

Abstract
Background and purpose:
Ginsenosides are used widely for medicinal purposes, but the mechanisms of their action are still unclear, although there is some evidence that these effects are mediated by nuclear receptors. Here we examined whether two metabolites of ginsenoside, protopanaxadiol (g-PPD) and protopanaxatriol (g-PPT), could modulate endothelial cell functions through the glucocorticoid receptor (GR) and oestrogen receptor (ER).
Experiment approaches:
The effects of g-PPD and g-PPT on intracellular calcium ion concentration ([Ca2+]i) and nitric oxide (NO) production in human umbilical vein endothelial cells (HUVECs) were measured using Fura-2-acetoxymethyl ester, 4-amino-5-methylamino-2′,7′-difluorofluorescein and Griess reagent. Effects on expression of GR and ER isoforms in HUVECs were determined using reverse transcriptase-/real-time PCR and immunocytochemistry. Phosphorylation of endothelial NO synthase (eNOS) was assessed by Western blotting.
Results:
Ginsenoside protopanaxadiol and g-PPT increased [Ca2+]i, eNOS phosphorylation and NO production in HUVECs, which were inhibited by the GR antagonist, RU486, the ER antagonist, ICI 182,780 and siRNA targeting GR or ERβ. The NO production was Ca2+-dependent and the [Ca2+]i elevation in HUVECs resulted from both intracellular Ca2+ release and extracellular Ca2+ influx.
Conclusions and implications:
Ginsenoside protopanaxadiol and g-PPT were functional ligands for both GR and ERβ, through which these ginsenoside metabolites exerted rapid, non-genomic effects on endothelial cells.
Keywords: angiogenesis, endothelial cell, ginsenoside, intracellular calcium concentration, nitric oxide, nuclear receptor
Introduction
Angiogenesis, the growth of new blood vessels from pre-existing vasculature, is an important physiological process in development and wound healing. Dysregulation of angiogenesis that promotes neovascular growth underlies the progression of many pathologies, including ocular disease and cancer. On the other hand, a lack of appropriate neovascularization can prevent wound healing and is associated with occurrence of ulcers, coronary artery disease and strokes.
For these reasons, modulation of the process of neovessel formation is clinically relevant to a wide range of disorders. Recombinant human platelet-derived growth factor, a pro-angiogenic agent, was the first to be introduced for treating diabetic foot ulcers in 1999 (Steed, 2006), followed by clinical trials for vascular endothelial growth factor (VEGF) gene therapy in patients with cardiovascular disease (Losordo et al., 1998; Rosengart et al., 1999). This approach reduced myocardial ischemia and improved angina (Losordo et al., 1998; Rosengart et al., 1999). Several drugs have also been developed to combat excessive angiogenesis. For example, bevacizumab (Avastin) and ranibizumab are antibodies targeting VEGF, which inhibit VEGF-induced endothelial cell growth. These drugs are widely used in treatments for cancer and more recently for age-related macular degeneration and glaucoma (Andreoli and Miller, 2007; Ichhpujani et al., 2007; Khosravi Shahi and Fernández Pineda, 2008). Soluble VEGF receptor and neuropilin receptor are used clinically to suppress neovascularization (Baka et al., 2006).
Pro-angiogenic molecules, including VEGF and bFGF, promote neovascularization by increasing intracellular calcium ion concentration ([Ca2+]i) and nitric oxide (NO) levels in endothelial cells (Morbidelli et al., 1996; Babaei et al., 1998). Calcium channel blockers, like nifedipine, verapamil, amlodipine and diltiazem, are now under clinical trials as angiogenesis regulatory agents, complementing cancer therapy (Munaron, 2006). In mice deficient in endothelial NO synthase (eNOS), there is an impaired intrinsic angiogenic response to hindlimb ischemia, and a lack of responsiveness in these mice to treatment with VEGF (Murohara et al., 1998). Treatment with the NO precursor, L-arginine, potentiated post-surgical angiogenesis and accelerated wound healing in patients (Ruel et al., 2008). Thus, [Ca2+]i and NO represent key determinants of angiogenesis.
In addition to the classical pro-angiogenic agents, some steroids like the glucocorticoids and oestrogens, also play a part in modulating vascular activities. Both steroids can regulate angiogenic (Iwai et al., 2004; Kasselman et al., 2007), and cell adhesion molecules (Burke-Gaffney and Hellewell, 1996; Simoncini et al., 2000; Groten et al., 2005), as well as affecting the expression of junctional proteins (Forster et al., 2005) in endothelial cells through their respective receptors, glucocorticoid receptor (GR) and oestrogen receptor (ER) (Simoncini et al., 2000; Groten et al., 2005; Kasselman et al., 2007). Insufficient glucocorticoid or over-expression of GR in endothelial cells is associated with vascular malformation and infant hypotension (Friedman et al., 1996). Also, oestrogen and its receptors have a role in atheroprotective actions (Hodgin et al., 2001).
A recently described agent shown to have beneficial effects in the cardiovascular, endocrine, immune and central nervous systems is Panax ginseng, although the actual mechanisms of action for this drug are largely undefined. Ginsenosides are the pharmacologically active ingredients of ginseng, which appear in two major variants: 20(S)-protopanaxadiol and 20(S)-protopanaxatriol. All ginsenosides have the basic steroidal backbone (Figure 1). Upon oral consumption, the 20(S)-protopanaxadiol and 20(S)-protopanaxatriol ginsenosides transform into their respective end metabolites, protopanaxadiol (g-PPD) and protopanaxatriol (g-PPT), respectively, by acid hydrolysis and a series of deglycosylations by the intestinal microflora (Hasegawa et al., 1996; Bae et al., 2002; Chi et al., 2005). Over 80% of g-PPD and g-PPT enter the circulation (Chen and Staba, 1980; Cui et al., 1997) and both metabolites possess anti-oxidative (Wang et al., 1995; Jiang et al., 1996) and GR-dependent anti-tumour activities (Park et al., 1999; Li et al., 2006; Wang et al., 2007). Based on the structural similarity of these molecules to steroid hormones, several reports suggest that the actions of these compounds are mediated through nuclear receptors (Attele et al., 1999).
Figure 1.
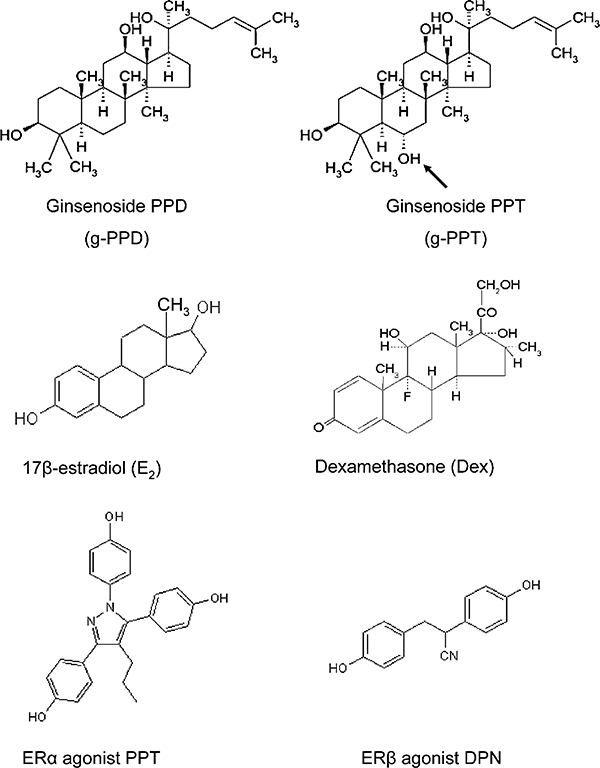
Chemical structures of ginsenoside protopanaxadiol (g-PPD), ginsenoside protopanaxatriol (g-PPT), 17β-estradiol (E2), dexamethasone (Dex), ERα agonist, PPT, and ERβ agonist, DPN. g-PPD, g-PPT, E2 and Dex possess a basic steroidal triterpene core with various sugar side-chains at C-3 and C-20 positions. Arrow shows the C-6 hydroxyl group unique to g-PPT. DPN, 2-diarylpropionitrile; ER, oestrogen receptor.
We have recently demonstrated that ginsenoside Rb1, a derivative of 20(S)-protopanaxadiol, is a functional ligand of ERβ (Leung et al., 2007a), whereas ginsenosides Rg1 and Re, which belong to the 20(S)-protopanaxatriol family, are functional ligands of GR (Leung et al., 2006a; 2007b). In this study, we examined the hypothesis that g-PPD, the end metabolite of Rb1, and g-PPT, the end metabolite of Rg1 and Re, like their precursors, could exert similar rapid, non-genomic actions by elevating [Ca2+]i and NO production in endothelial cells through activating GR or/and ER.
Methods
Cell culture and treatments
Primary human umbilical vein endothelial cells (HUVECs) were obtained from Clonetics (CC-2517, San Diego, CA, USA) and cultured in medium 199 supplemented with 20% FBS, 20 µg·mL−1 endothelial cell growth supplement (ECGS), 90 U·mL−1 heparin and 1% penicillin-streptomycin-neomycin (PSN) in a humidified incubator at 37°C with 5% CO2. HUVECs between passages 3 and 5 were used in these studies. The cells were incubated with phenol red-free medium 199 supplemented with 20% charcoal/dextran-treated FBS and 1% PSN for 24 h before the following experiments were carried out.
For functional assays, the cells were pretreated for 30 min with each of the following inhibitors: RU486 (10 µmol·L−1), ICI 182,780 (10 µmol·L−1), 2-aminoethyldiphenylborate (2-APB) (10 µmol·L−1) and thapsigargin (10 µmol·L−1) in phenol red-free assay medium (Medium 199 containing 1% PSN). In some experiments, cells were transfected with the Smart-Pool siRNA for silencing GR (M-003424-02) or ERβ (M-003402-02) (Dharmacon, Lafayette, CO, USA). Transfected cells were incubated in phenol red-free assay medium for 24 h prior to the treatment with g-PPD (1 µmol·L−1) or g-PPT (1 µmol·L−1). Knockdown efficacy of GR and ERβ was verified in our previous reports (Leung et al., 2006a,b).
Measurement of [Ca2+]i
Intracellular calcium ion concentration was determined using the fluorescent Ca2+ indicator, Fura-2-acetoxymethyl ester (AM) as described before (Leung et al., 2007b). Cells were incubated for 30 min with Fura-2 AM (10 µmol·L−1) in the presence of 0.0025% Pluronic F-127 at room temperature. After removal of the excessive dye, cells were then treated with dexamethasone (Dex; 10 nmol·L−1), 17β-estradiol (E2; 10 nmol·L−1), g-PPD (100–1000 nmol·L−1), g-PPT (100–1000 nmol·L−1), propylpyrazole-triol (PPT; 100 nmol·L−1), or 2-diarylpropionitrile (DPN; 100 nmol·L−1) for 4 min. Within this 4 min, the cells were excited with an alternating ultraviolet light at 340 and 380 nm wavelength, and the emission at 510 nm wavelength were collected in 2 s intervals using InCytIm2 ratio ion measurement program (version 5.27A, Intracellular Imaging Inc., Cincinnati, OH, USA). To create a zero extracellular Ca2+ environment, the assay medium was replaced by Ca2+-PSS buffer containing 140 mmol·L−1 NaCl, 5 mmol·L−1 KCl, 1 mmol·L−1 MgCl2, 10 mmol·L−1 glucose, 2 mmol·L−1 EGTA, 5 mmol·L−1 HEPES (pH 7.4) in some experiments. Three individual samples were analysed in each trial and each data point is the mean of three independent trials.
Measurement of NO
Intracellular levels of NO were monitored using the fluorescent NO indicator, 4-amino-5-methylamino-2′,7′-difluorofluorescein (DAF-FM) diacetate as described (Leung et al., 2007b). Cells were incubated for 30 min with DAF-FM (1 µmol·L−1) at room temperature. After removal of excess dye, the cells were treated with g-PPD (1 µmol·L−1), or g-PPT (1 µmol·L−1), and were excited at a wavelength of 495 nm. The emission at 515 nm wavelength was captured in 2 s intervals for 8 min. Three individual samples were analysed in each trial and each data point is the mean of three independent trials.
The amount of NO in the conditioned media was also determined using the Griess reagent (Assay Designs, Ann Arbor, MI, USA). Conditioned media obtained after 24 h treatment with Dex (10 nmol·L−1), E2 (10 nmol·L−1), g-PPD (1 µmol·L−1), g-PPT (1 µmol·L−1), PPT (100 nmol·L−1) or DPN (100 nmol·L−1) were incubated with nitrate reductase for 1 h at 37°C to reduce nitrate to nitrite. An equal volume of Griess reagent was then added and the optical density of the samples measured at a wavelength of 540 nm. Data were obtained from triplicate experiments and are expressed as fold change over untreated controls (±SD).
Reverse transcriptase (RT)- and real-time PCR
Total HUVECs mRNA was isolated using the RNeasy kit (Qiagen, Valencia, CA, USA) according to the manufacturer's protocol. First-strand cDNA was synthesized using iScript cDNA synthesis kit (BioRad, Hercules, CA, USA) and RT-PCR performed using iTaq polymerase (BioRad) at an annealing temperature of 58°C for 35 cycles for all primers. The accession number, primer sequence and PCR amplification product size for each gene are listed in Table 1. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the internal RNA loading control and samples where no reverse transcriptase was added to the PCR experiments were used as negative controls to ensure that amplification was RNA-dependent. PCR products were resolved by 1% agarose gel electrophoresis. For quantitative real-time PCR, the two-step amplifying protocol was used with iQ SYBR green supermix solution (BioRad). Both melting curve and gel electrophoretic analyses were used to determine amplicon homogeneity and quality of the data.
Table 1.
The Genbank accession numbers, primer sequences and amplicon sizes for GR, ERα and ERβ
| Gene | Accession no. | Forward | Reverse | Amplicon size |
|---|---|---|---|---|
| GR | NM_001018077 | TGGTTCCGAAAATTGGAATA | AAGCTTCATCAGAGCACACC | 209 bp |
| ERα | NM_000125 | AACACAAGCGCCAGAGAGAT | AGGATCTCTAGCCAGGCACA | 364 bp |
| ERβ | NM_001437 | CGAAGTGGGAATGGTGAAGT | ACAAAGCCGGGAATCTTCTT | 330 bp |
These primers were all used in RT-PCR experiments at an annealing temperature of 58°C for 35 cycles to confirm expression of the genes in HUVECs.
ER, oestrogen receptor; GR, glucocorticoid receptor; HUVECs, human umbilical vein endothelial cells; RT, reverse transcriptase.
Immunocytochemistry
Human umbilical vein endothelial cells were grown on coverslips, fixed with 4% paraformaldehyde, permeabilized with Triton-X and then labeled with a polyclonal antibody for GR (1:500; sc-8992; Santa Crutz, CA, USA), ERα (1:500; H-184; Santa Crutz, CA, USA), or for ERβ (1:500; H-150; Santa Crutz, CA, USA) for 3 h in the presence of normal goat serum. Samples were washed with PBS and then incubated with Alexa-Fluor 568 IgG antibody (1:1000; Invitrogen) and FITC-conjugated phalloidin (1:1000; Invitrogen) for 1 h. The coverslips were then washed and mounted with medium containing 2-(4-amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI) (Calbiochem, San Diego, CA, USA). Images were captured by confocal microscopy (Fluoview 300, Olympus America Inc., Center Valley, PA, USA).
Western blot analysis
Human umbilical vein endothelial cells were lysed using Cytobuster lysis buffer (Novagen, Gibbstown, NJ, USA) and protein concentrations in the supernatant estimated using Dc Protein Assay Kit (BioRad). 45 µg of protein was separated by SDS-PAGE and transferred onto nitrocellulose membrane (BioRad). After blocking with 5% (w/v) dried milk, the membrane was incubated with polyclonal antibody against phospho-eNOS (1:1000; BD Transduction Laboratories, Franklin Lakes, NJ, USA) or eNOS (1:1000; Upstate, Billerica, MA, USA) for 3 h, followed by several washes with TBS buffer containing 0.1% Tween 20 and subsequent incubation with the corresponding horseradish peroxidase (HRP)-conjugated IgG secondary antibody (1:1000) for 1 h. Protein bands were detected using Bio-Rad ECL detection system.
Statistical analysis
Numerical results were analysed using one-way ANOVA with Duncan post hoc test. For [Ca2+]i and NO measurement, nonparametric analysis with Prism Software was employed. Values shown are means of at least n = 3 experiments with ± standard deviation (SD). Differences were considered statistically significant at a value of P < 0.05.
Chemical and reagents
Ginsenoside protopanaxadiol and g-PPT (purity >98%) were purchased from the Division of Chinese Materia Medica and Natural Products, National Institute for the Control of Pharmaceutical and Biological Products, Ministry of Public Health, China, and were dissolved in sterile dimethyl sulphoxide (DMSO) for tissue culture purposes. The chemical structures of both agents are shown in Figure 1.
Phenol red-free culture medium 199, ECGS, Dex, RU486, E2, 2-APB and thapsigargin (Sigma, St. Louis, MO, USA). ICI 182,780, PPT and DPN were obtained from Tocris Biosciences, Ellisville, MI, USA; L-NG-monomethyl arginine (L-NMMA) (Cayman Chemical, Ann Arbor, MI, USA); fetal bovine serum (FBS, Gibco Carlsbad, CA, USA); Fura-2 AM, Pluronic F127 and NO sensitive fluorescent dye DAF-FM diacetate (Molecular Probes, Leiden, Netherlands).
Results
g-PPD and g-PPT increases [Ca2+]i in HUVECs
Exposure of HUVECs to g-PPD and g-PPT resulted in an increase in [Ca2+]i with EC50 values of 425 nmol·L−1 and 482 nmol·L−1 respectively (Figure 2A,B). [Ca2+]i peaked at 60 s after the addition of g-PPD and at 85 s after the addition of g-PPT (Figure 2A,B). Blocking calcium influx with the non-selective cation channel blocker, 2-APB (10 µmol·L−1); inhibiting the endoplasmic reticulum Ca2+-ATPase pump with thapsigargin (10 µmol·L−1); or removal of extracellular Ca2+, inhibited but could not abolish g-PPD- and g-PPT-induced rises in [Ca2+]i, indicating that both intracellular release and extracellular influx contributed to [Ca2+]i levels (Figure 2C).
Figure 2.
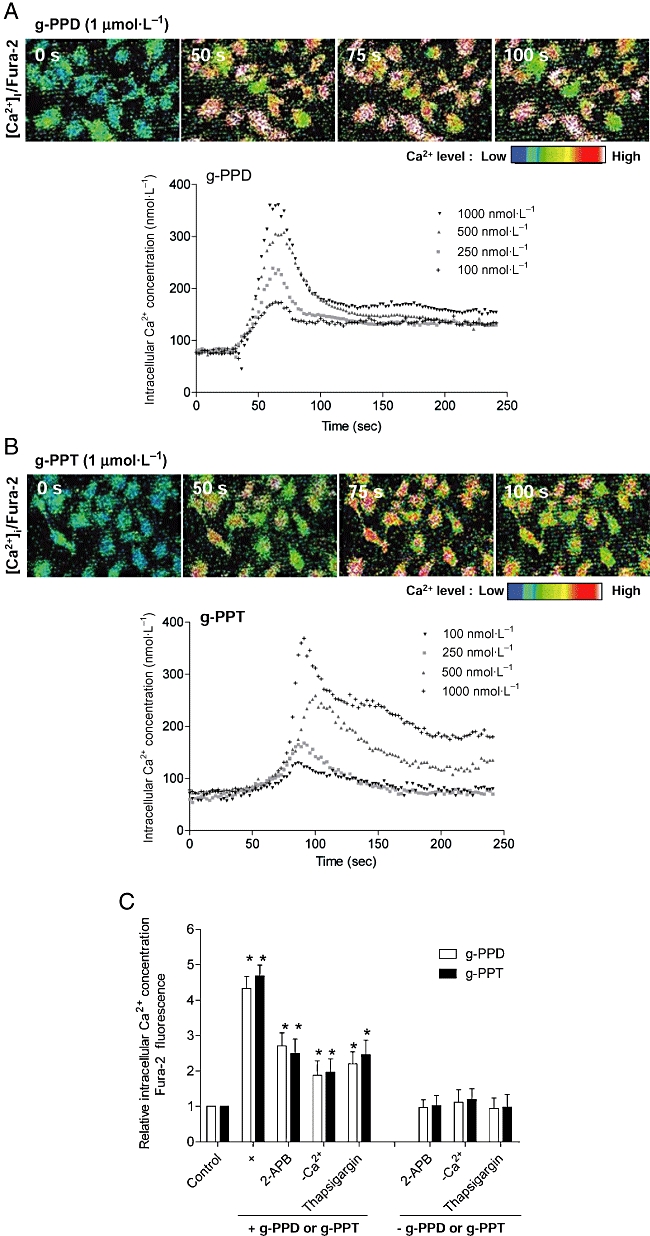
Time- and concentration-dependent increases of [Ca2+]i levels in HUVECs after stimulation with (A) g-PPD and (B) g-PPT. The cells were loaded with the fluorescent Ca2+ indicator, Fura-2, and the fluorescence intensity was measured at 2 s intervals for 4 min. The [Ca2+]i was estimated using internal standard curve. (C) The histogram shows fold changes in [Ca2+]i over control following the addition of g-PPD (1 µmol·L−1), g-PPT (1 µmol·L−1), or the treatment of each drug with one of the following calcium channel inhibitors: 2-APB (10 µmol·L−1), Ca2+-free solution, or thapsigargin (1 µmol·L−1). Bars represent area under the curve, indicative of the total free [Ca2+]i in a duration of 4 min. Data are mean ± SD of three experiments. Asterisk (*) indicates a significant difference between control and treatment groups (P ≤ 0.05). 2-APB, 2-aminoethyldiphenylborate; [Ca2+]i, intracellular calcium ion concentration; g-PPD, ginsenoside protopanaxadiol; g-PPT, ginsenoside protopanaxatriol; HUVECs, human umbilical vein endothelial cells.
NO production is elevated in HUVECs after treatment with g-PPD and g-PPT
Increased [Ca2+]i is known to stimulate the generation of NO from the activated form of eNOS in endothelial cells. We used the fluorescent dye, DAF-FM diacetate, to determine the effects of g-PPD and g-PPT on NO production in endothelial cells (Figure 3A). The fluorescence signal accumulated gradually in cells and reached a plateau 100 s after the addition of g-PPD or g-PPT (Figure 3A). Inhibition of the NOS activity by L-NMMA blocked the effect of g-PPD and g-PPT on NO production (Figure 3B). The g-PPD- and g-PPT-induced increase in the NO production was partially inhibited by 2-APB (10 µmol·L−1), thapsigargin (10 µmol·L−1), or by removal of extracellular Ca2+, suggesting that the induction of NO was Ca2+-dependent (Figure 3B).
Figure 3.
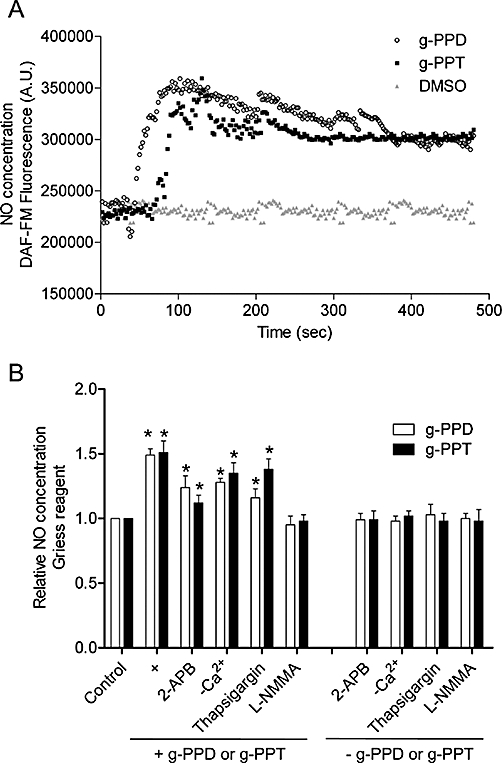
Time-dependent increases in NO generation after stimulation with (A) g-PPD (1 µmol·L−1) and g-PPT (1 µmol·L−1). NO production was determined after loading the cells with the DAF-FM diacetate fluorescent dye, and the fluorescence intensity was measured at 2 s intervals for 8 min. DMSO was used as the solvent control. NO concentrations in the conditioned media of HUVECs were also estimated using (B) Greiss reagents. HUVECs were treated with g-PPD (1 µmol·L−1), g-PPT (1 µmol·L−1), or co-treatment of each drug with one of the following: 2-APB (10 µmol·L−1), Ca2+-free solution, thapsigargin (1 µmol·L−1), or L-NMMA (10 µmol·L−1) for 24 h. Bars represent the fold changes in NO production over the controls. Data are mean ± SD of three experiments. Asterisk (*) indicates a significant difference between control and treatment groups (P ≤ 0.05). 2-APB, 2-aminoethyldiphenylborate; DAF-FM, 4-amino-5-methylamino-2′,7′-difluorofluorescein; DMSO, dimethyl sulphoxide; g-PPD, ginsenoside protopanaxadiol; g-PPT, ginsenoside protopanaxatriol; HUVECs, human umbilical vein endothelial cells; L-NMMA, L-NG-monomethyl arginine; NO, nitric oxide.
g-PPD and g-PPT bind to GR
Glucocorticoid receptor was found in HUVECs as detected by RT- and real-time PCR (Figure 4A). Triple immunofluorescence staining of GR (red), α-actin (green) and nuclei (DAPI, blue) in HUVEC, confirmed the expression of GR in the cytoplasm and nuclei (Figure 4B). Stimulation of the GR with Dex (10 nmol·L−1) increased [Ca2+]i and NO production in HUVECs (Figure 4C,D).
Figure 4.
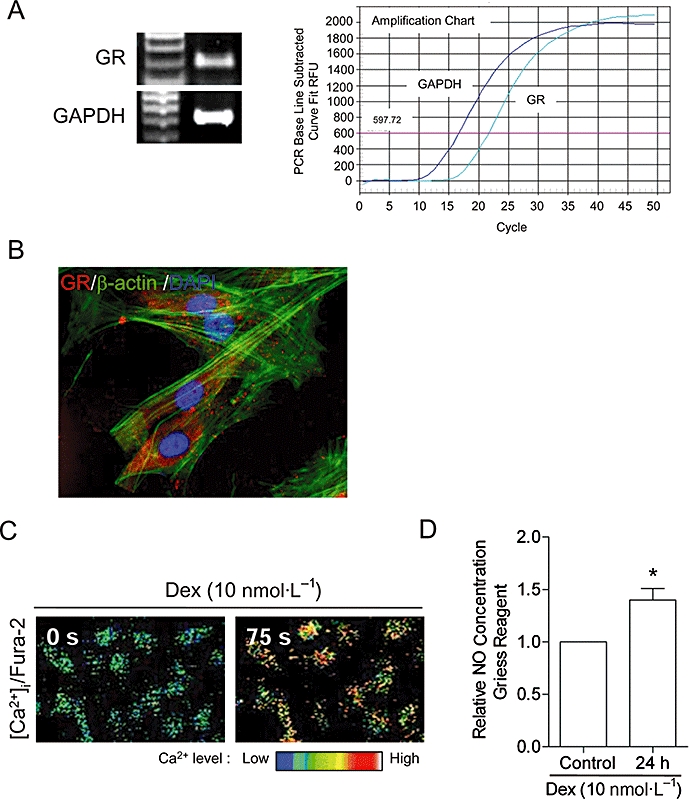
Ginsenoside protopanaxadiol and g-PPT bind to GR. (A) The mRNA of GR was detected in HUVECs using RT- and real-time PCR. (B) Triple fluorescence staining of GR (red), F-actin (green) and nuclei (DAPI, blue) in HUVECs. The GR agonist, Dex (10 nmol·L−1), increased (C) [Ca2+]i in 75 s as illustrated by the Fura-2; and (D) NO production in 24 h as measured by the Griess reagent. Bars represent the fold change in the NO production over control. Data are mean ± SD of three experiments. Asterisk (*) indicates a significant difference between control and treatment groups (P ≤ 0.05). [Ca2+]i, intracellular calcium ion concentration; DAPI, 2-(4-amidinophenyl)-6-indolecarbamidine dihydrochloride; Dex, dexamethasone; GAPDH, Glyceraldehyde-3-phosphate dehydrogenase; g-PPT, ginsenoside protopanaxatriol; GR, glucocorticoid receptor; HUVECs, human umbilical vein endothelial cells; NO, nitric oxide; RT, reverse transcriptase.
GR mediates the actions of ginsenoside metabolites
Next, we tested if the GR is involved in the action of the ginsenoside metabolites on HUVECs. RU486 (10 µmol·L−1), or knockdown of GR using siRNA partially attenuated g-PPD- and g-PPT-induced rises in [Ca2+]i and NO production (Figure 5A,B). DMSO, which was used as a solvent control for RU486, and non-specific siRNA had no effect (Figure 5A,B).
Figure 5.
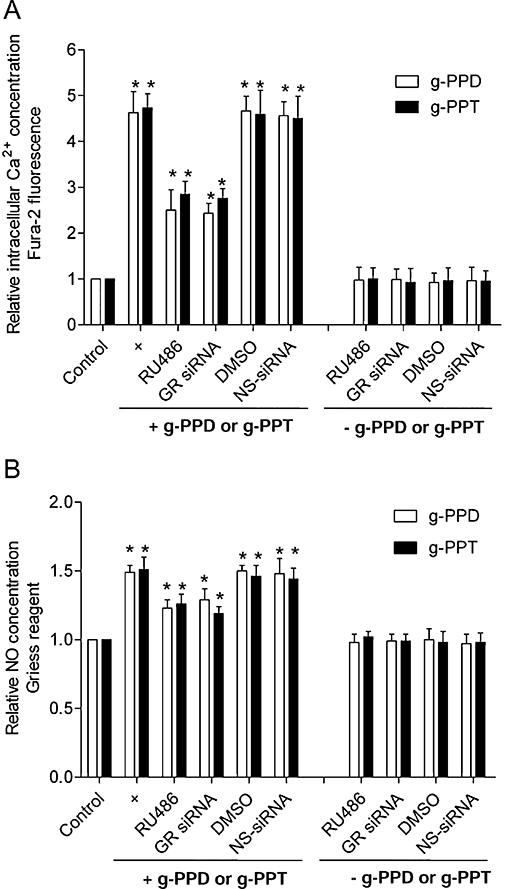
Glucocorticoid receptor (GR) antagonist or knockdown blocks the actions of g-PPD and g-PPT on HUVECs. Treatment with the GR antagonist, RU486 (10 µmol·L−1) or transfection with a siRNA that silences GR, partially abolishes g-PPD (1 µmol·L−1)- and g-PPT (1 µmol·L−1)-stimulated increases in (A) [Ca2+]i and (B) NO generation. DMSO is used here as a solvent control for RU486 and the non-specific (NS)-siRNA, as a control for the siRNA transfection studies. Data are a mean ± SD of three experiments. Asterisk (*) indicates a significant difference between control and treatment groups (P < 0.05). [Ca2+]i, intracellular calcium ion concentration; DMSO, dimethyl sulphoxide; g-PPD, ginsenoside protopanaxadiol; g-PPT, ginsenoside protopanaxatriol; HUVECs, human umbilical vein endothelial cells; NO, nitric oxide.
g-PPD and g-PPT bind to ER
17β-estradiol (10 nmol·L−1) also increased [Ca2+]i and NO production, thus confirming that the ER is functional in HUVECs (Figure 6A,B).
Figure 6.
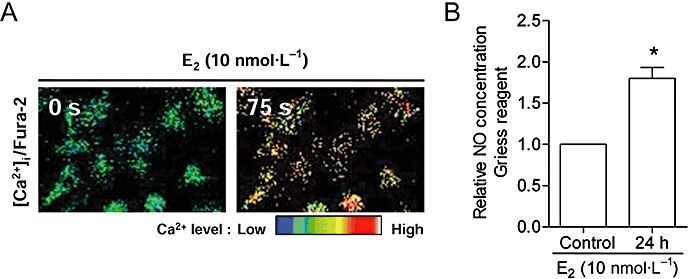
Oestrogen receptor agonist, E2 (10 nmol·L−1), increased (A) [Ca2+]i in 75 s as illustrated by Fura-2; and (B) NO production in 24 h as measured by the Griess reagent. Bars represent the fold change in the NO production over control. Data are mean ± SD of three experiments. Asterisk (*) indicates a significant difference between control and treatment groups (P ≤ 0.05). [Ca2+]i, intracellular calcium ion concentration; E2, 17β-estradiol; NO, nitric oxide.
ERβ is the dominant ER in HUVECs
The presence of ER isoforms was detected using RT- and real time PCR (Figure 7A). Triple immunofluorescence staining of ERα/ERβ (red), α-actin (green) and nucleus (DAPI, blue) in HUVEC confirmed the expression of ERα and ERβ in both cytoplasm and nuclei (Figure 7B). To distinguish between the action of the ERβ and that of the ERα, we used the specific ERα agonist, PPT (100 nmol·L−1), and the specific ERβ agonist, DPN (100 nmol·L−1). Compared with PPT, DPN was more effective in inducing rapid [Ca2+]i elevation (Figure 7C), eNOS phosphorylation in 60 min (Figure 7D,E), and increasing NO production in 24 h (Figure 7F), indicating that ERβ is the dominant ER isoform in HUVECs.
Figure 7.
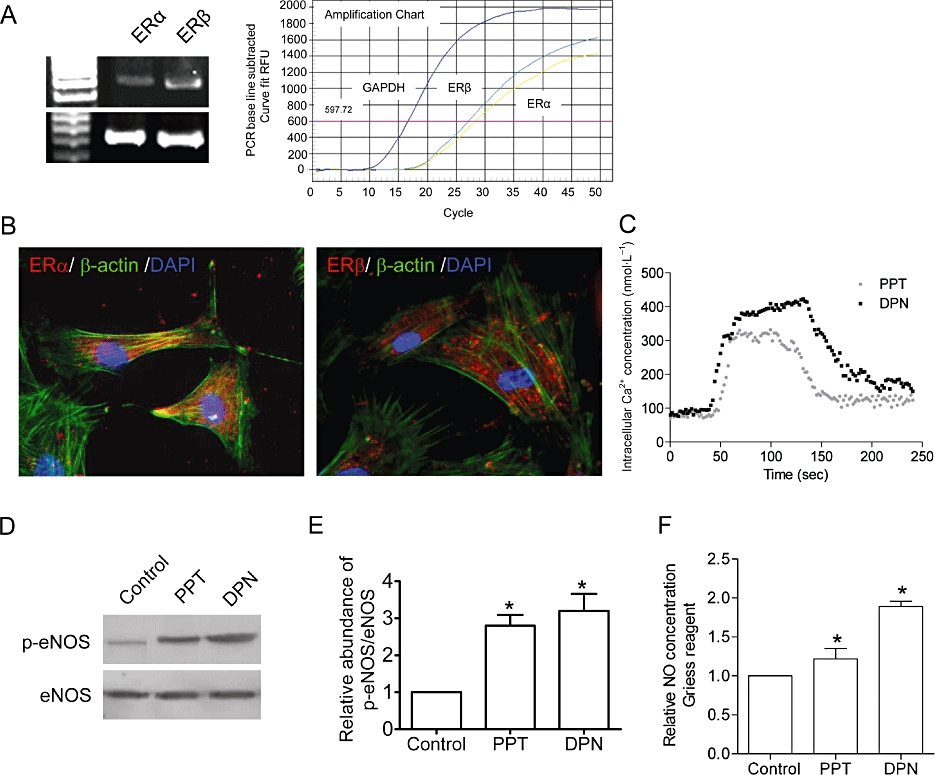
Both ERα and ERβ are expressed in HUVECs as determined using (A) RT- and real time-PCR. There is approximately two fold more ERβ than ERα. (B) Triple fluorescence staining of ERα (red) and ERβ (red), α-actin (green) and nuclei (DAPI, blue) in HUVECs. To examine the activities of these ER isoforms, HUVECs were treated with the specific ERα agonist, PPT (100 nmol·L−1), and the specific ERβ agonist, DPN (100 nmol·L−1). DPN causes greater [Ca2+]i elevation (C); Western blot analysis of (D) eNOS phosphorylated at Ser-1177 (p-eNOS); (E) summarized data of eNOS phosphorylation (p-eNOS, n = 3); (F) eNOS phosphorylation (p-eNOS); and (F) NO production when compared with PPT, indicating that ERβ plays a more dominant role in regulating the [Ca2+]i and NO levels in HUVECs. Data are mean ± SD of three experiments. Asterisk (*) indicates a significant difference between control and treatment groups (P ≤ 0.05). DAPI, 2-(4-amidinophenyl)-6-indolecarbamidine dihydrochloride; DPN, 2-diarylpropionitrile; eNOS, endothelial nitric oxide synthase; ER, oestrogen receptor; HUVECs, human umbilical vein endothelial cells; PPT, propylpyrazole-triol; RT, reverse transcriptase.
ERβ mediates g-PPD- and g-PPT-induced [Ca2+]i elevation, eNOS phosphorylation and NO production
Treatment with ICI 182,780 or transfection of siRNA targeting ERβ, partially suppressed the [Ca2+]i elevation (Figure 8A), eNOS phosphorylation (Figure 8B,C) and NO production (Figure 8D) elicited by g-PPD or g-PPT. Non-specific ER inhibition with ICI 182,780 (10 µmol·L−1) and knockdown of ERβ generated similar inhibition of the actions of the ginsenoside metabolites, supporting the suggestion that the ERα isoform was minimally involved.
Figure 8.
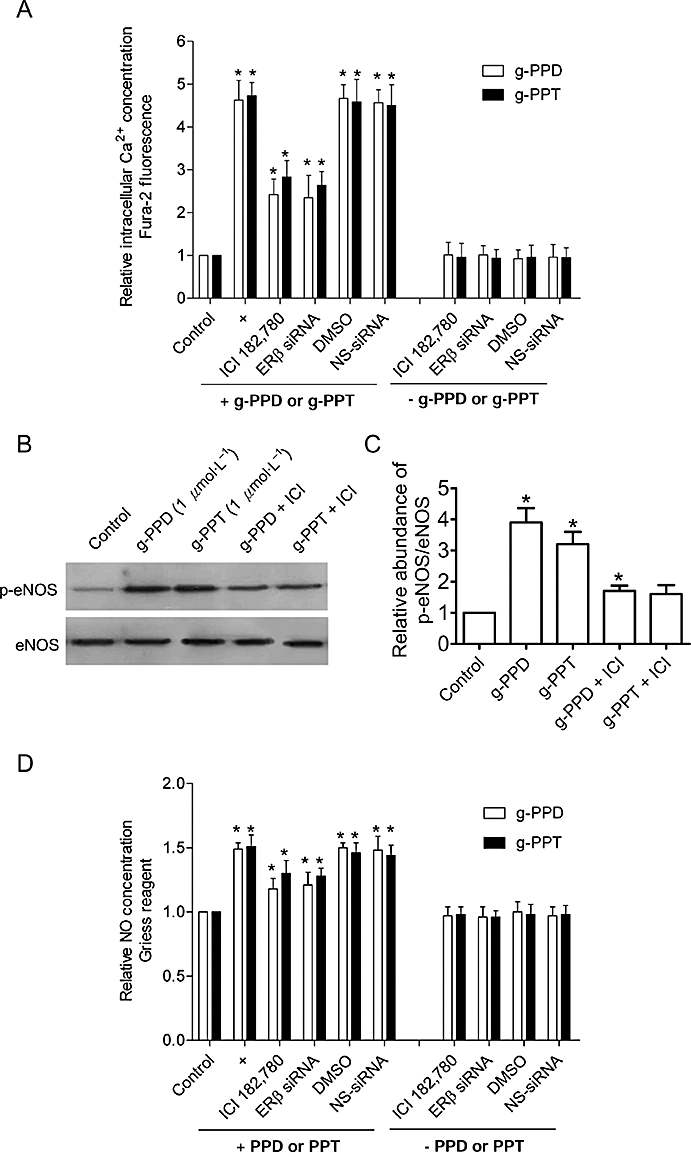
Oestrogen receptor (ER) antagonist and knockdown of ERβ block the action of g-PPD and g-PPT on HUVECs. Treatment with the ER antagonist, ICI 182,780 (ICI; 10 µmol·L−1), or transfection with a siRNA that silences the ERβ, partially inhibits g-PPD (1 µmol·L−1)- and g-PPT (1 µmol·L−1)-stimulated increases in (A) [Ca2+]i, Western blot analysis of (B) eNOS phosphorylated at Ser-1177 (p-eNOS); (C) summarized data of eNOS phosphorylation (p-eNOS, n = 3); (D) NO generation. DMSO is used here as a solvent control for ICI 182,780 and the non-specific (NS)-siRNA as a control for the siRNA transfection studies. Data are mean ± SD of three experiments. Asterisk (*) indicates a significant difference between control and treatment groups (P ≤ 0.05). [Ca2+]i, intracellular calcium ion concentration; DMSO, dimethyl sulphoxide; eNOS, endothelial nitric oxide synthase; g-PPD, ginsenoside protopanaxadiol; g-PPT, ginsenoside protopanaxatriol; HUVECs, human umbilical vein endothelial cells.
GR and ER have equal contribution to the actions of the ginsenoside metabolites
A complete inhibition of [Ca2+]i elevation and NO production were observed in a combined treatment with RU486 and ICI 182,780. DMSO, which was used as a solvent control for RU486 and ICI 182,780, had no effect (Figure 9A,B). These findings suggest that both GR and ER are involved in the rapid response of endothelial cells to g-PPD and g-PPT.
Figure 9.
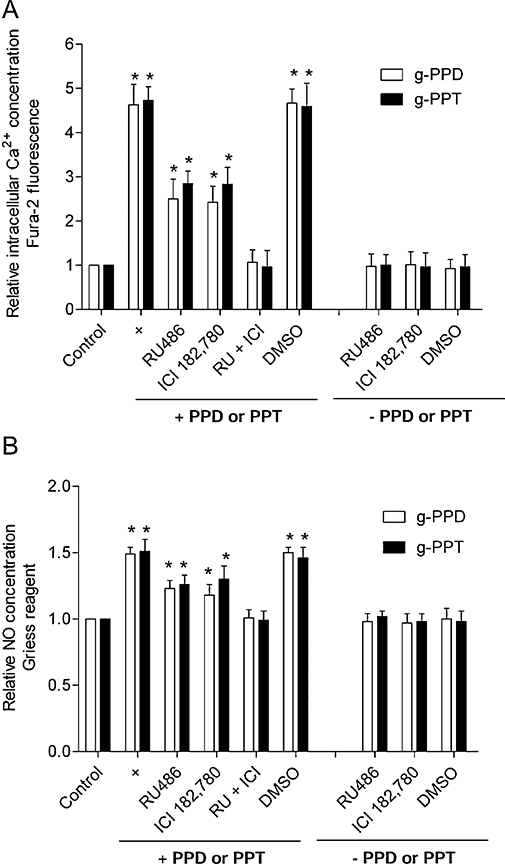
Glucocorticoid receptor (GR) and ER are both involved in the action of ginsenoside metabolites. Co-treatment with the GR antagonist, RU486 (10 µmol·L−1) and the ER antagonist, ICI 182,780 (10 µmol·L−1) (RU + ICI), causes complete inhibition of g-PPD (1 µmol·L−1)- and g-PPT (1 µmol·L−1)-induced increase in (A) [Ca2+]i and (B) NO generation, confirming that both GR and ER play a part in the responses of the cells to the g-PPD and g-PPT treatments. DMSO was used here as a solvent control for RU486 and ICI 182,780. Data are mean ± SD of three experiments. Asterisk (*) indicates a significant difference between control and treatment groups (P ≤ 0.05). DMSO, dimethyl sulphoxide; ER, oestrogen receptor; g-PPD, ginsenoside protopanaxadiol; g-PPT, ginsenoside protopanaxatriol; NO, nitric oxide.
Discussion
Ca2+ and NO are important signalling molecules in the physiological regulation of vascular function. Insufficient bioavailable NO is a pivotal event in the development of hypertension, heart failure and coronary artery disease (Lapu-Bula and Ofili, 2007). NO is produced when eNOS converts L-arginine to L-citrulline. There are three NOS isoforms and eNOS is the predominantly expressed isoform in the vasculature (Förstermann et al., 1995). Calcium-calmodulin activation of eNOS accounts for the critical early phase of NO production. Studies have shown that calcium removal or calmodulin antagonism inhibits NO production through eNOS (Busse and Mülsch, 1990; Förstermann et al., 1991). The calmodulin-eNOS complex then binds to heat shock protein 90 (HSP90), which allows eNOS to be phosphorylated by PKB/Akt and the late phase of the NO production starts (Fulton et al., 1999). Apart from vasodilatation, NO is also involved in angiogenesis mediated by VEGF (Ku et al., 1993; Dawson et al., 2006). Inhibiting NO production leads to a reduced growth of blood vessels, a condition associated with difficulties in wound healing and tissue repair (Schwentker et al., 2002). NO exerts anti-inflammatory effects in the blood vessel wall by inhibiting adhesion of leukocytes to endothelium (Kubes et al., 1991), and an anti-thrombotic action through inactivating platelet adhesion (Radomski et al., 1987). The present study provides evidence that ginsenosides alter the levels of Ca2+ and NO in endothelial cells and they do so through the GR and ERs.
Ginsenosides are triterpene saponins that contain a planar steroidal backbone and various sugar side-chains at C-3, C-6 or C-20 positions. The variations in sugar side-chains determine the binding preference of ginsenosides to nuclear receptors (Leung et al., 2007c). Generally, ginsenosides of the 20(S)-protopanaxatriol family are functional GR ligands, while those of the 20(S)-protopanaxadiol family are ER ligands. g-PPD and g-PPT are end metabolites of major ginsenosides without a sugar moiety at C-3, C-6 and C-20 positions. The specificity of action of these ginseng metabolites on GR and ER was evident after their effects were attenuated by GR or ER antagonists. However, no synergism between GR and ER was observed. Because of the steroidal skeleton of g-PPD and g-PPT and the absence of steric hindrance from the sugar side-chains, g-PPD and g-PPT may also bind to other hormonal receptors, such as androgen, progesterone and minerocorticoid receptors. The interaction with multiple hormonal receptors may help explain the wide range of actions of ginseng in the body and the historical reputation of ginseng as a panacea.
Ligand-activated ERα and ERβ function differently in endothelial cells. In transgenic animals, ERα mediates both the increase in basal NO production and vascular protective effects (Brouchet et al., 2001; Hisamoto et al., 2001; Pendaries et al., 2002; Bolego et al., 2005; Chambliss et al., 2005), while ERβ controls systemic blood pressure partly through favourable modulation of endothelial-independent vasodilation (Zhu et al., 2002). Our findings showed that ERβ had a higher expression than ERα in HUVECs and played a major role in regulating Ca2+ and NO levels in HUVECs (Figure 7). Although these ginsenoside metabolites displayed a comparable affinity for ERα in the in vitro binding assay (data not shown), transfection with siRNA that targeted ERβ produced the same inhibitory effect as the ER specific antagonist ICI 182,780, indicating a relatively small role for ERα in mediating the actions of g-PPD and g-PPT. The present results showed that the activation of ERβ by g-PPD and g-PPT produced a rapid synthesis of NO by endothelial cell which may be of therapeutic interest in the prevention and treatment of vascular diseases associated with endothelial cell dysfunction.
We have recently reported that ginsenoside Re, a member of the protopanaxatriol family, like g-PPT, specifically activates GR and triggers increases in endothelial cell [Ca2+]i, an event solely caused by influx of extracellular Ca2+ ions as removal of extracellular Ca2+ ions abolishes this response (Leung et al., 2007b). However, the mechanisms involved in the increased [Ca2+]i in response to g-PPD and g-PPT may be more complex, as a small increase in [Ca2+]i remained when endothelial cells were tested in a Ca2+-free solution. The increase in [Ca2+]i appeared to involve both extracellular and intracellular sources based on the following observations: 2-APB, the non-selective cation channel blocker, which inhibits Ca2+ influx (Gregory et al., 2001) and thapsigargin, the endoplasmic reticulum Ca2+-ATPase pump inhibitor, which depletes intracellular Ca2+ stores (Sagara et al., 1992) caused partial inhibition of g-PPD- and g-PPT-stimulated increase in [Ca2+]i. Although the actual reasons for such difference in the effect of Re and ginsenoside metabolites on endothelial cell [Ca2+]i are still unknown, our observations support the previous findings by Brailoiu et al. (2007) that activation of G protein-coupled ER augments [Ca2+]i, and that this response is partially inhibited by removal of extracellular Ca2+ in central neurons. Thus, it is possible that g-PPT and g-PPD may interact with membrane-bound binding sites to release second messengers that mobilize Ca2+ release from internal stores. The partial inhibition of [Ca2+]i by 2-APB or thapsigargin parallels a partial reduction in g-PPD- or g-PPT-induced NO production, suggesting that NO production is likely to be a downstream event, causally associated with the increased [Ca2+]i in endothelial cells when treated with these molecules.
Although ginsenosides have been widely used for thousands of years and have shown strong health benefits for a wide range of physiological ailments, there is surprisingly little known about the molecular effects or biochemical mechanisms of actions for these drugs. The rapid stimulatory effects of g-PPD and g-PPT on [Ca2+]i and NO production that we have shown here on human endothelial cells suggest a potential value of ginsenosides and their metabolites as angiotherapeutic agents.
Acknowledgments
This study is supported by Research Grants Council of Hong Kong (HKBU1/06C) and CUHK Focused Investment Scheme.
Glossary
Abbreviations:
- 2-APB
2-aminoethyldiphenylborate
- [Ca2+]i
intracellular calcium ion concentration
- DAF-FM
4-amino-5-methylamino-2′,7′-difluorofluorescein
- Dex
dexamethasone
- DPN
2-diarylpropionitrile
- E2
17β-estradiol
- ECGS
endothelial cell growth supplement
- eNOS
endothelial nitric oxide synthase
- g-PPD
ginsenoside protopanaxadiol
- g-PPT
ginsenoside protopanaxatriol
- GR
glucocorticoid receptor
- HUVECs
human umbilical vein endothelial cells
- L-NMMA
L-NG-monomethyl arginine
- PPT
propylpyrazole-triol
Conflict of interest
None.
References
- Andreoli CM, Miller JW. Anti-vascular endothelial growth factor therapy for ocular neovascular disease. Curr Opin Ophthalmol. 2007;18:502–508. doi: 10.1097/ICU.0b013e3282f0ca54. [DOI] [PubMed] [Google Scholar]
- Attele AS, Wu JA, Juan CS. Ginseng pharmacology: multiple constituents and multiple actions. Biochem Pharmaco. 1999;58:1685–1693. doi: 10.1016/s0006-2952(99)00212-9. [DOI] [PubMed] [Google Scholar]
- Babaei S, Teichert-Kuliszewska K, Monge JC, Mohamed F, Bendeck MP, Stewart DJ. Role of nitric oxide in the angiogenic response in vitro to basic fibroblast growth factor. Circ Res. 1998;82:1007–1015. doi: 10.1161/01.res.82.9.1007. [DOI] [PubMed] [Google Scholar]
- Bae EA, Han MJ, Choo MK, Park SY, Kim DH. Metabolism of 20(S)- and 20(R)-ginsenoside Rg3 by human intestinal bacteria and its relation to in vitro biological activities. Biol Pharm Bull. 2002;25:58–63. doi: 10.1248/bpb.25.58. [DOI] [PubMed] [Google Scholar]
- Baka S, Clamp AR, Jayson GC. A review of the latest clinical compounds to inhibit VEGF in pathological angiogenesis. Expert Opin Ther Targets. 2006;10:867–876. doi: 10.1517/14728222.10.6.867. [DOI] [PubMed] [Google Scholar]
- Bolego C, Cignarella A, Sanvito P, Pelosi V, Pellegatta F, Puglisi L, et al. The acute estrogenic dilation of rat aorta is mediated solely by selective estrogen receptoralpha agonists and is abolished by estrogen deprivation. J Pharmacol Exp Ther. 2005;313:1203–1208. doi: 10.1124/jpet.104.082867. [DOI] [PubMed] [Google Scholar]
- Brailoiu E, Dun SL, Brailoiu GC, Mizuo K, Sklar LA, Opera TI, et al. Distribution and characterization of estrogen receptor G protein-coupled receptor 30 in the rat central nervous system. J Endocrinol. 2007;193:311–321. doi: 10.1677/JOE-07-0017. [DOI] [PubMed] [Google Scholar]
- Brouchet L, Krust A, Dupont S, Chambon P, Bayard F, Arnal JF. Estradiol accelerates reendothelialization in mouse carotid artery through estrogen receptoralpha but not estrogen receptor-beta. Circulation. 2001;103:423–428. doi: 10.1161/01.cir.103.3.423. [DOI] [PubMed] [Google Scholar]
- Burke-Gaffney A, Hellewell PG. Tumour necrosis factor-alpha-induced ICAM-1 expression in human vascular endothelial and lung epithelial cells: modulation by tyrosine kinase inhibitors. Br J Pharmacol. 1996;119:1149–1158. doi: 10.1111/j.1476-5381.1996.tb16017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busse R, Mülsch A. Calcium-dependent nitric oxide synthesis in endothelial cytosol is mediated by calmodulin. FEBS Lett. 1990;265:133–136. doi: 10.1016/0014-5793(90)80902-u. [DOI] [PubMed] [Google Scholar]
- Chambliss KL, Simon L, Yuhanna IS, Mineo C, Shaul PW. Dissecting the basis of nongenomic activation of endothelial nitric oxide synthase by estradiol: role of ERalpha domains with known nuclear functions. Mol Endocrinol. 2005;19:277–289. doi: 10.1210/me.2004-0008. [DOI] [PubMed] [Google Scholar]
- Chen SE, Staba EJ. American ginseng. II. Analysis of ginsenosides and their sapogenins in biological fluids. J Nat Prod. 1980;43:463–466. doi: 10.1021/np50010a005. [DOI] [PubMed] [Google Scholar]
- Chi H, Kim DH, Ji GE. Transformation of ginsenosides Rb2 and Rc from Panax ginseng by food microorganisms. Biol Pharm Bull. 2005;28:2102–2105. doi: 10.1248/bpb.28.2102. [DOI] [PubMed] [Google Scholar]
- Cui JF, Bjorkhem I, Eneroth P. Gas chromatographic-mass spectrometric determination of 20(S)-protopanaxadiol and 20(S)-protopanaxatriol for study on human urinary excretion of ginsenosides after ingestion of ginseng preparations. J Chromatogr B Biomed Sci Appl. 1997;689:349–355. doi: 10.1016/s0378-4347(96)00304-0. [DOI] [PubMed] [Google Scholar]
- Dawson NS, Zewieja DC, Wu MH, Granger HJ. Signaling pathways mediating VEGF165-induced calcium transients and membrane depolarization in human endothelial cells. FASEB J. 2006;20:991–993. doi: 10.1096/fj.05-3923fje. [DOI] [PubMed] [Google Scholar]
- Forster C, Silwedel C, Golenhofen N, Burek M, Kietz S, et al. Occludin as direct target for glucocorticoid-induced improvement of blood-brain barrier properties in murine in vitro system. J Physiol. 2005;565:475–486. doi: 10.1113/jphysiol.2005.084038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Förstermann U, Pollock JS, Schmidt HH, Heller M, Murad F. Calmodulin-dependent endothelium-derived relaxing factor/nitric oxide synthase activity is present in the particulate and cytosolic fractions of bovine aortic endothelial cells. Proc Natl Acad Sci USA. 1991;88:1788–1792. doi: 10.1073/pnas.88.5.1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Förstermann U, Gath I, Schwarz P, Closs EI, Kleinert H. Isoforms of nitric oxide synthase. Properties, cellular distribution and expressional control. Biochem Pharmacol. 1995;50:1321–1332. doi: 10.1016/0006-2952(95)00181-6. [DOI] [PubMed] [Google Scholar]
- Friedman TC, Mastorakos G, Newman TD, Mullen NM, Horton EG, Costello R, et al. Carbohydrate and lipid metabolism in endogenous hypercortisolism: shared features with metabolic syndrome X and NIDDM. Endocr J. 1996;43:645–655. doi: 10.1507/endocrj.43.645. [DOI] [PubMed] [Google Scholar]
- Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, et al. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory RB, Rychkov G, Barritt GJ. Evidence that 2-aminoethoxydiphenyl borate is a novel inhibitor of store-operated Ca2+ channels in liver cells and acts through a mechanism which does not involve inositol triphosphate receptors. Biochem J. 2001;354:285–290. doi: 10.1042/0264-6021:3540285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groten T, Pierce AA, Huen AC, Schnaper HW. 17 beta-estradiol transiently disrupts adherens junctions in endothelial cells. FASEB J. 2005;19:1368–1370. doi: 10.1096/fj.04-2558fje. [DOI] [PubMed] [Google Scholar]
- Hasegawa H, Sung JH, Matsumiya S, Uchiyama M. Main ginseng saponin metabolites formed by intestinal bacteria. Planta Med. 1996;62:453–457. doi: 10.1055/s-2006-957938. [DOI] [PubMed] [Google Scholar]
- Hisamoto K, Ohmichi M, Kurachi H, Hayakawa J, Kanda Y, Nishio Y, et al. Estrogen induces the Akt dependent activation of endothelial nitric oxide synthase in vascular endothelial cells. J Biol Chem. 2001;276:3459–3467. doi: 10.1074/jbc.M005036200. [DOI] [PubMed] [Google Scholar]
- Hodgin JB, Krege JH, Reddick RL, Korach KS, Smithies O, Maeda N. Estrogen receptor alpha is a major mediator of 17beta-estradiol's atheroprotective effects on lesion size in Apoe-/- mice. J Clin Invest. 2001;107:333–340. doi: 10.1172/JCI11320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichhpujani P, Ramasubramanian A, Kaushik S, Pandav SS. Bevacizumab in glaucoma: a review. Can J Ophthalmol. 2007;42:812–815. doi: 10.3129/i07-160. [DOI] [PubMed] [Google Scholar]
- Iwai A, Fuji Y, Kawakami S, Takazawa R, Kageyama Y, Yoshida MA, et al. Down-regulation of vascular endothelial growth factor in renal cell carcinoma cells by glucocorticoids. Mol Cell Endocrinol. 2004;226:11–17. doi: 10.1016/j.mce.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Liu W, Wang XM, Zhong GG, Zhang WJ, Chen L, et al. Calcium channel blockade and anti-free-radical actions of panaxatriol saponins in cultured myocardiocytes. Zhongguo Yao Li Xue Bao. 1996;17:138–141. [PubMed] [Google Scholar]
- Kasselman LJ, Kintner J, Sideris A, Pasnikowski E, Krellman JW, Shah S, et al. Dexamethasone treatment and ICAM-1 deficiency impair VEGF-induced angiogenesis in adult brain. J Vasc Res. 2007;44:283–291. doi: 10.1159/000101450. [DOI] [PubMed] [Google Scholar]
- Khosravi Shahi P, Fernández Pineda I. Tumoral angiogenesis: review of the literature. Cancer Invest. 2008;26:104–108. doi: 10.1080/07357900701662509. [DOI] [PubMed] [Google Scholar]
- Ku DD, Zaleski JK, Liu S, Brock TA. Vascular endothelial growth factor induces EDRF-dependent relaxation in coronary arteries. Am J Physiol. 1993;265:H586–H592. doi: 10.1152/ajpheart.1993.265.2.H586. [DOI] [PubMed] [Google Scholar]
- Kubes P, Suzuki M, Granger DN. Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci USA. 1991;88:4651–4655. doi: 10.1073/pnas.88.11.4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapu-Bula R, Ofili E. From hypertension to heart failure: role of nitric oxide-mediated endothelial dysfunction and emerging insights from myocardial contrast echocardiography. Am J Cardiol. 2007;99:7D–14D. doi: 10.1016/j.amjcard.2006.12.014. [DOI] [PubMed] [Google Scholar]
- Leung KW, Cheng YK, Mak NK, Chan KK, Fan TP, Wong RN. Signaling pathway of ginsenoside-Rg1 leading to nitric oxide production in endothelial cells. FEBS Lett. 2006a;580:3211–3216. doi: 10.1016/j.febslet.2006.04.080. [DOI] [PubMed] [Google Scholar]
- Leung KW, Pon YL, Wong RN, Wong AS. Ginsenoside-Rg1 induces vascular endothelial growth factor expression through the glucocorticoid receptor-related phosphatidylinositol 3-kinase/Akt and beta-catenin/T-cell factor-dependent pathway in human endothelial cells. J Biol Chem. 2006b;281:36280–36288. doi: 10.1074/jbc.M606698200. [DOI] [PubMed] [Google Scholar]
- Leung KW, Cheung LW, Pon YL, Wong RN, Mak NK, Fan TP, et al. Ginsenoside Rb1 inhibits tube-like structure formation of endothelial cells by regulating pigment epithelium-derived factor through the oestrogen beta receptor. Br J Pharmacol. 2007a;152:207–215. doi: 10.1038/sj.bjp.0707359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung KW, Leung FP, Huang Y, Mak NK, Wong RN. Non-genomic effects of ginsenoside-Re in endothelial cells via glucocorticoid receptor. FEBS Lett. 2007b;581:2423–2428. doi: 10.1016/j.febslet.2007.04.055. [DOI] [PubMed] [Google Scholar]
- Leung KW, Yung KK, Mak NK, Yue PY, Luo HB, Cheng YK, et al. Angiomodulatory and neurological effects of ginsenosides. Curr Med Chem. 2007c;14:1371–1380. doi: 10.2174/092986707780597916. [DOI] [PubMed] [Google Scholar]
- Li G, Wang Z, Sun Y, Liu K, Wang Z. Ginsenoside 20(S)-protopanaxadiol inhibits the proliferation and invasion of human fibrosarcoma HT1080 cells. Basic Clin Pharmacol Toxicol. 2006;98:588–592. doi: 10.1111/j.1742-7843.2006.pto_415.x. [DOI] [PubMed] [Google Scholar]
- Losordo DW, Vale PR, Symes JF, Dunnington CH, Esakof DD, Maysky M, et al. Gene therapy for myocardial angiogenesis: initial clinical results with direct muocardial injection of phVEGF165 as sole therapy for myocardial ischemia. Circulation. 1998;98:2800–2804. doi: 10.1161/01.cir.98.25.2800. [DOI] [PubMed] [Google Scholar]
- Morbidelli L, Chang CH, Douglas JG, Granger HJ, Ledda F, Ziche M. Nitric oxide mediates mitogenic effect of VEGF on coronary venular endothelium. Am J Physiol. 1996;270:H411–H415. doi: 10.1152/ajpheart.1996.270.1.H411. [DOI] [PubMed] [Google Scholar]
- Munaron L. Intracellular calcium, endothelial cells and angiogenesis. Recent Patents Anticancer Drug Discov. 2006;1:105–119. doi: 10.2174/157489206775246502. [DOI] [PubMed] [Google Scholar]
- Murohara T, Asahara T, Silver M, Bauters C, Masuda H, Kalka C, et al. Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. J Clin Invest. 1998;101:2567–2578. doi: 10.1172/JCI1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park MT, Cha HJ, Jeong JW, Kim SI, Chung HY, Kim ND, et al. Glucocorticoid receptor-induced down-regulation of MMP-9 by ginseng components, PD and PT contributes to inhibition of the invasive capacity of HT1080 human fibrosarcoma cells. Mol Cells. 1999;9:476–483. [PubMed] [Google Scholar]
- Pendaries C, Darblade B, Rochaix P, Krust A, Chambon P, Korach KS, et al. The AF-1 activation-function of ERalpha may be dispensable to mediate the effect of estradiol on endothelial NO production in mice. Proc Natl Acad Sci USA. 2002;99:2205–2210. doi: 10.1073/pnas.042688499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radomski MW, Palmer RM, Mancada S. The role of nitric oxide and cGMP in platelet adhesion to vascular endothelium. Biochem Biophys Res Commun. 1987;148:1482–1489. doi: 10.1016/s0006-291x(87)80299-1. [DOI] [PubMed] [Google Scholar]
- Rosengart TK, Lee LY, Patel SR, Sanborn TA, Parikh M, Bergman GW, et al. Angiogenesis gene therapy: phase I assessment of direct intramyocardial administration of an adenovirus vector expressing VEGF121 cDNA to individuals with clinically significant severe coronary artery disease. Circulation. 1999;100:468–474. doi: 10.1161/01.cir.100.5.468. [DOI] [PubMed] [Google Scholar]
- Ruel M, Beanlands RS, Lortie M, Chan V, Camack N, deKemp RA, et al. Concomitant treatment with oral L-arginine improves the efficacy of surgical angiogenesis in patients with severe diffuse coronary artery disease: the Endothelial Modulation in Angiogenic Therapy randomized controlled trial. J Thorac Cardiovasc Surg. 2008;135:762–770. doi: 10.1016/j.jtcvs.2007.09.073. [DOI] [PubMed] [Google Scholar]
- Sagara Y, Fernandez-Belda F, de Meis L, Inesi G. Characterization of the inhibition of intracellular Ca2+ transport ATPases by thapsigargin. J Biol Chem. 1992;267:12606–12613. [PubMed] [Google Scholar]
- Schwentker A, Vodovotz Y, Weller R, Billiar TR. Nitric oxide and wound repair: role of cytokines? Nitric Oxide. 2002;7:1–10. doi: 10.1016/s1089-8603(02)00002-2. [DOI] [PubMed] [Google Scholar]
- Simoncini T, Maffei S, Basta G, Barsacchi G, Genazzani AR, Liao JK, et al. Estrogens and glucocorticoids inhibit endothelial vascular cell adhesion molecule-1 expression by different transcriptional mechanisms. Circ Res. 2000;87:19–25. doi: 10.1161/01.res.87.1.19. [DOI] [PubMed] [Google Scholar]
- Steed DL. Clinical evaluation of recombinant human platelet-derived growth factor for the treatment of lower extremity ulcers. Plast Reconstr Surg. 2006;117:143S–149S. doi: 10.1097/01.prs.0000222526.21512.4c. [DOI] [PubMed] [Google Scholar]
- Wang W, Zhao Y, Rayburn ER, Hill DL, Wang H, Zhang R. In vitro anti-cancer and structure-activity relationship of natural products isolated form fruits of Panax ginseng. Cancer Chemother Pharmacol. 2007;59:589–601. doi: 10.1007/s00280-006-0300-z. [DOI] [PubMed] [Google Scholar]
- Wang ZF, Xiao JS, Yan SZ, Wan ZB. Protective effects of panaxadiol saponins on cardiac functions in burned rats. Zhongguo Yao Li Xue Bao. 1995;16:345–348. [PubMed] [Google Scholar]
- Zhu Y, Bian Z, Lu P, Karas RH, Bao L, Cox D, et al. Abnormal vascular function and hypertension in mice deficient in estrogen receptor β. Science. 2002;295:505–508. doi: 10.1126/science.1065250. [DOI] [PubMed] [Google Scholar]