi: 10.1161/01.cir.101.22.2645. [DOI] [PubMed] [Google Scholar]
- Lepran I, Koltai M, Siegmund W, Szekeres L. Coronary artery ligation, early arrhythmias, and determination of the ischemic area in conscious rats. J Pharmacol Methods. 1983;9(3):219–230. doi: 10.1016/0160-5402(83)90041-4. [DOI] [PubMed] [Google Scholar]
- Maestroni GJ, Conti A, Pedrinis E. Effect of adrenergic agents on hematopoiesis after syngeneic bone marrow transplantation in mice. Blood. 1992;80(5):1178–1182. [PubMed] [Google Scholar]
- Mainland D, Herrera L, Sutcliffe MI. Statistical Tables for Use with Binomial Samples – Contingency Tests, Confidence Limits and Sample Size Estimates. New York: University College of Medicine Publications; 1956. [Google Scholar]
- Milmer KE, Clough DP. Optimum ventilation levels for maintenance of normal arterial blood pO2, pC02, and pH in the pithed rat preparation. J Pharmacol Methods. 1983;10(3):185–192. doi: 10.1016/0160-5402(83)90029-3. [DOI] [PubMed] [Google Scholar]
- Mullane KM, Read N, Salmon JA, Moncada S. Role of leukocytes in acute myocardial infarction in anesthetized dogs: relationship to myocardial salvage by anti-inflammatory drugs. J Pharmacol Exp Ther. 1984;228(2):510–522. [PubMed] [Google Scholar]
- Mullane KM, Kraemer R, Smith B. Myeloperoxidase activity as a quantitative assessment of neutrophil infiltration into ischemic myocardium. J Pharmacol Methods. 1985;14(3):157–167. doi: 10.1016/0160-5402(85)90029-4. [DOI] [PubMed] [Google Scholar]
- Nielson CP. Beta-adrenergic modulation of the polymorphonuclear leukocyte respiratory burst is dependent upon the mechanism of cell activation. J Immunol. 1987;139(7):2392–2397. [PubMed] [Google Scholar]
- Opitz CF, Mitchell GF, Pfeffer MA, Pfeffer JM. Arrhythmias and death after coronary artery occlusion in the rat. Continuous telemetric ECG monitoring in conscious, untethered rats. Circulation. 1995;92(2):253–261. doi: 10.1161/01.cir.92.2.253. [DOI] [PubMed] [Google Scholar]
- Pitchford SC, Yano H, Lever R, Riffo-Vasquez Y, Ciferri S, Rose MJ, et al. Platelets are essential for leukocyte recruitment in allergic inflammation. J Allergy Clin Immunol. 2003;112(1):109–118. doi: 10.1067/mai.2003.1514. [DOI] [PubMed] [Google Scholar]
- Pratt CM, Moye LA. The cardiac arrhythmia suppression trial. Casting suppression in a different light. Circulation. 1995;91(1):245–247. doi: 10.1161/01.cir.91.1.245. [DOI] [PubMed] [Google Scholar]
- Ravingerova T, Tribulova N, Slezak J, Curtis MJ. Brief, intermediate and prolonged ischemia in the isolated crystalloid perfused rat heart: relationship between susceptibility to arrhythmias and degree of ultrastructural injury. J Mol Cell Cardiol. 1995;27(9):1937–1951. doi: 10.1016/0022-2828(95)90016-0. [DOI] [PubMed] [Google Scholar]
- Ridley PD, Yacoub MH, Curtis MJ. A modified model of global ischaemia: application to the study of syncytial mechanisms of arrhythmogenesis. Cardiovasc Res. 1992;26(4):309–315. doi: 10.1093/cvr/26.4.309. [DOI] [PubMed] [Google Scholar]
- Sasaki K, Ueno A, Katori M, Kikawada R. Detection of leukotriene B4 in cardiac tissue and its role in infarct extension through leucocyte migration. Cardiovasc Res. 1988;22(2):142–148. doi: 10.1093/cvr/22.2.142. [DOI] [PubMed] [Google Scholar]
- Schömig A, Dart AM, Dietz R, Mayer E, Kubler W. Release of endogenous catecholamines in the ischemic myocardium of the rat. Part A: locally mediated release. Circ Res. 1984;55(5):689–701. doi: 10.1161/01.res.55.5.689. [DOI] [PubMed] [Google Scholar]
- Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, et al. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150(1):76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- Smith TW, Cain ME. Sudden cardiac death: epidemiologic and financial worldwide perspective. J Interv Card Electrophysiol. 2006;17(3):199–203. doi: 10.1007/s10840-006-9069-6. [DOI] [PubMed] [Google Scholar]
- Walker MJ, Curtis MJ, Hearse DJ, Campbell RW, Janse MJ, Yellon DM, et al. The Lambeth Conventions: guidelines for the study of arrhythmias in ischaemia infarction, and reperfusion. Cardiovasc Res. 1988;22(7):447–455. doi: 10.1093/cvr/22.7.447. [DOI] [PubMed] [Google Scholar]
- Zurier RB, Weissmann G, Hoffstein S, Kammerman S, Tai HH. Mechanisms of lysosomal enzyme release from human leukocytes. II. Effects of cAMP and cGMP, autonomic agonists, and agents which affect microtubule function. J Clin Invest. 1974;53(1):297–309. doi: 10.1172/JCI107550. [DOI] [PMC free article] [PubMed] [Google Scholar]