

Abstract
Background and purpose:
Adenylyl cyclase sensitization occurs on chronic agonist activation of µ-opioid receptors and is manifested by an increase in cAMP levels (overshoot) on challenge with antagonist. It has been proposed that a long lasting constitutively active receptor is formed on chronic µ-opioid exposure and that antagonists with inverse agonist activity rapidly return the receptor to a basal state causing a cAMP overshoot and a more severe withdrawal response in vivo. This hypothesis depends on an accurate characterization of neutral and inverse agonist properties of opioid antagonists.
Experimental approach:
C6 glioma and HEK293 cells expressing µ-opioid receptors were used. Opioid antagonists were examined for their ability to induce a cAMP overshoot following chronic treatment with the agonist DAMGO ([D-Ala2,N-Me-Phe4,Glyol5]-enkephalin). The compounds were also characterized as agonists, inverse agonists or neutral antagonists by using assays for competitive binding, [35S]GTPγS (guanosine-5′-O-(3-[35S]thio)triphosphate) binding and changes in cell surface receptor expression.
Key results:
Naltrexone, 6β-naltrexol and naloxone were indistinguishable to the µ-opioid receptor in the opioid-naïve or dependent state and acted as neutral antagonists. The δ-opioid receptor inverse agonist RTI-5989-25 [(+)-N-[trans-4′-(2-methylphenyl)-2′-butenyl]-(3R,4R)-dimethyl-4-(3-hydroxyphenyl)piperidine], a 3,4-dimethyl-4-(3-hydroxyphenyl)-piperidine, was an inverse agonist at the µ-opioid receptor, and the peptide antagonist CTAP (H-D-Phe-Cys-Tyr-D-Trp-Arg-Thr-Pen-Thr-NH2) showed variable, assay-dependent properties. All the antagonists precipitated the same degree of cAMP overshoot in opioid-dependent cells.
Conclusions and implications:
Antagonists at the µ-opioid receptor may be neutral or show inverse agonist activity. Formation of a constitutively active µ-opioid receptor is not a requirement for the development or expression of adenylyl cyclase sensitization.
Keywords: µ-opioid receptor, constitutive activity, inverse agonism, protean agonism, adenylyl cyclase, cAMP overshoot, naltrexone, 6β-naltrexol, RTI-5989-25, CTAP
Introduction
The µ-opioid receptor belongs to the class of G-protein coupled receptors (GPCRs) that activate Gαi/o proteins and inhibit the family of adenylyl cyclase (AC) enzymes. Chronic exposure of these receptors, including the µ-opioid receptor, to agonist results in sensitization of AC as an adaptive cellular response (Watts and Neve, 2005). This occurs to maintain homeostatic control of AC activity and may be a cellular model of dependence (Christie, 2008). Following challenge with antagonist there is an expression of the developed sensitization, resulting in an increased accumulation of cAMP, so-called ‘cAMP overshoot’. This cAMP overshoot is seen not only in cultured cells exposed to µ-opioids (Clark et al., 2004; Zhao et al., 2006; Wang et al., 2007b) but also in vitro in CNS tissues from µ-opioid-dependent animals (Bohn et al., 2000). AC sensitization has been shown to be isoform-dependent, Pertussis toxin sensitive with an important role for Gβγ subunits and may involve an increased interaction between AC and Gαs (Watts and Neve, 2005). The mechanism behind this AC adaptation is not known, but kinase enzymes such as Raf-1 and PKC that could phosphorylate AC have been implicated.
Numerous studies have shown the µ-opioid receptor itself is a target for phosphorylation leading to desensitization (Johnson et al., 2005). Additionally, phosphorylation of the receptor has been implicated in AC sensitization (Wang et al., 1994; Wang et al.,1999; Sadee and Wang, 1995). Consistent with this, AC sensitization is abolished by mutation of Tyr394 in the C-terminal tail of the µ-opioid receptor expressed in CHO cells (Wang et al., 2007b). Phosphorylation has been suggested to convert the receptor to a persistent constitutively active (R*) state that develops on chronic opioid exposure and continuously signals even in the absence of agonist. Rapid reversal of the persistent R* state back to a basal resting state (R) is then postulated to cause the observed cAMP overshoot (Wang et al., 1994; Sadee and Wang, 1995; Liu and Prather, 2001). Ligands that inhibit constitutive activity are known as inverse agonists or are said to have negative intrinsic activity and bind preferentially to R, thus shifting the equilibrium in favour of this state. In contrast, neutral antagonists bind equally to R and R* and do not alter the equilibrium between receptor states (Milligan, 2003; Kenakin, 2004). Consequently, opioid antagonists that cause a cAMP overshoot have been characterized as inverse agonists (Wang et al., 1994; Sadee and Wang, 1995; Liu and Prather, 2001; Szucs et al., 2004). A role for a constitutively active µ-opioid receptor has been extended to dependence and withdrawal in vivo, and again antagonists that cause a more severe withdrawal have been identified as inverse agonists (Bilsky et al., 1996; Wang et al., 2001; Wang et al.2004; Raehal et al., 2005).
Based on their ability to induce a cAMP overshoot and precipitate a severe withdrawal in opioid-dependent systems, the opioid antagonists naloxone and naltrexone have been characterized as inverse agonists, while the naltrexone metabolite 6β-naltrexol, and the peptide CTAP (H-D-Phe-Cys-Tyr-D-Trp-Arg-Thr-Pen-Thr-NH2) were perceived to be neutral antagonists (Wang et al., 1994;Wang et al., 2001; Bilsky et al., 1996; Liu and Prather, 2001; Raehal et al., 2005). In support of this, a continuum of negative efficacies has been reported for µ-opioid antagonists, determined by their relative abilities to precipitate acute withdrawal in the mouse (Walker and Sterious, 2005). In contrast, we have shown in both the mouse (Divin et al., 2008) and the monkey (Ko et al., 2006) that there is not a qualitative difference between naltrexone and 6β-naltrexol to precipitate morphine withdrawal, but rather a quantitative difference that may have a pharmacokinetic basis.
The characterization of a role for a persistent, constitutively active µ-opioid receptor as a contributing factor in AC sensitization and opioid dependence in vivo relies on the correct definition of antagonists as neutral antagonists or inverse agonists. To test this in the absence of interference due to distribution and metabolism we have used a heterologous expression system of C6 glioma cells (C6µ), together with HEK293 cells expressing a FLAG-tagged µ-opioid receptor to study cell surface receptor levels. We have previously used C6µ cells in studies of opioid signalling including AC sensitization (Clark et al., 2004; Clark and Traynor, 2006) and have shown similar µ-opioid-mediated sensitization in HEK cells (Clark and Traynor, 2006). We have compared the ability to precipitate expression of AC sensitization and the pharmacological profiles of naltrexone and 6β-naltrexol, along with the standard opioid antagonist naloxone, the peptidic antagonist CTAP and the known δ-opioid inverse agonist (+)-N-[trans-4′-(2-methylphenyl)-2′-butenyl]-(3R,4R)-dimethyl-4-(3-hydroxyphenyl)piperidine (RTI-5989-25; Zaki et al., 2001). The results show that there is no inherent efficacy difference between 6β-naltrexol and naltrexone under the conditions studied and furthermore that development and manifestation of AC sensitization is not dependent on the formation of a constitutively active µ-opioid receptor.
Methods
Cell culture and treatments
C6 rat glioma cells stably transfected with the rat µ-opioid receptor (C6µ) or HEK293 cells stably transfected with the FLAG-tagged mouse µ-opioid receptor were grown to confluence in Dulbecco's modified Eagle's medium (DMEM) containing 0.5 mg·mL−1 or 0.8 mg·mL−1 Geneticin respectively. Cells were grown in the presence of 10% fetal bovine serum at 37°C in 5% CO2. For chronic opioid treatment, cells were incubated overnight with 10 µmol·L−1 DAMGO ([D-Ala2,N-Me-Phe4,Glyol5]-enkephalin). C6µ cells were used for all experiments except for the determination of cell surface receptor number, which utilized HEK cells expressing a FLAG-tagged µ-opioid receptor. C6µ cells expressed 3.2 ± 0.2 pmol·mg−1 protein receptor and HEK cells 9.7 ± 1.3 pmol·mg−1 protein receptor, determined by [3H]diprenorphine binding.
Membrane preparation
Cells were washed twice with ice cold phosphate-buffered saline (0.9% NaCl, 0.61 mmol·L−1 Na2HPO4 and 0.38 mmol·L−1 KH2PO4, pH 7.4), detached from the plate by incubation in harvesting buffer (20 mmol·L−1 HEPES, pH 7.4, 150 mmol·L−1 NaCl and 0.68 mmol·L−1 EDTA) and pelleted by centrifugation. The resulting pellet was suspended in cold 50 mmol·L−1 Tris buffer, pH 7.4 and homogenized with a Tissue Tearor (Biospec Products Inc., Bartlesville, OK). The homogenate was centrifuged at 18 000× g at 4°C for 20 min, and the pellet resuspended in 50 mmol·L−1 Tris, homogenized with a Tissue Tearor and recentrifuged. The final pellet was resuspended in 50 mmol·L−1 Tris, aliquoted and stored at −80°C until use. Protein concentration was measured by the method of Bradford (1976).
[3H]Diprenorphine binding
For competitive binding, cell membranes were incubated for 75 min at 25°C with varying concentrations (0.1 nmol·L−1–1 µmol·L−1) of ligand and 0.2 nmol·L−1[3H]diprenorphine in 50 mmol·L−1 Tris, pH 7.4 with and without the presence of 100 mmol·L−1 NaCl and 10 µmol·L−1 GTPγS. Non-specific binding was determined in the presence of 10 µmol·L−1 naloxone. Assays were stopped by rapid filtration through glass microfiber filtermats, type GF/C (Whatman, Clifton, NJ) by using a Brandell harvester (Gaithersburg, MD) followed by washing with cold 50 mmol·L−1 Tris buffer. Filtermats were dried, and 0.1 mL Ecolume was added to each sample. Filtermats were heat sealed in polyethylene bags, and radioactivity retained on the filters was measured by liquid scintillation counting in a Wallac 1450 MicroBeta Liquid Scintillation and Luminescence Counter (Perkin Elmer, Boston, MA).
[35S]GTPγS [Guanosine-5′-O-(3-[35S]thio)triphosphate] binding
C6 glioma cell membranes were incubated for 60 min at 25°C with 0.1 nmol·L−1[35S]GTPγS and with ligand (DAMGO, morphine, 6β-naltrexol, CTAP, naltrexone, naloxone or RTI-5989-25; 10 µmol·L−1) or vehicle (H2O) in GTPγS Buffer [50 mmol·L−1 Tris, pH 7.4, 1 mmol·L−1 EDTA, 5 mmol·L−1 MgCl2, 100 mmol·L−1 NaCl, 2.4 mmol·L−1 dithiothreitol (DTT), 30 µmol·L−1 GDP, 1 mU adenosine deaminase] or GTPγS buffer in which NaCl was replaced with KCl. In certain experiments with CTAP, the DTT was omitted. Alternatively, membranes were incubated with varying concentrations of morphine (1 nmol·L−1–0.1 mmol·L−1) with and without the presence of antagonist (10, 30 or 100 nmol·L−1) in GTPγS Buffer. Reactions were terminated by rapidly filtering samples through glass microfiber filtermats mounted in a Brandell harvester and rinsing three times with wash buffer (50 mmol·L−1 Tris, pH 7.4, 5 mmol·L−1 MgCl2 and 100 mmol·L−1 NaCl or KCl as appropriate). Bound [35S]GTPγS retained on the filtermats was determined as described for binding assays.
cAMP accumulation
Cells were grown in 24-well plates to reach confluence on the day of the assay. To measure AC inhibition cells were treated with varying concentrations of DAMGO (1 nmol·L−1–10 µmol·L−1) in DMEM for 15 min in the presence of 10 µmol·L−1 forskolin and 1 mmol·L−1 phosphodiesterase inhibitor IBMX (3-isobutyl-1-methylxanthine), without or with the presence of 6β-naltrexol or naltrexone (100 nmol·L−1). To measure AC sensitization, cells were treated overnight with the opioid agonist DAMGO (10 µmol·L−1). To begin the assay, media containing the opioid agonist was removed, and replaced with media containing 10 µmol·L−1 forskolin representing an approximately EC30 concentration (Clark et al., 2004), 1 mmol·L−1 IBMX unless otherwise stated and opioid antagonist (6β-naltrexol, CTAP, naltrexone, naloxone or RTI-5989-25). Alternatively, cells were washed by quickly removing and replacing media three times to remove the opioid agonist. Cells were incubated at 37°C for 5 min, and the assay was stopped with ice cold 0.1 mol·L−1 HCl. After 30 min at 4°C, cAMP accumulation was measured by using a cAMP enzyme immunoassay kit (Assay Designs, Ann Arbor, MI) following the manufacturer's instructions.
Cell surface receptor levels
HEK293-FLAG-µ cells were seeded onto poly-D-lysine coated plates (BD Biosciences, San Jose, CA) and incubated with or without 10 µmol·L−1 antagonist (6β-naltrexol, naltrexone, CTAP or RTI-5989-25) for 24 h. Cells were fixed with 3.7% formaldehyde in Tris-buffered saline, washed and blocked with 1% non-fat dry milk. The cells were then washed and incubated with monoclonal anti-FLAG-M2 alkaline phosphatase antibody (Sigma) followed by incubation with p-nitrophenyl-phosphate. At the end of the incubation each sample was added to 3 N NaOH in a 96-well plate, and absorbance at 405 nm was measured. Background absorbance was obtained from similarly treated untransfected HEK293 cells and subtracted from the absorbance of stable HEK293-FLAG-µ cells.
Data analysis and statistics
Data were analysed by using GraphPad Prism 4.0 (San Diego, CA). Antagonist binding affinities derived from competition curves were calculated as Ki (nmol·L−1) values and as their negative logarithm (pKi). Antagonist binding affinities from pharmacological experiments were also determined from antagonist-induced shifts in µ-opioid agonist concentration–effect curves as pKB or pA2 values. These values are the negative logarithm of the dissociation constant of an antagonist determined under equilibrium conditions and are a measure of an antagonist's affinity for its receptors. pKB values were calculated from shifts in µ-opioid agonist concentration–effect curves caused by a single (100 nmol·L−1) concentration of antagonist in the cAMP accumulation assays according to the equation pKB = −log[B/(dose-ratio − 1)], where B equals the concentration of opioid receptor antagonist and dose-ratio represents the EC50 concentration in the presence of antagonist divided by the EC50 concentration in the absence of antagonist (Divin et al., 2008). pA2 values were determined from shifts in the DAMGO concentration–effect curves in the [35S]GTPγS assay experiments in response to three different concentrations of the antagonists according to the Schild method (Arunlakshana and Schild, 1959). The data presented are from at least three experiments performed in duplicate, with results presented as mean ± SEM. Data were compared by using a two-tailed t-test, or two-way anova to compare concentration–response curves. Differences were considered significant if P < 0.05.
Drugs and reagents
Tissue culture media, Geneticin, fetal bovine serum and trypsin were from Invitrogen (Carlsbad, CA). [35S]GTPγS (1250 Ci·mmol−1) and [3H]diprenorphine (50 Ci·mmol−1) were obtained from Perkin-Elmer Life Sciences (Boston, MA). Adenosine deaminase was obtained from CalBiochem (San Diego, CA). Ecolume scintillation fluid was from ICN (Aurora, OH). Morphine sulphate, 6β-naltrexol, naltrexone and naloxone were obtained through the Narcotic Drug and Opioid Peptide Basic Research Center at the University of Michigan (Ann Arbor, MI). DAMGO, CTAP, GDP, GTPγS, forskolin, IBMX and all other biochemicals were from Sigma (St. Louis, MO) and were of analytical grade. RTI-5989-25 was prepared as previously described (Zaki et al., 2001). FLAG-tagged mouse µ-opioid receptor was a kind gift from Dr Lakshmi Devi, Mt. Sinai School of Medicine, New York, NY.
Results
Adenylyl cyclase sensitization
On chronic treatment and subsequent rapid removal of opioid agonist, cells expressing µ-opioid receptors exhibit an enhanced cAMP accumulation (overshoot) above untreated forskolin-stimulated controls (Watts and Neve, 2005). To assess cAMP overshoot in C6µ cells an approximately EC30 concentration of 10 µmol·L−1 forskolin was employed (Clark et al., 2004). At maximal concentration (10 µmol·L−1) the antagonists, 6β-naltrexol, CTAP, naltrexone, naloxone or RTI-5989-25, were all able to induce a cAMP overshoot following overnight treatment of C6µ cells with the high-efficacy µ-opioid agonist DAMGO (10 µmol·L−1; Figure 1A). All antagonists induced the same degree of cAMP overshoot that was the same as that obtained by washing cells by removing and replacing media to dissociate bound opioid agonist from the receptor (P > 0.05). Using morphine (10 µmol·L−1) to induce AC sensitization gave a lower percentage of cAMP overshoot compared with DAMGO across the antagonists, as previously reported (Liu and Prather, 2001), but the antagonists all gave a similar results with the putative inverse agonist naltrexone giving the same degree of overshoot (225 ± 20%) as 6β-naltrexol (248 ± 16%), CTAP (277 ± 14%) or RTI-5989-25 (202 ± 13%). In addition, the phosphodiesterase inhibitor IBMX present in our assays to prevent cAMP breakdown has been reported to block the inverse agonist effect of naltrexone (Wang et al., 1999). In the absence of IBMX the percentage of cAMP overshoot in cells treated overnight with 10 µmol·L−1 DAMGO was reduced, but removing IBMX from the assay did not reveal any significant difference between the degree of overshoot seen with naltrexone (275 ± 13%) compared with 6β-naltrexol (274 ± 16%), RTI (266 ± 68%) or CTAP (313 ± 34%).
Figure 1.
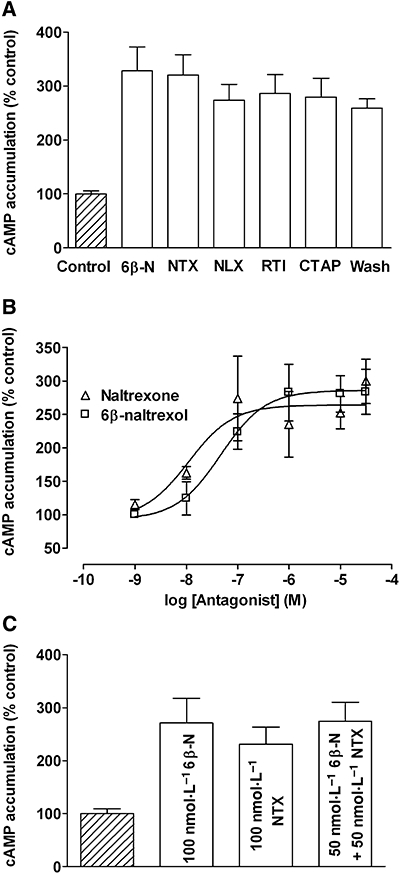
Adenylyl cyclase sensitization in C6 glioma (C6µ) cells. C6µ cells were treated overnight with 10 µmol·L−1 DAMGO, and cAMP overshoot was precipitated in the presence of 10 µmol·L−1 forskolin and 1 mmol·L−1 IBMX to prevent hydrolysis of cAMP. (A) cAMP overshoot was induced with 10 µmol·L−1 6β-naltrexol (6β-N), naltrexone (NTX), naloxone (NLX), RTI-5989-25 (RTI), CTAP or by washing. (B) cAMP overshoot was precipitated with 1 nmol·L−1–10 µmol·L−1 6β-naltrexol or naltrexone. (C) cAMP overshoot was precipitated with 100 nmol·L−1 6β-naltrexol, 100 nmol·L−1 naltrexone or 50 nmol·L−1 6β-naltrexol with 50 nmol·L−1 naltrexone. cAMP accumulation is expressed as a percentage of 10 µmol·L−1 forskolin-stimulated cAMP levels in vehicle-treated control cells (4.7 ± 0.5 pmol·µg−1 protein). Values represent mean ± SEM of three to five experiments performed in duplicate. CTAP, H-D-Phe-Cys-Tyr-D-Trp-Arg-Thr-Pen-Thr-NH2; DAMGO, [D-Ala2,N-MePhe4,Glyol5]-enkephalin; IBMX, 3-isobutyl-1-methylxanthine; RTI-5989-25, (+)-N2-[trans-4′-(2-methylphenyl)-2′-butenyl]2-2(3R,4R)-dimethyl-4-(3-hydroxyphenyl)piperidine.
As these results contrasted with previous reports, we further compared the ability of 6β-naltrexol and naltrexone to induce a cAMP overshoot in chronic DAMGO-treated cells (Figure 1B). Both compounds concentration-dependently induced an increase in cAMP in treated cells over control, vehicle-treated cells with similar EC50 values of 28.2 ± 5.2 nmol·L−1 and 22.4 ± 6.0 nmol·L−1 (P > 0.05) respectively. This was confirmed when cAMP overshoot was precipitated with a combination of the two antagonists. Thus, following overnight DAMGO, 100 nmol·L−1 6β-naltrexol, 100 nmol·L−1 naltrexone or 50 nmol·L−1 6β-naltrexol with 50 nmol·L−1 naltrexone precipitated the same level of cAMP overshoot (P > 0.05; Figure 1C).
These results demonstrate that the loss of a constitutively active receptor is not required for cAMP overshoot but that removing the chronic µ-opioid agonist, either by challenge with antagonist or by washing, is sufficient. Consequently in this assay all the antagonists appeared operationally the same, and so we undertook a more comprehensive pharmacological analysis to better define their relative efficacies as neutral antagonists or inverse agonists.
Receptor affinity
The affinity of ligands for GPCRs is greatly influenced by the presence or absence of Na+ and guanine nucleotides in the assay buffer that inhibit the formation of activated, R* receptor conformations and inhibit coupling to G-proteins respectively, to promote the basal, R state of the receptor. In contrast, in low ionic strength buffers, high affinity G-protein coupled states (R*G) exist (Strange, 2008). The ability of 6β-naltrexol, CTAP, naltrexone, naloxone and RTI-5989-25 to concentration-dependently displace the binding of the non-selective opioid antagonist [3H]diprenorphine in membranes from C6µ cells was measured in Tris-HCl buffer without and with 100 mmol·L−1 NaCl and 10 µmol·L−1 GTPγS. All compounds showed high µ-opioid receptor affinity in the order RTI-5989-25 > naltrexone > 6β-naltrexol > CTAP > naloxone (Table 1). The affinities of 6β-naltrexol, naltrexone and naloxone in the presence or absence of NaCl and GTPγS were not significantly different (P > 0.05), indicating an inability to distinguish R and R*G states of the µ-opioid receptor. However, CTAP was shifted to a lower affinity in a buffer containing NaCl and GTPγS (**P < 0.01), showing preferable binding to R*G states suggesting a compound with agonist activity in this assay. In contrast, RTI-5989-25 had a higher affinity in the NaCl and GTPγS containing buffer (*P < 0.05) showing preference for the basal R state as expected for an inverse agonist.
Table 1.
Opioid antagonist affinities for the µ-opioid receptor in C6 glioma cells
| Opioid |
Competitive binding |
[35S]GTPγS |
cAMP |
|
|---|---|---|---|---|
| Tris buffer+ Na+, GTPγS | Ki(nmol·L−1) (pKi) | pA2 | KB(nmol·L−1) (pKB) | |
| 6β-naltrexol | 0.93 ± 0.04 (9.03) | 1.26 ± 0.13 (8.90) | 8.91 ± 0.32 | 0.63 ± 0.19 (9.20) |
| Naltrexone | 0.38 ± 0.08 (9.42) | 0.46 ± 0.21 (9.33) | 9.35 ± 0.36 | 0.52 ± 0.12 (9.28) |
| RTI-5989-25 | 0.062 ± 0.024 (10.21) | 0.011 ± 0.005* (10.96) | 11.26 ± 0.17a | ND |
| CTAP | 1.52 ± 0.31 (8.82) | 7.00 ± 1.59** (8.15) | 7.96 ± 0.30 | ND |
| Naloxone | 2.44 ± 0.49 (8.61) | 1.62 ± 0.47 (8.79) | ND | ND |
Ki values were determined by competitive displacement of [3H]diprenorphine (0.2 nmol·L−1) binding in 50 mmol·L−1 Tris buffer, pH 7.4, in the presence and absence of 100 mmol·L−1 NaCl and 10 µmol·L−1 GTPγS. pA2 values were determined by Schild analysis of antagonism of morphine-stimulated [35S]GTPγS binding. The 95% confidence interval of all slopes in the Schild analysis contained unity. KB values were determined by measurement of the ability of 100 nmol·L−1 antagonist to shift the concentration–response curve for DAMGO-induced inhibition of forskolin-stimulated cAMP accumulation. Details of all assays are in the Methods. For comparison, pKi and pKB values were calculated as −log(K). Values represent means ± SEM of three experiments performed in duplicate.
[35S]GTPγS, guanosine-5′-O-(3-[35S]thio)triphosphate; CTAP, H-D-Phe-Cys-Tyr-D-Trp-Arg-Thr-Pen-Thr-NH2; DAMGO, [D-Ala2,N-MePhe4,Glyol5]-enkephalin; ND, not determined.
Affinity determined by single concentration of antagonist.
P < 0.05,
P < 0.01, compared with Tris buffer.
Antagonist affinity was also determined in a functional assay by measuring the ability of the antagonists to inhibit morphine-stimulated binding of [35S]GTPγS to G-protein (Table 1). All of the antagonists concentration-dependently induced parallel rightward shifts in the morphine concentration–response curve. Analysis of these results showed that the affinity values determined by Schild analysis (pA2) for naltrexone and 6β-naltrexol in the [35S]GTPγS assay were similar to their affinity values (pKi) determined in competition binding assays in Tris-HCl buffer in the absence or presence of NaCl and GTPγS, confirming equivalent affinity for basal and active states of the receptor. With CTAP, the pA2 matched its pKi in the presence of NaCl and GTPγS because of the predominance of low affinity (R) states of the receptor in the [35S]GTPγS assay. In contrast to results obtained for naltrexone and 6β-naltrexol, the affinity of RTI-5989-25 measured in the [35S]GTPγS assay matched the competitive binding affinity values in Tris-HCl buffer in the presence of NaCl and GTPγS (Table 1), but not in Tris-HCl buffer alone, suggesting a higher affinity for the basal R state of the receptor indicating inverse agonism. Additionally, using acute DAMGO-mediated inhibition of forskolin-stimulated cAMP formation as a measure of agonism, 100 nmol·L−1 6β-naltrexol or 100 nmol·L−1 naltrexone resulted in approximately the same degree of rightward shift in the DAMGO concentration–effect curve, inducing a 196 ± 62-fold shift and a 218 ± 36-fold shift respectively. These data yielded a similar affinity value (KB or pKB) for both antagonists (Table 1) again confirming 6β-naltrexol and naltrexone were indistinguishable to the µ-opioid receptor.
Binding affinities in buffers promoting high or low affinity states of the receptor are not necessarily indicative of agonism or inverse agonism at a receptor. For example, the highly efficacious opioid agonists etorphine and BW373U86 bind no differently in buffers promoting high and low affinity states of their respective receptors (Childers et al., 1993; Lee et al., 1999). Moreover, the antagonists 7-benzylidenenaltrexone and naltriben that show inverse agonism at the δ-opioid receptor do not bind preferentially to low affinity states (Neilan et al., 1999). Therefore, additional measures of ligand efficacy were examined.
Efficacy measures using the [35S]GTPγS binding assay
DAMGO (10 µmol·L−1) stimulated [35S]GTPγS binding in C6µ cell membranes by approximately sixfold (Table 2), indicating very efficient receptor–G-protein coupling. At a maximal concentration of 10 µmol·L−1, 6β-naltrexol, CTAP, naltrexone, naloxone and RTI-5989-25 alone did not significantly alter G-protein activation from basal values. However, there was a small, but non-significant increase in [35S]GTPγS binding for naloxone, naltrexone and CTAP as previously reported (Wang et al., 2001; Wang et al., 2007a), indicating a very high sensitivity to agonist stimulation in this system.
Table 2.
Opioid effects on [35S]GTPγS binding in membranes from C6 glioma cells without or with overnight pretreatment with 10 µmol·L−1 DAMGO
| Opioid (10 µmol·L−1) |
% Basal [35S]GTPγS binding |
|||
|---|---|---|---|---|
|
Na+ containing buffer |
K+ containing buffer |
|||
| Vehicle-treated | DAMGO-treated | Vehicle-treated | DAMGO-treated | |
| DAMGO | 580 ± 59** | 330 ± 21** | 226 ± 11** | 170 ± 11** |
| 6β-Naltrexol | 92.4 ± 3.2 | 114 ± 9.1 | 104 ± 3.8 | 103 ± 6.6 |
| Naltrexone | 115 ± 6.9 | 106 ± 5.6 | 111 ± 6.7 | 97.7 ± 4.0 |
| RTI-5989-25 | 96 ± 3.1 | 107 ± 7.6 | 73.9 ± 5.8** | 90.6 ± 5.2 |
| CTAP | 105 ± 6.4 | 101 ± 6.7 | 80.3 ± 5.2** | 82.9 ± 5.3* |
| CTAP (−DTT) | 132 ± 9.1* | 103 ± 13 | 108 ± 3.4 | 95.5 ± 3.2 |
| Naloxone | 107 ± 4.1 | 98.4 ± 7.0 | 102 ± 3.8 | 99.2 ± 3.7 |
| Basal binding | ||||
| +DTT | 14.9 ± 1.1 | 10.8 ± 0.8 | 27.2 ± 1.7 | 13.0 ± 1.2 |
| −DTT | 56.3 ± 3.9 | 57.8 ± 13.9 | 42.6 ± 7.0 | 31.9 ± 4.7 |
Assays were performed in the presence of 100 mmol·L−1 NaCl or 100 mmol·L−1 KCl as described in the Methods. All assays were performed in the presence of 2.4 mmol·L−1 dithiothreitol with the exception of CTAP where noted. Values represent means ± SEM for three to five experiments performed in duplicate. Basal binding values are given as fmol·mg−1 protein.
[35S]GTPγS, guanosine-5′-O-(3-[35S]thio)triphosphate; CTAP, H-D-Phe-Cys-Tyr-D-Trp-Arg-Thr-Pen-Thr-NH2; DAMGO, [D-Ala2,N-MePhe4,Glyol5]-enkephalin; DTT, dithiothreitol; RTI-5989-25, (+)-N-[trans-4′-(2-methylphenyl)-2′-butenyl]-(3R,4R)-dimethyl-4-(3-hydroxyphenyl)piperidine.
P < 0.05,
P < 0.001, significantly different from basal values.
Sodium ions by decreasing the level of active R* receptor also decrease basal G-protein activation. Consequently, basal signalling can be increased by replacing Na+ ions with K+ ions (Szekeres and Traynor, 1997; Selley et al., 2000). Under these conditions, basal [35S]GTPγS stimulation was almost doubled (14.9 fmol·mg−1 in NaCl, 27.2 fmol·mg−1 in KCl). Due to this increased basal activity, DAMGO stimulation, measured as percentage increase over basal, was reduced to approximately one-third of its level in the presence of Na+ ions. Even under these conditions of enhanced basal signalling, 6β-naltrexol, naltrexone and naloxone did not alter G-protein activation from basal values. In contrast, RTI-5989-25 and CTAP significantly decreased basal binding of [35S]GTPγS (***P < 0.001), suggesting inverse agonist activity in this assay.
CTAP is a cyclic peptide constrained by a disulphide bridge, and so the integrity of this structure may be compromised by the presence of the disulphide reducing agent, DTT present in the [35S]GTPγS assay buffer. Indeed, in the absence of DTT, CTAP no longer reduced [35S]GTPγS binding below basal values, but rather showed partial agonist activity that was significant in the presence of Na+ ions (Table 2). This reversal of CTAP efficacy in the absence of DTT was not, however, due to breaking of the disulphide bond of CTAP, which was stable to incubation with 2.5 mmol·L−1 DTT for 1 h at 25°C as determined by mass spectrometry (data not shown), in agreement with the stability of this compound in vivo (Abbruscato et al., 1997). Additionally, the receptor binding affinity for CTAP was not significantly different in the presence or absence of DTT (Ki: 1.52 ± 0.31 nmol·L−1 in the absence of DTT; 1.75 ± 0.41 nmol·L−1 in the presence of DTT) confirming stability of the peptide.
Chronic agonist treatment has been reported to reveal inverse agonist activity at the level of [35S]GTPγS binding in HEK293 cells stably expressing the µ-opioid receptor (Burford et al., 2000), in GH3 cells (Liu and Prather, 2001) and in brain membranes from chronically morphine-treated mice (Wang et al., 2004). Although our findings with cAMP overshoot do not support this, we examined [35S]GTPγS binding after chronic agonist treatment. C6µ cells were treated overnight with 10 µmol·L−1 DAMGO, which causes an eightfold shift in the potency of DAMGO and a 50% reduction in maximal effect of DAMGO to stimulate [35S]GTPγS binding in these cells (Yabaluri and Medzihradsky, 1997). [35S]GTPγS binding was then examined in the presence of either 100 mmol·L−1 NaCl or KCl (Table 2). There was no change in the basal level of [35S]GTPγS binding suggesting no increase in active states of the receptor. However, the effect of DAMGO (10 µmol·L−1) to stimulate G-protein activation was markedly reduced in both NaCl (by 43%) and KCl (by 25%) containing buffers, confirming tolerance. Neither 6β-naltrexol, naltrexone nor naloxone significantly altered G-protein activation from basal values. The ability of RTI-5989-25 to reduce basal levels of [35S]GTPγS binding was lost following DAMGO pretreatment, although the effect of CTAP in the presence of DTT to decrease basal signalling activity in Na+ free buffer was unchanged.
Cell surface receptor expression
Chronic treatment with inverse agonists increases GPCR cell surface receptor expression, possibly by inhibiting constitutive recycling (Zaki et al., 2001; Miserey-Lenkei et al., 2002). To further compare antagonists, changes in cell surface receptor expression following chronic antagonist exposure were determined in HEK293 cells stably expressing a FLAG-tagged µ-opioid receptor. Cells were treated for 24 h with 10 µmol·L−1 6β-naltrexol, naltrexone, CTAP or RTI-5989-25 (Figure 2). Neither 6β-naltrexol nor naltrexone treatment resulted in a change in the number of cell surface µ-opioid receptors, while treatment with RTI-5989-25 increased cell surface receptor levels by 41.5 ± 6.9% (**P < 0.01) and CTAP increased cell surface receptors by 11.3 ± 2.5% (*P < 0.05).
Figure 2.
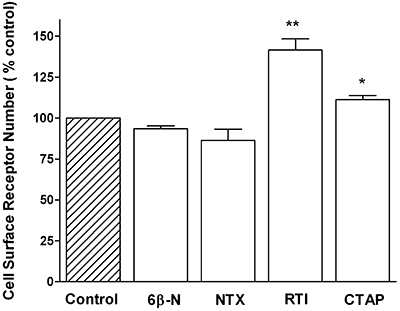
Cell surface receptor levels in HEK293-FLAG-µ cells treated for 24 h with 10 µmol·L−1 6β-naltrexol, naltrexone, RTI-5989-25 (RTI) or CTAP. Values are expressed as percentage of control, vehicle-treated cells and represent mean ± SEM of three experiments performed in duplicate. *P < 0.05, **P < 0.01, significantly different from vehicle. 6β-N, 6β-naltrexol; CTAP, H-D-Phe-Cys-Tyr-D-Trp-Arg-Thr-Pen-Thr-NH2; NTX, naltrexone RTI-5989-25, (+)-N-[Trans-4′-(2-methylphenyl)-2′-butenyl]-(3R,4R)dimethyl-4-(3-hydroxyphenyl) piperidine.
Antagonists in combination
Neutral antagonists inhibit the observable effects of inverse agonists (Costa and Herz, 1989; Neilan et al., 1999; Milligan, 2003). If antagonists have different degrees of efficacy then they should compete; alternatively if they have the same efficacy their effects should be additive. The ability of a combination of 6β-naltrexol and naltrexone to inhibit agonist action in the [35S]GTPγS binding assay was measured (Figure 3A). Morphine concentration-dependently stimulated [35S]GTPγS binding in C6µ cell membranes. Antagonist treatment resulted in rightward shifts of the morphine concentration–response curve with 10 nmol·L−1 6β-naltrexol inducing a 13.7 ± 4.9-fold shift, 10 nmol·L−1 naltrexone inducing a 14.7 ± 2.0-fold shift and a combination of 5 nmol·L−1 6β-naltrexol and 5 nmol·L−1 naltrexone inducing a similar 11.9 ± 2.8-fold shift in the morphine concentration–effect curve (P > 0.05) (Figure 3A), showing the compounds are indistinguishable to the receptor. In support of this, treatment with 100 nmol·L−1 6β-naltrexol, 100 nmol·L−1 naltrexone or a combination of 50 nmol·L−1 6β-naltrexol and 50 nmol·L−1 naltrexone antagonized maximal DAMGO-induced inhibition of forskolin-stimulated cAMP accumulation, resulting in 47.3 ± 4.4%, 42.7 ± 8.5% and 48.0 ± 7.9% inhibition respectively (P > 0.05; Figure 3B).
Figure 3.
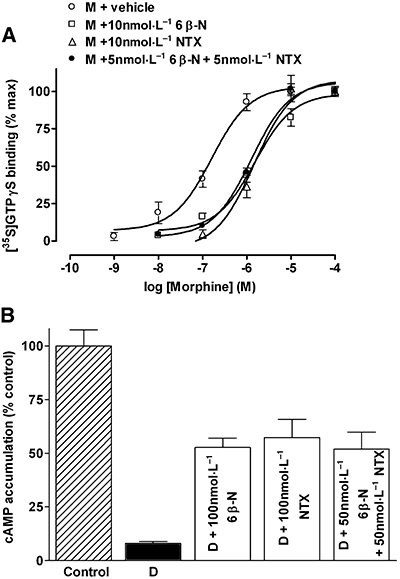
Effects of opioid antagonists in combination. (A) Morphine (M)-induced [35S]GTPγS binding in C6 µ glioma cell membranes in the absence and presence of 10 nmol·L−1 6β-naltrexol (6β-N), 10 nmol·L−1 naltrexone (NTX) or 5 nmol·L−1 6β-naltrexol and 5 nmol·L−1 naltrexone in combination. [35S]GTPγS binding is expressed as percentage maximal. (B) Inhibition of forskolin-stimulated cAMP accumulation by 1 µmol·L−1 DAMGO (D) in the absence and presence of 100 nmol·L−1 6β-naltrexol, 100 nmol·L−1 naltrexone or 50 nmol·L−1 6β-naltrexol and 50 nmol·L−1 naltrexone in combination. Accumulation of cAMP is expressed as percentage of vehicle-treated cells. Values represent mean ± SEM of three experiments performed in duplicate. [35S]GTPγS, guanosine-5′-O-(3-[35S]thio)triphosphate; DAMGO, [D-Ala2,N-MePhe4,Glyol5]-enkephalin.
Discussion
The present results suggest that, at least in C6µ cells, RTI-5989-25 is an inverse agonist at the µ-opioid receptor; CTAP has variable efficacy that depends on the assay conditions and naltrexone; naloxone and 6β-naltrexol are all neutral antagonists. Moreover, all of the antagonists examined, including the inverse agonist RTI-5989-25, promoted the same level of cAMP overshoot in cells chronically treated with µ-opioid agonist. This indicates that rapid formation of R from a putatively phosphorylated, constitutively active R* form was not involved in the development or expression of AC sensitization.
The putative inverse agonist naltrexone and the putative neutral antagonist 6β-naltrexol appeared indistinguishable to the µ-opioid receptor in vitro and were operationally the same in precipitation of cAMP overshoot, supporting our findings in the mouse (Divin et al., 2008), reinforced by our data in the monkey (Ko et al., 2006), that differences between the antagonists may not be pharmacodynamic, but rather due to differential access to µ-opioid receptors in the CNS. Opioid withdrawal is rapidly induced following administration of an opioid antagonist before steady-state concentrations are likely to be established. Thus, a differential rate of access will result in non-equivalent concentrations of antagonists at the receptor, resulting in different degrees of agonist displacement and consequently differences in the severity of the observed withdrawal behaviours. This idea is substantiated by in vitro findings from Zhao et al. (2006) who reported differences between µ-opioid agonists to induce AC sensitization are not due to agonist-dependent effects in the development of sensitization, but rather due to variation in the expression of AC sensitization caused by the ability of antagonists to displace agonist from the receptor.
Constitutive activity and increased basal signalling of the µ-opioid receptor in naïve cells has been difficult to detect (Neilan et al., 1999), but has been observed in HEK293 cells (Burford et al., 2000), in CHO cells (Szucs et al., 2004) and in dorsal root ganglion neurons from β-arrestin2 knockout mice (Walwyn et al., 2007). However, constitutive activity of µ-opioid receptors and the inverse agonist activity of naltrexone or naloxone has been reported following chronic pretreatment with the µ-opioid agonists morphine or DAMGO in several systems including GH3 cells (Liu and Prather, 2001), HEK293 cells (Wang et al., 1999; Wang et al., 2001), SH-SY5Y cells (Wang et al., 1994) and mouse brain homogenates (Wang et al., 2004). Our results suggest this does not occur in C6 cells. Similarly, an inverse agonist effect of naloxone was not seen in morphine-treated CHO cells (Wang et al., 1999), and no development of constitutive µ-opioid signalling has been observed at the level of whole cell calcium currents in locus ceruleus or periaqueductal grey neurons from chronically morphine-treated rodents (Connor et al., 1999; Bagley et al., 2005). Consequently, the ability to observe the development of constitutive activity of the µ-opioid receptor on chronic opioid treatment and an inverse action of naltrexone or naloxone appears to be highly system-and/or assay-dependent.
It is possible that, in systems where an inverse agonist effect of naloxone or naltrexone is not seen, the level of µ-opioid receptor constitutive activity is low (Neilan et al., 1999), even in the opioid-dependent state and consequently ligands that differentiate only weakly between R and R* appear as neutral antagonists, except under particular conditions. For example, our assays use 5 mmol·L−1 Mg2+, but inhibition of basal µ-opioid signalling, as measured by inhibition of basal [35S]GTPγS binding by β-chlornaltrexamine is seen in naïve CHO cells only at low levels of Mg2+, although the level of Mg2+ is not important to observe this response in naïve GH3 cells (Wang et al., 2001). Thus, specific environments, interacting proteins and receptor conformations, perhaps including distinctive receptor phosphorylation, may be needed to show the inverse agonist properties of naltrexone and naloxone. Indeed, Li et al. (2001) using a mutation in the DRY (Asp-Arg-Tyr) region of the second intracellular loop to give a constitutively active µ-opioid receptor, suggested naloxone and naltrexone to have inverse agonist activity. However, at another constitutively active µ-opioid receptor mutant formed by alanine replacement of two cysteine residues in the C-terminal tail, naloxone and naltrexone were neutral antagonists (Brillet et al., 2003). In the current study using wild-type µ-opioid receptors, naloxone, naltrexone and 6β-naltrexol behaved as neutral antagonists but RTI-5989-25 and CTAP did show inverse agonist properties confirming the cells can distinguish between antagonists on the basis of the presence or absence of negative efficacy and therefore the effects of antagonists on the expression of AC sensitization.
The variable properties of CTAP support the highly situation-sensitive nature of inverse agonism. CTAP acted as an inverse agonist in the [35S]GTPγS assay when performed in the presence of the reducing agent DTT, and CTAP increased µ-opioid receptor cell surface expression. On the other hand, CTAP stimulated [35S]GTPγS binding in the absence of DTT indicating partial agonist activity, and bound preferentially to the µ-opioid receptor in Tris-HCl buffer that promotes high agonist-affinity (R*) states. Condition-dependent properties of CTAP can also be inferred from other reports on this compound. CTAP did not precipitate withdrawal in mice following a single injection of a high dose of morphine (Bilsky et al., 1996) yet, precipitated withdrawal symptoms in chronically morphine-pelleted rats (Maldonado et al., 1992) and evoked contractions in guinea-pig ilea treated overnight with morphine (Mundey et al., 2000). The differential ability of CTAP to induce withdrawal in these situations may be a consequence of the severity of dependence. On the other hand, CTAP did not precipitate a cAMP overshoot in SH-SY5Y cells (Wang et al., 1994) or GH3 cells (Liu and Prather, 2001), treated for long periods with high concentrations of morphine and/or DAMGO but showed inverse agonist properties in both naïve and chronic morphine-treated CHO cells expressing a µ-opioid receptor, possibly through a mechanism involving Gαs (Szucs et al., 2004). CTAP has been shown to antagonize DAMGO in vivo much more efficiently than other peptides and non-peptides and may non-competitively interact with the alkaloids etorphine and morphine and the antagonist naltrexone (Sterious and Walker, 2003; Walker, 2006), so it is possible that this these varied reports are due to an unusual mode of binding to the µ-opioid receptor. Overall, CTAP appears to be a protean ligand, and it can behave as a positive and inverse agonist on the same receptor (Kenakin, 2004; Neubig, 2007), with properties highly dependent on the assay conditions.
Our assay-dependent results with CTAP are not due to instability of the peptide so may be caused by the presence of alternative conformational states of the receptor under the different assay conditions. The µ-opioid receptor is not very sensitive to the reducing action of DTT (Shahrestanifar et al., 1996). Nonetheless, the increased basal [35S]GTPγS binding and the loss of effect of Na+ suggests that the receptor itself might be involved. Like other GPCRs, the µ-opioid receptor contains two conserved cysteine residues in the first and second extracellular loops that form a disulphide bond. The integrity of this disulphide bond controls receptor conformation of GPCRs (Pedersen and Ross, 1985; Lin et al., 1996) and so could alter the properties of CTAP, in particular if this compound does have an atypical interaction with the µ-opioid receptor (Sterious and Walker, 2003; Walker, 2006). Studies with purified receptors may be needed to explain these observations.
RTI-5989-25 has been previously identified as an inverse agonist at the δ-opioid receptor (Zaki et al., 2001), and this study has characterized RTI-5989-25 as an inverse agonist at the µ-opioid receptor. This definition is based on a greater affinity for the µ-opioid receptor in a buffer system that promotes low affinity (R) states of the receptor and a decrease in [35S]GTPγS binding below basal levels when constitutive signalling is enhanced in Na+-free buffer by formation of R* and R*G. RTI-5989-25 treatment also resulted in an increase in cell surface µ-opioid receptor expression in HEK293-FLAG-µ cells. A surprising finding of the present study was the loss of negative intrinsic activity of RTI-5989-25 in cells chronically treated with the µ-opioid agonist DAMGO. This suggests that rather than being more active in the dependent state compounds with negative intrinsic activity lose inverse agonist activity. This could be due to a reduction in the level of µ-opioid receptors (Yabaluri and Medzihradsky, 1997) and/or desensitization of the receptor (Johnson et al., 2005), thus reducing the chance of receptor–G-protein collisions. It is unclear why this is opposite to effects seen in other systems, but this situation may predominate in the absence of factors that provide for constitutive activity. However, this observation does not support the need for formation of a constitutively active receptor in AC sensitization.
In summary, the results show that in systems that are capable of identifying compounds with inverse agonist activity, naltrexone and 6β-naltrexol are neutral antagonists that are indistinguishable to the µ-opioid receptor. The degree of cAMP overshoot following chronic opioid sensitization of AC precipitated by opioid antagonists, whether characterized as neutral, inverse or protean, was the same as that seen by washing cells with buffer to dissociate receptor-bound agonist. AC sensitization is a highly complex process that is likely to depend on a variety of cell-specific factors including the G-protein and AC isoform profile (Watts and Neve, 2005), which may determine whether constitutive activity and inverse agonism are involved. However, the findings from the present study, together with our previous in vivo studies (Ko et al., 2006; Divin et al., 2008) indicate that formation of a stable constitutively active (R*) state of the µ-opioid receptor is not a necessary prerequisite for the development of AC sensitization or µ-opioid dependence and withdrawal.
Acknowledgments
We thank Lauren Purington for assistance with the stability analysis of CTAP and Lewis Hicks (Undergraduate Research Opportunities Program) for performing some of the [35S]GTPγS assays. This study was funded by NIH grants DA 04087 and DA19276 (JRT, MFD) and DA09045 (FIC). MFD and FAB were also supported by NIH training grants GM07767 and DA07261.
Glossary
Abbreviations
- [35S]GTPγS
guanosine-5′-O-(3-[35S]thio)triphosphate
- CTAP
H-D-Phe-Cys-Tyr-D-Trp-Arg-Thr-Pen-Thr-NH2
- DAMGO
[D-Ala2,N-MePhe4,Glyol5]-enkephalin
- DTT
dithiothreitol
- HEK
human embryonic kidney
- IBMX
3-isobutyl-1-methylxanthine
- RTI-5989-25
(+)-N-[trans-4′-(2-methylphenyl)-2′-butenyl]-(3R,4R)-dimethyl-4-(3-hydroxyphenyl)piperidine
Conflict of interest
The authors state no conflict of interest.
References
- Abbruscato TJ, Thomas SA, Hruby VJ, Davis TP. Blood-brain barrier permeability and bioavailability of a highly potent and mu-selective opioid antagonist, CTAP: comparison with morphine. J Pharmacol Exp Ther. 1997;280:402–409. [PubMed] [Google Scholar]
- Arunlakshana O, Schild HO. Some quantitative uses of drug antagonists. Br J Pharmacol Chemother. 1959;15:48–58. doi: 10.1111/j.1476-5381.1959.tb00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagley EE, Chieng BCH, Christie MJ, Connor M. Opioid tolerance in periaqueductal gray mouse neurons isolated from mice chronically treated with morphine. Br J Pharmacol. 2005;146:68–76. doi: 10.1038/sj.bjp.0706315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilsky EJ, Bernstein RN, Wang Z, Sadee W, Porreca F. Effects of naloxone and D-Phe-Cys-Tyr-D-Trp-Arg-Thr-Pen-Thr-NH2 and the protein kinase inhibitors H7 and H8 on acute morphine dependence and antinociceptive tolerance in mice. J Pharmacol Exp Ther. 1996;277:484–490. [PubMed] [Google Scholar]
- Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG. Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature. 2000;408:720–723. doi: 10.1038/35047086. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Brillet K, Kieffer KL, Massotte D. Enhanced spontaneous activity of the mu opioid receptor by cysteine mutations: characterization of a tool for inverse agonist screening. BMC Pharmacol. 2003;3:14. doi: 10.1186/1471-2210-3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burford NT, Wang D, Sadee W. G-protein coupling of µ-opioid receptors (OP3): elevated basal signaling activity. Biochem J. 2000;348:531–537. [PMC free article] [PubMed] [Google Scholar]
- Childers SR, Fleming LM, Selley DE, McNutt RW, Chang KJ. BW373U86: a nonpeptidic delta-opioid agonist with novel receptor G-protein mediated actions in rat brain membranes and neuroblastoma cells. Mol Pharmacol. 1993;44:827–834. [PubMed] [Google Scholar]
- Christie MJ. Cellular adaptations to chronic opioids: tolerance, withdrawal and addiction. Br J Pharmacol. 2008;154:384–396. doi: 10.1038/bjp.2008.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MJ, Traynor JR. Mediation of adenylyl cyclase sensitization by PTX-insensitive GαoA, Gαi1, Gαi2 or Gαi3. J Neurochem. 2006;99:1494–1504. doi: 10.1111/j.1471-4159.2006.04176.x. [DOI] [PubMed] [Google Scholar]
- Clark MJ, Neubig RR, Traynor JR. Endogenous regulator of G protein signaling protein suppress Go-dependent, µ-opioid agonist-mediated adenylyl cyclase supersensitization. J Pharmacol Exp Ther. 2004;310:215–222. doi: 10.1124/jpet.103.064824. [DOI] [PubMed] [Google Scholar]
- Connor M, Borgland SL, Christie MJ. Continued morphine modulation of calcium channel currents in acutely isolated locus coeruleus neurons from morphine-dependent rats. Br J Pharmacol. 1999;128:1561–1569. doi: 10.1038/sj.bjp.0702922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa T, Herz A. Antagonists with negative intrinsic activity at δ-opioid receptors coupled to GTP-binding proteins. Proc Natl Acad Sci USA. 1989;86:7321–7325. doi: 10.1073/pnas.86.19.7321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divin MF, Ko MC, Traynor JR. Comparison of the opioid antagonist properties of naltrexone and 6β-naltrexol in morphine-naïve and morphine-dependent mice. Eur J Pharmacol. 2008;583:48–55. doi: 10.1016/j.ejphar.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EE, Christie MJ, Connor M. The role of opioid receptor phosphorylation and trafficking in adaptations to persistent opioid treatment. Neurosignals. 2005;14:290–302. doi: 10.1159/000093044. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Efficacy as a vector: the relative prevalence and paucity of inverse agonism. Mol Pharmacol. 2004;65:2–11. doi: 10.1124/mol.65.1.2. [DOI] [PubMed] [Google Scholar]
- Ko MC, Divin MF, Lee H, Woods JH, Traynor JR. Differential in vivo potencies of naltrexone and 6β-naltrexol in the monkey. J Pharmacol Exp Ther. 2006;316:772–779. doi: 10.1124/jpet.105.094409. [DOI] [PubMed] [Google Scholar]
- Lee KO, Akil H, Woods JH, Traynor JR. Differential binding properties of oripavines at cloned mu-and delta-opioid receptors. Eur J Pharmacol. 1999;378:323–330. doi: 10.1016/s0014-2999(99)00460-4. [DOI] [PubMed] [Google Scholar]
- Li J, Chen C, Huang P, Liu-Chen LY. Inverse agonist up-regulates the constitutively active D3.49(164)Q mutant of the rat µ-opioid receptor by stabilizing the structure and blocking constitutive internalization and down-regulation. Mol Pharmacol. 2001;60:1064–1075. doi: 10.1124/mol.60.5.1064. [DOI] [PubMed] [Google Scholar]
- Lin S, Gether U, Kobilka BK. Ligand stabilization of the β2-adrenergic receptor: effect of DTT on receptor conformation monitored by circular dichroism and fluorescence spectroscopy. Biochemistry. 1996;35:14445–14451. doi: 10.1021/bi961619+. [DOI] [PubMed] [Google Scholar]
- Liu JG, Prather PL. Chronic exposure to µ-opioid agonists produces constitutive activation of the µ-opioid receptors in direct proportion to the efficacy of the agonist used for pretreatment. Mol Pharmacol. 2001;60:53–62. doi: 10.1124/mol.60.1.53. [DOI] [PubMed] [Google Scholar]
- Maldonado R, Negus S, Koob GF. Precipitation of morphine withdrawal syndrome in rats by administration of mu-, delta-and kappa-selective opioid antagonists. Neuropharmacology. 1992;31:1231–1241. doi: 10.1016/0028-3908(92)90051-p. [DOI] [PubMed] [Google Scholar]
- Milligan G. Constitutive activity and inverse agonists of G protein coupled receptors: a current perspective. Mol Pharm. 2003;64:1271–1276. doi: 10.1124/mol.64.6.1271. [DOI] [PubMed] [Google Scholar]
- Miserey-Lenkei S, Parnot C, Bardin S, Corvol P, Clauser E. Constitutive internalization of constitutively active angiotensin II AT1A receptor mutants is blocked by inverse agonists. J Biol Chem. 2002;277:5891–5901. doi: 10.1074/jbc.M108398200. [DOI] [PubMed] [Google Scholar]
- Mundey MK, Ali A, Wilson VG. Pharmacological examination of contractile responses of the guinea-pig isolated ileum produced by µ-opioid receptor antagonists in the presence of, and following exposure to, morphine. Br J Pharmacol. 2000;131:893–902. doi: 10.1038/sj.bjp.0703659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neilan CL, Akil H, Woods JH, Traynor JR. Constitutive activity of the δ-opioid receptor expressed in C6 glioma cells: identification of non-peptide δ-inverse agonists. Br J Pharmacol. 1999;128:556–562. doi: 10.1038/sj.bjp.0702816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubig RR. Missing links: mechanisms of protean agonism. Mol Pharmacol. 2007;71:1200–1202. doi: 10.1124/mol.107.034926. [DOI] [PubMed] [Google Scholar]
- Pedersen SE, Ross EM. Functional activation of β-adrenergic receptors by thiols in the presence or absence of agonist. J Biol Chem. 1985;260:14150–14157. [PubMed] [Google Scholar]
- Raehal KM, Lowery JJ, Bhamidipati CM, Paolino RM, Blair JR, Wang D, et al. In vivo characterization of 6β-naltrexol, an opioid ligand with less inverse agonist activity compared with naltrexone and naloxone in opioid-dependent mice. J Pharmacol Exp Ther. 2005;313:1150–1162. doi: 10.1124/jpet.104.082966. [DOI] [PubMed] [Google Scholar]
- Sadee W, Wang Z. Agonist induced constitutive receptor activation as a novel regulatory mechanism. In: Sharp BM, Friedman H, Eisenstein TK, Madden JJ, editors. The Brain Immune Axis and Substance Abuse. New York: Plenum Press; 1995. pp. 85–90. [Google Scholar]
- Selley DE, Cao CC, Liu Q, Childers SR. Effects of sodium on agonist efficacy for G-protein activation in µ-opioid receptor-transfected CHO cells and rat thalamus. Br J Pharmacol. 2000;130:987–996. doi: 10.1038/sj.bjp.0703382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahrestanifar M, Wang WW, Howells RD. Studies on inhibition of µ and δ opioid receptor binding by dithiothreitol and N-ethylmaleimide. J Biol Chem. 1996;271:5505–5512. doi: 10.1074/jbc.271.10.5505. [DOI] [PubMed] [Google Scholar]
- Sterious SN, Walker EA. Potency differences for D-Phe-Cys-Tyr-D-Trp-Arg-Thr-Pen-Thr-NH2 as an antagonist of peptide and alkaloid µ-agonists in an antinociception assay. J Pharmacol Exp Ther. 2003;304:301–309. doi: 10.1124/jpet.102.042093. [DOI] [PubMed] [Google Scholar]
- Strange PG. Agonist binding, agonist affinity and agonist efficacy at G protein-coupled receptors. Br J Pharmacol. 2008;153:1353–1363. doi: 10.1038/sj.bjp.0707672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szekeres PG, Traynor JR. Delta opioid modulation of the binding of guanosine-5′-O-(3-[35S]thio)triphosphate to NG108-15 cell membranes: characterization of agonist and inverse agonist effects. J Pharmacol Exp Ther. 1997;283:1276–1284. [PubMed] [Google Scholar]
- Szucs M, Boda K, Gintzler AR. Dual effects of DAMGO [D-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin and CTAP (D-Phe-Cys-Tyr-D-Trp-Arg-Thr-Pen-Thr-NH2) on adenylyl cyclase activity: implications for µ-opioid receptor Gs coupling. J Pharmacol Exp Ther. 2004;310:256–262. doi: 10.1124/jpet.104.066837. [DOI] [PubMed] [Google Scholar]
- Walker EA. In vivo pharmacological resultant analysis reveals noncompetitive interactions between opioid antagonists in the rat tail-withdrawal assay. Br J Pharmacol. 2006;149:1071–1082. doi: 10.1038/sj.bjp.0706946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker EA, Sterious SN. Opioid antagonists differ according to negative intrinsic efficacy in a mouse model of acute dependence. Br J Pharmacol. 2005;145:975–983. doi: 10.1038/sj.bjp.0706247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walwyn W, Evans CJ, Hales TG. β-arrestin2 and cSrc regulate the constitutive activity and recycling of µ opioid receptors in dorsal root ganglion neurons. J Neurosci. 2007;27:5092–5102. doi: 10.1523/JNEUROSCI.1157-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Raehal KM, Bilsky EJ, Sadee W. Inverse agonists and neutral antagonists at µ-opioid receptor (MOR): possible role of basal receptor signaling in narcotic dependence. J Neurochem. 2001;77:1590–1600. doi: 10.1046/j.1471-4159.2001.00362.x. [DOI] [PubMed] [Google Scholar]
- Wang D, Raehal KM, Lin ET, Lowery JJ, Kieffer BL, Bilsky EJ, et al. Basal signaling activity of µ-opioid receptor in mouse brain: role in narcotic dependence. J Pharmacol Exp Ther. 2004;308:512–520. doi: 10.1124/jpet.103.054049. [DOI] [PubMed] [Google Scholar]
- Wang D, Sun X, Sadee W. Different effects of opioid antagonists on µ-, δ-, and κ-opioid receptors with and without agonist pretreatment. J Pharmacol Exp Ther. 2007a;321:544–552. doi: 10.1124/jpet.106.118810. [DOI] [PubMed] [Google Scholar]
- Wang H, Guang W, Barbier E, Shapiro P, Wang JB. Mu opioid receptor mutant, T394A, abolishes opioid-mediated adenylyl cyclase superactivation. Neuroreport. 2007b;18:1969–1973. doi: 10.1097/WNR.0b013e3282f228b2. [DOI] [PubMed] [Google Scholar]
- Wang Z, Bilsky EJ, Porreca F, Sadee W. Constitutive mu opioid receptor activation as a regulatory mechanism underlying narcotic tolerance and dependence. Life Sci. 1994;54:PL339–PL350. doi: 10.1016/0024-3205(94)90022-1. [DOI] [PubMed] [Google Scholar]
- Wang Z, Bilsky EJ, Xang D, Porreca F, Sadee W. 3-Isobutyl-1-methylxanthine inhibits basal µ-opioid receptor phosphorylation and reverses acute morphine tolerance and dependence in mice. Eur J Pharmacol. 1999;371:1–9. doi: 10.1016/s0014-2999(99)00131-4. [DOI] [PubMed] [Google Scholar]
- Watts VJ, Neve KA. Sensitization of adenylate cyclase by Gαi/o-coupled receptors. Pharmacol Ther. 2005;106:405–421. doi: 10.1016/j.pharmthera.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Yabaluri N, Medzihradsky F. Down-regulation of the µ-opioid receptor by full but not partial agonists is independent of G protein coupling. Mol Pharmacol. 1997;52:896–902. doi: 10.1124/mol.52.5.896. [DOI] [PubMed] [Google Scholar]
- Zaki PA, Keith DE, Jr, Thomas JB, Carroll FI, Evans CJ. Agonist-, antagonist-, and inverse agonist-regulated trafficking of the δ-opioid receptor correlates with, but does not require, G protein activation. J Pharmacol Exp Ther. 2001;298:1015–1020. [PubMed] [Google Scholar]
- Zhao H, Loh H, Law PY. Adenylyl cyclase superactivation induced by long-term treatment with opioid agonists is dependent on receptor localization within lipid rafts and is independent of receptor internalization. Mol Pharmacol. 2006;69:1421–1432. doi: 10.1124/mol.105.020024. [DOI] [PubMed] [Google Scholar]