

Abstract
Medicines that activate cannabinoid CB1 and CB2 receptor are already in the clinic. These are Cesamet® (nabilone), Marinol® (dronabinol; Δ9-tetrahydrocannabinol) and Sativex® (Δ9-tetrahydrocannabinol with cannabidiol). The first two of these medicines can be prescribed to reduce chemotherapy-induced nausea and vomiting. Marinol® can also be prescribed to stimulate appetite, while Sativex® is prescribed for the symptomatic relief of neuropathic pain in adults with multiple sclerosis and as an adjunctive analgesic treatment for adult patients with advanced cancer. One challenge now is to identify additional therapeutic targets for cannabinoid receptor agonists, and a number of potential clinical applications for such agonists are mentioned in this review. A second challenge is to develop strategies that will improve the efficacy and/or the benefit-to-risk ratio of a cannabinoid receptor agonist. This review focuses on five strategies that have the potential to meet either or both of these objectives. These are strategies that involve: (i) targeting cannabinoid receptors located outside the blood-brain barrier; (ii) targeting cannabinoid receptors expressed by a particular tissue; (iii) targeting up-regulated cannabinoid receptors; (iv) targeting cannabinoid CB2 receptors; or (v) ‘multi-targeting’. Preclinical data that justify additional research directed at evaluating the clinical importance of each of these strategies are also discussed.
Keywords: cannabis, Δ9-tetrahydrocannabinol, CB1 receptors, CB2 receptors, cannabinoid receptor agonism, clinical applications, clinical strategies, blood-brain barrier, synergistic interactions, endocannabinoid system
Introduction
There are at least two types of cannabinoid receptor, CB1 and CB2, each of which is G protein-coupled. These receptors can be activated not only by cannabis-derived and synthetic agonists but also by endogenous cannabinoids produced in mammalian tissues and often referred to as ‘endocannabinoids’. The first endocannabinoids to be discovered were N-arachidonoyl ethanolamine (anandamide) and 2- arachidonoyl glycerol (Devane et al., 1992; Mechoulam et al., 1995; Sugiura et al., 1995). These can activate both CB1 and CB2 receptors and are synthesized on demand in response to elevations of intracellular calcium (reviewed in Howlett et al., 2002; Di Marzo et al., 2005). Other ligands that may be endocannabinoids have also been identified (reviewed in Pertwee, 2005a). This system of cannabinoid receptors and endocannabinoids together with the enzymes and processes responsible for endocannabinoid biosynthesis, cellular uptake and metabolism constitutes the ‘endocannabinoid system’ (reviewed in Howlett et al., 2002; Pertwee, 2005b; 2006).
Cannabinoid CB1 receptors are located primarily at the terminals of central and peripheral neurons where they mediate inhibition of ongoing release of various neurotransmitters that include acetylcholine, noradrenaline, dopamine, 5-hydroxytryptamine, γ-aminobutyric acid, glutamate, D-aspartate and cholecystokinin (reviewed in Howlett et al., 2002; Pertwee and Ross, 2002; Szabo and Schlicker, 2005). Cannabinoid CB2 receptors are expressed mainly by immune cells. When activated, they too can affect the release of chemical messengers, in this case the secretion of cytokines by immune cells, and can in addition modulate immune cell trafficking (Ni et al., 2004; Xu et al., 2007; Zhang et al., 2007; see Walter and Stella, 2004; Cabral and Staab, 2005; Pertwee, 2005b for reviews). CB1 receptors are also present in some non-neuronal cells, including immune cells, and CB2 receptors in some central and peripheral neurons (Skaper et al., 1996; Ross et al., 2001; Van Sickle et al., 2005; Wotherspoon et al., 2005; Beltramo et al., 2006; Gong et al., 2006; Baek et al., 2008; see Onaivi, 2006 for a review). However, the role of neuronal CB2 receptors has still to be established. As well as orthosteric site(s), the CB1 receptor possesses one or more allosteric sites that can be targeted by ligands in a manner that enhances or inhibits the activation of this receptor by both exogenously administered and endogenously released direct agonists (Price et al., 2005; Adam et al., 2007; Horswill et al., 2007).
The discovery of cannabinoid receptors prompted the development of CB1- and CB2-selective agonists and antagonists and of these, those that have most often been used as research tools are listed in Figure 1. Included in this list are the antagonists, N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide hydrochloride (SR141716A), N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (AM251), N-(morpholin-4-yl)-1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-1H-pyrazole-3-carboxamide (AM281), N-[(1S)-endo-1,3,3-trimethyl bicyclo [2.2.1] heptan-2-yl]-5-(4-chloro-3-methylphenyl)-1-(4-methylbenzyl)-pyrazole-3-carboxamide (SR144528) and 6-iodo-2-methyl-1-[2-(4-morpholinyl)ethyl]-1H-indol-3-yl](4-methoxyphenyl)methanone (AM630). These all behave as inverse agonists rather than as neutral antagonists, one indication that CB1 and CB2 receptors can exist in a constitutively active state (reviewed in Pertwee, 2005b,c). Certain agonists that bind more or less equally well to CB1 and CB2 receptors are also widely used as research tools (Figure 1). They include the main psychoactive constituent of cannabis, Δ9-tetrahydrocannabinol (Δ9-THC) and the endocannabinoid, anandamide, each of which behaves as a partial agonist at both CB1 and CB2 receptors (reviewed in Howlett et al., 2002; Pertwee, 2005b; 2008a). The other CB1/CB2 receptor agonists listed in Figure 1 all display relatively high CB1 and CB2 efficacy (reviewed in Howlett et al., 2002; Pertwee, 2005b). These are the endocannabinoid, 2-arachidonoyl glycerol, and the synthetic compounds, 11-hydroxy-Δ8-THC-dimethylheptyl [(6aR)-trans-3-(1,1-dimethylheptyl)-6a,7,10,10a-tetrahydro-1-hydroxy-6,6-dimethyl-6H-dibenzo[b,d]pyran-9-methanol (HU-210)], (−)-cis-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-trans-4-(3-hydroxypropyl)cyclohexanol (CP55940) and (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo-[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone [R-(+)-WIN55212].
Figure 1.
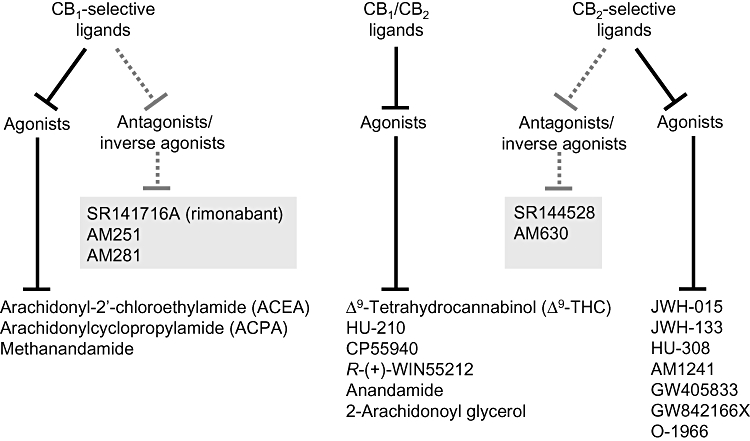
Cannabinoid receptor ligands that are often used as research tools. The structures of these ligands and descriptions of their pharmacological properties can be found elsewhere (Howlett et al., 2002; Wiley et al., 2002; Pertwee, 2005b; Whiteside et al., 2007; Zhang et al., 2007; Guindon and Hohmann, 2008). AM1241, (2-iodo-5-nitrophenyl)-[1-(1-methylpiperidin-2-ylmethyl)-1H-indol-3-yl]-methanone; AM251, N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide; AM281, N-(morpholin-4-yl)-1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-1H-pyrazole-3-carboxamide; AM630, 6-iodo-2-methyl-1-[2-(4-morpholinyl)ethyl]-1H-indol-3-yl](4-methoxyphenyl)methanone; anandamide, N-arachidonoyl ethanolamine; CP55940, (−)-cis-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-trans-4-(3-hydroxypropyl)cyclohexanol; HU-210, (6aR)-trans-3-(1,1-dimethylheptyl)-6a,7,10,10a-tetrahydro-1-hydroxy-6,6-dimethyl-6H-dibenzo[b,d]pyran-9-methanol; JWH-015, (2-methyl-1-propyl-1H-indol-3-yl)-1-naphthalenylmethanone; JWH-133, 3-(1,1-dimethylbutyl)-6,6,9-trimethyl-6α,7,10,10α-tetrahydro-6H-benzo[c]chromene; R-(+)-WIN55212, (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo-[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone; SR141716A, N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide hydrochloride; SR144528, N-[(1S)-endo-1,3,3-trimethyl bicyclo [2.2.1] heptan-2-yl]-5-(4-chloro-3-methylphenyl)-1-(4-methylbenzyl)-pyrazole-3-carboxamide.
Licensed medicines that target cannabinoid receptors
Unlike cannabis, which has been used as a medicine over many centuries (reviewed in Mechoulam, 1986), individual cannabinoid receptor agonists made their first entry into the clinic less than 30 years ago (reviewed in Pertwee and Thomas, 2009). The first of these was the CB1/CB2 receptor agonist, nabilone (Cesamet®), which is a synthetic analogue of Δ9-THC and was licensed in 1981 for the suppression of nausea and vomiting produced by chemotherapy. Δ9-THC is also a licensed medicine. It initially entered the clinic as Marinol® (dronabinol), in 1985 as an anti-emetic and in 1992 as an appetite stimulant, for example for AIDS patients experiencing excessive loss of body weight. In 2005, nabilone and dronabinol were joined by the cannabis-based medicine, Sativex®. This contains approximately equal amounts of Δ9-THC and the non-psychoactive plant cannabinoid, cannabidiol, and is prescribed for the symptomatic relief of neuropathic pain in adults with multiple sclerosis and as an adjunctive analgesic treatment for adult patients with advanced cancer. Cannabinoid CB1/CB2 receptor agonists also have a number of potential therapeutic targets (Bambico et al., 2007; Rubio-Araiz et al., 2008; see Izzo and Camilleri, 2008; Pertwee, 2008b; Pertwee and Thomas, 2009 for reviews). These include the
relief of pain induced by certain disorders or conditions in addition to cancer and multiple sclerosis;
management of some gastrointestinal disorders;
management of atherosclerosis and of certain other cardiovascular disorders;
inhibition of angiogenesis and growth of malignant tumours;
relief from various symptoms of multiple sclerosis, spinal cord injury, Alzheimer's disease and amyotrophic lateral sclerosis;
relief from tics and behavioural problems experienced by patients with Tourette's syndrome;
management of anxiety disorders, attention-deficit hyperactivity disorder, depression and brain repair;
management of tardive dyskinesia induced in psychiatric patients by neuroleptic drugs;
management of glaucoma, cough and cholestatic pruritus.
CB1/CB2 receptor agonists can of course produce adverse effects in patients, and many of these are probably caused by the activation of central CB1 receptors rather than of CB2 or peripheral CB1 receptors. Adverse effects most often observed, at least in clinical trials with multiple sclerosis patients, have been dizziness/light-headedness, dry mouth, tiredness/fatigue, muscle weakness, myalgia (muscle pain) and palpitations (reviewed in Pertwee, 2007a). Other less frequently reported side effects of CB1/CB2 agonists include disorientation, feeling of drunkenness, ‘high sensation’, mental clouding and/or altered time perception, impairment of memory or ability to concentrate, tremor, balance impairment or lack of coordination, nausea/feeling sick, hypotension, blurred vision, constipation or diarrhoea, confusion, dysphoria/depression, disorientation, paranoia and hallucinations (reviewed in Pertwee, 2007a; Pertwee and Thomas, 2009). It is possible, however, for patients being treated with a cannabinoid receptor agonist to optimize the benefit-to-risk ratio by downward self-titration of the dose they take (Brady et al., 2004; Svendsen et al., 2004; Wade et al., 2004). It is also noteworthy that there is evidence that tolerance develops more readily to some of the unwanted effects of a CB1/CB2 receptor agonist than to some of its sought-after therapeutic effects. This comes both from a clinical trial in which multiple sclerosis patients had been repeatedly treated with Δ9-THC (dronabinol) (Svendsen et al., 2004) and from experiments in which rats had been repeatedly injected with CP55940 (De Vry et al., 2004).
The only cannabinoid receptor antagonist to have been licensed as a medicine to-date is the CB1 antagonist/inverse agonist, SR141716A (rimonabant; Acompliat®) (Després et al., 2006). This was introduced into European clinics in 2006 for the management of obesity. Adversere actions experienced by some patients in clinical trials with rimonabant, mainly during the first few months, included nausea, vomiting, diarrhoea, headache, dizziness, arthralgia (pain in a joint), insomnia, influenza, feelings of anxiety and depression (Després et al., 2005; Van Gaal et al., 2005; 2008). Unfortunately, safety concerns about the adverse effects of rimonabant in patients taking it as an anti-obesity agent, particularly an increased incidence of depression and suicidality, recently prompted the European Medicines Agency to recommend sales of this drug to be halted. Rimonabant does of course remain an extremely valuable experimental tool. However, it is likely that most pharmaceutical companies will be deterred, at least for the time being, from developing a drug that displays rimonabant-like CB1 receptor inverse agonist/antagonist activity for the management of any disorders. In addition to obesity and type-2 diabetes, these disorders include some that are discussed in this review and also nicotine dependence, impaired fertility in some women, stroke, hypotension resulting from endotoxaemic shock triggered by advanced liver cirrhosis and intestinal hypomotility in paralytic ileus (Izzo and Coutts, 2005; Le Foll and Goldberg, 2005; Pertwee, 2005a; Di Marzo, 2008; Pertwee and Thomas, 2009). Clearly then there is an urgent need for a new strategy for blocking CB1 receptors that shares the generally acknowledged effectiveness of rimonabant against obesity, type-2 diabetes and associated cardiometabolic risk factors (reviewed in Di Marzo, 2008) but not its apparent ability to produce signs of anxiety and marked depression/suicidal ideation in some patients. Possible solutions to this problem include administering a neutral antagonist because it lacks the CB1 inverse agonist activity of rimonabant (reviewed in Pertwee 2005b,c), developing a peripherally restricted drug that selectively blocks CB1 receptors expressed outside the brain or an allosteric antagonist that selectively blocks the CB1 receptor-mediatedactions of just one of the endocannabinoids, or applying an adjunctive strategy that exploits synergism between a low dose of a CB1 receptor antagonist and some other type of anti-obesity agent.
Emerging strategies for targeting cannabinoid receptors in the clinic
There is currently tremendous interest in the possibility of developing a second generation of medicines that will activate or block the endocannabinoid system with improved efficacy and/or selectivity. This interest has been stimulated both by the successful development of cannabinoid receptor ligands as medicines and by the likelihood that such compounds have a number of as yet unexploited but significant clinical applications. No doubt it has also been sparked by the discovery that one important role of the endocannabinoid system is to maintain homeostasis in health and disease (reviewed in Pertwee, 2005a). Thus, it has been found that there are certain disorders, including several that are in urgent need of better therapies, in which the endocannabinoid system seems to up-regulate in particular cells or tissues through increased endocannabinoid release, increased expression of CB1 or CB2 receptors and/or an increase in the coupling efficiency of these receptors. This up-regulation appears to lead in some instances to ‘autoprotection’ through a suppression of unwanted signs and symptoms or even a slowing of disease progression, and in other instances to ‘autoimpairment’ through the production or exacerbation of undesirable effects. The ever-growing list of disorders in which the endocannabinoid system seems to have autoprotective or autoimpairing roles has provided additional rationale for the accepted uses of cannabinoid receptor ligands as medicines and is, in addition, helping to identify or justify new therapeutic uses for such ligands that include the potential therapeutic applications listed in the previous section (Licensed medicines that target cannabinoid receptors).
Knowledge gained about the endocannabinoid system has revealed several potential strategies either for mimicking its autoprotective effects in patients with improved selectivity or for enhancing these autoprotective effects in the clinic. Some of these can be termed ‘direct strategies’ because they rely on the administration of a direct agonist and have in common the objective of restricting the activation of cannabinoid receptors by such a ligand as far as possible to subpopulations of these receptors that, when activated, generate sought-after rather than unwanted effects. The other strategies can be termed ‘indirect’ because their purpose is to augment sought-after effects resulting from the endogenous release of endocannabinoids onto certain of their receptors. These are strategies that exploit the ability of an ‘indirect agonist’ either to inhibit the cellular uptake or enzymatic degradation of endocannabinoids and so delay their removal from their sites of action or to target the allosteric sites on CB1 receptors in a manner that will enhance the ability of released endocannabinoids to activate these receptors.
The therapeutic potential of inhibitors of the cellular uptake or enzymatic degradation of endocannabinoids and, indeed, of CB1 receptor antagonists was the subject of a recent review (Di Marzo, 2008), as were the potential clinical applications both of CB1 receptor allosteric modulators (Ross, 2007) and of CB2 receptor inverse agonists (Lunn et al., 2008). Consequently, the remainder of this review focuses solely on potential ‘direct strategies’ for improving the selectivity and/or efficacy as medicines of cannabinoid receptor agonists. These are strategies that involve
targeting cannabinoid receptors located outside the blood-brain barrier;
targeting cannabinoid receptors expressed by a particular tissue;
targeting up-regulated cannabinoid receptors;
targeting cannabinoid CB2 receptors;
‘multi-targeting’.
Targeting cannabinoid receptors located outside the blood-brain barrier
CB1 and/or CB2 receptors expressed outside the central nervous system appear to mediate a number of sought-after effects that include pain relief, amelioration of certain intestinal, cardiovascular and hepatic disorders and inhibition of cancer cell proliferation and spread (reviewed in Pertwee, 2005a). Consequently, because many of the unwanted effects of cannabinoid receptor agonists are probably mediated by CB1 receptors located within the brain, interest is growing in the possibility of developing a direct CB1/CB2 cannabinoid receptor agonist that on the one hand is excluded from much of the brain and spinal cord because it does not readily cross the blood-brain barrier but on the other hand still retains an ability to produce sought-after clinical effects by activating peripherally located cannabinoid receptors.
Preclinical proof of principal for this approach was recently provided by Dziadulewicz et al. (2007) of Novartis Pharma AG who have developed a potent, high-efficacy, orally bioavailable CB1/CB2 receptor agonist that displays both marked antihyperalgesic activity in the Seltzer (sciatic nerve partial ligation) rat model of neuropathic pain and limited brain penetration. That this compound, naphthalen-1-yl-(4-pentyloxynaphthalen-1-yl)methanone, was producing its antihyperalgesic effect by targeting CB1 receptors outside the blood-brain barrier is supported both by pharmacokinetic data and by the observations first, that its antihyperalgesic effect was attenuated by the CB1-selective antagonist, SR141716A, but not by the CB2-selective antagonist, SR144528 and second, that it could produce this effect without inducing any sign of catalepsy. More recently, the development of another peripherally restricted, potent, orally active CB1/CB2 receptor agonist (compound A) was announced by Organon/Schering Plough (Boyce, 2008). This produces antihyperalgesic effects both in a rat spinal nerve ligation model of neuropathic pain and in the mouse formalin paw model of inflammatory pain at doses well below any that cause catalepsy, at least in rats.
Another compound that may ameliorate neuropathic pain mainly by targeting cannabinoid receptors located outside the blood-brain barrier is ajulemic acid (CT-3), a synthetic analogue of Δ8-THC (Dyson et al., 2005; Mitchell et al., 2005). More specifically, this compound has been reported to behave as a potent, high-efficacy CB1 and CB2 receptor agonist and to penetrate rat brain less readily than Δ9-THC or R-(+)-WIN55212. It has also been reported to reverse signs of hyperalgesia in the Seltzer rat model of neuropathic pain and signs of inflammatory pain induced in rats by intraplantar injection of Freund's complete adjuvant in a manner that can be blocked by SR141716A but not by SR144528, and to exhibit greater potency in these pain models than in inducing effects such as catalepsy that are thought to require the activation of central CB1 receptors. In addition, ajulemic acid has been found to display analgesic efficacy in a randomized, placebo-controlled, double-blind cross-over clinical trial performed with patients with chronic neuropathic pain. However although significant efficacy was observed for one primary outcome measure (visual analogue scale score) it was not observed for another (verbal rating scale score) or, indeed, when the measured response was mechanical hypersensitivity to von Frey hairs (Karst et al., 2003; Salim et al., 2005). Reported adverse reactions to ajulemic acid were dry mouth, tiredness, dizziness, limited power of concentration, sweating and more pain, suggesting that it did enter the brain in an amount sufficient to trigger some CB1-mediated side effects.
When considering the merits of developing a peripherally restricted cannabinoid receptor agonist, it is important to bear in mind evidence that the permeability of the blood-brain barrier may increase in some disorders that, for example, include Alzheimer's disease and Parkinson's disease (Desai et al., 2007). There is, however, another possible strategy for targeting cannabinoid receptors expressed outside the brain that does not rely on the integrity of the blood-brain barrier and this is described in next section.
Targeting cannabinoid receptors expressed by a particular tissue
Some tissues located outside the brain express cannabinoid receptors that appear to mediate effects that are mainly beneficial. Consequently, another strategy for reducing the unwanted effects of CB1 receptor agonism might be to limit the distribution of active concentrations of an administered cannabinoid receptor agonist to one or more tissues of this kind.
One possibility, at least for the relief of some types of pain, would be to inject a cannabinoid receptor agonist intrathecally as there is good evidence that activation of cannabinoid CB1 receptors within the spinal cord can induce antinociceptive effects in preclinical models of acute, inflammatory and neuropathic pain (reviewed in Pertwee, 2001; Walker and Hohmann, 2005). CB2 receptors within the spinal cord are also thought to mediate antinociception, for example in animal models of neuropathic pain. These may be CB2 receptors expressed by microglial cells or even by neurons. Thus, there is evidence first, that after peripheral nerve injury CB2 receptors appear on sensory neurons and/or activated microglia in mouse or rat spinal cord, and second, that CB2 receptors are expressed by neonatal rat dorsal root ganglion neurons (Ross et al., 2001; Zhang et al., 2003; Walczak et al., 2005; 2006; Wotherspoon et al., 2005). It will be important to establish the extent to which a CB1 or CB1/CB2 receptor agonist produces off-target effects in the clinic when given to patients intrathecally at doses that relieve unwanted symptoms.
For the management of symptoms that are generated in specific regions of the body surface, another possibility would be to limit the distribution of active concentrations of an administered cannabinoid receptor agonist to these regions by applying it directly to the skin, for example by skin patch. Thus there is evidence first, that cannabinoid CB1 and CB2 receptors are present in the skin, for example in cutaneous nerve fibres, mast cells, macrophages and epidermal keratinocytes and also in the epithelial cells of hair follicles, sebocytes and eccrine sweat glands, and second, that cutaneous CB1 and CB2 receptor activation produces antinociception in preclinical models of acute, inflammatory and neuropathic pain (Ständer et al., 2005; see Pertwee, 2001; Fox and Bevan, 2005; Whiteside et al., 2007; Guindon and Hohmann, 2008 for reviews). In addition, it has been found that pretreatment of human volunteers with HU-210 by skin patch (50 mmol·L−1) or dermal microdialysis (5 mmol·L−1) can significantly decrease mechanical and thermal hyperalgesia induced by capsaicin application to the skin and the perception of itch induced by topically applied histamine (Dvorak et al., 2003; Rukwied et al., 2003). Skin blood flow and neurogenic flare responses to histamine were also attenuated by HU-210. Importantly, no psychological side effects were detected in response to HU-210 in these experiments. It has also been found that topical administration of the CB1/CB2 receptor agonist, R-(+)-WIN55212, to mice can induce antinociception in the radiant heat tail-flick test at a dose that does not impair rotarod performance and in a manner that is susceptible to antagonism by AM251 (Dogrul et al., 2003; Yesilyurt et al., 2003). Similarly, Potenzieri et al. (2008) have found that injection of R-(+)-WIN55212 into tumour-bearing hindpaws of mice can induce signs of reduced hyperalgesia without also producing catalepsy. This antinociceptive effect could be blocked both by AM251 and by the CB2-selective antagonist, AM630.
Clearly then, it may well prove possible to relieve the unwanted symptoms of some disorders and yet avoid major off-target CB1-mediated effects by administering a cannabinoid CB1 or CB1/CB2 receptor agonist directly into tissues such as the skin or spinal cord.
Targeting up-regulated cannabinoid receptors
It is possible that some disorders could be treated with improved selectivity by administering a cannabinoid receptor partial agonist rather than a full agonist. These would be disorders that trigger a ‘protective’ up-regulation of subpopulations of cannabinoid CB1 and/or CB2 receptors that can mediate symptom relief or oppose disease progression. Evidence that such ‘protective’ up-regulation occurs has, for example, been obtained for
CB1 receptors in the brain in rodent models of stroke (Jin et al., 2000) and temporal lobe epilepsy (Wallace et al., 2003);
CB1 receptors in the upper intestinal tract in mouse models of intestinal inflammation and diarrhoea (Izzo et al., 2001; 2003; see Izzo and Camilleri, 2008 for a review);
CB2 receptors in the upper intestinal tract in a rat model of lipopolysaccharide-enhanced gastrointestinal transit (Duncan et al., 2008; see Izzo and Camilleri, 2008 for a review);
CB1 receptors in enteric neurons and CB2 receptors in colonic infiltrated immune cells in mouse models of colitis (Massa et al., 2004; Kimball et al., 2006);
CB1 and CB2 receptors in human hepatocellular carcinoma tumours, non-Hodgkin lymphomas and human prostate cancer cells (Sarfaraz et al., 2005; Xu et al., 2006; Gustafsson et al., 2008);
CB1 and/or CB2 receptors in brain, spinal cord, dorsal root ganglia and skin in rat or mouse models of neuropathic pain (Siegling et al., 2001; Lim et al., 2003; Zhang et al., 2003; Walczak et al., 2005; 2006; Wotherspoon et al., 2005; Beltramo et al., 2006; Mitrirattanakul et al., 2006);
CB2 receptors in macrophages and T lymphocytes located in atherosclerotic plaques (Steffens et al., 2005);
CB2 receptors in microglial cells and macrophages in regions of human post mortem spinal cord affected by multiple sclerosis or amyotrophic lateral sclerosis (Yiangou et al., 2006) and in the central nervous systems of mice with experimental autoimmune encephalomyelitis (Maresz et al., 2005).
Intriguingly, there is also evidence, first that CB1 receptor expression in human colorectal tumours is down-regulated due to methylation of the CB1 promoter, and second that it might be possible to enhance the ability of a CB1 receptor agonist to inhibit the growth of these tumours by first boosting CB1 receptor levels in human colorectal tumours with a demethylating agent (Wang et al., 2008).
Provided that there is no concomitant increase in the expression of CB1 or CB2 receptors that mediate adverse effects, such seemingly ‘protective’ up-regulation should according to the receptor occupation theory of drug action improve the benefit-to-risk ratio of a CB1/CB2 receptor agonist by selectively increasing its potency for producing sought-after effects while leaving its potency for the production of unwanted effects unchanged. Importantly, up-regulation of this kind can also augment the size of the maximal response to a partial agonist while producing little or no increase in the maximal response to a full agonist. Consequently, any increase in the benefit-to-risk ratio that results from receptor up-regulation could well be greater when the administered drug is a partial CB1/CB2 receptor agonist such as Δ9-THC than when it is a high-efficacy agonist such as CP55940. Support for this hypothesis comes from the finding that an increase in CB1 expression level in the mouse small intestine was accompanied not only by a leftward shift in the in vivo log dose–response curve of the CB1 receptor partial agonist, cannabinol, for inhibition of intestinal transit but also by an increase in the size of its maximal effect (Izzo et al., 2001). CP55940, which has higher CB1 efficacy than cannabinol (reviewed in Pertwee, 1999), exhibited a potency increase but no change in its maximal effect. Still lacking, however, are any in vivo human data indicating whether or not the benefit-to-risk ratio for the management of any disorder in which there is an up-regulation of cannabinoid receptors that is both protective and selective would be significantly higher for a CB1/CB2 receptor partial agonist than for a full agonist. It should also be borne in mind that there are certain brain areas in which CB1 receptors are particularly highly expressed and that consequently there will most likely be some unwanted effects that are produced in patients no less effectively by a low-efficacy CB1 receptor agonist than by a high-efficacy CB1 agonist (Gifford et al., 1999; see Howlett et al., 2002 for a review).
It is noteworthy that both Δ9-THC and a Δ9-THC-containing cannabis extract have been reported to be ineffective against acute pain in human volunteers when administered at doses that did not produce serious side effects (Naef et al., 2003; Roberts et al., 2006; Kraft et al., 2008). This finding could possibly be an indication that the expression level of cannabinoid receptors in these healthy human subjects was not high enough for a partial agonist such as Δ9-THC to produce CB1 receptor-mediated analgesia, at least at an acceptable dose. It would then be consistent with the hypothesis that Δ9-THC relieves some types of chronic pain in humans with reasonable selectivity because this is pain that has been caused by disorders that selectively up-regulate the cannabinoid receptors that mediate this pain relief. As to the ability of Δ9-THC to reduce signs of acute pain in some animals, this could possibly be an indication that the expression or coupling efficiency of cannabinoid receptors capable of mediating relief from this kind of pain is relatively high in these animals. However, it could also be that Δ9-THC appears to be particularly effective in inducing signs of antinociception in animal models of acute pain because these models rely on the ability of an animal to exhibit a motor response to a noxious stimulus. Thus, some species, including rodents, seem to be more sensitive than humans to the motor impairing effects of a CB1 receptor agonist (reviewed in Pertwee, 2008a).
Targeting cannabinoid CB2 receptors
Evidence obtained mainly from experiments with rats or mice suggests that another strategy for circumventing the unwanted consequences of cannabinoid CB1 receptor activation, for example for the amelioration of some types of pain, may be to target CB2 receptors. More specifically, it has been possible to demonstrate that CB2-selective agonists display antinociceptive activity in well-validated models of acute pain, persistent inflammatory pain, post-operative pain, cancer pain and neuropathic pain (reviewed in Whiteside et al., 2007; Guindon and Hohmann, 2008). That this antinociceptive activity is CB2 receptor-mediated is supported by reports that it can be induced by CB2-selective agonists, that it is susceptible to antagonism by CB2- but not CB1-selective antagonists and/or that it can be abolished by genetic deletion of the CB2 receptor but not of the CB1 receptor. Importantly, it has also been possible to demonstrate that systemically administered CB2-selective agonists can induce antinociception in rodents at doses that do not also produce effects known to result from the activation of central CB1 receptors (reviewed in Whiteside et al., 2007; Guindon and Hohmann, 2008).
The precise locations of the CB2 receptors that mediate antinociception in these animal models remain to be established. However there is evidence that these could include brain areas such as the thalamus as well as skin tissue, dorsal root ganglion neurons and microglia or neurons in the spinal cord (Jhaveri et al., 2008; Yamamoto et al., 2008; see Whiteside et al., 2007; Guindon and Hohmann, 2008 for reviews).
Other potential therapeutic targets for CB2-selective agonists include multiple sclerosis (reviewed in Pertwee, 2007a; Dittel, 2008), amyotrophic lateral sclerosis (Kim et al., 2006; Shoemaker et al., 2007), Huntington's disease (reviewed in Sagredo et al., 2007), stroke (Zhang et al., 2007; see Pacher and Hasko, 2008 for a review), atherosclerosis (reviewed in Mach et al., 2008), gastrointestinal inflammatory states (reviewed in Izzo and Camilleri, 2008; Wright et al., 2008), chronic liver diseases (reviewed in Mallat et al., 2007; Izzo and Camilleri, 2008; Lotersztajn et al., 2008) and cancer (reviewed in Guzmán, 2003; Izzo and Camilleri, 2008; Wright et al., 2008).
When developing novel CB2-selective agonists and attempting to predict their in vivo efficacies from in vitro data it is important to bear in mind that some of these compounds may interact with CB2 receptors in a manner that varies both with species and with the constitutive activity of these receptors. Thus, for example, Bingham et al. (2007) found one such compound, R,S-AM1241 [(2-iodo-5-nitrophenyl)-[1-(1-methylpiperidin-2-ylmethyl)-1H-indol-3-yl]-methanone], to behave in vitro as an agonist at human CB2 receptors but as an inverse agonist at mouse and rat CB2 receptors, and Yao et al. (2006) have found that R,S-AM1241 switches from producing agonism to producing inverse agonism at human CB2 receptors in vitro when the constitutive activity of these receptors is increased, suggesting that this compound may be a ‘protean agonist’.
It should also be borne in mind that none of the CB2-selective agonists that have been developed to date are completely CB2-specific. Thus they are all expected to display CB2-selectively only within a finite dose range and to target CB1 receptors as well when administered at a dose that lies above this range. For any particular agonist, the width of its CB2-selectivity window will of course be affected by the CB1 to CB2 receptor ratios of the tissues in which sought-after (or unwanted) effects are induced by CB1 or CB2 receptor activation. It is noteworthy, therefore, that not all tissues express CB1 and CB2 receptors in equal numbers in health and that there is also evidence that disparate changes in CB1 and CB2 expression levels may be induced in some cells or tissues either pathologically or pharmacologically (reviewed in Pertwee, 2005a). Consequently, when an agonist that binds more readily to CB2 receptors is administered to patients, it is likely that the CB2 selectivity of this ligand will not be the same in all tissues that express both types of cannabinoid receptor and also that any selectivity will be lost when a certain dose level is exceeded.
Multi-targeting
One other strategy for increasing the benefit-to-risk ratio of a CB1 or CB1/CB2 receptor agonist could be to rely on its ability to interact additively or synergistically with another type of ligand for the production of a sought-after effect. Attention has focused particularly on numerous reports that Δ9-THC can interact additively or synergistically with opioid analgesics such as morphine, codeine or fentanyl in the production of combined cannabinoid CB1 and opioid receptor-mediated antinociception in mouse, rat, guinea-pig and monkey models of acute pain (Cichewicz and McCarthy, 2003; Cichewicz et al., 2005; Tham et al., 2005; Williams et al., 2006; Smith et al., 2007; Li et al., 2008; see Pertwee, 2001; Cichewicz, 2004 for reviews), in the rat formalin paw model of inflammatory pain (Finn et al., 2004) and in a rat model of arthritic pain (Cox et al., 2007). Importantly, in some of these investigations, clear evidence was obtained through the construction of isobolograms that cannabinoid and opioid receptor agonists can undergo synergistic rather than just additive interactions in their production of antinociception (Cichewicz and McCarthy, 2003; Tham et al., 2005; Cox et al., 2007). Other important findings have been first, that signs of marked analgesia can be induced by co-administering Δ9-THC and an opioid each at a dose that by itself is sub-effective, and second that low-dose Δ9-THC can restore the efficacy of codeine and morphine at a time when antinociception in the absence of Δ9-THC has worn off (Reche et al., 1996; Williams et al., 2006). Also noteworthy is the finding that repeated administration of a low-dose combination of Δ9-THC and morphine to rats can reduce signs of pain without producing antinociceptive tolerance (Smith et al., 2007). This observation was made in a rat model of acute pain in which such tolerance was induced when Δ9-THC or morphine was administered repeatedly by itself at an antinociceptive dose.
There is also evidence that combined administration of cannabis or Δ9-THC and an opioid can produce beneficial effects in patients experiencing chronic pain. Holdcroft et al. (1997) found that oral administration of a cannabis extract significantly reduced the amount of morphine required for pain relief by a patient with familial Mediterranean fever experiencing chronic relapsing gastrointestinal pain and inflammation. More recently, Narang et al. (2008) carried out a randomized single-dose, double-blinded, placebo-controlled, crossover trial with Δ9-THC (Marinol®) followed by an open-label multi-dose extension study. They found that oral administration of Marinol® produced a significant degree of additional analgesia in 30 patients with chronic non-cancer pain who were taking stable doses of opioid analgesics. Sleep quality was also improved by this adjunctive treatment. Side effects most commonly reported in the initial trial were drowsiness, sleepiness, dizziness and dry mouth. Importantly, the patients in this study preferred Δ9-THC to placebo in spite of these side effects. It is noteworthy, however, that although Δ9-THC appears to interact synergistically with opioid analgesics in animals models of acute pain, no sign of such an interaction was detected either in human volunteers subjected to noxious electrical or thermal stimulation of the skin or to painful digital pressure (Naef et al., 2003; Roberts et al., 2006), or in patients experiencing post-operative pain (Seeling et al., 2006). Interestingly, however, morphine and Δ9-THC, together but not separately, did reduce the affective response to cutaneous thermal stimulation in one of these investigations (Roberts et al., 2006).
Additional research is required to establish the extent to which cannabinoid and opioid receptor agonists interact additively or synergistically in man to produce unwanted effects in addition to those described by Narang et al. (2008), or indeed, to produce sought-after effects in addition to analgesia. In the meantime it is worth noting that there is evidence that cannabinoid receptor agonists may increase opioid-dependence liability. Thus, for example, Manzanedo et al. (2004) have detected additive/synergistic interactions between R-(+)-WIN55212 and morphine in mice for the production of signs of reward in the conditioned place preference paradigm, and Ellgren et al. (2007) have found that repeated pretreatment of rats with Δ9-THC can augment the acquisition and maintenance of heroin self-administration behaviour. It has also been found that repeated Δ9-THC pretreatment can enhance heroin-induced stimulation of rat locomotor activity (Singh et al., 2005). However, there is evidence too that after single administration, R-(+)-WIN55212 and morphine interact additively/synergistically in rats to depress rearing, grooming and locomotor activity (Finn et al., 2004). For reasons already given (section on Targeting up-regulated cannabinoid receptors), such a depressant effect on motor function could explain why Δ9-THC appears to interact additively or synergistically with opioids against signs of acute pain in rats and mice but has so far been found not to undergo an interaction of this kind in human subjects.
Cannabinoid CB1 receptor agonists have also been found to interact additively or synergistically with certain non-opioid, non-cannabinoid compounds in the production of antinociception in animal models of acute or inflammatory pain. More specifically, interactions of this kind have been detected between Δ9-THC or the selective CB1 receptor agonist, arachidonylcyclopropylamide (ACPA), and nicotine, and also between R-(+)-WIN55212 and clonidine, neostigmine, bupivacaine and cyclooxygenase (COX)-2 inhibitors (Table 1). Such interactions have been detected as well in animal models of anxiety, depression and emesis (Table 2), suggesting that it might be possible to exploit interactions between CB1 receptor agonists and non-cannabinoids in the clinic not only for pain relief but also for the management of other unwanted symptoms. Importantly, although the selective 5-HT1A receptor agonist, 8-hydroxy-2-(di-n-propylamino) tetralin hydrobromide (8-OH-DPAT) and Δ9-THC have been reported to interact synergistically in rats for the production of an anxiolytic effect (Table 2), there are reports too that 8-OH-DPAT can oppose both Δ9-THC-induced catalepsy in mice (Egashira et al., 2006) and Δ9-THC-induced memory impairment and hypothermia in rats (Malone and Taylor, 2001; Inui et al., 2004).
Table 1.
Cannabinoid receptor agonists interact additively or synergistically with certain non-opioids in the production of antinociception in animal models of pain
| Cannabinoid receptor agonist | Co-administered compound | Measured effect | Reference |
|---|---|---|---|
| •low-dose R-(+)-WIN55212 (intracisternal) | •COX-1/2 inhibitor (indomethacin)b or selective COX-2 inhibitor (NS-398)b (intracisternal) | •reduction of nociceptive scratching behaviour induced in rats by injection of formalin into the temporomandibular joint | •Ahn et al. (2007) |
| •R-(+)-WIN55212 (intrathecal) | •α2-adrenoceptor agonist (clonidine)c, cholinesterase inhibitor (neostigmine)c or local anaesthetic (bupivacaine)c (intrathecal) | •antinociception in the rat formalin paw model of inflammatory pain | •Yoon and Choi (2003); Kang et al. (2007) |
| •ACPAa (intracerebroventricular) | •nicotine (intracerebroventricular) | •antinociception in the mouse formalin paw model of inflammatory pain | •Jafari et al. (2008) |
| •CP55940 (subcutaneous) | •α2-adrenoceptor agonist (dexmedetomidine)c (subcutaneous) | •antinociception in a mouse model of acute pain | •Tham et al. (2005) |
| •low-dose Δ9-THC (intraperitoneal) | •low-dose nicotine (subcutaneous) | •antinociception enhanced and THC tolerance onset slowed in mouse models of acute pain | •Valjent et al. (2002) |
| •R-(+)-WIN55212 (intravenous) | •ultra-low dose SR141716Ad (intravenous) | •rat model of acute pain: (a) duration of antinociceptive effect of agonist (single injection), (b) tolerance to agonist (repeated injections) | •Paquette et al. (2007) |
See Figure 1 for further information about the cannabinoid receptor ligands mentioned in this table.
ACPA, arachidonylcyclopropylamide; COX, cyclooxygenase; CP55940, (−)-cis-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-trans-4-(3-hydroxypropyl)cyclohexanol; R-(+)-WIN55212, (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo-[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone; SR141716A, N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide hydrochloride; THC, tetrahydrocannabinol.
CB1-selective agonist.
Neither of these compounds displayed antinociceptive activity by itself and their ability to potentiate R-(+)-WIN55212 did not extend to the COX-1-selective inhibitor, SC-560.
Isobolographic analysis indicated that the interaction between the cannabinoid receptor agonist and each of these compounds was super-additive (synergistic).
CB1-selective antagonist/inverse agonist.
Table 2.
Cannabinoid receptor agonists interact additively or synergistically with other types of compound in animal models of anxiety, depression, emesis, epilepsy and stroke
| Cannabinoid receptor agonist | Co-administered compound | Measured effect | Reference |
|---|---|---|---|
| •low-dose Δ9-THC (intraperitoneal) | •low-dose nicotine (subcutaneous) | •anxiolytic effects in mouse light-dark box and open-field tests | •Valjent et al. (2002) |
| •low-dose Δ9-THC (intraperitoneal) | •low-dose nicotine (subcutaneous) | •anxiolytic effect in mouse elevated plus-maze | •Balerio et al. (2006) |
| •R-(+)-WIN55212a (intraperitoneal) | •diazepama (intraperitoneal) | •anxiolytic effects in mouse elevated plus-maze and hole-board tests | •Naderi et al. (2008) |
| •low-dose Δ9-THC (intraperitoneal) | •5-HT1A-selective agonist (low-dose 8-OH-DPAT) (intraperitoneal) | •anxiolytic effect in rat elevated plus-maze test | •Braida et al. (2007) |
| •low-dose CP55940 (intraperitoneal) | •low-dose imipramine (intraperitoneal) | •antidepressant effect in rat forced swim test | •Adamczyk et al. (2008) |
| •low-dose Δ9-THC (intraperitoneal) | •5-HT3 antagonist (low-dose ondansetron) (intraperitoneal) | •inhibition of vomiting and retching induced in house musk shrews by cisplatin | •Kwiatkowska et al. (2004) |
| •ACEAb (intraperitoneal) | •ultra-low dose AM251d (intraperitoneal) | •protection of mice from pentylenetetrazole-induced seizures as indicated by changes in clonic seizure threshold (PTZ i.v.) and in tonic-clonic-generalized seizure latency and mortality (PTZ i.p.) | •Gholizadeh et al. (2007) |
| •O-1966c (intravenous) | •SR141716Ad (intraperitoneal) | •protection of mice from cerebral ischaemic/reperfusion injury | •Zhang et al. (2008) |
See Figure 1 for further information about the cannabinoid receptor ligands mentioned in this table.
8-OH-DPAT, 8-hydroxy-2-(di-n-propylamino) tetralin hydrobromide; ACEA, arachidonyl-2′-chloroethylamide; AM251, N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide; CP55940, (−)-cis-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-trans-4-(3-hydroxypropyl)cyclohexanol; i.p., intraperitoneal; i.v., intravenous; PTZ, pentylenetetrazole; R-(+)-WIN55212, (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo-[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone; SR141716A, N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide hydrochloride; THC, tetrahydrocannabinol.
Isobolographic analysis indicated that the interaction between these two compounds observed in the elevated plus-maze experiments was super-additive (synergistic).
CB1-selective agonist.
CB2-selective agonist.
CB1-selective antagonist/inverse agonist.
Δ9-THC also undergoes additive or synergistic interactions in mice with nicotine for the production of conditioned place preference, hypothermia and hypolocomotion (Valjent et al., 2002), with 3,4-methylenedioxymethamphetamine (MDMA) in conditioned place preference, self-administration and working memory paradigms (Young et al., 2005; Robledo et al., 2007) and, for the production of catalepsy or hypothermia in mice or rats, with a range of non-cannabinoids that in addition to opioids, nicotine, and MDMA include benzodiazepines, prostaglandins, reserpine and ligands that target muscarinic cholinoceptors or some types of dopamine, noradrenaline, 5-hydroxytryptamine or γ-aminobutyric acid receptors as agonists or antagonists (Marchese et al., 2003; review by Pertwee, 1992).
Intriguingly, one other potential strategy for the management of pain, or of disorders such as epilepsy or stroke, may be to co-administer a CB1 or CB2 receptor agonist and a selective CB1 receptor antagonist/inverse agonist (Tables 1 and 2). The combined administration of such compounds may also constitute a new effective treatment for chronic liver diseases (reviewed in Mallat et al., 2007; Lotersztajn et al., 2008).
Combining strategies
Consideration should be given to the potential benefits of combining some of the strategies mentioned in this review. For the relief of chronic pain, one possible way of reducing the incidence of unwanted off-target effects would be to administer a cannabinoid CB1/CB2 receptor agonist transdermally or intrathecally together with a transdermally administered opioid instead of co-administering these drugs orally. Thus, transdermal co-administration of R-(+)-WIN55212 and morphine at doses that did not affect rotarod performance has been found by Yesilyurt et al. (2003) to induce significantly greater and longer-lasting antinociception in a mouse model of acute pain than that induced by transdermal administration of either compound alone. They also found that intrathecal administration of an ineffective dose of R-(+)-WIN55212 markedly potentiated the antinociceptive effect of transdermally administered morphine. Similarly, Cichewicz et al. (2005) have found the potencies of fentanyl and buprenorphine for the production of antinociception in a guinea-pig model of acute pain to be greater when either of these opioids is co-administered transdermally with Δ9-THC. There may also be some therapeutic advantage to be gained from administering a single cannabinoid receptor agonist concomitantly to skin and spinal cord as Dogrul et al. (2003) have obtained evidence for an antinociceptive synergism between transdermally absorbed and intrathecally administered R-(+)-WIN55212 in a mouse model of acute pain. A third possible way of relieving pain with improved selectivity might be to target the spinal cord with a CB2-selective agonist rather than with CB1 or CB1/CB2 receptor agonist. Thus, Romero-Sandoval and Eisenach (2007) have recently reported that some intrathecal doses of the CB1/CB2 receptor agonist, CP55940, that reduced signs of post-operative hypersensitivity in rats also induced catalepsy, vocalization and a decrease in exploratory activity, effects that all appeared to be at least partly CB1 receptor-mediated. In contrast, no such effects on motor function or vocalization were induced by the CB2-selective agonist, (2-methyl-1-propyl-1H-indol-3-yl)-1-naphthalenylmethanone (JWH-015), when this was injected intrathecally at doses reducing post-operative hypersensitivity. It has also been found that intrathecal administration of a CB2-selective agonist [3-(1,1-dimethylbutyl)-6,6,9-trimethyl-6α,7,10,10α-tetrahydro-6H-benzo[c]chromene (JWH-133)] can produce antinociception in the mouse partial sciatic nerve ligation model of neuropathic pain (Yamamoto et al., 2008), although not in the rat formalin paw model of inflammatory pain (Yoon and Choi, 2003). Another possibility, again for pain relief, would be to administer a CB1 agonist intradermally or intrathecally together with an orally administered CB2-selective agonist. Also worth exploring is the potential benefit of administering a CB2-selective agonist together with CB1 receptor allosteric enhancer to patients with disorders that provoke release of endocannabinoids onto CB1 receptors that then mediate symptom relief.
Future directions
The need for clinical research
There is clearly already strong preclinical evidence in support of the hypothesis that it should be possible to improve the efficacy and/or benefit-to-risk ratio of a cannabinoid receptor agonist as a medicine by adopting one or more of the five strategies described in this review alone or in combination. The need now is for clinical research in which the effectiveness of each of these strategies is tested in patients. Given the preclinical data described in this review, these strategies should perhaps be evaluated initially by focusing on patients with disorders that give rise to chronic inflammatory or neuropathic pain. However, in the longer term it will be important to match any disorder that is treatable with a cannabinoid receptor agonist with the strategy that works best for that particular disorder. Because the consequences of chronic treatment with a CB2-selective agonist have been little investigated, it will also be important when evaluating the strategy of repeatedly administering such an agonist as a medicine to seek out any serious unwanted effects that it produces, particularly in patients with disorders that affect the immune system, for example multiple sclerosis.
There are currently no clinical human data indicating whether or not the efficacy or benefit-to-risk ratio of a full or partial cannabinoid receptor agonist increases when it is given to patients with a disorder that provokes a selective up-regulation of cannabinoid receptors that may mediate symptom relief or slow disease progression. Consequently as a first step in evaluating the strategy of targeting up-regulated cannabinoid receptors in patients with such a disorder, an assessment should be made of the extent to which there is any correlation between (i) cannabinoid receptor density in a tissue in which these receptors mediate a sought-after effect and (ii) the efficacy/benefit-to-risk ratio of a cannabinoid receptor agonist. It would also be of interest to monitor cannabinoid receptor coupling efficiency in an investigation of this kind and to compare a cannabinoid CB1/CB2 partial agonist such as THC with a higher efficacy agonist. As to the disorder that should be investigated in such a project, the animal data described in this review suggest that this could be one that gives rise to chronic inflammatory or neuropathic pain. Another possibility would be to focus on some type of cancer as there is evidence first that cannabinoid receptor expression is higher in some human cancer cells than in normal cells and second that cannabinoid receptor agonists can inhibit human tumour cell growth (Sarfaraz et al., 2005; Xu et al., 2006; Gustafsson et al., 2008; see Guzmán, 2003; Pertwee, 2005a for reviews). If such research does yield positive results, it would justify the development of a diagnostic test that could use cannabinoid receptor expression level or coupling efficiency in particular tissues as a biomarker for selecting patient subpopulations that would benefit most from treatment with a CB1/CB2 receptor partial agonist.
Multi-targeting: what next?
To ensure that the strategy of multi-targeting is fully exploited, it will be important to seek out any as yet undiscovered interactions between cannabinoid receptor agonists and other types of ligand that have therapeutic potential. The possible advantages of multi-targeting by co-administering more than two compounds should be explored as well. So too should the likely advantage of multi-targeting with a single ‘multiple ligand’ that possesses two or more actions that interact additively or synergistically in the production of a beneficial effect. One such compound may be a ligand (compound 12) that has been found to behave both as a CB2 receptor inverse agonist and as a TRPV1 receptor agonist and that, because it has these two actions, has been postulated to be a potential anti-inflammatory agent (Appendino et al., 2006). Another compound that may have clinical applications or serve as a template for a new medicine because it is a multiple ligand is the plant cannabinoid, Δ9-tetrahydrocannabivarin. Thus, there is evidence that this compound can both activate CB2 receptors and block CB1 receptors (reviewed in Pertwee, 2008a), a combination of actions that may render it a particularly effective medicine for the treatment chronic liver diseases or stroke (section on Multi-targeting). Because Δ9-tetrahydrocannabivarin is a CB1 receptor antagonist, it may share the ability of rimonabant (SR141716A) to enhance monoamine release in the brain and so, like rimonabant and AM251 (Takahashi et al., 2008), also have the potential to improve the benefit-to-risk ratio of a selective serotonin reuptake inhibitor when this is being used to treat affective disorders. However, any interest in such a therapeutic strategy is of course likely to be affected by the recent decision to halt sales of rimonabant because of evidence that it increases the incidence of depression and suicidality (section on Licensed medicines that target cannabinoid receptors).
To facilitate the exploitation of multi-targeting in the clinic as fully as possible, it will be important to explore the mechanisms that underlie synergistic interactions between cannabinoid receptor agonists and other types of ligand. One possible way forward would be to seek out evidence for functional interactions between cannabinoid and non-cannabinoid receptors that are expressed by the same cells and then to dissect out any crosstalk between these co-expressed receptors that results in a synergistic interaction from any that leads to mutual antagonism. It is noteworthy, therefore, that there is already evidence for neuronal co-expression of CB1 receptors with certain non-cannabinoid receptors that include µ-opioid, 5-HT1B, 5-HT3A, dopamine D1, dopamine D2 and GABAB receptors, and for the occurrence of functional interactions between some of these co-expressed receptors (Hermann et al., 2002; Morales et al., 2004; Cinar et al., 2008; see Wager-Miller et al., 2002 for a review). Such crosstalk may sometimes involve competition between cannabinoid receptors and co-expressed G protein-coupled non-cannabinoid receptors for the same signalling pathway(s) and a resultant switch in the preferred G-protein subtypes of these receptors, a mechanism that might for example underlie the functional interactions that have been reported to take place between co-expressed CB1 and dopamine D2 receptors (reviewed in Wager-Miller et al., 2002; Pertwee, 2005b). It is also possible that an ultra-low dose of a CB1 receptor antagonist can oppose CB1 agonist-induced antinociceptive tolerance by preventing an agonist-induced G protein coupling switch from Gi to Gs protein (Paquette et al., 2007). For compounds that interact synergistically in the production of a clinically beneficial effect, the extent to which this relies on other kinds of pharmacodynamic interaction or, indeed, on interactions that are pharmacokinetic or metabolic in nature should also be established.
CB1 and CB2 receptor agonists have diverse off-target actions
There is evidence that some cannabinoid CB1 and/or CB2 receptor agonists have one or more non-CB1, non-CB2 pharmacological targets (reviewed in Pertwee, 2008b). This should be borne in mind when selecting a cannabinoid receptor agonist for use as a pharmacological tool or potential medicine. Thus, CB1/CB2 receptor agonists do not all interact with the same non-CB1, non-CB2 pharmacological targets and will therefore most likely differ from each other in the kinds of effect that they produce at doses/concentrations at which they activate or block CB1 or CB2 receptors to the same extent. Attracting particular attention at the moment is the manner in which some cannabinoid receptor ligands interact with GPR55. More specifically, it has been reported that this orphan receptor is activated by the CB1-selective agonist, methanandamide, by the CB2-selective agonist, JWH-015, and by certain CB1/CB2 receptor agonists that include Δ9-THC, HU-210 and anandamide (Ryberg et al., 2007; Lauckner et al., 2008; reviewed in Pertwee, 2007b). Two other CB1/CB2 receptor agonists, CP55940 and 2-arachidonoyl glycerol, were found to activate GPR55 in one of these investigations (Ryberg et al., 2007) but not in another (Lauckner et al., 2008). In addition, it has been found that GPR55 is activated by one CB1-selective antagonist (AM251) but not by another (AM281), and that it is not activated by the CB1/CB2 agonist R-(+)-WIN55212 (Johns et al., 2007; Ryberg et al., 2007; Lauckner et al., 2008). There has also been one report, however, that neither Δ9-THC, CP55940, HU-210, anandamide nor 2-arachidonoyl glycerol activate GPR55, when the measured response is extracellular signal-regulated kinase (ERK) phosphorylation (Oka et al., 2007). Because of these conflicting pharmacological data, the question of whether GPR55 should or should not be regarded as a cannabinoid receptor has still to be resolved, as indeed has the question of what roles this receptor plays in health or disease.
Acknowledgments
The writing of this review was supported by a grant from the National Institute on Drug Abuse (NIDA) (DA-03672) and funding from GW Pharmaceuticals.
Glossary
Abbreviations:
- 8-OH-DPAT
8-hydroxy-2-(di-n-propylamino) tetralin hydrobromide
- ACEA
arachidonyl-2′-chloroethylamide
- ACPA
arachidonylcyclopropylamide
- AM251
N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide
- AM281
N-(morpholin-4-yl)-1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-1H-pyrazole-3-carboxamide
- AM630
6-iodo-2-methyl-1-[2-(4-morpholinyl)ethyl]-1H-indol-3-yl](4-methoxyphenyl)methanone
- AM1241
(2-iodo-5-nitrophenyl)-[1-(1-methylpiperidin-2-ylmethyl)-1H-indol-3-yl]-methanone
- anandamide
N-arachidonoyl ethanolamine
- COX
cyclooxygenase
- CP55940
(−)-cis-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-trans-4-(3-hydroxypropyl)cyclohexanol
- HU-210
(6aR)-trans-3-(1,1-dimethylheptyl)-6a,7,10,10a-tetrahydro-1-hydroxy-6,6-dimethyl-6H-dibenzo[b,d]pyran-9-methanol
- JWH-015
(2-methyl-1-propyl-1H-indol-3-yl)-1-naphthalenylmethanone
- JWH-133
3-(1,1-dimethylbutyl)-6,6,9-trimethyl-6α,7,10,10α-tetrahydro-6H-benzo[c]chromene
- MDMA
3,4-methylenedioxymethamphetamine
- R-(+)-WIN55212
(R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo-[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone
- SR141716A
N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide hydrochloride
- SR144528
N-[(1S)-endo-1,3,3-trimethyl bicyclo [2.2.1] heptan-2-yl]-5-(4-chloro-3-methylphenyl)-1-(4-methylbenzyl)-pyrazole-3-carboxamide
- THC
tetrahydrocannabinol
Conflict of interest
The author has formal links with GW Pharmaceuticals that funds some of his research.
References
- Adam L, Salois D, Rihakova L, Lapointe S, St-Onge S, Labrecque J, et al. Positive allosteric modulators of CB1 receptors. Symposium on the Cannabinoids. Burlington, Vermont, USA. International Cannabinoid Research Society, p. 86.
- Adamczyk P, Golda A, McCreary AC, Filip M, Przegalnski E. Activation of endocannabinoid transmission induces antidepressant-like effects in rats. J Physiol Pharmacol. 2008;59:217–228. [PubMed] [Google Scholar]
- Ahn DK, Choi HS, Yeo SP, Woo YW, Lee MK, Yang GY, et al. Blockade of central cyclooxygenase (COX) pathways enhances the cannabinold-induced antinociceptive effects on inflammatory temporomandibular joint (TMJ) nociception. Pain. 2007;132:23–32. doi: 10.1016/j.pain.2007.01.015. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC) Br J Pharmacol. 2008;153(Suppl.)(2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appendino G, Cascio MG, Bacchiega S, Moriello AS, Minassi A, Thomas A, et al. First ‘hybrid’ ligands of vanilloid TRPV1 and cannabinoid CB2 receptors and non-polyunsaturated fatty acid-derived CB2-selective ligands. FEBS Lett. 2006;580:568–574. doi: 10.1016/j.febslet.2005.12.069. [DOI] [PubMed] [Google Scholar]
- Baek J-H, Zheng Y, Darlington CL, Smith PF. Cannabinoid CB2 receptor expression in the rat brainstem cochlear and vestibular nuclei. Acta Oto-Laryngol. 2008;128:961–967. doi: 10.1080/00016480701796944. [DOI] [PubMed] [Google Scholar]
- Balerio GN, Aso E, Maldonado R. Role of the cannabinoid system in the effects induced by nicotine on anxiety-like behaviour in mice. Psychopharmacology. 2006;184:504–513. doi: 10.1007/s00213-005-0251-9. [DOI] [PubMed] [Google Scholar]
- Bambico FR, Katz N, Debonnel G, Gobbi G. Cannabinoids elicit antidepressant-like behavior and activate serotonergic neurons through the medial prefrontal cortex. J Neurosci Res. 2007;27:11700–11711. doi: 10.1523/JNEUROSCI.1636-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltramo M, Bernardini N, Bertorelli R, Campanella M, Nicolussi E, Fredduzzi S, et al. CB2 receptor-mediated antihyperalgesia: possible direct involvement of neural mechanisms. Eur J Neurosci. 2006;23:1530–1538. doi: 10.1111/j.1460-9568.2006.04684.x. [DOI] [PubMed] [Google Scholar]
- Bingham B, Jones PG, Uveges AJ, Kotnis S, Lu P, Smith VA, et al. Species-specific in vitro pharmacological effects of the cannabinoid receptor 2 (CB2) selective ligand AM1241 and its resolved enantiomers. Br J Pharmacol. 2007;151:1061–1070. doi: 10.1038/sj.bjp.0707303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce S. Targeting peripheral cannabinoid 1 (CB1) receptors for chronic pain. Fundam Clin Pharmacol. 2008;22(Suppl.)(2):12. [Google Scholar]
- Brady CM, DasGupta R, Dalton C, Wiseman OJ, Berkley KJ, Fowler CJ. An open-label pilot study of cannabis-based extracts for bladder dysfunction in advanced multiple sclerosis. Mult Scler. 2004;10:425–433. doi: 10.1191/1352458504ms1063oa. [DOI] [PubMed] [Google Scholar]
- Braida D, Limonta V, Malabarba L, Zani A, Sala M. 5-HT1A receptors are involved in the anxiolytic effect of Δ9-tetrahydrocannabinol and AM 404, the anandamide transport inhibitor, in Sprague-Dawley rats. Eur J Pharmacol. 2007;555:156–163. doi: 10.1016/j.ejphar.2006.10.038. [DOI] [PubMed] [Google Scholar]
- Cabral GA, Staab A. Effects on the immune system. In: Pertwee RG, editor. Cannabinoids. Handbook of Experimental Pharmacology. Heidelberg: Springer-Verlag; 2005. pp. 385–423. Vol. 168. [DOI] [PubMed] [Google Scholar]
- Cichewicz DL. Synergistic interactions between cannabinoid and opioid analgesics. Life Sci. 2004;74:1317–1324. doi: 10.1016/j.lfs.2003.09.038. [DOI] [PubMed] [Google Scholar]
- Cichewicz DL, McCarthy EA. Antinociceptive synergy between Δ9-tetrahydrocannabinol and opioids after oral administration. J Pharmacol Exp Ther. 2003;304:1010–1015. doi: 10.1124/jpet.102.045575. [DOI] [PubMed] [Google Scholar]
- Cichewicz DL, Welch SP, Smith FL. Enhancement of transdermal fentanyl and buprenorphine antinociception by transdermal Δ9-tetrahydrocannabinol. Eur J Pharmacol. 2005;525:74–82. doi: 10.1016/j.ejphar.2005.09.039. [DOI] [PubMed] [Google Scholar]
- Cinar R, Freund TF, Katona I, Mackie K, Szucs M. Reciprocal inhibition of G-protein signaling is induced by CB1 cannabinoid and GABAB receptor interactions in rat hippocampal membranes. Neurochem Int. 2008;52:1402–1409. doi: 10.1016/j.neuint.2008.02.005. [DOI] [PubMed] [Google Scholar]
- Cox ML, Haller VL, Welch SP. Synergy between Δ9-tetrahydrocannabinol and morphine in the arthritic rat. Eur J Pharmacol. 2007;567:125–130. doi: 10.1016/j.ejphar.2007.04.010. [DOI] [PubMed] [Google Scholar]
- De Vry J, Jentzsch KR, Kuhl E, Eckel G. Behavioral effects of cannabinoids show differential sensitivity to cannabinoid receptor blockade and tolerance development. Behav Pharmacol. 2004;15:1–12. doi: 10.1097/00008877-200402000-00001. [DOI] [PubMed] [Google Scholar]
- Desai BS, Monahan AJ, Carvey PM, Hendey B. Blood-brain barrier pathology in Alzheimer's and Parkinson's disease: implications for drug therapy. Cell Transplant. 2007;16:285–299. doi: 10.3727/000000007783464731. [DOI] [PubMed] [Google Scholar]
- Després J-P, Golay A, Sjöström L. Effects of rimonabant on metabolic risk factors in overweight patients with dyslipidemia. N Engl J Med. 2005;353:2121–2134. doi: 10.1056/NEJMoa044537. [DOI] [PubMed] [Google Scholar]
- Després J-P, Lemieux I, Alméras N. Contribution of CB1 blockade to the management of high-risk abdominal obesity. Int J Obes. 2006;30:S44–S52. doi: 10.1038/sj.ijo.0803278. [DOI] [PubMed] [Google Scholar]
- Devane WA, Hanuš L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- Di Marzo V. Targeting the endocannabinoid system: to enhance or reduce? Nat Rev Drug Discov. 2008;7:438–455. doi: 10.1038/nrd2553. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, De Petrocellis L, Bisogno T. The biosynthesis, fate and pharmacological properties of endocannabinoids. In: Pertwee RG, editor. Cannabinoids. Handbook of Experimental Pharmacology. Heidelberg: Springer-Verlag; 2005. pp. 147–185. Vol. 168. [DOI] [PubMed] [Google Scholar]
- Dittel BN. Direct suppression of autoreactive lymphocytes in the central nervous system via the CB2 receptor. Br J Pharmacol. 2008;153:271–276. doi: 10.1038/sj.bjp.0707493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogrul A, Gul H, Akar A, Yildiz O, Bilgin F, Guzeldemir E. Topical cannabinoid antinociception: synergy with spinal sites. Pain. 2003;105:11–16. doi: 10.1016/s0304-3959(03)00068-x. [DOI] [PubMed] [Google Scholar]
- Duncan M, Mouihate A, Mackie K, Keenan CM, Buckley NE, Davison JS, et al. Cannabinoid CB2 receptors in the enteric nervous system modulate gastrointestinal contractility in lipopolysaccharide-treated rats. Am J Physiol Gastrointest Liver Physiol. 2008;295:G78–G87. doi: 10.1152/ajpgi.90285.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvorak M, Watkinson A, McGlone F, Rukwied R. Histamine induced responses are attenuated by a cannabinoid receptor agonist in human skin. Inflamm Res. 2003;52:238–245. doi: 10.1007/s00011-003-1162-z. [DOI] [PubMed] [Google Scholar]
- Dyson A, Peacock M, Chen A, Courade J-P, Yaqoob M, Groarke A, et al. Antihyperalgesic properties of the cannabinoid CT-3 in chronic neuropathic and inflammatory pain states in the rat. Pain. 2005;116:129–137. doi: 10.1016/j.pain.2005.03.037. [DOI] [PubMed] [Google Scholar]
- Dziadulewicz EK, Bevan SJ, Brain CT, Coote PR, Culshaw AJ, Davis AJ, et al. Naphthalen-1-yl-(4-pentyloxynaphthalen-1-yl)methanone: a potent, orally bioavailable human CB1/CB2 dual agonist with antihyperalgesic properties and restricted central nervous system penetration. J Med Chem. 2007;50:3851–3856. doi: 10.1021/jm070317a. [DOI] [PubMed] [Google Scholar]
- Egashira N, Matsuda T, Koushi E, Mishima K, Iwasaki K, Shoyama Y, et al. Involvement of 5-hydroxytryptamine1A receptors in Δ9-tetrahydrocannabinol-induced catalepsy-like immobilization in mice. Eur J Pharmacol. 2006;550:117–122. doi: 10.1016/j.ejphar.2006.08.051. [DOI] [PubMed] [Google Scholar]
- Ellgren M, Spano SM, Hurd YL. Adolescent cannabis exposure alters opiate intake and opioid limbic neuronal populations in adult rats. Neuropsychopharmacology. 2007;32:607–615. doi: 10.1038/sj.npp.1301127. [DOI] [PubMed] [Google Scholar]
- Finn DP, Beckett SRG, Roe CH, Madjd A, Fone KCF, Kendall DA, et al. Effects of coadministration of cannabinoids and morphine on nociceptive behaviour, brain monoamines and HPA axis activity in a rat model of persistent pain. Eur J Neurosci. 2004;19:678–686. doi: 10.1111/j.0953-816x.2004.03177.x. [DOI] [PubMed] [Google Scholar]
- Fox A, Bevan S. Therapeutic potential of cannabinoid receptor agonists as analgesic agents. Expert Opin Investig Drugs. 2005;14:695–703. doi: 10.1517/13543784.14.6.695. [DOI] [PubMed] [Google Scholar]
- Gholizadeh S, Shafaroodi H, Ghasemi M, Bahremand A, Sharifzadeh M, Dehpour AR. Ultra-low dose cannabinoid antagonist AM251 enhances cannabinoid anticonvulsant effects in the pentylenetetrazole-induced seizure in mice. Neuropharmacology. 2007;53:763–770. doi: 10.1016/j.neuropharm.2007.08.005. [DOI] [PubMed] [Google Scholar]
- Gifford AN, Bruneus M, Gatley SJ, Lan R, Makriyannis A, Volkow ND. Large receptor reserve for cannabinoid actions in the central nervous system. J Pharmacol Exp Ther. 1999;288:478–483. [PubMed] [Google Scholar]
- Gong J-P, Onaivi ES, Ishiguro H, Liu Q-R, Tagliaferro PA, Brusco A, et al. Cannabinoid CB2 receptors: immunohistochemical localization in rat brain. Brain Res. 2006;1071:10–23. doi: 10.1016/j.brainres.2005.11.035. [DOI] [PubMed] [Google Scholar]
- Guindon J, Hohmann AG. Cannabinoid CB2 receptors: a therapeutic target for the treatment of inflammatory and neuropathic pain. Br J Pharmacol. 2008;153:319–334. doi: 10.1038/sj.bjp.0707531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson K, Wang X, Severa D, Eriksson M, Kimby E, Merup M, et al. Expression of cannabinoid receptors type 1 and type 2 in non-Hodgkin lymphoma: growth inhibition by receptor activation. Int J Cancer. 2008;123:1025–1033. doi: 10.1002/ijc.23584. [DOI] [PubMed] [Google Scholar]
- Guzmán M. Cannabinoids: potential anticancer agents. Nat Rev Cancer. 2003;3:745–755. doi: 10.1038/nrc1188. [DOI] [PubMed] [Google Scholar]
- Hermann H, Marsicano G, Lutz B. Coexpression of the cannabinoid receptor type 1 with dopamine and serotonin receptors in distinct neuronal subpopulations of the adult mouse forebrain. Neuroscience. 2002;109:451–460. doi: 10.1016/s0306-4522(01)00509-7. [DOI] [PubMed] [Google Scholar]
- Holdcroft A, Smith M, Jacklin A, Hodgson H, Smith B, Newton M, et al. Pain relief with oral cannabinoids in familial Mediterranean fever. Anaesthesia. 1997;52:483–488. doi: 10.1111/j.1365-2044.1997.139-az0132.x. [DOI] [PubMed] [Google Scholar]
- Horswill JG, Bali U, Shaaban S, Keily JF, Jeevaratnam P, Babbs AJ, et al. PSNCBAM-1, a novel allosteric antagonist at cannabinoid CB1 receptors with hypophagic effects in rats. Br J Pharmacol. 2007;152:805–814. doi: 10.1038/sj.bjp.0707347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev. 2002;54:161–202. doi: 10.1124/pr.54.2.161. [DOI] [PubMed] [Google Scholar]
- Inui K, Egashira N, Mishima K, Yano A, Matsumoto Y, Hasebe N, et al. The serotonin1A receptor agonist 8-OHDPAT reverses Δ9-tetrahydrocannabinol-induced impairment of spatial memory and reduction of acetylcholine release in the dorsal hippocampus in rats. Neurotox Res. 2004;6:153–158. doi: 10.1007/BF03033218. [DOI] [PubMed] [Google Scholar]
- Izzo AA, Camilleri M. Emerging role of cannabinoids in gastrointestinal and liver diseases: basic and clinical aspects. Gut. 2008;57:1140–1155. doi: 10.1136/gut.2008.148791. [DOI] [PubMed] [Google Scholar]
- Izzo AA, Coutts AA. Cannabinoids and the digestive tract. In: Pertwee RG, editor. Cannabinoids. Handbook of Experimental Pharmacology. Heidelberg: Springer-Verlag; 2005. pp. 573–598. Vol. 168. [DOI] [PubMed] [Google Scholar]
- Izzo AA, Fezza F, Capasso R, Bisogno T, Pinto L, Iuvone T, et al. Cannabinoid CB1-receptor mediated regulation of gastrointestinal motility in mice in a model of intestinal inflammation. Br J Pharmacol. 2001;134:563–570. doi: 10.1038/sj.bjp.0704293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzo AA, Capasso F, Costagliola A, Bisogno T, Marsicano G, Ligresti A, et al. An endogenous cannabinoid tone attenuates cholera toxin-induced fluid accumulation in mice. Gastroenterology. 2003;125:765–774. doi: 10.1016/s0016-5085(03)00892-8. [DOI] [PubMed] [Google Scholar]
- Jafari MR, Ghiasvand F, Golmohammadi S, Zarrindast MR, Djahanguiri B. Influence of central nicotinic receptors on arachidonylcyclopropylamide (ACPA)-induced antinociception in mice. Int J Neurosci. 2008;118:531–543. doi: 10.1080/00207450701239467. [DOI] [PubMed] [Google Scholar]
- Jhaveri MD, Elmes SJR, Richardson D, Barrett DA, Kendall DA, Mason R, et al. Evidence for a novel functional role of cannabinoid CB2 receptors in the thalamus of neuropathic rats. Eur J Neurosci. 2008;27:1722–1730. doi: 10.1111/j.1460-9568.2008.06162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin KL, Mao XO, Goldsmith PC, Greenberg DA. CB1 cannabinoid receptor induction in experimental stroke. Ann Neurol. 2000;48:257–261. [PubMed] [Google Scholar]
- Johns DG, Behm DJ, Walker DJ, Ao Z, Shapland EM, Daniels DA, et al. The novel endocannabinoid receptor GPR55 is activated by atypical cannabinoids but does not mediate their vasodilator effects. Br J Pharmacol. 2007;152:825–831. doi: 10.1038/sj.bjp.0707419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S, Kim CH, Lee H, Kim DY, Han JI, Chung RK, et al. Antinociceptive synergy between the cannabinoid receptor agonist WIN 55,212-2 and bupivacaine in the rat formalin test. Anesth Analg. 2007;104:719–725. doi: 10.1213/01.ane.0000255291.38637.26. [DOI] [PubMed] [Google Scholar]
- Karst M, Salim K, Burstein S, Conrad I, Hoy L, Schneider U. Analgesic effect of the synthetic cannabinoid CT-3 on chronic neuropathic pain. A randomized controlled trial. J Am Med Assoc. 2003;290:1757–1762. doi: 10.1001/jama.290.13.1757. [DOI] [PubMed] [Google Scholar]
- Kim K, Moore DH, Makriyannis A, Abood ME. AM1241, a cannabinoid CB2 receptor selective compound, delays disease progression in a mouse model of amyotrophic lateral sclerosis. Eur J Pharmacol. 2006;542:100–105. doi: 10.1016/j.ejphar.2006.05.025. [DOI] [PubMed] [Google Scholar]
- Kimball ES, Schneider CR, Wallace NH, Hornby PJ. Agonists of cannabinoid receptor 1 and 2 inhibit experimental colitis induced by oil of mustard and by dextran sulfate sodium. Am J Physiol Gastrointest Liver Physiol. 2006;291:G364–G371. doi: 10.1152/ajpgi.00407.2005. [DOI] [PubMed] [Google Scholar]
- Kraft B, Frickey NA, Kaufmann RM, Reif M, Frey R, Gustorff B, et al. Lack of analgesia by oral standardized cannabis extract on acute inflammatory pain and hyperalgesia in volunteers. Anesthesiology. 2008;109:101–110. doi: 10.1097/ALN.0b013e31817881e1. [DOI] [PubMed] [Google Scholar]
- Kwiatkowska M, Parker LA, Burton P, Mechoulam R. A comparative analysis of the potential of cannabinoids and ondansetron to suppress cisplatin-induced emesis in the Suncus murinus (house musk shrew) Psychopharmacology. 2004;174:254–259. doi: 10.1007/s00213-003-1739-9. [DOI] [PubMed] [Google Scholar]
- Lauckner JE, Jensen JB, Chen H-Y, Lu H-C, Hille B, Mackie K. GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc Natl Acad Sci USA. 2008;105:2699–2704. doi: 10.1073/pnas.0711278105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Foll B, Goldberg SR. Cannabinoid CB1 receptor antagonists as promising new medications for drug dependence. J Pharmacol Exp Ther. 2005;312:875–883. doi: 10.1124/jpet.104.077974. [DOI] [PubMed] [Google Scholar]
- Li J-X, McMahon LR, Gerak LR, Becker GL, France CP. Interactions between Δ9-tetrahydrocannabinol and µ opioid receptor agonists in rhesus monkeys: discrimination and antinociception. Psychopharmacology. 2008;199:199–208. doi: 10.1007/s00213-008-1157-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim G, Sung B, Ji R-R, Mao J. Upregulation of spinal cannabinoid-1-receptors following nerve injury enhances the effects of Win 55,212-2 on neuropathic pain behaviors in rats. Pain. 2003;105:275–283. doi: 10.1016/s0304-3959(03)00242-2. [DOI] [PubMed] [Google Scholar]
- Lotersztajn S, Teixeira-Clerc F, Julien B, Deveaux V, Ichigotani Y, Manin S, et al. CB2 receptors as new therapeutic targets for liver diseases. Br J Pharmacol. 2008;153:286–289. doi: 10.1038/sj.bjp.0707511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunn CA, Reich E-P, Fine JS, Lavey B, Kozlowski JA, Hipkin RW, et al. Biology and therapeutic potential of cannabinoid CB2 receptor inverse agonists. Br J Pharmacol. 2008;153:226–239. doi: 10.1038/sj.bjp.0707480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mach F, Montecucco F, Steffens S. Cannabinoid receptors in acute and chronic complications of atherosclerosis. Br J Pharmacol. 2008;153:290–298. doi: 10.1038/sj.bjp.0707517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallat A, Teixeira-Clerc F, Deveaux V, Lotersztajn S. Cannabinoid receptors as new targets of antifibrosing strategies during chronic liver diseases. Expert Opin Ther Targets. 2007;11:403–409. doi: 10.1517/14728222.11.3.403. [DOI] [PubMed] [Google Scholar]
- Malone DT, Taylor DA. Involvement of somatodendritic 5-HT1A receptors in Δ9-tetrahydrocannabinol-induced hypothermia in the rat. Pharmacol Biochem Behav. 2001;69:595–601. doi: 10.1016/s0091-3057(01)00567-6. [DOI] [PubMed] [Google Scholar]
- Manzanedo C, Aguilar MA, Rodríguez-Arias M, Navarro M, Miñarro J. Cannabinoid agonist-induced sensitisation to morphine place preference in mice. Neuroreport. 2004;15:1373–1377. doi: 10.1097/01.wnr.0000126217.87116.8c. [DOI] [PubMed] [Google Scholar]
- Marchese G, Casti P, Ruiu S, Saba P, Sanna A, Casu G, et al. Haloperidol, but not clozapine, produces dramatic catalepsy in Δ9-THC-treated rats: possible clinical implications. Br J Pharmacol. 2003;140:520–526. doi: 10.1038/sj.bjp.0705478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maresz K, Carrier EJ, Ponomarev ED, Hillard CJ, Dittel BN. Modulation of the cannabinoid CB2 receptor in microglial cells in response to inflammatory stimuli. J Neurochem. 2005;95:437–445. doi: 10.1111/j.1471-4159.2005.03380.x. [DOI] [PubMed] [Google Scholar]
- Massa F, Marsicano G, Hermann H, Cannich A, Monory K, Cravatt BF, et al. The endogenous cannabinoid system protects against colonic inflammation. J Clin Invest. 2004;113:1202–1209. doi: 10.1172/JCI19465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechoulam R. The pharmacohistory of Cannabis sativa. In: Mechoulam R, editor. Cannabinoids as Therapeutic Agents. Boca Raton: CRC Press; 1986. pp. 1–19. [Google Scholar]
- Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol. 1995;50:83–90. doi: 10.1016/0006-2952(95)00109-d. [DOI] [PubMed] [Google Scholar]
- Mitchell VA, Aslan S, Safaei R, Vaughan CW. Effect of the cannabinoid ajulemic acid on rat models of neuropathic and inflammatory pain. Neurosci Lett. 2005;382:231–235. doi: 10.1016/j.neulet.2005.03.019. [DOI] [PubMed] [Google Scholar]
- Mitrirattanakul S, Ramakul N, Guerrero AV, Matsuka Y, Ono T, Iwase H, et al. Site-specific increases in peripheral cannabinoid receptors and their endogenous ligands in a model of neuropathic pain. Pain. 2006;126:102–114. doi: 10.1016/j.pain.2006.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales M, Wang S-D, Diaz-Ruiz O, Jho DH-J. Cannabinoid CB1 receptor and serotonin 3 receptor subunit A (5-HT3A) are co-expressed in GABA neurons in the rat telencephalon. J Comp Neurol. 2004;468:205–216. doi: 10.1002/cne.10968. [DOI] [PubMed] [Google Scholar]
- Naderi N, Haghparast A, Saber-Tehrani A, Rezaii N, Alizadeh A-M, Khani A, et al. Interaction between cannabinoid compounds and diazepam on anxiety-like behaviour of mice. Pharmacol Biochem Behav. 2008;89:64–75. doi: 10.1016/j.pbb.2007.11.001. [DOI] [PubMed] [Google Scholar]
- Naef M, Curatolo M, Petersen-Felix S, Arendt-Nielsen L, Zbinden A, Brenneisen R. The analgesic effect of oral delta-9-tetrahydrocannabinol (THC), morphine, and a THC-morphine combination in healthy subjects under experimental pain conditions. Pain. 2003;105:79–88. doi: 10.1016/s0304-3959(03)00163-5. [DOI] [PubMed] [Google Scholar]
- Narang S, Gibson D, Wasan AD, Ross EL, Michna E, Nedelikovic SS, et al. Efficacy of dronabinol as an adjuvant treatment for chronic pain patients on opioid therapy. J Pain. 2008;9:254–264. doi: 10.1016/j.jpain.2007.10.018. [DOI] [PubMed] [Google Scholar]
- Ni X, Geller EB, Eppihimer MJ, Eisenstein TK, Adler MW, Tuma RF. Win 55212-2, a cannabinoid receptor agonist, attenuates leukocyte/endothelial interactions in an experimental autoimmune encephalomyelitis model. Mult Scler. 2004;10:158–164. doi: 10.1191/1352458504ms1009oa. [DOI] [PubMed] [Google Scholar]
- Oka S, Nakajima K, Yamashita A, Kishimoto S, Sugiura T. Identification of GPR55 as a lysophosphatidylinositol receptor. Biochem Biophys Res Commun. 2007;362:928–934. doi: 10.1016/j.bbrc.2007.08.078. [DOI] [PubMed] [Google Scholar]
- Onaivi ES. Neuropsychobiological evidence for the functional presence and expression of cannabinoid CB2 receptors in the brain. Neuropsychobiology. 2006;54:231–246. doi: 10.1159/000100778. [DOI] [PubMed] [Google Scholar]
- Pacher P, Hasko G. Endocannabinoids and cannabinoid receptors in ischaemia-reperfusion injury and preconditioning. Br J Pharmacol. 2008;153:252–262. doi: 10.1038/sj.bjp.0707582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquette JJ, Wang H-Y, Bakshi K, Olmstead MC. Cannabinoid-induced tolerance is associated with a CB1 receptor G protein coupling switch that is prevented by ultra-low dose rimonabant. Behav Pharmacol. 2007;18:767–776. doi: 10.1097/FBP.0b013e3282f15890. [DOI] [PubMed] [Google Scholar]
- Pertwee RG. In vivo interactions between psychotropic cannabinoids and other drugs involving central and peripheral neurochemical mediators. In: Murphy L, Bartke A, editors. Marijuana/Cannabinoids. Neurobiology and Neurophysiology. Boca Raton: CRC Press; 1992. pp. 165–218. [Google Scholar]
- Pertwee RG. Pharmacology of cannabinoid receptor ligands. Curr Med Chem. 1999;6:635–664. [PubMed] [Google Scholar]
- Pertwee RG. Cannabinoid receptors and pain. Prog Neurobiol. 2001;63:569–611. doi: 10.1016/s0301-0082(00)00031-9. [DOI] [PubMed] [Google Scholar]
- Pertwee RG. The therapeutic potential of drugs that target cannabinoid receptors or modulate the tissue levels or actions of endocannabinoids. AAPS J. 2005a;7:E625–E654. doi: 10.1208/aapsj070364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG. Pharmacological actions of cannabinoids. In: Pertwee RG, editor. Cannabinoids. Handbook of Experimental Pharmacology. Heidelberg: Springer-Verlag; 2005b. pp. 1–51. Vol. 168. [DOI] [PubMed] [Google Scholar]
- Pertwee RG. Inverse agonism and neutral antagonism at cannabinoid CB1 receptors. Life Sci. 2005c;76:1307–1324. doi: 10.1016/j.lfs.2004.10.025. [DOI] [PubMed] [Google Scholar]
- Pertwee RG. Cannabinoid pharmacology: the first 66 years. Br J Pharmacol. 2006;147:S163–S171. doi: 10.1038/sj.bjp.0706406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG. Cannabinoids and multiple sclerosis. Mol Neurobiol. 2007a;36:45–59. doi: 10.1007/s12035-007-0005-2. [DOI] [PubMed] [Google Scholar]
- Pertwee RG. GPR55: a new member of the cannabinoid receptor clan? Br J Pharmacol. 2007b;152:984–986. doi: 10.1038/sj.bjp.0707464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: Δ9-tetrahydrocannabinol, cannabidiol and Δ9-tetrahydrocannabivarin. Br J Pharmacol. 2008a;153:199–215. doi: 10.1038/sj.bjp.0707442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG. Ligands that target cannabinoid receptors in the brain: from THC to anandamide and beyond. Addict Biol. 2008b;13:147–159. doi: 10.1111/j.1369-1600.2008.00108.x. [DOI] [PubMed] [Google Scholar]
- Pertwee RG, Ross RA. Cannabinoid receptors and their ligands. Prostaglandins Leukot Essent Fatty Acids. 2002;66:101–121. doi: 10.1054/plef.2001.0341. [DOI] [PubMed] [Google Scholar]
- Pertwee RG, Thomas A. Therapeutic applications for agents that act at CB1 and CB2 receptors. In: Reggio P, editor. The Cannabinoid Receptors. Totowa: Humana Press; 2009. pp. 361–392. [Google Scholar]
- Potenzieri C, Harding-Rose C, Simone DA. The cannabinoid receptor agonist, WIN 55, 212-2, attenuates tumor-evoked hyperalgesia through peripheral mechanisms. Brain Res. 2008;1215:69–75. doi: 10.1016/j.brainres.2008.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MR, Baillie GL, Thomas A, Stevenson LA, Easson M, Goodwin R, et al. Allosteric modulation of the cannabinoid CB1 receptor. Mol Pharmacol. 2005;68:1484–1495. doi: 10.1124/mol.105.016162. [DOI] [PubMed] [Google Scholar]
- Reche I, Fuentes JA, Ruiz Gayo M. Potentiation of Δ9-tetrahydrocannabinol-induced analgesia by morphine in mice: involvement of µ- and κ-opioid receptors. Eur J Pharmacol. 1996;318:11–16. doi: 10.1016/s0014-2999(96)00752-2. [DOI] [PubMed] [Google Scholar]
- Roberts JD, Gennings C, Shih M. Synergistic affective analgesic interaction between delta-9-tetrahydrocannabinol and morphine. Eur J Pharmacol. 2006;530:54–58. doi: 10.1016/j.ejphar.2005.11.036. [DOI] [PubMed] [Google Scholar]
- Robledo P, Trigo JM, Panayi F, de la Torre R, Maldonado R. Behavioural and neurochemical effects of combined MDMA and THC administration in mice. Psychopharmacology. 2007;195:255–264. doi: 10.1007/s00213-007-0879-8. [DOI] [PubMed] [Google Scholar]
- Romero-Sandoval A, Eisenach JC. Spinal cannabinoid receptor type 2 activation reduces hypersensitivity and spinal cord glial activation after paw incision. Anesthesiology. 2007;106:787–794. doi: 10.1097/01.anes.0000264765.33673.6c. [DOI] [PubMed] [Google Scholar]
- Ross RA. Allosterism and cannabinoid CB1 receptors: the shape of things to come. Trends Pharmacol Sci. 2007;28:567–572. doi: 10.1016/j.tips.2007.10.006. [DOI] [PubMed] [Google Scholar]
- Ross RA, Coutts AA, McFarlane SM, Anavi-Goffer S, Irving AJ, Pertwee RG, et al. Actions of cannabinoid receptor ligands on rat cultured sensory neurones: implications for antinociception. Neuropharmacology. 2001;40:221–232. doi: 10.1016/s0028-3908(00)00135-0. [DOI] [PubMed] [Google Scholar]
- Rubio-Araiz A, Arévalo-Martín A, Gómez-Torres O, Navarro-Galve B, García-Ovejero D, Suetterlin P, et al. The endocannabinoid system modulates a transient TNF pathway that induces neural stem cell proliferation. Mol Cell Neurosci. 2008;38:374–380. doi: 10.1016/j.mcn.2008.03.010. [DOI] [PubMed] [Google Scholar]
- Rukwied R, Watkinson A, McGlone F, Dvorak M. Cannabinoid agonists attenuate capsaicin-induced responses in human skin. Pain. 2003;102:283–288. doi: 10.1016/S0304-3959(02)00401-3. [DOI] [PubMed] [Google Scholar]
- Ryberg E, Larsson N, Sjögren S, Hjorth S, Hermansson N-O, Leonova J, et al. The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol. 2007;152:1092–1101. doi: 10.1038/sj.bjp.0707460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagredo O, García-Arencibia M, de Lago E, Finetti S, Decio A, Fernández-Ruiz J. Cannabinoids and neuroprotection in basal ganglia disorders. Mol Neurobiol. 2007;36:82–91. doi: 10.1007/s12035-007-0004-3. [DOI] [PubMed] [Google Scholar]
- Salim K, Schneider U, Burstein S, Hoy L, Karst M. Pain measurements and side effect profile of the novel cannabinoid ajulemic acid. Neuropharmacology. 2005;48:1164–1171. doi: 10.1016/j.neuropharm.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Sarfaraz S, Afaq F, Adhami VM, Mukhtar H. Cannabinoid receptor as a novel target for the treatment of prostate cancer. Cancer Res. 2005;65:1635–1641. doi: 10.1158/0008-5472.CAN-04-3410. [DOI] [PubMed] [Google Scholar]
- Seeling W, Kneer L, Büchele B, Gschwend J, Maier L, Nett C, et al. 9-tetrahydrocannabinol and the opioid receptor agonist piritramide do not act synergistically in postoperative pain. Anaesthesist. 2006;55:391–400. doi: 10.1007/s00101-005-0963-6. [DOI] [PubMed] [Google Scholar]
- Shoemaker JL, Seely KA, Reed RL, Crow JP, Prather PL. The CB2 cannabinoid agonist AM-1241 prolongs survival in a transgenic mouse model of amyotrophic lateral sclerosis when initiated at symptom onset. J Neurochem. 2007;101:87–98. doi: 10.1111/j.1471-4159.2006.04346.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegling A, Hofmann HA, Denzer D, Mauler F, De Vry J. Cannabinoid CB1 receptor upregulation in a rat model of chronic neuropathic pain. Eur J Pharmacol. 2001;415:R5–R7. doi: 10.1016/s0014-2999(01)00798-1. [DOI] [PubMed] [Google Scholar]
- Singh ME, McGregor IS, Mallet PE. Repeated exposure to Δ9-tetrahydrocannabinol alters heroin-induced locomotor sensitisation and Fos-immunoreactivity. Neuropharmacology. 2005;49:1189–1200. doi: 10.1016/j.neuropharm.2005.07.008. [DOI] [PubMed] [Google Scholar]
- Skaper SD, Buriani A, Dal Toso R, Petrelli L, Romanello S, Facci L, et al. The ALIAmide palmitoylethanolamide and cannabinoids, but not anandamide, are protective in a delayed postglutamate paradigm of excitotoxic death in cerebellar granule neurons. Proc Natl Acad Sci USA. 1996;93:3984–3989. doi: 10.1073/pnas.93.9.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PA, Selley DE, Sim-Selley LJ, Welch SP. Low dose combination of morphine and Δ9-tetrahydrocannabinol circumvents antinociceptive tolerance and apparent desensitization of receptors. Eur J Pharmacol. 2007;571:129–137. doi: 10.1016/j.ejphar.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ständer S, Schmelz M, Metze D, Luger T, Rukwied R. Distribution of cannabinoid receptor 1 (CB1) and 2 (CB2) on sensory nerve fibers and adnexal structures in human skin. J Dermatol Sci. 2005;38:177–188. doi: 10.1016/j.jdermsci.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Steffens S, Veillard NR, Arnaud C, Pelli G, Burger F, Staub C, et al. Low dose oral cannabinoid therapy reduces progression of atherosclerosis in mice. Nature. 2005;434:782–786. doi: 10.1038/nature03389. [DOI] [PubMed] [Google Scholar]
- Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, et al. 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem Biophys Res Comm. 1995;215:89–97. doi: 10.1006/bbrc.1995.2437. [DOI] [PubMed] [Google Scholar]
- Svendsen KB, Jensen TS, Bach FW. Does the cannabinoid dronabinol reduce central pain in multiple sclerosis? Randomised double blind placebo controlled crossover trial. Br Med J. 2004;329:253–257. doi: 10.1136/bmj.38149.566979.AE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo B, Schlicker E. Effects of cannabinoids on neurotransmission. In: Pertwee RG, editor. Cannabinoids. Heidelberg: Springer-Verlag; 2005. pp. 327–365. [DOI] [PubMed] [Google Scholar]
- Takahashi E, Katayama M, Niimi K, Itakura C. Additive subthreshold dose effects of cannabinoid CB1 receptor antagonist and selective serotonin reuptake inhibitor in antidepressant behavioral tests. Eur J Pharmacol. 2008;589:149–156. doi: 10.1016/j.ejphar.2008.05.020. [DOI] [PubMed] [Google Scholar]
- Tham SM, Angus JA, Tudor EM, Wright CE. Synergistic and additive interactions of the cannabinoid agonist CP55,940 with µ opioid receptor and α2-adrenoceptor agonists in acute pain models in mice. Br J Pharmacol. 2005;144:875–884. doi: 10.1038/sj.bjp.0706045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Mitchell JM, Besson M-J, Caboche J, Maldonado R. Behavioural and biochemical evidence for interactions between Δ9-tetrahydrocannabinol and nicotine. Br J Pharmacol. 2002;135:564–578. doi: 10.1038/sj.bjp.0704479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gaal LF, Rissanen AM, Scheen AJ, Ziegler O, Rössner S. Effects of the cannabinoid-1 receptor blocker rimonabant on weight reduction and cardiovascular risk factors in overweight patients: 1-year experience from the RIO-Europe study. Lancet. 2005;365:1389–1397. doi: 10.1016/S0140-6736(05)66374-X. [DOI] [PubMed] [Google Scholar]
- Van Gaal LF, Scheen AJ, Rissanen AM, Rössner S, Hanotin C, Ziegler O. Long-term effect of CB1 blockade with rimonabant on cardiometabolic risk factors: two year results from the RIO-Europe Study. Eur Heart J. 2008;29:1761–1771. doi: 10.1093/eurheartj/ehn076. [DOI] [PubMed] [Google Scholar]
- Van Sickle MD, Duncan M, Kingsley PJ, Mouihate A, Urbani P, Mackie K, et al. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science. 2005;310:329–332. doi: 10.1126/science.1115740. [DOI] [PubMed] [Google Scholar]
- Wade DT, Makela P, Robson P, House H, Bateman C. Do cannabis-based medicinal extracts have general or specific effects on symptoms in multiple sclerosis? A double-blind, randomized, placebo-controlled study on 160 patients. Mult Scler. 2004;10:434–441. doi: 10.1191/1352458504ms1082oa. [DOI] [PubMed] [Google Scholar]
- Wager-Miller J, Westenbroek R, Mackie K. Dimerization of G protein-coupled receptors: CB1 cannabinoid receptors as an example. Chem Phys Lipids. 2002;121:83–89. doi: 10.1016/s0009-3084(02)00151-2. [DOI] [PubMed] [Google Scholar]
- Walczak J-S, Pichette V, Leblond F, Desbiens K, Beaulieu P. Behavioral, pharmacological and molecular characterization of the saphenous nerve partial ligation: a new model of neuropathic pain. Neuroscience. 2005;132:1093–1102. doi: 10.1016/j.neuroscience.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Walczak J-S, Pichette V, Leblond F, Desbiens K, Beaulieu P. Characterization of chronic constriction of the saphenous nerve, a model of neuropathic pain in mice showing rapid molecular and electrophysiological changes. J Neurosci Res. 2006;83:1310–1322. doi: 10.1002/jnr.20821. [DOI] [PubMed] [Google Scholar]
- Walker JM, Hohmann AG. Cannabinoid mechanisms of pain suppression. In: Pertwee RG, editor. Cannabinoids. Handbook of Experimental Pharmacology. Heidelberg: Springer-Verlag; 2005. pp. 509–554. [DOI] [PubMed] [Google Scholar]
- Wallace MJ, Blair RE, Falenski KW, Martin BR, DeLorenzo RJ. The endogenous cannabinoid system regulates seizure frequency and duration in a model of temporal lobe epilepsy. J Pharmacol Exp Ther. 2003;307:129–137. doi: 10.1124/jpet.103.051920. [DOI] [PubMed] [Google Scholar]
- Walter L, Stella N. Cannabinoids and neuroinflammation. Br J Pharmacol. 2004;141:775–785. doi: 10.1038/sj.bjp.0705667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Wang H, Ning W, Backlund MG, Dey SK, DuBois RN. Loss of cannabinoid receptor 1 accelerates intestinal tumor growth. Cancer Res. 2008;68:6468–6476. doi: 10.1158/0008-5472.CAN-08-0896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteside GT, Lee GP, Valenzano KJ. The role of the cannabinoid CB2 receptor in pain transmission and therapeutic potential of small molecule CB2 receptor agonists. Curr Med Chem. 2007;14:917–936. doi: 10.2174/092986707780363023. [DOI] [PubMed] [Google Scholar]
- Wiley JL, Beletskaya ID, Ng EW, Dai Z, Crocker PJ, Mahadevan A, et al. Resorcinol derivatives: a novel template for the development of cannabinoid CB1/CB2 and CB2-selective agonists. J Pharmacol Exp Ther. 2002;301:679–689. doi: 10.1124/jpet.301.2.679. [DOI] [PubMed] [Google Scholar]
- Williams IJ, Edwards S, Rubo A, Haller VL, Stevens DL, Welch SP. Time course of the enhancement and restoration of the analgesic efficacy of codeine and morphine by Δ9-tetrahydrocannabinol. Eur J Pharmacol. 2006;539:57–63. doi: 10.1016/j.ejphar.2006.04.003. [DOI] [PubMed] [Google Scholar]
- Wotherspoon G, Fox A, McIntyre P, Colley S, Bevan S, Winter J. Peripheral nerve injury induces cannabinoid receptor 2 protein expression in rat sensory neurons. Neuroscience. 2005;135:235–245. doi: 10.1016/j.neuroscience.2005.06.009. [DOI] [PubMed] [Google Scholar]
- Wright KL, Duncan M, Sharkey KA. Cannabinoid CB2 receptors in the gastrointestinal tract: a regulatory system in states of inflammation. Br J Pharmacol. 2008;153:263–270. doi: 10.1038/sj.bjp.0707486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Cheng CL, Chen M, Manivannan A, Cabay L, Pertwee RG, et al. Anti-inflammatory property of the cannabinoid receptor-2-selective agonist JWH-133 in a rodent model of autoimmune uveoretinitis. J Leukoc Biol. 2007;82:532–541. doi: 10.1189/jlb.0307159. [DOI] [PubMed] [Google Scholar]
- Xu X, Liu Y, Huang S, Liu G, Xie C, Zhou J, et al. Overexpression of cannabinoid receptors CB1 and CB2 correlates with improved prognosis of patients with hepatocellular carcinoma. Cancer Genet Cytogenet. 2006;171:31–38. doi: 10.1016/j.cancergencyto.2006.06.014. [DOI] [PubMed] [Google Scholar]
- Yamamoto W, Mikami T, Iwamura H. Involvement of central cannabinoid CB2 receptor in reducing mechanical allodynia in a mouse model of neuropathic pain. Eur J Pharmacol. 2008;583:56–61. doi: 10.1016/j.ejphar.2008.01.010. [DOI] [PubMed] [Google Scholar]
- Yao BB, Mukherjee S, Fan Y, Garrison TR, Daza AV, Grayson GK, et al. In vitro pharmacological characterization of AM1241: a protean agonist at the cannabinoid CB2 receptor? Br J Pharmacol. 2006;149:145–154. doi: 10.1038/sj.bjp.0706838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yesilyurt O, Dogrul A, Gul H, Seyrek M, Kusmez O, Ozkan Y, et al. Topical cannabinoid enhances topical morphine antinociception. Pain. 2003;105:303–308. doi: 10.1016/s0304-3959(03)00245-8. [DOI] [PubMed] [Google Scholar]
- Yiangou Y, Facer P, Durrenberger P, Chessell IP, Naylor A, Bountra C, et al. COX-2, CB2 and P2X7-immunoreactivities are increased in activated microglial cells/macrophages of multiple sclerosis and amyotrophic lateral sclerosis spinal cord. [accessed 19 January 2009];BMC Neurol. 2006 6 doi: 10.1186/1471-2377-6-12. Available at: http://www.biomedcentral.com/1471-2377/6/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon MH, Choi JI. Pharmacologic interaction between cannabinoid and either clonidine or neostigmine in the rat formalin test. Anesthesiology. 2003;99:701–707. doi: 10.1097/00000542-200309000-00027. [DOI] [PubMed] [Google Scholar]
- Young JM, McGregor IS, Mallet PE. Co-administration of THC and MDMA (‘Ecstasy’) synergistically disrupts memory in rats. Neuropsychopharmacology. 2005;30:1475–1482. doi: 10.1038/sj.npp.1300692. [DOI] [PubMed] [Google Scholar]
- Zhang J, Hoffert C, Vu HK, Groblewski T, Ahmad S, O'Donnell D. Induction of CB2 receptor expression in the rat spinal cord of neuropathic but not inflammatory chronic pain models. Eur J Neurosci. 2003;17:2750–2754. doi: 10.1046/j.1460-9568.2003.02704.x. [DOI] [PubMed] [Google Scholar]
- Zhang M, Martin BR, Adler MW, Razdan RK, Jallo JI, Tuma RF. Cannabinoid CB2 receptor activation decreases cerebral infarction in a mouse focal ischemia/reperfusion model. J Cereb Blood Flow Metab. 2007;27:1387–1396. doi: 10.1038/sj.jcbfm.9600447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Martin BR, Adler MW, Razdan RK, Ganea D, Tuma RF. Modulation of the balance between cannabinoid CB1 and CB2 receptor activation during cerebral ischemic/reperfusion injury. Neuroscience. 2008;152:753–760. doi: 10.1016/j.neuroscience.2008.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]