

Abstract
Background and purpose:
Reactive oxygen species (ROS) derived from Nox2-containing reduced form of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity is reportedly detrimental in cerebrovascular disease. However, ROS generation by other Nox isoforms may have a physiological role. No Nox2-selective inhibitors have yet been identified, and thus it is unclear whether isoform non-selective Nox inhibitors would necessarily improve outcome after stroke. We assessed the effect of apocynin on cerebrovascular ROS production and also on outcome following cerebral ischaemia when administered either before ischaemia or after cerebral reperfusion. The involvement of Nox2-containing NADPH oxidase in the effects of apocynin was assessed using Nox2−/− mice.
Experimental approach:
Transient cerebral ischaemia was induced by 0.5 h middle cerebral artery occlusion followed by 23.5 h reperfusion. Mice received apocynin (2.5 mg·kg−1, i.p.) either 0.5 h before ischaemia or 1 h after reperfusion. In situ superoxide production after cerebral ischaemia-reperfusion was measured in brain sections of wild-type mice at 24 h using dihydroethidium fluorescence.
Key results:
Treatment with apocynin 0.5 h before ischaemia reduced total infarct volume, neurological impairment and mortality in wild-type but not Nox2−/− mice. Conversely, treatment with apocynin 1 h after initiation of reperfusion had no protective effect. Cerebral ischaemia and reperfusion increased superoxide production in the brain at 24 h, and pretreatment but not posttreatment with apocynin reduced superoxide levels.
Conclusions and implications:
Apocynin improves outcome following stroke when administered before ischaemia in wild-type but not Nox2−/− mice.
Keywords: cerebral ischaemia, NADPH oxidase, Nox2, apocynin, superoxide
Introduction
Emerging evidence suggests that reduced form of nicotinamide adenine dinucleotide phosphate (NADPH) oxidases are the predominant source of cardiovascular reactive oxygen species (ROS) (Mohazzab et al., 1994; Griendling, 2004). NADPH oxidase-derived ROS are thought to play a detrimental role in the brain following stroke (Chan, 2001; Miller et al., 2006a; Muralikrishna Adibhatla and Hatcher, 2006) and in other vascular beds during diseases associated with oxidative stress, including hypertension and atherosclerosis (Brandes and Kreuzer, 2005; Miller et al., 2005). There is therefore much interest in the potential use of NADPH oxidase inhibitors as cardiovascular therapeutics.
Apocynin is currently the most selective commercially available inhibitor of the NADPH oxidase family of enzymes. It is thought to inhibit activation of the enzyme by preventing translocation of p47phox from the cytosol to the Nox1/2 catalytic membrane domain (Stolk et al., 1994); however, there is little direct evidence that inhibition of ROS levels by apocynin in fact requires this action.
Apocynin has been shown to reduce lipid peroxidation, oxidative DNA damage, glial cell activation and hippocampal neuronal degeneration following 3 h global cerebral ischaemia in gerbils (Wang et al., 2006). Similarly, apocynin has been reported to inhibit blood–brain barrier disruption (Kahles et al., 2007), improve neurological function and reduce both infarct volume and cerebral haemorrhage when administered before induction of cerebral ischaemia in mice, albeit within a narrow therapeutic window (Tang et al., 2008). There is currently no information regarding the contribution of specific isoforms of NADPH oxidase to beneficial effects of apocynin in cerebral ischaemia. Most importantly, it remains to be tested whether similar benefits can be observed when apocynin is given after induction of cerebral ischaemia, which represents a more clinically relevant scenario.
Interestingly, it is possible that NADPH oxidases play a physiological role in the brain. Nox1, Nox2 and Nox4 containing isoforms of NADPH oxidase are expressed at higher levels in cerebral versus systemic arteries in rats (Miller et al., 2005) and other animal species including mice (CG Sobey and AA Miller, unpublished data). Production of NADPH oxidase-derived ROS is also significantly greater in rat (and mouse) cerebral versus systemic arteries and is associated with powerful cerebral vasorelaxant responses to NADPH and increased flow, and attenuated vasoconstrictor responses to angiotensin II (Miller et al., 2005). Interestingly, these vasorelaxant effects of NADPH oxidase activation can be blocked by apocynin (Miller et al., 2005). While Nox2 expression and activity results in high levels of superoxide and is likely to be detrimental in stroke (Walder et al., 1997; Kahles et al., 2007; Kunz et al., 2007), it is not yet clear whether activity of Nox1 (which may be expected to be inhibited by apocynin) and/or Nox4 is similarly harmful, or is even beneficial, following cerebral vascular disease. For example, vasodilatation by NADPH oxidase-derived hydrogen peroxide (H2O2) may support cerebral blood flow during chronic hypertension which is associated with higher Nox4 mRNA levels in cerebral vessels of spontaneously hypertensive rat compared with Wistar-Kyoto (Paravicini et al., 2004). Thus, it is unclear whether NADPH oxidase inhibitors that do not distinguish between isoforms of NADPH oxidase would necessarily be beneficial for the treatment of cerebrovascular diseases, such as stroke.
The aims of this study were, therefore, to test the effect of apocynin on outcome following transient cerebral ischaemia in mice when administered either before ischaemia or after induction of cerebral reperfusion. In addition, we assessed the contribution of Nox2-containing NADPH oxidase to these effects using Nox2−/− mice. Brain superoxide production following induction of cerebral ischaemia/reperfusion, and the effect of pretreatment or posttreatment with apocynin, was also assessed using dihydroethidium fluorescence.
Methods
A total of 108 male C57Bl/6J and 22 male Nox2−/− mice were used in this study. Our colony of Nox2−/−mice were originally obtained from Professor Mary Dinauer and have been back-crossed to the C57Bl6/J strain for at least 10 generations.
Lucigenin-enhanced chemiluminescence
Lucigenin (5 µmol·L−1)-enhanced chemiluminescence was used to test the effect of apocynin on NADPH oxidase-derived superoxide production by isolated cerebral (pooled basilar and middle cerebral) arteries from naïve wild-type mice. Mice (nine male C57Bl/6J: 6–8 weeks old, 24.7 ± 0.9 g; six male Nox2−/−: 6–8 weeks old, 23.1 ± 0.5 g) were killed by exposure to isoflurane followed by decapitation. Cerebral arteries were excised and incubated at 37°C in a Krebs-HEPES solution containing apocynin (300 µmol·L−1) or dimethyl sulphoxide (DMSO; 0.03%) vehicle for 60 min. NADPH (100 µmol·L−1) and diethyldithiocarbamate (300 mmol·L−1) were then added to the solution for 30 min. Counts were measured using a Hidex Plate Chameleon. Background counts were subtracted and superoxide production was normalized for dry artery weight.
Amplex red fluorescence
Amplex red fluorescence assay was used to test the effect of apocynin on H2O2 production by isolated wild-type mouse cerebral arteries. Mice (six male C57Bl/6J: 8 weeks old, 24.02 ± 0.24 g) were killed by exposure to isoflurane followed by decapitation. Cerebral arteries were excised. H2O2 standards (0–0.125 µmol·L−1) and cerebral arteries (pooled basilar and middle cerebral) were incubated at 37°C in a Krebs-HEPES solution containing apocynin (300 µmol·L−1) or DMSO (0.03%) vehicle for 60 min and then transferred to a 96-well plate. Fluorescence was determined in 100 µL incubations containing 15 µmol·L−1 Amplex red reagent (10-acetyl-3,7-dihydroxyphenoxazine) and 0.1 u·mL−1 horseradish peroxidase in the absence and presence of apocynin (300 µmol·L−1) or DMSO (0.03%). Fluorescence was measured in a fluorimeter using an excitation filter of 530 nm, and an emission filter of 590 nm. Background fluorescence was subtracted and total H2O2 (µmol) at 120 min normalized for dry tissue weight.
Cerebral ischaemia in mice
Focal cerebral ischaemia was induced by transient intraluminal occlusion of the middle cerebral artery (MCA). Mice (96 male C57Bl6/J: 5–9 weeks old, 23.0 ± 0.2 g; 16 male Nox2−/−: 8–12 weeks old, 25.6 ± 0.8 g) were anaesthetized with a mixture of ketamine (80 mg·kg−1 i.p.) and xylazine (10 mg·kg−1 i.p.). Body temperature was maintained with a heat lamp throughout the procedure and until animals regained consciousness. The right proximal common carotid artery was ligated, and a 6-0 nylon monofilament with silicone-coated tip was introduced into the distal internal carotid artery. It was advanced 11–12 mm distal to the carotid bifurcation, occluding the MCA at the junction of the Circle of Willis. Severe (∼80%) reduction in regional cerebral blood flow (rCBF) was confirmed using trans-cranial laser-Doppler (Perimed, SE) in the area of cerebral cortex supplied by the MCA. Occlusion of the MCA was maintained for 0.5 h, and the monofilament was then retracted to allow reperfusion (for 23.5 h). The wound was then closed and the animal was allowed to recover. Mice were treated i.p. with either vehicle (0.1% DMSO) or apocynin (2.5 or 5 mg·kg−1) 0.5 h prior to ischaemia or with vehicle or apocynin (2.5 mg·kg−1) 1 h after initiation of reperfusion.
Neurological score
At the end of the experiment (24 h), neurological assessment was performed using a 5-point scoring system commonly used in mice (Iadecola et al., 1997; 2001): 0, normal motor function; 1, flexion of torso and contralateral forelimb when mouse is lifted by the tail; 2, circling to the contralateral side when mouse held by the tail on a flat surface but normal posture at rest; 3, leaning to the contralateral side at rest; 4, no spontaneous motor activity. Neurological assessment was performed by an investigator blinded to the experimental treatment.
Evaluation of cerebral infarct size
Mice were killed at 24 h by inhalation of CO2 (80%)/O2 (20%), followed by decapitation. The brain was immediately removed and frozen with liquid nitrogen. Coronal sections (30 µm) were obtained and stained with thionin (0.1%) to delineate the infarct and images of the sections were captured with a CCD camera mounted above a light box. Infarct volume (total, cortical and subcortical) was quantified using image analysis software, and corrected for brain swelling, estimated using the following formula: CIV = [LHA − (RHA − RIA)] × diameter of slice, where CIV is corrected infarct volume, LHA is left hemisphere area, RHA is right hemisphere area and RIA is right hemisphere infarct area (Tsuchiya et al., 2003; Xia et al., 2006). Corrected infarct volumes of individual brain sections were then added giving a three-dimensional approximation of the total infarct volume. Animals were randomly assigned to surgery, and infarct analysis was performed by an operator blinded to treatment group.
Detection of superoxide production in situ using dihydroethidium
Upon reaction with superoxide, dihydroethidium (hydroethidine) is oxidized to ethidium and 2-hydroxyethidium. Ethidium exhibits a peak absorbance at 510 nm and an emission maximum at 590 nm while 2-hydroxyethidium exhibits a peak absorbance at 490 nm and emission maximum of 560–570 nm (Zhao et al., 2005). Dihydroethidium (10 mg·kg−1) was administered i.v. (jugular vein) into some ketamine/xylazine-anaesthetized wild-type mice (n = 3 per group) 23 h after induction of middle cerebral artery occlusion (MCAO) or sham MCAO and was allowed to circulate for 1 h. At 24 h, these mice were transcardially perfused with phosphate buffered saline, killed and the brain removed and frozen. Five serial 30 µm sections were obtained per animal, at the levels of +1.6, +1.0, +0.4, −0.2 and −0.8 mm from bregma. Images were acquired at 10X magnification using a dotSlide microscope system using an excitation filter or 530–550 nm and emission filter of 575 nm. Semiquantitative fluorescence (i.e. n = 5 slices per brain from n = 3 mice per group) was measured in the ipsilateral hemisphere and cortex by calculating the sum integral intensity using image analysis software. Integral intensity of sham MCAO sections was then subtracted to correct for background.
Drugs and materials
All drugs were purchased from Sigma (St. Louis, MO, USA), except the Amplex red fluorescence assay and dihydroethidium which were purchased from Molecular Probes and Invitrogen respectively. Apocynin was dissolved in DMSO and diluted with 0.9% saline, achieving a final concentration of 0.1% DMSO.
Hidex Plate Chameleon was obtained from Hidex (Finland); the fluorimeter, Flexstation (Molecular Devices); 6-0 nylon monofilament with silicone-coated tip, Doccol Co. (Redlands, CA, USA); CCD camera, Cohu Inc. (San Diego, CA, USA); light box, Biotec-Fischer Colour Control 5000 (Reiskirchin, Germany); image analysis software, ImageJ (NIH); dotSlide microscope system, Olympus (USA); image analysis software (analySIS 5 Life Science Professional), Olympus Soft Imaging Solutions (USA); GraphPad Prism version 4, Graph Pad Software Inc. (San Diego, CA).
Statistical analysis
All data are presented as the mean ± s.e.mean. Statistical analyses were performed using GraphPad Prism version 4. Between-group comparisons of infarct volume and swelling were compared using one-way anova or Student's unpaired t-test, as appropriate. Survival data were analysed using a logrank test. Neurological score was compared using a Kruskal-Wallis test, followed by Dunn's post test and logrank test. Group numbers are shown in parentheses. P represents < 0.05.
Results
Effect of apocynin on ROS production by cerebral arteries
Apocynin (300 µmol·L−1) inhibited NADPH-induced superoxide production by ∼40% in cerebral arteries isolated from C57Bl/6J mice (n = 9, control, 10 055 ± 2136; apocynin, 5917 ± 1307 counts·mg−1; P < 0.05). In contrast, apocynin had no significant effect on NADPH-induced superoxide production by cerebral arteries isolated from Nox2−/− mice (n = 6, control, 23 224 ± 5045; apocynin, 19 553 ± 5080 counts·mg−1). Apocynin also inhibited H2O2 production by ∼75% in cerebral arteries isolated from wild-type mice (control, 0.17 ± 0.01; apocynin, 0.04 ± 0.003 µmol·mg−1 tissue; n = 6, P < 0.05).
Effect of treatment with apocynin 0.5 h pre-ischaemia on outcome after MCAO in wild-type mice
rCBF
Regional cerebral blood flow was reduced by ∼80% following insertion of the monofilament and increased upon removal of the monofilament to a similar extent in mice treated with either vehicle (n = 23) or 2.5 mg·kg−1 apocynin (n = 10; Figure 1A). Mice pretreated with 5 mg·kg−1 apocynin had a similar profile of rCBF (n = 14; data not shown).
Figure 1.
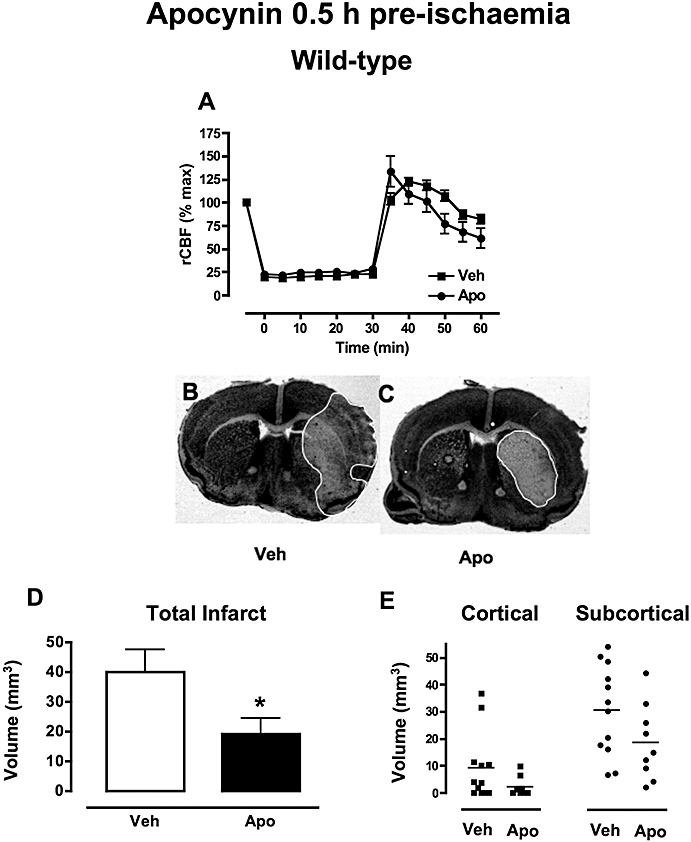
The effect of apocynin 0.5 h pre-ischaemia in wild-type mice. (A) Regional cerebral blood flow (rCBF) decreased by ∼80% upon insertion of the monofilament, remained low throughout the ischaemic period (0.5 h) and increased upon reperfusion to a similar extent in mice treated with either vehicle (0.1% DMSO, n = 23) or apocynin (2.5 mg·kg−1, n = 10). (B-C) Representative coronal brain sections from mice treated with vehicle (B) or apocynin (C) showing a smaller infarct area in apocynin (2.5 mg·kg−1)-treated mice outlined in white. (D-E) Mice treated with apocynin (2.5 mg·kg−1; n = 8, *P < 0.05) had smaller total, cortical and subcortical infarct volumes following MCAO than vehicle-treated mice (0.1% DMSO, n = 10). DMSO, dimethyl sulphoxide; MCAO, middle cerebral artery occlusion.
Mortality and neurological impairment
Mortality during the 23.5 h reperfusion period following MCAO occurred in 12% (3/26) of vehicle-treated mice. There was no mortality (0%; 0/10) in wild-type mice treated with 2.5 mg·kg−1 apocynin (P = 0.20 vs. vehicle-treated group). By contrast, mice treated with a higher dose of apocynin (5 mg·kg−1) had substantially greater mortality than the vehicle-treated group (41%; 14/34, P < 0.05). There were no deaths among seven additional mice treated with the 5 mg·kg−1 apocynin and subjected to sham MCAO surgery, suggesting that apocynin is not toxic at this dose in mice unless cerebral ischaemia is induced.
Twenty-four hours after MCAO, vehicle-treated mice had a mean neurological score of 2.1 ± 0.3 (n = 22) with 91% (20/22) of mice having some neurological impairment at 24 h (i.e. scores of 1 to 4). Treatment with 2.5 mg·kg−1 apocynin had no overall significant effect on mean neurological score following MCAO (1.8 ± 0.6, n = 10), but 50% of these mice were in fact without neurological impairment at 24 h (5/10; P < 0.05 vs. vehicle group, logrank test). In contrast, 5 mg·kg−1 apocynin had no effect on mean neurological score (1.8 ± 0.3, n = 20) and 85% (17/20) of mice had neurological impairment at 24 h.
Brain infarct and swelling
In vehicle-treated mice, cerebral infarcts were mostly subcortical (31 ± 5 mm3, n = 12; Figure 1B,E) with a variable degree of cortical involvement (9 ± 4 mm3; Figure 1B,E). Pretreatment with 2.5 mg·kg−1 apocynin significantly reduced total infarct volume following MCAO (n = 10, P < 0.05; Figure 1B–D), but had no effect on swelling volume (vehicle: 24 ± 4 mm3, apocynin: 18 ± 4 mm3). Apocynin (2.5 mg·kg−1) tended to reduce both subcortical (19 ± 5 mm3; Figure 1E) and especially cortical (2 ± 1 mm3; Figure 1E) infarct volume, although these individual volume differences did not reach statistical significance versus the vehicle-treated group. Pretreatment with the higher dose of apocynin (5 mg·kg−1) had no effect on either total infarct (38 ± 7 mm3, n = 13) or swelling (22 ± 4 mm3) volume.
Effect of treatment with apocynin 0.5 h pre-ischaemia on outcome after MCAO in Nox2−/− mice
rCBF
Regional cerebral blood flow was reduced by ∼80% following insertion of the monofilament and increased upon removal of the monofilament to a similar extent in Nox2−/− treated with either vehicle (n = 6) or 2.5 mg·kg−1 apocynin (n = 5; Figure 2A).
Figure 2.
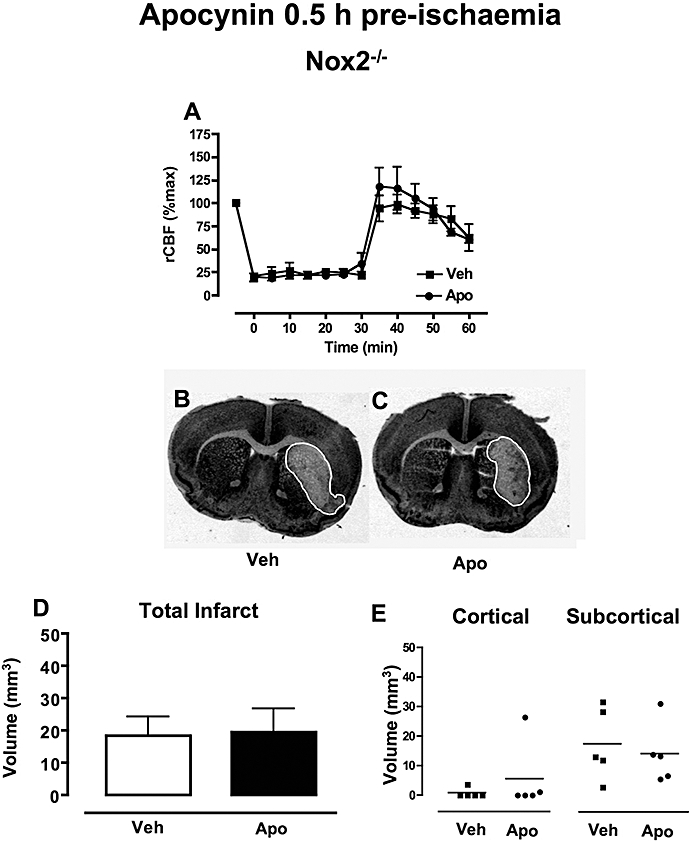
The effect of apocynin 0.5 h pre-ischaemia in Nox2−/− mice. (A) Regional cerebral blood flow (rCBF) during MCAO in Nox2−/− mice treated with either vehicle (0.1% DMSO, n = 6) or apocynin (2.5 mg·kg−1, n = 5). (B-C) Representative coronal brain sections from Nox2−/− mice treated with either vehicle (B) or apocynin (C) showing similar infarct areas outlined in white. (D-E) Mice treated with either vehicle (0.1% DMSO, n = 5) or apocynin (2.5 mg·kg−1, n = 5) had similar total, cortical and subcortical infarct volumes following MCAO. DMSO, dimethyl sulphoxide; MCAO, middle cerebral artery occlusion.
Mortality and neurological impairment
Mortality during the 23.5 h reperfusion period occurred in 40% (4/11) of vehicle-treated Nox2−/− mice and tended to be lower in Nox2−/− mice treated with 2.5 mg·kg−1 apocynin (17%, 1/6; P = 0.4).
Mean neurological scores were similar for vehicle- (1.8 ± 0.7) and 2.5 mg·kg−1 apocynin- (2.8 ± 0.7) treated Nox2−/− mice. The frequency of Nox2−/− mice with neurological impairment was similar in apocynin- and vehicle-treated groups (20% vs. 33%; P = 0.2, logrank test).
Brain infarct and swelling
Vehicle-treated Nox2−/− mice had mainly subcortical infarcts with limited cortical involvement (Figure 2E, cortical: 1 ± 1 mm3; subcortical: 18 ± 5 mm3, n = 5; Figure 2B,D,E). Treatment of Nox2−/− mice with 2.5 mg·kg−1 apocynin had no effect on the volume or localization of infarct (Figure 2C–E) or on swelling volume (vehicle: 21 ± 8 mm3, apocynin: 21 ± 7 mm3).
Effect of treatment with apocynin 1 h post-reperfusion on outcome after MCAO in wild-type mice
rCBF
Regional cerebral blood flow was reduced by ∼80% following insertion of the filament in mice subsequently treated with either vehicle (n = 6) or 2.5 mg·kg−1 apocynin (n = 6; Figure 3A). Upon retraction of the filament and induction of reperfusion, rCBF increased to a similar extent in both groups.
Figure 3.
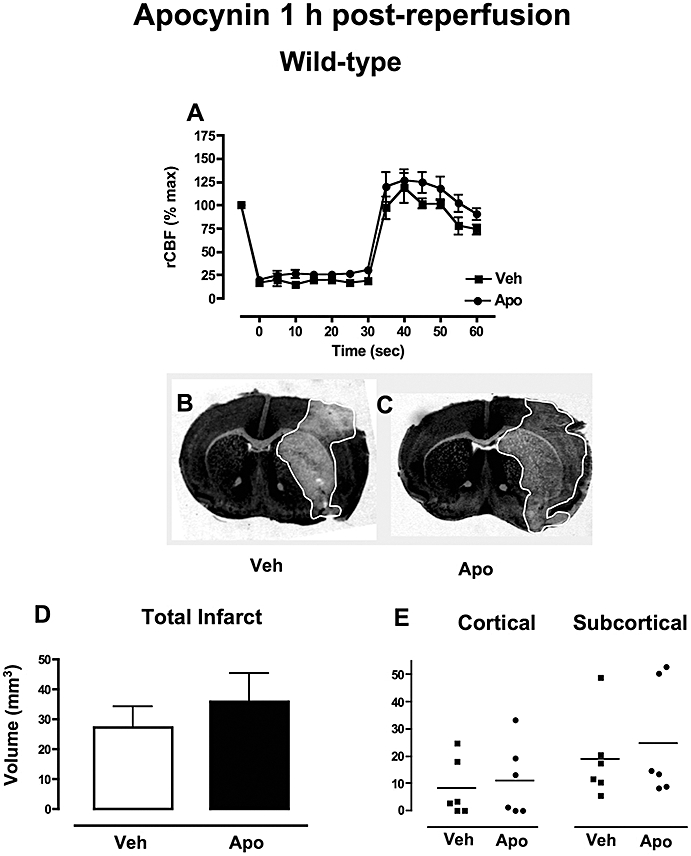
The effect of apocynin 1 h post-reperfusion in wild-type mice. (A) Regional cerebral blood flow (rCBF) during MCAO in Mice treated with either vehicle (0.1% DMSO, n = 6) or apocynin (2.5 mg·kg−1, n = 6). (B-C) Representative coronal brain sections from mice treated with either vehicle (B) or apocynin (C) showing similar infarct areas outlined in white. (D-E) Mice treated with either vehicle (0.1% DMSO, n = 6) or apocynin (2.5 mg·kg−1, n = 6) had similar total, cortical and subcortical infarct volumes following MCAO. DMSO, dimethyl sulphoxide; MCAO, middle cerebral artery occlusion.
Mortality and neurological impairment
Survival rate was 100% (0/6) in mice treated with either vehicle (0/6) or 2.5 mg·kg−1 apocynin (0/6) 1 h post-reperfusion.
Mice treated with vehicle had a mean neurological score of 1.3 ± 0.6 (n = 6) at 24 h. There was a trend for a greater percentage of mice with neurological impairment at 24 h (100% vs. 67%) and a greater mean neurological score in the mice treated with 2.5 mg·kg−1 apocynin after ischaemia-reperfusion (2.2 ± 0.5, n = 5; P = 0.3).
Brain infarct swelling
In these concurrently performed experimental groups, volume and localization of infarct (Figure 3B–E) and also swelling volume (vehicle: 29 ± 7 mm3, apocynin: 29 ± 5 mm3) were similar in mice treated with either vehicle (n = 6) or 2.5 mg·kg−1 apocynin after ischaemia-reperfusion (n = 6; Figure 3B–E).
In situ superoxide production using dihydroethidium fluorescence
Minimal fluorescence was observed in coronal sections from wild-type mice subjected to sham MCAO (Figure 4A). Fluorescence was visible in both the ipsilateral and contralateral hemispheres of coronal sections taken from wild-type mice subjected to MCAO, and especially in the striatum of the ipsilateral side (Figure 4B). Treatment with 2.5 mg·kg−1 apocynin 0.5 h before ischaemia substantially reduced the fluorescent intensity (Figure 4C), while treatment with 2.5 mg·kg−1 apocynin 1 h post-reperfusion appeared to be without effect (Figure 4D). Semi-quantitative group data (n = 5 slices per brain from n = 3 mice per group) from the total ipsilateral hemisphere and also the ipsilateral cortex confirmed that brain superoxide levels paralleled infarct volume (Figure 4E,F).
Figure 4.
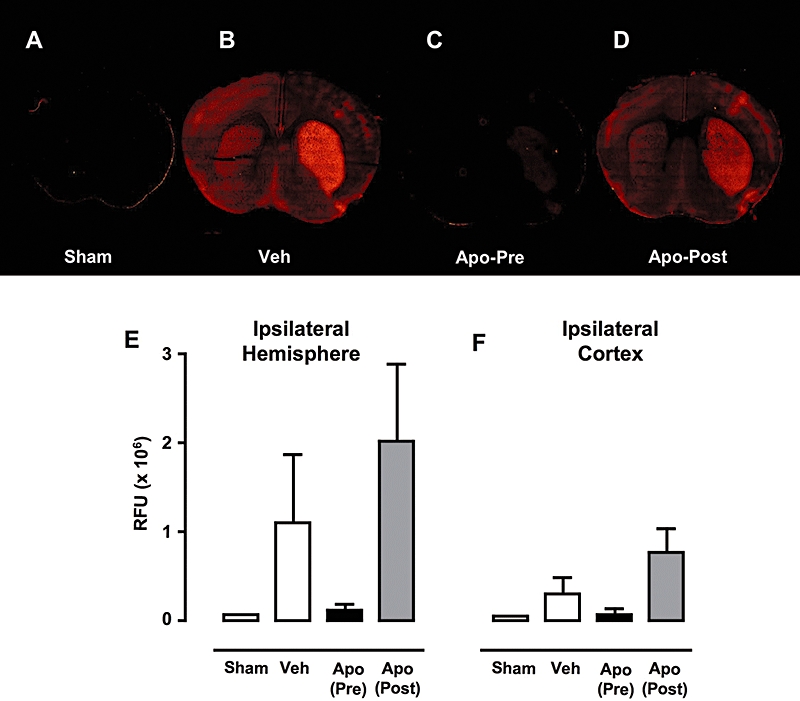
In situ cerebral superoxide production. Representative images of coronal brain sections (+1.0 mm from bregma) from mice infused with dihydroethidium. (A) Sham MCAO, (B) vehicle-treated MCAO, (C) 2.5 mg·kg−1 apocynin 0.5 h pre-ischaemia and (D) 2.5 mg·kg−1 apocynin 1 h post-reperfusion. (D) and (E) Semi-quantitative group data indicating relative fluorescence units (RFU; average of n = 5 slices per brain from n = 3 mice per group) presented as mean ± s.e.mean from the total ipsilateral hemisphere (E) and the ipsilateral cortex (F). MCAO, middle cerebral artery occlusion.
Discussion
NADPH oxidases are regarded as a major source of ROS during vascular diseases involving oxidative stress, including stroke (Brandes and Kreuzer, 2005; Miller et al., 2005; 2006a; Cave et al., 2006). Thus, there is significant interest in the development of NADPH oxidase inhibitors for cardiovascular therapy. This study examined the effect of the currently best available NADPH oxidase inhibitor, apocynin, on cerebral infarct size and other endpoints that determine outcome following ischaemic stroke. The major new findings are: (i) apocynin is protective in the setting of cerebral ischaemia-reperfusion only when administered before induction of ischaemia; (ii) this protection is associated with reduced brain ROS levels at 24 h; (iii) apocynin inhibits NADPH oxidase-derived superoxide production in mouse cerebral arteries; (iv) all these effects of apocynin are absent in Nox2−/− mice; and (v) both apocynin and Nox2 deletion appear to reduce infarct volume similarly in both cortex and subcortex.
Generation of ROS by NADPH oxidases is mostly regarded as being non-physiological and toxic (Griendling et al., 2000), particularly following cerebral ischaemia when very high levels of superoxide are produced in bursts by phagocytic-like Nox2-containing NADPH oxidases in endothelium and glial cells (Walder et al., 1997; Miller et al., 2006b; Kunz et al., 2007). However, additionally, ROS appear to be continuously generated in cerebral vessels under normal conditions at lower levels than in phagocytes but at much higher levels than in systemic arteries, leading to the suggestion that ROS normally have key physiological roles particularly in the regulation of cerebral vascular tone (Miller et al., 2005). In non-diseased arteries, NADPH oxidase-derived ROS have been shown to contribute to cerebral vasodilatation and modulation of cerebral vasoconstriction (Didion and Faraci, 2002; Paravicini et al., 2004; 2006; Park et al., 2004; Miller et al., 2005). Three isoforms of NADPH oxidase – containing Nox1, Nox2 or Nox4 – are known to be expressed in the brain, with Nox1 and/or Nox4 thought to largely account for physiological ROS production (Ago et al., 2005; Miller et al., 2005; 2007) and Nox2 expression and activity more likely to be important during oxidative stress, including after ischaemic stroke (Walder et al., 1997; Kahles et al., 2007; Kunz et al., 2007).
We found administration of apocynin 0.5 h before induction of cerebral ischaemia to be protective, reducing total infarct volume, neurological impairment and mortality. However, in Nox2−/− mice, apocynin had no such effects, raising the possibility that the protection afforded by apocynin pretreatment in this model of stroke occurred via inhibition of Nox2-containing NADPH oxidase. Compared with control mice, we found a similarly smaller mean infarct volume in both the cortex and subcortex of either 2.5 mg·kg−1 apocynin-pretreated or Nox2-deficient mice. Hence, although these data did not reach statistical significance (P = 0.09 for both), one plausible interpretation is that Nox2 is targeted by apocynin in both the cortex and the subcortex. These overall findings are also consistent with studies reporting mice lacking Nox2−/− to have smaller infarct volumes following MCAO (Walder et al., 1997; Kahles et al., 2007; Kunz et al., 2007). In rats, infarct volume following 30 min ischaemia does not reach maximum until 48 h (Li et al., 2000). Therefore, the possibility that apocynin slowed the evolution of ischaemic damage at 24 h, rather than reducing the final infarct volume, cannot be excluded. Our further finding that a twofold higher dose of apocynin increased mortality while having no beneficial effect on other measures of post-stroke outcome is consistent with another recent finding (Tang et al., 2008) and suggests that apocynin has quite a narrow therapeutic window and thus may represent a challenge for therapeutic development. We found that the increased mortality with the higher dose of apocynin was dependent on the mouse receiving MCAO. It is therefore plausible that greater scavenging of H2O2in vivo by this dose of apocynin might compromise NADPH oxidase-dependent cerebral vasodilatation in ischaemic brain regions, which is thought to be mediated by H2O2 (Paravicini et al., 2004; 2006; Miller et al., 2005; 2006b). Interestingly, when apocynin was administered 1 h after induction of reperfusion, it failed to improve outcome, with no reduction in infarct or swelling volume and no improvement in neurological function. Our findings suggest that the timing of administration of apocynin is critical to its neuroprotective effects and casts further doubt on the suitability of apocynin for therapeutic development for the treatment of stroke.
This is the first study to examine the effects of apocynin on ROS production by mouse cerebral arteries. In isolated cerebral arteries, we found that at the commonly used concentration of 300 µmol·L−1, apocynin inhibits generation of superoxide (by ∼40%) in wild-type but not Nox2-deficient mice. Apocynin was also found to reduce H2O2 levels (by ∼75%) in isolated cerebral arteries from wild-type mice which may either be a direct consequence of reduced superoxide production or due to its known action as a scavenger of H2O2 (Stolk et al., 1994). Interestingly, the finding from the superoxide assay suggests that at this concentration apocynin may indeed be relatively Nox2-selective in that it appears to only inhibit superoxide production by Nox2-containing NADPH oxidases in mouse cerebral arteries. While we did not perform a direct within-assay comparison of levels of superoxide production by cerebral arteries from wild-type versus Nox2−/− mice, our data suggest that Nox2 deletion may lead to a higher level of superoxide production. The reason for this is presently unclear, but could reflect a compensatory upregulation of Nox1 or Nox4 and warrants further investigation. As Nox2−/− mice have virtually no cortical infarct in this model, it could be speculated that this is related to a higher basal level of ROS supporting flow through collaterals during MCAO that is deeper than can be detected by transcranial laser Doppler flowmetry.
The dose of 2.5 mg·kg−1 apocynin was selected for in vivo studies of cerebral ischaemia, as we estimated that it would achieve a similar concentration in plasma to that used in our in vitro studies. Similar to our findings in isolated cerebral arteries, treatment with 2.5 mg·kg−1 apocynin 0.5 h before ischaemia reduced brain superoxide levels, measured in situ using dihydroethedium fluorescence, in parallel with the changes in infarct volume and consistent with its ability to improve outcome following cerebral ischaemia. By contrast, treatment with 2.5 mg·kg−1 apocynin after induction of reperfusion, which failed to improve measures of outcome following cerebral ischaemia, similarly failed to reduce brain superoxide levels, suggesting not only a key role of superoxide in brain injury following cerebral ischaemia but also that the ability of apocynin to reduce superoxide levels at 24 h after cerebral ischaemia is highly dependent on the timing of administration. This latter finding, as well as the lack of effect on vascular superoxide generation from Nox2−/− mice, would suggest that apocynin is not merely acting as a scavenger of superoxide in this setting, as has been suggested recently in the vascular system (Heumuller et al., 2008). One possibility is that when administered prior to ischaemia apocynin effectively inhibits phagocytic (Nox2-containing) NADPH oxidase, thus reducing either leukocyte infiltration or activity and markedly reducing the extent of consequent downstream free radical production and brain injury. It could also be speculated that the time-dependence of apocynin's protective effects could relate to the presence and localization of neutrophil-derived myeloperoxidase, which has been shown to be critical to activation of apocynin (Simons et al., 1990). Finally, apocynin has recently been shown to inhibit rho kinase (Schlüter et al., 2008), an enzyme known to contribute to infarct volume after experimental stroke (Shin et al., 2007), and so at present we cannot rule out the possibility that such an action of apocynin played a role in the protection observed in this study. The tendency for a lower mortality in apocynin-treated Nox2−/− mice might also be consistent with an action on other systems (e.g. rho kinase) in the ischaemic brain. Notwithstanding our findings in Nox2−/− mice, the fact that administration of apocynin relatively early after ischaemia had no protective effect after 24 h may argue against NADPH oxidase-derived ROS being a target of its protective effects.
The key findings of this study are that administration of apocynin before cerebral ischaemia improves outcome in wild-type but not Nox2-deficient mice. This protective effect of apocynin is lost if it is administered even relatively early after cerebral reperfusion.
Acknowledgments
This study was supported by funds from project grants from the National Health and Medical Research Council of Australia (NHMRC ID 208969, 350477) and from the University of Melbourne Research Grant Scheme. KAJ is supported by an NHMRC Dora Lush Biomedical Research Scholarship. AAM is a High Blood Pressure Research Council Of Australia Postdoctoral Fellow. TMD is supported by an Australian Postgraduate Award. GRD is an RD Wright Career Development Award Fellow of the NHMRC. CGS is a Senior Research Fellow of the NHMRC.
Glossary
Abbreviations:
- DMSO
dimethyl sulphoxide
- H2O2
hydrogen peroxide
- MCA
middle cerebral artery
- MCAO
middle cerebral artery occlusion
- NADPH
reduced form of nicotinamide adenine dinucleotide phosphate
- rCBF
regional cerebral blood flow
- ROS
reactive oxygen species
Conflict of interest
CGS and GRD have potential conflicts of interest in that they are consultants for, and have significant ownership interests in, Radical Biotechnology Pty Ltd. of Australia.
References
- Ago T, Kitazono T, Kuroda J, Kumai Y, Kamouchi M, Ooboshi H, et al. NAD(P)H oxidases in rat basilar arterial endothelial cells. Stroke. 2005;36:1040–1046. doi: 10.1161/01.STR.0000163111.05825.0b. [DOI] [PubMed] [Google Scholar]
- Brandes RP, Kreuzer J. Vascular NADPH oxidases: molecular mechanisms of activation. Cardiovasc Res. 2005;65:16–27. doi: 10.1016/j.cardiores.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Cave AC, Brewer AC, Narayanapanicker A, Ray R, Grieve DJ, Walker S, et al. NADPH oxidases in cardiovascular health and disease. Antioxid Redox Signal. 2006;8:691–728. doi: 10.1089/ars.2006.8.691. [DOI] [PubMed] [Google Scholar]
- Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab. 2001;21:2–14. doi: 10.1097/00004647-200101000-00002. [DOI] [PubMed] [Google Scholar]
- Didion SP, Faraci FM. Effects of NADH and NADPH on superoxide levels and cerebral vascular tone. Am J Physiol Heart Circ Physiol. 2002;282:H688–H695. doi: 10.1152/ajpheart.00576.2001. [DOI] [PubMed] [Google Scholar]
- Griendling KK. Novel NAD(P)H oxidases in the cardiovascular system. Heart. 2004;90:491–493. doi: 10.1136/hrt.2003.029397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- Heumuller S, Wind S, Barbosa-Sicard E, Schmidt HH, Busse R, Schroder K, et al. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension. 2008;51:211–217. doi: 10.1161/HYPERTENSIONAHA.107.100214. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Zhang F, Casey R, Nagayama M, Ross ME. Delayed reduction of ischemic brain injury and neurological deficits in mice lacking the inducible nitric oxide synthase gene. J Neurosci. 1997;17:9157–9164. doi: 10.1523/JNEUROSCI.17-23-09157.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, Sugimoto K, Niwa K, Kazama K, Ross ME. Increased susceptibility to ischemic brain injury in cyclooxygenase-1-deficient mice. J Cereb Blood Flow Metab. 2001;21:1436–1441. doi: 10.1097/00004647-200112000-00008. [DOI] [PubMed] [Google Scholar]
- Kahles T, Luedike P, Endres M, Galla HJ, Steinmetz H, Busse R, et al. NADPH oxidase plays a central role in blood-brain barrier damage in experimental stroke. Stroke. 2007;38:3000–3006. doi: 10.1161/STROKEAHA.107.489765. [DOI] [PubMed] [Google Scholar]
- Kunz A, Anrather J, Zhou P, Orio M, Iadecola C. Cyclooxygenase-2 does not contribute to postischemic production of reactive oxygen species. J Cereb Blood Flow Metab. 2007;27:545–551. doi: 10.1038/sj.jcbfm.9600369. [DOI] [PubMed] [Google Scholar]
- Li F, Silva MD, Sotak CH, Fisher M. Temporal evolution of ischemic injury evaluated with diffusion-, perfusion-, and T2-weighted MRI. Neurology. 2000;54:689–696. doi: 10.1212/wnl.54.3.689. [DOI] [PubMed] [Google Scholar]
- Miller AA, Drummond GR, Schmidt HH, Sobey CG. NADPH oxidase activity and function are profoundly greater in cerebral versus systemic arteries. Circ Res. 2005;97:1055–1062. doi: 10.1161/01.RES.0000189301.10217.87. [DOI] [PubMed] [Google Scholar]
- Miller AA, Drummond GR, Sobey CG. Novel isoforms of NADPH-oxidase in cerebral vascular control. Pharmacol Ther. 2006a;111:928–948. doi: 10.1016/j.pharmthera.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Miller AA, Dusting GJ, Roulston CL, Sobey CG. NADPH-oxidase activity is elevated in penumbral and non-ischemic cerebral arteries following stroke. Brain Res. 2006b;1111:111–116. doi: 10.1016/j.brainres.2006.06.082. [DOI] [PubMed] [Google Scholar]
- Miller AA, Drummond GR, Mast AE, Schmidt HH, Sobey CG. Effect of gender on NADPH-oxidase activity, expression, and function in the cerebral circulation: role of estrogen. Stroke. 2007;38:2142–2149. doi: 10.1161/STROKEAHA.106.477406. [DOI] [PubMed] [Google Scholar]
- Mohazzab KM, Kaminski PM, Wolin MS. NADH oxidoreductase is a major source of superoxide anion in bovine coronary artery endothelium. Am J Physiol. 1994;266:H2568–H2572. doi: 10.1152/ajpheart.1994.266.6.H2568. [DOI] [PubMed] [Google Scholar]
- Muralikrishna Adibhatla R, Hatcher JF. Phospholipase A2, reactive oxygen species, and lipid peroxidation in cerebral ischemia. Free Radic Biol Med. 2006;40:376–387. doi: 10.1016/j.freeradbiomed.2005.08.044. [DOI] [PubMed] [Google Scholar]
- Paravicini TM, Chrissobolis S, Drummond GR, Sobey CG. Increased NADPH-oxidase activity and Nox4 expression during chronic hypertension is associated with enhanced cerebral vasodilatation to NADPH in vivo. Stroke. 2004;35:584–589. doi: 10.1161/01.STR.0000112974.37028.58. [DOI] [PubMed] [Google Scholar]
- Paravicini TM, Miller AA, Drummond GR, Sobey CG. Flow-induced cerebral vasodilatation in vivo involves activation of phosphatidylinositol-3 kinase, NADPH-oxidase, and nitric oxide synthase. J Cereb Blood Flow Metab. 2006;26:836–845. doi: 10.1038/sj.jcbfm.9600235. [DOI] [PubMed] [Google Scholar]
- Park L, Anrather J, Zhou P, Frys K, Wang G, Iadecola C. Exogenous NADPH increases cerebral blood flow through NADPH oxidase-dependent and -independent mechanisms. Arterioscler Thromb Vasc Biol. 2004;24:1860–1865. doi: 10.1161/01.ATV.0000142446.75898.44. [DOI] [PubMed] [Google Scholar]
- Schlüter T, Steinbach AC, Steffen A, Rettig R, Grisk O. Apocynin-induced vasodilation involves Rho kinase inhibition but not NADPH oxidase inhibition. Cardiovasc Res. 2008;80:271–279. doi: 10.1093/cvr/cvn185. [DOI] [PubMed] [Google Scholar]
- Shin HK, Salomone S, Potts EM, Lee SW, Millican E, Noma K, et al. Rho-kinase inhibition acutely augments blood flow in focal cerebral ischemia via endothelial mechanisms. J Cereb Blood Flow Metab. 2007;27:998–1009. doi: 10.1038/sj.jcbfm.9600406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons JM, Hart BA, Ip Vai Ching TR, Van Dijk H, Labadie RP. Metabolic activation of natural phenols into selective oxidative burst agonists by activated human neutrophils. Free Radic Biol Med. 1990;8:251–258. doi: 10.1016/0891-5849(90)90070-y. [DOI] [PubMed] [Google Scholar]
- Stolk J, Hiltermann TJ, Dijkman JH, Verhoeven AJ. Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am J Respir Cell Mol Biol. 1994;11:95–102. doi: 10.1165/ajrcmb.11.1.8018341. [DOI] [PubMed] [Google Scholar]
- Tang XN, Cairns B, Cairns N, Yenari MA. Apocynin improves outcome in experimental stroke with a narrow dose range. Neuroscience. 2008;154:556–562. doi: 10.1016/j.neuroscience.2008.03.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya D, Hong S, Kayama T, Panter SS, Weinstein PR. Effect of suture size and carotid clip application upon blood flow and infarct volume after permanent and temporary middle cerebral artery occlusion in mice. Brain Res. 2003;970:131–139. doi: 10.1016/s0006-8993(03)02300-x. [DOI] [PubMed] [Google Scholar]
- Walder CE, Green SP, Darbonne WC, Mathias J, Rae J, Dinauer MC, et al. Ischemic stroke injury is reduced in mice lacking a functional NADPH oxidase. Stroke. 1997;28:2252–2258. doi: 10.1161/01.str.28.11.2252. [DOI] [PubMed] [Google Scholar]
- Wang Q, Tompkins KD, Simonyi A, Korthuis RJ, Sun AY, Sun GY. Apocynin protects against global cerebral ischemia-reperfusion-induced oxidative stress and injury in the gerbil hippocampus. Brain Res. 2006;1090:182–189. doi: 10.1016/j.brainres.2006.03.060. [DOI] [PubMed] [Google Scholar]
- Xia CF, Smith RS, Jr, Shen B, Yang ZR, Borlongan CV, Chao L, et al. Postischemic brain injury is exacerbated in mice lacking the kinin B2 receptor. Hypertension. 2006;47:752–761. doi: 10.1161/01.HYP.0000214867.35632.0e. [DOI] [PubMed] [Google Scholar]
- Zhao H, Joseph J, Fales HM, Sokoloski EA, Levine RL, Vasquez-Vivar J, et al. Detection and characterization of the product of hydroethidine and intracellular superoxide by HPLC and limitations of fluorescence. Proc Natl Acad Sci USA. 2005;102:5727–5732. doi: 10.1073/pnas.0501719102. [DOI] [PMC free article] [PubMed] [Google Scholar]