

Abstract
Family C of human G-protein-coupled receptors (GPCRs) is constituted by eight metabotropic glutamate receptors, two γ-aminobutyric acid type B (GABAB1–2) subunits forming the heterodimeric GABAB receptor, the calcium-sensing receptor, three taste1 receptors (T1R1–3), a promiscuous L-α-amino acid receptor G-protein-coupled receptor family C, group 6, subtype A (GPRC6A) and seven orphan receptors. Aside from the orphan receptors, the family C GPCRs are dimeric receptors characterized by a large extracellular Venus flytrap domain which bind the endogenous agonists. Except from the GABAB1–2 and T1R2–3 receptor, all receptors are either activated or positively modulated by amino acids. In this review, we outline mutational, biophysical and structural studies which have elucidated the interaction of the amino acids with the Venus flytrap domains, molecular mechanisms of receptor selectivity and the initial steps in receptor activation.
Keywords: G-protein-coupled receptors, family C, metabotropic glutamate receptors, calcium-sensing receptor, GABAB receptor, T1R1 taste receptor, GPRC6A receptor, amino acid sensing, mutations
Introduction
The superfamily of G-protein-coupled receptors (GPCRs) contains seven transmembrane (7TM) segments and constitute a large superfamily of cell-surface proteins that are activated by a broad range of ligands (Pierce et al., 2002) and are known to be implicated in many important physiological processes making them targets for approximately 40% of marketed drugs and >60% of drugs in development (Lundstrom, 2005). In the human genome, about 800 genes encode GPCRs of which 400 are olfactory receptors (Bjarnadóttir et al., 2006). The non-olfactory receptors have been classified into three families (A, B and C) based on phylogenetic analysis of the 7TM domain (Kolakowski, 1994). Family C of human GPCRs contains 22 receptor subtypes including eight metabotropic glutamate (mGlu) receptors, the calcium-sensing receptor (CaR), two γ-aminobutyric acid type B (GABAB) receptors, three taste1 receptors (T1R1–3), the G-protein-coupled receptor family C, group 6, subtype A (GPRC6A) and seven orphan receptors (Bjarnadóttir et al., 2005; Bräuner-Osborne et al., 2007)1. As seen in Figure 1A, the receptors cluster into four groups containing (i) the mGlu receptors; (ii) CaR, GPRC6A and T1Rs; (iii) the GABAB receptors including three orphan receptors; and (iv) a group of four RAIG1-like orphan receptors. The mGlu receptors further cluster into three subgroups (termed Group I–III) which correlates with their signal transduction pathway and orthostatic ligand pharmacology (Bräuner-Osborne et al., 2007).
Figure 1.
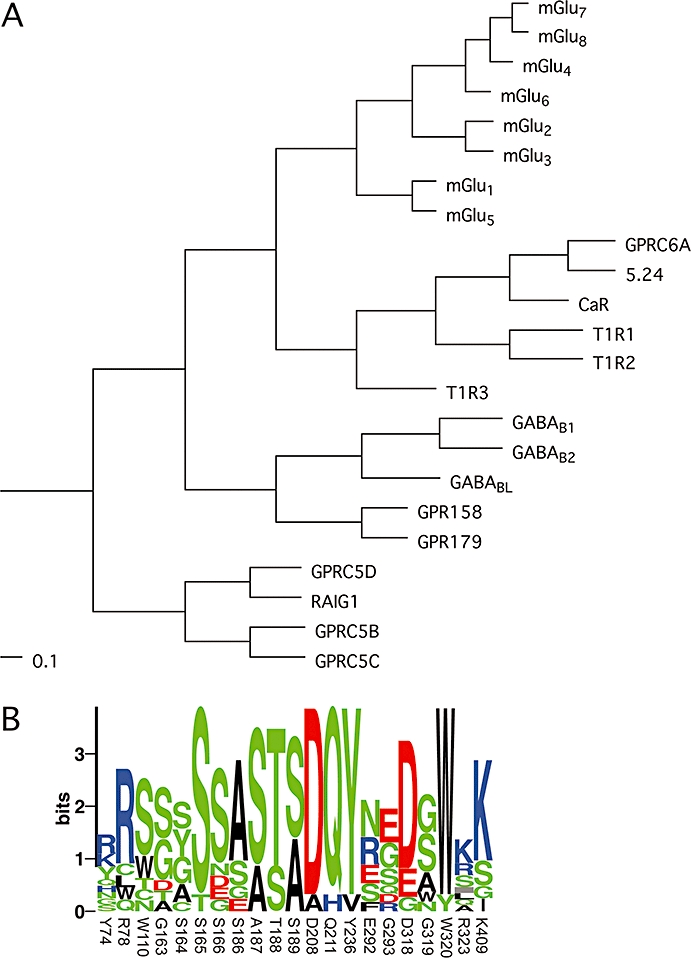
(A) Phylogenetic analysis of family C GPCRs based on their seven transmembrane domains. A multiple sequence alignment of the predicted seven transmembrane domains was generated using the program ClustalX 2.0.9, and the shown phylograms were generated using the unweighted pair group method with arithmetic mean (UPGMA) algorithm and viewed with the program TreeviewX 0.5.0. The scale bars are a function of amino acid substitutions based on the Gonnet series substitution matrix. (B) Sequence logo of binding pocket residues in family C receptors, displaying the degree of conservation of each amino acid. Residues submitted are identical to those listed in Table 1 and numbers refer to the residue numbers in mGlu1. The height of each symbol is proportional to its frequency and colour coded according to polarity. The logo was generated using the server at the Center for Biological Sequence Analysis (http://www.cbs.dtu.dk/~gorodkin/appl/plogo.html) (Schneider and Stephens, 1990; Gorodkin et al., 1997). CaR, calcium-sensing receptor; GABAB, γ-aminobutyric acid type B; GPCR, G-protein-coupled receptor; GPRC6A, G-protein coupled receptor family C, group 6, subtype A; mGlu, metabotropic glutamate; T1R, taste1 receptor.
In the present review, we will focus on the structure, function and molecular pharmacology of the family C receptors with particular focus on the orthosteric binding pocket of the amino acid-binding subtypes. For the topic of allosteric modulation and the physiological and therapeutical roles of family C receptors, the reader is referred to recent reviews in the field (Bräuner-Osborne et al., 2007; Gasparini and Spooren, 2007; Jensen and Bräuner-Osborne, 2007; Pin and Prézeau, 2007; Trivedi et al., 2008).
Structure and function of family C GPCRs
Apart from the orphan receptors, all family C GPCRs have a very large extracellular Venus flytrap (VFT) domain which contains a dimerization interface and an orthosteric binding site for the endogenous agonist (Kunishima et al., 2000; Tsuchiya et al., 2002; Muto et al., 2007). Apart from the orphan and GABAB receptors, all family C receptors also contain a cysteine-rich domain (CRD), of unknown function, which links the VFT and 7TM domains (Figure 2). Interestingly, none of the orphan family C receptors contain a VFT domain, harbouring the orthosteric binding site, which has made ligand predictions based on modelling impossible. Their endogenous ligands, if any, thus remain to be discovered (Cheng and Lotan, 1998; Bräuner-Osborne and Krogsgaard-Larsen, 2000; Robbins et al., 2000; Bräuner-Osborne et al., 2001; Calver et al., 2003). It is also interesting to note that the three orphan receptors, GABABL, GPR158 and GPR179, clustering with the GABAB receptors, contain very long C-terminal domains of unknown function.
Figure 2.

Model of a dimeric family C GPCR in its open–open/resting (left) and open–closed/active (right) conformations. The two conformations are in equillibrium with each other and additional conformations (not shown). Agonists and antagonists will shift the equillibrium towards the active or resting conformation respectively. The localizations of the Venus flytrap (VFT) domain, cysteine-rich domain (CRD) and seven transmembrane domain (7TM) are indicated. The models were constructed with the program MacPyMol using coordinates from PDB files 1EWT (mGlu1 open–open/resting VFT), 1EWK (mGlu1 open–closed/active VFT), 2E4U (mGlu3 CRR) and 2R4S (β2-adrenergic receptor 7TM). GPCR, G-protein-coupled receptor; mGlu, metabotropic glutamate.
Receptor structure
The structure of the extracellular domain(s) of mGlu1, mGlu3 and mGlu7 has been solved by X-ray crystallography, which has shown that the VFT domain consists of two distinct globular domains (termed LB1 and 2), arranged as a central ß-sheet flanked on both sides by α-helices, connected by a hinge region and separated by a cleft (Figure 2) (Kunishima et al., 2000; Tsuchiya et al., 2002; Muto et al., 2007). The CRD consists of three ß-sheets each composed of two antiparallel ß-strands. The CRD contains nine conserved cysteines which form four intradomain disulfide bridges and a disulfide bridge to the VFT domain (Rondard et al., 2006; Muto et al., 2007). All nine cysteines have been shown to be imperative for receptor expression and/or activation and it is thus evident that the rigid structure of the CRD and its rigid connection to the VFT domain is important for family C receptor function (Fan et al., 1998; Rondard et al., 2006). The structure of a 7TM domain of a family C receptor has yet to be elucidated, but biochemical evidence point to a similar topology as the recently crystallized β2-adrenergic family A receptor (Bhave et al., 2003; Rasmussen et al., 2007).
The family C GPCRs exist as constitutive dimeric receptor complexes in the cell membrane (Figure 2). Whereas the GABAB and taste1 receptors exist as heterodimeric receptors composed of two different subunits, CaR and the mGlu receptors form homodimeric complexes via several covalent and non-covalent interactions between the two subunits (Pace et al., 1999; Kunishima et al., 2000; Tsuji et al., 2000; Romano et al., 2001; Zhang et al., 2001; Nomura et al., 2008). The conserved inter-receptor covalent disulfide bridge (e.g. Cys140 in mGlu1) is located in a loop in the VFT – a loop which has been shown by a random saturation mutagenesis study to be involved in keeping the receptor in its inactive conformation (Jensen et al., 2000). In addition, the loop is a hotspot for naturally occurring activating mutations in CaR causing autosomal dominant hypocalcaemia (Hu and Spiegel, 2007). The non-covalent dimer interactions occur via a non-polar interface in LB1, whose integrity is also important for proper receptor function as mutations or introduction of an N-glycan in the interface disrupt function (Tsuji et al., 2000; Rondard et al., 2008).
Activation mechanism
The orthosteric binding site is located in the cleft between LB domain 1 and 2. X-ray crystallography of the mGlu1 receptor has revealed that the initial event in receptor activation is closing of at least one of the VFT domains around the agonist leading to a 70° twist in the dimer interface and thus contraction of the CRD and 7TM domains (termed the ‘open–closed/active conformation’, Figure 2) (Kunishima et al., 2000). So far, crystals of the closed–closed/active conformation has only been obtained with mGlu1 in the presence of Gd3+ which binds to acidic residues between the LB2 domains of the dimer (Tsuchiya et al., 2002; Abe et al., 2003). Recently, crystals of a closed–closed/resting mGlu3 VFT domain was reported (Muto et al., 2007) casting doubt on the dimer interface twisting as the main mechanism of receptor activation. However, several lines of biochemical evidence support a role of the dimer interface twist as necessary for receptor activation such as an engineered antagonistic zinc-site in the interface of the two LB2 domains (in the Gd3+-binding site), which prevent activation when Zn2+ is bound (Jensen et al., 2001b), and an engineered N-glycan in the LB2 domain, which prevent activation but not dimerization of the GABAB heterodimer (Rondard et al., 2008). Several biochemical studies, using fluorescence resonance energy transfer (FRET) (Tateyama et al., 2004) or rescue of activity in dimers consisting of inactivated monomers which each have been made non-functional due to mutations in either the VFT or 7TM domains (Bai et al., 1999; Brock et al., 2007), have also demonstrated that rearrangement of the 7TM domains are required for receptor activation. However, it remains unclear whether the rearrangement is a contraction of the 7TM domains as shown in Figure 2 or a more subtle reorientation of the 7TM domains/helices.
The crystallography studies have shown (S)-glutamic acid (L-Glu) binding to both open and closed VFT domains, raising the question whether closure of the VFT domain(s) is indeed the initial event in receptor activation. However, several lines of studies have pointed to this mechanism-of-activation. First of all, the X-ray structure of a mGlu1 VFT with the antagonist (S)-(α)-methyl-4-carboxyphenylglycine [(S)-MCPG] shows that the antagonist serves as a wedge in the binding pocket preventing closure of the VFT (Figure 3) (Tsuchiya et al., 2002). Second, this study was elaborated by an elegant study in which mutations enlarging the VFT-binding pocket of mGlu8 converted the antagonists (1R,3R,4S)-1-aminocyclopentane-1,3,4-tricarboxylic acid and (S)-2-amino-2-methyl-4-phosphonobutanoic acid into agonists (due to the additional space permitting VFT closure) (Bessis et al., 2002). Third, introduction of cysteines in the LB1 and LB2 domains have created constitutively activated mutant GABAB receptors that presumably are locked in the closed VFT conformation by a disulfide bridge (Kniazeff et al., 2004b). However, it remains debated whether closing of one (Kniazeff et al., 2004a) or both (Kammermeier and Yun, 2005) VFT domains in a dimer is required for activation. Recently, X-ray crystallography and mutational studies have also shown that the low potency of L-Glu on mGlu7 most likely is caused by steric hindrance among the residues surrounding the orthosteric binding pocket, thus preventing establishment of a stable, fully closed VFT domain (Rosemond et al., 2004; Muto et al., 2007).
Figure 3.
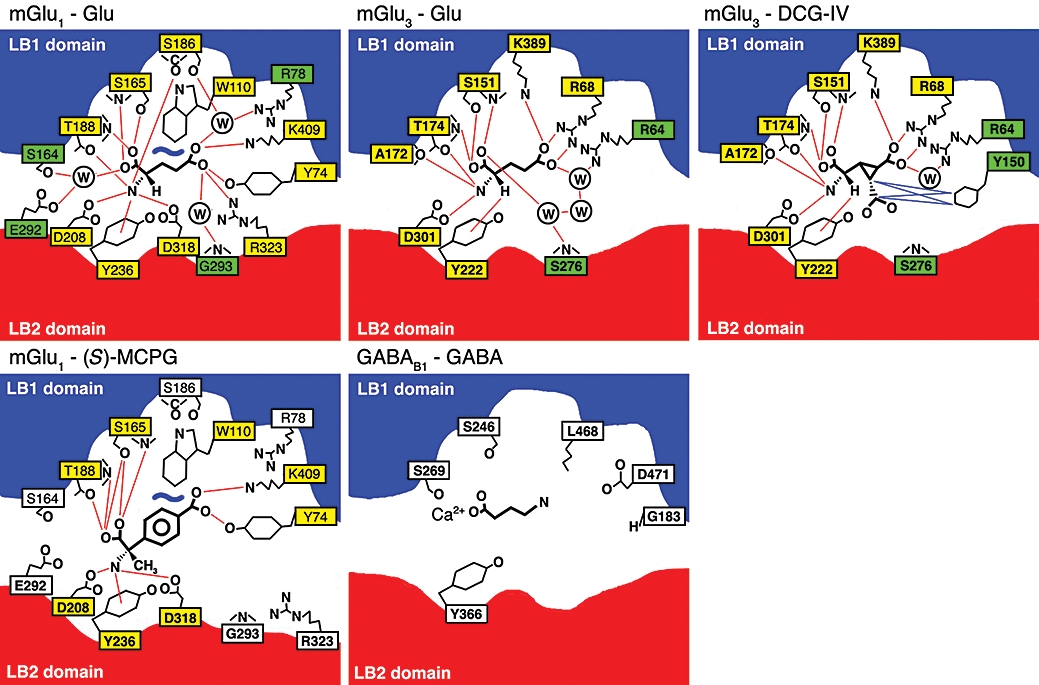
The orthosteric binding site in mGlu and GABAB receptors. Schematic drawings of the binding of agonists (Glu, DCG-IV or GABA) or antagonist [(S)-MCPG] to mGlu1, mGlu3 or GABAB1. The mGlu1 and mGlu3 drawings are based on X-ray crystallographic structures (Kunishima et al., 2000; Tsuchiya et al., 2002; Muto et al., 2007) whereas the GABAB drawing is based on molecular modelling and mutational data (Galvez et al., 1999; 2000a; Jensen et al., 2001a). Red and blue lines indicates hydrogen-bonding and van der Wahls contacts respectively. Yellow and green filled boxes indicates direct or indirect (via water) contacts respectively. Redrawn from the listed references. DCG-IV, (2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine; GABAB, γ-aminobutyric acid type B; mGlu, metabotropic glutamate; (S)-MCPG, (S)-(α)-methyl-4-carboxyphenylglycine.
Collectively, the structural and molecular pharmacology evidence points towards an activation model in which the VFT is in equilibrium between an open and closed form, and a resting and active/twisted conformation. Agonists will shift the equilibrium towards the closed VFT conformation, which in turn will shift the interface twist equilibrium towards the active conformation. Conversely, antagonists will shift the equilibrium towards the open VFT conformation, which in turn will shift the interface twist equilibrium towards the resting conformation (Figure 2). The CRD domain acts as a rigid lever to transfer the energy of the conformational change in the VFT domains to the 7TM domains, but the exact mechanism of activation of the 7TM domains remains to be fully elucidated.
The orthosteric binding site in the VFT domain
An alignment of the amino acids residues in close (6 Å) proximity to L-Glu in the closed mGlu1 VFT with the other amino acid-binding family C receptors is shown in Table 1. From this alignment (Table 1) and the X-ray crystal structures of the closed mGlu1 and mGlu3 L-Glu-bound VFTs (Figure 3), it is clear that particularly five residues (S165, T188, D208, Y236 and D318 in mGlu1) are vital for binding of the α-amino acid moiety and that two basic residues (R78 and K409 in mGlu1) are vital for binding of the distal carboxylic acid of L-Glu. The former residues have also been identified as an important motif of α-amino acid recognition in a large database mining study (Acher and Bertrand, 2005), which also identified three additional residues (R203, Q211 and G237 in mGlu1) as part of the motif, which however do not make direct contacts with the amino acid (Figure 3). Mutational, modelling and phylogenetic studies have also suggested the existence of a Ca2+-binding site adjacent to the amino acid-binding site in the VFT domain, which will be discussed in further detail below.
Table 1.
Orthosteric binding site homology of family C receptor subtypesa
| mGlu1 | Y74 | R78 | W110 | G163 | S164 | S165 | S166 | S186 | A187 | T188 | S189 | D208 | Q211 | Y236 | E292 | G293 | D318 | G319 | W320 | R323 | K409 |
| mGlu5 | Y64 | R68 | W100 | G149 | S150 | S151 | S152 | S172 | A173 | T174 | S175 | D194 | Q197 | Y222 | E278 | G279 | D304 | G305 | W306 | R309 | K395 |
| mGlu2 | R57 | R61 | S93 | S143 | Y144 | S145 | D146 | A166 | S167 | T168 | S169 | D188 | Q191 | Y226 | R271 | S272 | D295 | G296 | W297 | L300 | K377 |
| mGlu3 | R64 | R68 | S100 | S149 | Y150 | S151 | S152 | A172 | S173 | T174 | S175 | D194 | Q197 | Y222 | R277 | S278 | D301 | G303 | W303 | Q306 | K389 |
| mGlu4 | K74 | R78 | S110 | S157 | G158 | S159 | S160 | A180 | S181 | T182 | A183 | D202 | Q205 | Y230 | N286 | E287 | D312 | S313 | W314 | K317 | K405 |
| mGlu6 | Q58 | R62 | S94 | S146 | A147 | S148 | S149 | A169 | S170 | T171 | A172 | D191 | Q194 | Y219 | N275 | E276 | D301 | S302 | W303 | K306 | K394 |
| mGlu7 | N74 | R78 | S110 | S157 | G158 | S159 | S160 | A180 | S181 | T182 | A183 | D202 | Q205 | Y230 | N288 | D289 | D314 | S315 | W316 | K319 | K407 |
| mGlu8 | K71 | R75 | S107 | A154 | A155 | S156 | S160 | A177 | S178 | T179 | A180 | D199 | Q202 | Y227 | N283 | E284 | D309 | S310 | W311 | K314 | K401 |
| CaR | R66 | W70 | N102 | T145 | G146 | S147 | G148 | A168 | S169 | S170 | S171 | D190 | Q193 | Y218 | S272 | G273 | E297 | A298 | W299 | S302 | I416 |
| GPRC6A | S69 | Q73 | T104 | G147 | Y148 | S149 | E150 | E170 | S171 | T172 | A173 | D192 | Q195 | Y220 | R279 | Q280 | D303 | N304 | W305 | A308 | L411 |
| T1R1 | H71 | L75 | S107 | D147 | S148 | T149 | N150 | A170 | A171 | S172 | S173 | D192 | Q195 | Y220 | S276 | R277 | E301 | A302 | W303 | S306 | S385 |
| GABAB1a | G183 | C187 | C220 | G244 | C245 | S246 | S247 | G267 | S268 | S269 | S270 | A289 | H292 | V317 | F365 | E367 | G393 | W394 | Y395 | – | G461 |
List of residues within 6 Å of l-Glu bound to mGlu1 (Madsen et al., 2005) based on previously published protein sequence alignments (Bräuner-Osborne et al., 1999; Galvez et al., 1999; Wellendorph and Bräuner-Osborne, 2004). Residues in bold are known to interact with l-Glu either directly or indirectly via water molecules in mGlu1 and mGlu3 respectively (Kunishima et al., 2000; Muto et al., 2007). Underlined and italic residues interact with the α-amino acid or distal carboxylic acid moiety of l-Glu respectively (Kunishima et al., 2000; Muto et al., 2007). Highly conserved residues involved in binding of the α-amino acid moiety or the distal carboxylic acid moiety of l-Glu are shown in red and green respectively.
CaR, calcium-sensing receptor; GABAB, γ-aminobutyric acid type B; GPRC6A, G-protein-coupled receptor family C, group 6, subtype A; mGlu, metabotropic glutamate; T1R, taste1 receptor.
Molecular pharmacology of mGlu receptors
Receptor subtypes
As previously noted, the mGlu receptors have been divided into three groups (Figure 1). Receptors within a group show more than 60% sequence identity whereas there is 40–50% sequence identity between the groups. The grouping also coincides with the signal transduction pathways used by the receptors. Thus, Group I receptors stimulate phospholipase C causing an increase in intracellular inositol phosphates and Ca2+ levels whereas both Group II and III inhibit adenylate cyclase causing a decrease in intracellular cyclic AMP levels (Bräuner-Osborne et al., 2000). Finally, receptors within the three groups also share pharmacological properties. Thus, as will be explained in greater detail in later sections, selective agonists and antagonists in most cases affect all receptors within a group. However, recently a number of ligands, in particular allosteric modulators, with specific activity for just one receptor subtype have been discovered. The extensive pharmacology of orthosteric and allosteric ligands has recently been reviewed in great detail (Schoepp et al., 1999; Bräuner-Osborne et al., 2000; 2007; Madsen et al., 2005; Ritzén et al., 2005; Gasparini and Spooren, 2007) and will thus not be reviewed in detail here.
Mechanism for agonist selectivity
The difficulties in designing subtype-specific orthosteric ligands are not surprising when taking a closer look at the orthosteric binding pocket. The 21 amino acid residues within 6 Å of L-Glu bound to the closed VFT of mGlu1 are given in Table 1 and the degree of conservation is shown in Figure 1B. Nine of these residues are identical in all eight mGlu receptors, which mainly participate in the previously discussed motifs recognizing the α-amino acid moiety and distal carboxylic acid of L-Glu. All 21 amino acid residues are identical between mGlu1 and the other Group I receptor mGlu5, only two of the 21 amino acid residues differ between the Group II receptors mGlu2 and mGlu3, and only four of the 21 amino acid residues differ between the Group III receptors mGlu4, mGlu6, mGlu7 and mGlu8 (Table 1). A large number of mutagenesis studies have investigated the importance of individual residues in the binding pocket (Table 2). Not surprisingly, these studies have pointed out the five highly conserved residues binding the α-amino acid moiety (S165, T188, D208, Y236 and D318 in mGlu1) and the two highly conserved basic residues binding the distal carboxylic acid of L-Glu (R78 and K409 in mGlu1) as being of particular importance for agonist binding (Tables 1 and 2, Figure 3). Several studies have addressed the basis for Group selectivity. Based on modelling and ligand docking, we suggested that differences in the shape and electronic environment of the binding pocket of mGlu1 and mGlu4 as the basis for selectivity of ibotenic acid for the former subtype (Hermit et al., 2004). Subsequent mutational studies showed that binding affinities of the Group I selective agonists ibotenic acid and quisqualic acid were dramatically increased at mGlu4 when three residues in the receptor concomitantly were mutated to the corresponding residues in mGlu1 (K74Y, E287G and K317R). The individual mutations had only minor effects which led to the conclusion that the subtype selectivity arises from a synergy of contributions and a complex interplay of residues shaping the binding pocket, rather than being attributable to a single specific ligand–receptor interaction. The recent X-ray crystallography structure of the mGlu3 VFT complexed with L-Glu or several Group II selective agonists has revealed a similar complex interplay of several residues providing subtype selectivity. Thus, it had previously been shown that mutation of Y150A, Y222A or R277A dramatically reduced the potency of (2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine (DCG-IV) but not (2S,1′S,2′S)-2-(carboxycyclopropyl)glycine at mGlu3 (Table 2), which led the authors to suggest that the extra carboxylic acid moiety of DCG-IV at position C3′ points towards these three residues (Yao et al., 2003). Y222 is identical in all eight mGlu receptors and can thus not contribute to the Group II selectivity of DCG-IV, whereas Y150 and R277 are indeed only present in Group II (Table 1). However, the recent X-ray crystallography structure of the mGlu3 VFT complexed with DCG-IV shows that only Y150 interacts directly with the agonist via van der Wahls contacts (Figure 3) (Muto et al., 2007). Based on this observation, it was suggested that the R227 residue played a role in supporting the rigid ligand-binding pocket, thus participating in ligand selectivity via an indirect mechanism rather than direct interaction with the ligand (Muto et al., 2007).
Table 2.
The effect on agonist potency of engineered mutations in or near the ligand-binding pocket of metabotropic glutamate receptors
| Residue | Potencya | Agonistb(assayc) | References |
|---|---|---|---|
| mGlu1 | |||
| Y74E | ↓↓↓ | Glu (IP), Quis (B) | (Sato et al., 2003) |
| R78A/L/E | ↓↓(↓) | Glu (IP), Quis (IP, B) | (Jensen et al., 2000; Sato et al., 2003) |
| S164A | = | Glu (IP, B), Quis (IP, B) | (O’Hara et al., 1993; Sato et al., 2003) |
| S165A | ↓↓(↓) | Glu (IP), Quis (IP, B) | (O’Hara et al., 1993; Sato et al., 2003) |
| S166A | = | Glu (EP) | (Kubo et al., 1998) |
| S186A | = | Glu (IP), Quis (IP, B) | (O’Hara et al., 1993; Sato et al., 2003) |
| T188A | ↓↓↓ | Glu (IP), Quis (IP,B) | (O’Hara et al., 1993; Sato et al., 2003) |
| D208A | ↓↓↓ | Glu (IP), Quis (B) | (Sato et al., 2003) |
| Y236A | ↓↓↓ | Glu (IP), Quis (B) | (Sato et al., 2003) |
| E292A | ↓↓↓ | Quis (B) | (Sato et al., 2003) |
| G293A | ↓↓↓ | Glu (IP), Quis (B) | (Sato et al., 2003) |
| D318A | ↓↓↓ | Glu (IP), Quis (B) | (Sato et al., 2003) |
| R323A | = | Glu/Quis (B) | (Sato et al., 2003) |
| K409A | = | Glu/Quis (B) | (Sato et al., 2003) |
| mGlu2 | |||
| R57A/Y | ↓↓(↓) | Glu (EP), LY354740 (EP, B) | (Malherbe et al., 2001) |
| Y144A/S/G/F | = (↓) | Glu (G, EP), LY354740 (G, EP, B) | (Malherbe et al., 2001) |
| S145A | ↓↓↓ | Glu (G), LY354740 (G, B) | (Malherbe et al., 2001) |
| D146S | = | Glu (G), LY354740 (G, B) | (Malherbe et al., 2001) |
| S148A | >↓ | Glu (G), LY354740 (G, B) | (Malherbe et al., 2001) |
| S164A | = | LY354740 (B) | (Malherbe et al., 2001) |
| A166S | = | Glu (G), LY354740 (G, B) | (Malherbe et al., 2001) |
| S167A | = | Glu (G), LY354740 (G, B) | (Malherbe et al., 2001) |
| T168A | ↓(↓↓) | Glu (G), LY354740 (G, B) | (Malherbe et al., 2001) |
| S169A | = | Glu (G), LY354740 (G, B) | (Malherbe et al., 2001) |
| R183A | ↓ | Glu (EP), LY354740 (EP, B) | (Malherbe et al., 2001) |
| D188A | ↓↓↓ | DCG-IV (B) | (Yao et al., 2003) |
| Y216A/F | ↓↓↓ | LY354740 (B) | (Malherbe et al., 2001) |
| R271A | = | LY354740 (B) | (Malherbe et al., 2001) |
| ↓↓ | DCG-IV (B) | (Yao et al., 2003) | |
| D295A/R | ↓↓↓ | Glu (EP), LY354740 (EP, B) | (Malherbe et al., 2001) |
| L300A | ↓ | DCG-IV (B) | (Yao et al., 2003) |
| K377A | ↓↓↓ | DCG-IV (B) | (Yao et al., 2003) |
| mGlu3 | |||
| R64A | ↓↓↓ | DCG-IV (C, B), L-CCG-I (C) | (Yao et al., 2003) |
| R68A | ↓↓↓ | DCG-IV (C, B), L-CCG-I (C) | (Yao et al., 2003) |
| Y150A | ↓↓(↓) | DCG-IV (C, B) | (Yao et al., 2003) |
| = | L-CCG-I (C) | (Yao et al., 2003) | |
| S151A | ↓↓↓ | DCG-IV (C, B), L-CCG-I (C) | (Yao et al., 2003) |
| S152D/H | = | LY354740 (B) | (Malherbe et al., 2001) |
| T174A | ↓↓↓ | DCG-IV (C, B), L-CCG-I (C) | (Yao et al., 2003) |
| D194A | ↓↓↓ | DCG-IV (C, B), L-CCG-I (C) | (Yao et al., 2003) |
| Y222A | ↓↓↓ | DCG-IV (C, B) | (Yao et al., 2003) |
| = | L-CCG-I (C) | (Yao et al., 2003) | |
| R277A | ↓↓(↓) | DCG-IV (C, B) | (Yao et al., 2003) |
| = | L-CCG-I (C) | (Yao et al., 2003) | |
| D301A | ↓↓↓ | DCG-IV (C, B), L-CCG-I (C) | (Yao et al., 2003) |
| Q306A | ↓↓↓ | DCG-IV (B) | (Yao et al., 2003) |
| K389A | ↓↓(↓) | DCG-IV (C, B), L-CCG-I (C) | (Yao et al., 2003) |
| mGlu4 | |||
| K74A/Y/N/Q | = | L-AP4 (B) | (Rosemond et al., 2002; Hermit et al., 2004) |
| K74Y | ↓↓↓ | L-AP4 (B) | (Rosemond et al., 2002) |
| H77Q | = | L-AP4 (B) | (Rosemond et al., 2002) |
| R78A | ↓↓↓ | L-AP4 (B) | (Hampson et al., 1999) |
| S157A | = | Glu (C), L-AP4 (B) | (Hampson et al., 1999; Frauli et al., 2007) |
| G158A | = | Glu (C), L-AP4 (B) | (Hampson et al., 1999; Frauli et al., 2007) |
| S159A | ↓↓↓ | L-AP4 (B) | (Hampson et al., 1999) |
| S160A | = | L-AP4 (B) | (Hampson et al., 1999) |
| S181A | = | L-AP4 (B) | (Hampson et al., 1999) |
| T182A | ↓↓↓ | L-AP4 (B) | (Hampson et al., 1999) |
| D202A | = | L-AP4 (B) | (Rosemond et al., 2002) |
| Y230A | = | L-AP4 (B) | (Rosemond et al., 2002) |
| R258A | = | L-AP4 (B) | (Rosemond et al., 2002) |
| N286A | = | L-AP4 (B) | (Rosemond et al., 2002) |
| E287A | ↓(↓↓) | L-AP4 (B) | (Rosemond et al., 2002; Hermit et al., 2004) |
| D312A | = | L-AP4 (B) | (Rosemond et al., 2002) |
| S313G | = | L-AP4 (B) | (Hermit et al., 2004) |
| K317A/R | = | L-AP4 (B) | (Rosemond et al., 2002; Hermit et al., 2004) |
| K405A | ↓↓↓ | L-AP4 (B) | (Rosemond et al., 2002) |
| mGlu7 | |||
| N74K | ↑ | L-AP4 (B) | (Rosemond et al., 2004) |
| mGlu8 | |||
| K71A | = | L-AP4 (B) | (Rosemond et al., 2002) |
| K71Y | ↓↓↓ | L-AP4 (B) | (Rosemond et al., 2002) |
| R75A | ↓↓↓ | L-AP4 (B) | (Rosemond et al., 2002) |
| A154S | = | Glu (C) | (Frauli et al., 2007) |
| A155S | = | Glu (C) | (Frauli et al., 2007) |
| Y227A/F | = (↓) | Glu/L-AP4 (IP) | (Bessis et al., 2002) |
| D309A | ↓↓(↓) | Glu/L-AP4 (IP) | (Bessis et al., 2002) |
| D309E | = (↓) | Glu (IP) | (Bessis et al., 2002) |
Potency of mutant compared with wild-type receptor: ↑, 10- to 50-fold increase; =, less than 10-fold difference; ↓, 10- to 50-fold decrease or reduced binding/response; ↓↓, 50- to 500-fold decrease or weak binding/response; ↓↓↓, >500-fold decrease or no binding/response.
DCG-IV, (2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine; Glu, L-glutamic acid; L-AP4, L-amino-4-phosphonobutyric acid; L-CCG-I, (2S,1'S,2'S)-2-(carboxycyclopropyl)glycine; LY354740, (+)-2-aminobicyclo-[3.1.0]-hexane-2,6-dicarboxylate; Quis, L-quisqualic acid.
B, binding; C, intracellular [Ca2+]; EP, electrophysiology; G, [35S]GTPγS binding; IP, inositol phosphate.
It is interesting to note the difference in the positions of the water molecules in the L-Glu-bound mGlu1 and mGlu3 VFTs (Figure 3). Likewise, it is interesting to note that mGlu3 accommodates the bulkier agonist DCG-IV by replacement of two water molecules by the additional carboxylic acid group rather than by repositioning of the amino acid side chains in the binding pocket (Figure 3). The higher potency of DCG-IV compared with L-Glu at mGlu3 is thus obtained by the lower cost of binding energy of the former ligand in terms of solvent entropy (Muto et al., 2007). Given the different positioning of the water molecules in mGlu1, DCG-IV cannot obtain a similar energy advantage at this subtype, which is one possible explanation for the Group II selectivity of the ligand.
Collectively, these studies show that several different molecular mechanisms play a role in subtype/group selectivity which makes structure-based ligand design very challenging.
Molecular pharmacology of the GABAB receptor
The GABAB receptor was first identified in the early eighties on the basis of pharmacological responses to the agonist baclofen and insensitivity to the GABAA antagonist bicuculline (Bowery et al., 1980; Hill and Bowery, 1981) but resisted cloning until the late nineties (Kaupmann et al., 1997). The GABAB receptor is coupled to Gαi proteins and activation causes a decrease in Ca2+ and an increase in K+ membrane conductance and inhibition of cyclic adenosine monophosphate (cAMP) formation (Bowery et al., 2002; Bettler et al., 2004). The GABAB receptor is a heterodimer consisting of the GABAB1 and GABAB2 subunits (Jones et al., 1998; Kaupmann et al., 1998; White et al., 1998; Kuner et al., 1999) of which the former contains the orthosteric GABA-binding site and the latter appears to be primarily involved in G-protein coupling (Pin et al., 2004). Interestingly, the GABAB2 subunit does contain a VFT domain but phylogenetic analysis has shown that it is unlikely to contain a ligand-binding site (Kniazeff et al., 2002). The heterodimerization is controlled by an RSRR motif in the C-terminal of GABAB1 which traps the subunit in the endoplasmatic reticulum unless it is masked by a coiled-coil interaction with the C-terminal of GABAB2 enabling the trafficking of both subunits to the plasma membrane (Pin et al., 2004). Initial cloning efforts revealed two major amino-terminal isoforms, GABAB1(a) and GABAB1(b). The former contains two amino-terminal sushi-repeats, which are protein–protein interaction motifs that are expected to serve as an extracellular targeting signal that dictates subcellular localization (Kaupmann et al., 1997).
Extensive mutational analysis of the GABAB1 VFT has revealed surprisingly few residues with substantial effects on GABA or baclofen potency (Table 3). Mutations of S246 and D471 lead to significantly lower potencies of both agonists while mutation of Y366 has a moderate effect (Galvez et al., 1999; 2000a; Jensen et al., 2001a). Ca2+ allosterically modulates the potency of GABA but not baclofen. Presumably Ca2+ interacts with S269 and stabilizes the active closed conformation of the VFT (Galvez et al., 2000b; Jensen et al., 2001a). Together with molecular modelling, these results have led to a model where the carboxylic acid of GABA binds to S246 and S269 (via Ca2+) and the amine of GABA binds to D471 (Figure 3) (Galvez et al., 2000a; Costantino et al., 2001).
Table 3.
The effect on agonist potency of engineered mutations in or near the ligand binding pocket of the GABAB1 receptor
| Residuea | Potencyb | Agonist (assayc) | References |
|---|---|---|---|
| C187A/S | = | GABA (B, IP) | (Galvez et al., 1999; 2000a) |
| Q188A | = | GABA (B) | (Galvez et al., 1999) |
| E192A | = | GABA (B) | (Galvez et al., 2000a) |
| S246A/P/T/N | ↓↓↓ | GABA/baclofen (IP, B) | (Galvez et al., 1999; 2000a; Jensen et al., 2001a) |
| S247A | = (↑) | GABA/baclofen (IP, B) | (Galvez et al., 1999; 2000b) |
| S249A | = | GABA (B) | (Galvez et al., 1999) |
| T250A | = | GABA (B) | (Galvez et al., 1999) |
| S265A | = | GABA (IP) | (Galvez et al., 2000a) |
| Y266F | = | GABA/baclofen (IP) | (Galvez et al., 2000a) |
| S268A | = | GABA/baclofen (B, IP) | (Galvez et al., 1999; 2000b) |
| S269A | = (↓) | GABA/baclofen (B, IP) | (Galvez et al., 1999; 2000b; Jensen et al., 2001a) |
| S270A | ↓ | GABA/baclofen (B, IP) | (Galvez et al., 1999; 2000a) |
| T310A | = | GABA (B) | (Galvez et al., 1999) |
| Q312A | = | GABA/baclofen (IP, B) | (Galvez et al., 1999; 2000a) |
| Q313A | = | GABA (B) | (Galvez et al., 1999) |
| T314A | = | GABA (B) | (Galvez et al., 1999) |
| T315A | = | GABA (B) | (Galvez et al., 1999) |
| E316A | = | GABA (B) | (Galvez et al., 1999) |
| F365A | = | GABA/baclofen (IP, B) | (Galvez et al., 2000a) |
| Y366A | ↓(↓) | GABA/baclofen (IP, B) | (Galvez et al., 2000a) |
| F367A | = | GABA/baclofen (IP, B) | (Galvez et al., 2000a) |
| E458A | = | GABA (B) | (Galvez et al., 1999) |
| E459A | = | GABA (B) | (Galvez et al., 1999) |
| T460A | = | GABA (B) | (Galvez et al., 1999) |
| F463A | = | GABA/baclofen (IP) | (Galvez et al., 2000a) |
| Q464A | = | GABA (B) | (Galvez et al., 1999) |
| E465A | = (↓) | GABA (B, IP) | (Galvez et al., 2000a) |
| Y470A/F | = | GABA (IP) | (Galvez et al., 2000a) |
| D471A/E | ↓↓↓ | GABA/baclofen (IP, B) | (Galvez et al., 2000a) |
Numbering according to the GABAB1a variant.
Potency of mutant compared to wild-type receptor: ↑, 10- to 50-fold increase; =, less than 10-fold difference; ↓, 10- to 50-fold decrease or reduced binding/response; ↓↓, 50- to 500-fold decrease or weak binding/response; ↓↓↓, >500-fold decrease or no binding/response.
B, binding; GABAB, γ-aminobutyric acid type B; IP, inositol phosphate.
Despite plenty of in vitro and in vivo evidence of distinct pharmacological GABAB receptor subtypes, the GABAB1(a) and GABAB1(b) isoforms display no differences in binding or functional experiments with native (Malitschek et al., 1998) or recombinant expression (Kaupmann et al., 1997; 1998; Bräuner-Osborne and Krogsgaard-Larsen, 1999). Together with the group of Bettler, we developed isoform-specific GABAB1(a) and GABAB1(b) knockout mice which have shown that the two isoforms display distinct expression patterns at the synaptic level via which the isoforms control different effector systems and physiological functions (Pérez-Garci et al., 2006; Vigot et al., 2006). It thus seems evident that the pharmacological subtypes arise from spatial control of expression, rather than a multitude of receptor subtypes.
Molecular pharmacology of the CaR
Orthosteric ligands
As the name implies, the CaR functions to sense Ca2+ and does so by responding to millimolar concentrations of Ca2+ present in the extracellular fluid, and hereby regulates the release of hormones important for maintaining calcium homeostasis of the organism [for reviews see (Tfelt-Hansen and Brown, 2005) and (Brown, 2007)]. In addition to Ca2+, CaR is also activated by other inorganic cations in the millimolar range, including Mg2+, Ba2+ and Sr2+, as well as Gd3+ in the micromolar range (Brown et al., 1993; Ruat et al., 1996; Coulombe et al., 2004). Both Ca2+ and Mg2+ exhibit cooperativity in binding, yielding Hill coefficients aroung 3–5 (Bai et al., 1996; Ruat et al., 1996; Bräuner-Osborne et al., 1999), which from a physiological point of view allows for differentiation of the CaR response by only minute fluctuations in agonist concentrations.
Similar to other family C receptors, CaR is also sensitive to L-α-amino acids. This is not surprising given the high degree of conservation of key residues known to be important in binding of L-Glu in mGlu1 (Table 1). However, whereas other family C receptors (mGlu, GABAB and GPRC6A receptors) are directly activated by amino acids and positively modulated by calcium, CaR operates in a reciprocal fashion, being directly activated by Ca2+ and positively modulated by amino acids. Furthermore, while mGlu receptors respond only to L-Glu (Frauli et al., 2006), the group of CaR, GPRC6A and T1R1+T1R3 are less selective and respond to several classes of amino acids. In fact, each of these receptors prefer different classes of amino acids and are able to cover a wide range of amino acid selectivities in concert (Figure 4), suggested to be of physiological relevance for instance in the gut (Conigrave and Brown, 2006; Conigrave et al., 2008). At CaR, the most potent are the aromatic amino acids L-Phe and L-Trp, followed by aliphatic and polar amino acids. The least potent are the branched-chain, basic and sulfur-containing amino acids (Conigrave et al., 2000). Specifically for CaR, the potential physiological relevance of the positive modulation of receptor activity by amino acids has been demonstrated in the parathyroid and the intestinal/digestive system (Conigrave et al., 2004; Busque et al., 2005; Hira et al., 2008), and suggested in several other tissues (Conigrave et al., 2007; 2008).
Figure 4.

L-Amino acid selectivity profiles at CaR, GPRC6A and the T1R1+T1R3 heterodimer. Amino acids are grouped according to side chain charge and polarity. Data have been normalized to allow for comparison of relative amino acid preferences. The profile for CaR was generated by normalizing reported EC50 values from seven amino acids (measured in human parathyroid cells in the presence of 2 mmol·L−1 Ca2+) to the response of L-Trp (set to 100%). Amino acids marked with asterices (*) were not included in the original study. The profile for GPRC6A is based on reported EC50 values from mouse GPRC6A measured in the presence of 1 mmol·L−1 Ca2+ and 1 mmol·L−1 Mg2+ (Christiansen et al., 2007) and here normalized to the L-Lys response (set to 100%). T1R1+T1R3 data originally in the form of ‘number of responsive cells’ measured in the presence of 2.5 mmol·L−1 IMP (Nelson et al., 2001) were converted to percentage normalized response by calculating the response relative to that of L-Cys (set to 100%). The symbol (#) denotes that L-Tyr was not tested at T1R1+T1R3 (due to insolubility). CaR, calcium-sensing receptor; GPRC6A, G-protein coupled receptor family C, group 6, subtype A; T1R, taste1 receptor.
Other positive modulators at CaR include polyvalent molecules (including spermine, spermidine), β-amyloid peptides, several aminoglycoside antibiotics (Ruat et al., 1996; Quinn et al., 1997; Ye et al., 1997; Brown and MacLeod, 2001) along with increases in pH and ionic strength (Quinn et al., 1998; Quinn et al., 2004). Furthermore, it was recently found that the binding pocket of CaR (and the related goldfish 5.24 receptor) is in fact large enough to accommodate small peptides such as glutathione (GSH), nicely illustrated by the binding of [3H]GSH to the soluble VFT domain of CaR and the ability of GSH to potentiate the Ca2+ response (Wang et al., 2006). In addition to allosteric modulation in the VFT domain, CaR exhibits an allosteric activator site for calcimimetics and an allosteric inhibitory site for calcilytics in the 7TM domain of the receptor (Nemeth et al., 1998; 2001).
The orthosteric binding site(s)
Based on studies using chimeric receptors, it has been firmly established that Ca2+ binding takes place in the CaR VFT domain (Bräuner-Osborne et al., 1999; Hammerland et al., 1999), although reports also point to the existence of separate Ca2+/Gd3+-binding sites in the 7TM domain (Hammerland et al., 1999; Hu et al., 2002; 2005; Ray and Northup, 2002). Focusing on the VFT domain-binding sites, mutagenesis studies have identified a number of residues important for Ca2+-activation (Table 4); however, as no crystal structure of CaR exists and as no binding assay is available to confirm if these residues directly contact the ligand, the exact residues involved in Ca2+ binding have not been unequivocally demonstrated. As given in Table 4, the five residues S147, S170, D190, Y218 and E297, conserved as the basic α-amino acid recognizing motif in family C GPCRs, yield significantly decreased sensitivity to Ca2+ (and other cations) when mutated, suggesting that these residues are involved in cation binding (Bai et al., 1996; Pearce et al., 1996; Bräuner-Osborne et al., 1999; Hauache et al., 2000; Zhang et al., 2002; Mun et al., 2005). From homology models of CaR using the mGlu1 crystal structure as template, these residues have as well been predicted to be part of the Ca2+-binding site. In one of the more recent modelling studies, Silve et al predicted and validated a Ca2+-binding site in the cleft of the two lobes, lying adjacent to the amino acid-binding site but being of smaller dimensions. In addition to the five participating residues already mentioned, they identified the three residues Q193, F270 and S296 to be part of the cation-binding site (Silve et al., 2005). More recently, another study homology-modelled CaR and identified three distinct Ca2+-binding sites, which correlates well with the experimentally determined Hill coefficient of 3–5. One of these corresponded with the one reported by Silve et al. Ligand binding to the two other predicted sites was validated by FRET-based methods by expressing the relevant sequences (up to 30 amino acids long) in scaffolding proteins, hereby demonstrating two novel cation-binding sites centred around E378 and E398 (Huang et al., 2007).
Table 4.
The effect of mutations in the predicted Ca2+ and/or L-amino acid-binding sites on the potency and efficacy of Ca2+ on the CaR
| CaR | Ca2+sensitivity | Amino acid sensitivity | ||||||
|---|---|---|---|---|---|---|---|---|
| Residue no. | Potency | Maximal response | Other observations | References | Ca2+potency | Ca2+max response | Other observations | References |
| R66C/H | ↓ | ↓ | Reduced Gd3+ response naturally occurring inactivating mutations (FHH/NSHPT) | (Bai et al., 1996; Pidasheva et al., 2006) | ||||
| T145A | = | = | (Mun et al., 2005) | (+) | NE | Mutation affects L/D-aa selectivity | (Mun et al., 2005) | |
| S147A | ↓ | ↓ | (Bräuner-Osborne et al., 1999; Zhang et al., 2002; Mun et al., 2005) | + | + | (Zhang et al., 2002; Mun et al., 2005) | ||
| S169A | = | = | (Bräuner-Osborne et al., 1999; Zhang et al., 2002) | + | NE | (Zhang et al., 2002) | ||
| S169T | ↓=/ | ↓=/ | Lower sensitivity in Ca2+ mobilization/unchanged sensitivity in ERK assays | (Lee et al., 2007) | +/+ | +/+ | Enhanced sensitivity in both Ca2+ mobilization/ERK assays | (Lee et al., 2007) |
| S170A | ↓ | ↓ | (Bräuner-Osborne et al., 1999; Zhang et al., 2002; Mun et al., 2005) | NE | (+) | (Zhang et al., 2002; Mun et al., 2005) | ||
| S170T | = | = | (Mun et al., 2005) | (+) | NE | (Mun et al., 2005) | ||
| T145A/S170T | = | = | (Mun et al., 2005) | NE | NE | (Mun et al., 2005) | ||
| S171A | (↓) | ↑ | (Bräuner-Osborne et al., 1999; Zhang et al., 2002 | |||||
| S169–171A | ↓ | ↓ | (Mun et al., 2005) | NE | NE | (Mun et al., 2005) | ||
| D190A/K | ↓ | ↓ | (Hauache et al., 2000; Zhang et al., 2002) | + | + | Zhang et al., 2002) | ||
| Q193A | ↓ | = | (Silve et al., 2005) | |||||
| Y218A/S/F | ↓ | ↓ | Mun et al., 2005; Zhang et al., 2002) | + | + | (Zhang et al., 2002; Mun et al., 2005) | ||
| Y218S | ↓ | ↓ | Only mild attenuation of Gd3+ response | (Pearce et al., 1996; Zhang et al., 2002) | + | + | ||
| Naturally occurring inactivating mutation (NSHPT) | ||||||||
| E224I E228/229I | (←) | ↓ | (Huang et al., 2007) | |||||
| F270A | ↓ | ↓ | (Silve et al., 2005) | |||||
| S296A | ↑ | ↑ | (Silve et al., 2005) | |||||
| E297K/I/Q | ↓ | ↓ | Reduced Gd3+ response naturally occurring inactivating mutation (FHH/NSHPT) | (Bai et al., 1996; Hauache et al., 2000; Zhang et al., 2002; Mun et al., 2005; Huang et al., 2007) | + | + | (Zhang et al., 2002; Mun et al., 2005) | |
| E297D | ↑ | = | Naturally occurring activating mutation (ADH) | (Silve et al., 2005 | ||||
| E378/379I | ↑ | ↑ | (Huang et al., 2007) | |||||
| E398/399I | ↓ | ↓ | (Huang et al., 2007) | |||||
ADH: autosomal dominant hypocalcaemia; CaR, calcium-sensing receptor; ERK, extra cellular signal regulated kinase; FHH: familial hypercalciuric hypocalcaemia; NE: no effect; NSPHT: neonatal severe hyperparathyroidism.
Based on the observation that the five residues known to bind the α-amino acid moiety of L-Glu in mGlu1 are conserved in CaR (Table 1), it is anticipated that an analogous amino acid-binding site exists in CaR. Mounting evidence for the location of the amino acid-binding site to the VFT domain has come from chimeric receptor studies (Mun et al., 2004), from the finding that L-Phe and the allosteric modulator, NPS R-467, known to bind in the 7TM domain of CaR, act synergistically and hence at distinct binding sites (Zhang et al., 2002), as well as from site-directed mutageneses (collected in Table 4). It has, however, been inherently difficult to pinpoint residues specifically involved in amino acid binding and activation, as the amino acids require the presence of Ca2+ to work and many of the examined mutations simultaneously reduce Ca2+ sensitivity. Mun et al. identified two mutations, T145A and S170T that specifically impair amino acid-sensing while leaving Ca2+ sensing intact (Mun et al., 2005). Others have also identified the three serines S169–171 as being important for amino acid binding (Zhang et al., 2002; Lee et al., 2007). Obviously, as CaR prefers aromatic amino acids and is not sensitive to L-Glu, the residues involved in binding of the distal end of L-Glu are not conserved in CaR (Table 1), exemplified by the lack of conservation of residues corresponding to the mGlu1 residues Y74 and R323 to CaR (Silve et al., 2005). It remains to be investigated which residues participate in binding of the aromatic moieties of L-Phe/L-Trp in the CaR VFT domain.
Molecular pharmacology of GPRC6A
GPRC6A was originally identified using bioinformatics based on sequence identity to known family C GPCRs and has subsequently been molecularly cloned from human, mouse and rat (Wellendorph and Bräuner-Osborne, 2004; Kuang et al., 2005; Wellendorph et al., 2005; 2007). As illustrated in the phylogenetic tree (Figure 1A), GPRC6A is most closely related to the goldfish 5.24 receptor, which based on ligand preferences for basic amino acids (Speca et al., 1999; Christiansen et al., 2006a) is believed to be the GPRC6A orthologue in this species. Of the human family C receptors, CaR is the closest homologue.
Orthosteric ligands
GPRC6A is stereoselectively activated by natural L-α-amino acids, preferentially basic amino acids L-Arg, L-Lys and L-ornithine (L-Orn), but also small and polar amino acids, whereas aromatic amino acids are inactive (Kuang et al., 2005; Wellendorph et al., 2005; 2007; Christiansen et al., 2007). Furthermore, we have identified a number of synthetic derivatives of L-Arg, L-Lys and L-Orn, otherwise known to be regulators of the nitric oxide synthase and arginase isoenzymes, to be GPRC6A agonists (Christiansen et al., 2006b; Hrabák, 2006). This sensitivity profile is complementary to CaR, and both GPRC6A and CaR also differ from T1R1+T1R3 with respect to amino acid preferences (Figure 4) (Conigrave and Brown, 2006; Bräuner-Osborne et al., 2007). Using heterologous expression systems, we and others have found that the L-α-amino acid response of GPRC6A is augmented by divalent cations Ca2+ and Mg2+ in physiological relevant concentrations (Kuang et al., 2005; Christiansen et al., 2007; Wellendorph et al., 2007) and one report has even demonstrated a direct activation of GPRC6A by Ca2+ (Pi et al., 2005). GPRC6A thus bears the general characteristic of a family C receptor in having a dual responsiveness to Ca2+ and amino acids. To date, no antagonists have been identified at GPRC6A.
The orthosteric binding site
So far, only few studies have addressed the molecular basis of amino acid activation of GPRC6A. We have generated a homology model of the human GPRC6A with L-Lys docked into the predicted binding pocket based on the mGlu1 crystal as template, and constructed under the assumption that the binding site orientation of L-Lys in GPRC6A substantially resembles that of L-Glu in the mGlu receptors (Wellendorph et al., 2005) – an assumption shared by Kuang et al. in their model of L-Lys bound to the goldfish 5.24 receptor (Kuang et al., 2003). From this homology modelling and from alignments with other family C GPCRs, the residues known to interact with the α-amino acid moiety of L-Glu in mGlu1 are clearly conserved in GPRC6A (Table 1). The modelled L-α-amino acid recognition site was validated as mutations S149A and T172A (corresponding to S165 and T188 in mGlu1) completely obliterated receptor activity in response to amino acids (Wellendorph et al., 2005). Importance of these residues is substantiated by mutagenesis studies conducted on the homologous goldfish receptor 5.24, in that mutation of all the five highly conserved residues known to bind to the α-amino acid moiety (Table 1) also dramatically disturbs activity of this receptor (Kuang et al., 2003; Luu et al., 2004). Furthermore, in the 5.24 receptor, two acidic residues (D388 and E47) have been confirmed to be directly involved in binding of the distal basic guanidinium group of L-Arg. The precise environment of the distal end of the GPRC6A-binding pocket is less clear and the residue(s) responsible for binding the positively charged distal end of the amino acid have not been identified. The equivalent of D388 is not conserved to GPRC6A and in general the sequences are quite divergent in this region. Possibly, this could reflect the fact that GPRC6A is less selective for basic amino acids than 5.24. Based on our homology model, residues E67, E170, D401 and D403 are potential candidates for interacting with the distal end of positively charged ligands (Wellendorph et al., 2005) but remain to be investigated by site-directed mutagenesis. Finally, alternative scenarios for the amino acid-binding pocket in GPRC6A might be relevant to consider, such as the use of the lysine-arginine-ornithine-binding protein (Kang et al., 1991; Oh et al., 1993) as template, in which L-Lys binds in a more folded conformation than L-Glu in mGlu1 (Wellendorph et al., 2005).
Molecular pharmacology of the T1R1+T1R3 taste receptor
The T1R class of GPCRs consists of three subunits: T1R1, T1R2 and T1R3 that all carry the typical family C characteristics of a large VFT domain, a CRD and a 7TM domain. T1R subunits are selectively expressed in taste buds (Hoon et al., 1999) where they combine to form heterodimeric taste receptors, responding to either sweet stimuli (T1R2+T1R3) or amino acids/umami taste (T1R1+T1R3) (Nelson et al., 2001; 2002; Li et al., 2002). In order to form functional receptors, the individual subunits T1R1 and T1R2 depend on co-expression and dimerization with T1R3, thus comparable to the GABAB receptor in being obligatory heterodimers. This functional prerequisite is underlined by studies using chimeric receptors and knockout animals (Zhao et al., 2003; Xu et al., 2004). Given the topic of this review, the T1R1+T1R3 amino acid receptor is highlighted. Being an L-amino acid-sensing receptor, the conservation of the five key residues involved in binding of the α-amino acid moiety of L-Glu in mGlu1 is not unexpected. The signalling of T1R1+T1R3 is augmented dramatically by 5′-inosine monophospate (IMP), which is a hallmark of umami taste (Yamaguchi, 1991). Indeed, in the presence of IMP, the receptor responds to all natural L-amino acids but L-Trp (Figure 4), as well as to the synthetic L-Glu analogue L-AP4 (Li et al., 2002; Nelson et al., 2002). Because L-Glu and IMP have no effect on the T1R2+T1R3 sweet taste receptor, it is inferred that these bind to the T1R1 subunit (Li et al., 2002; Xu et al., 2004).
Whereas the mGlu1 crystal has formed the basis for homology models of T1R2 and T1R3 (reviewed by Cui et al., 2006), no model exists for T1R1, which is the subunit believed to harbour the predicted amino acid-binding pocket. Consequently and additionally, no mutations in the presumed binding pocket have been reported and details about the molecular recognition of amino acids in the umami receptor remain to be addressed. From work by Silve et al., it seems plausible that T1R1 and/or T1R3 also contain a Ca2+-binding site as residues known to involve Ca2+ binding in CaR are conserved to these receptor subunits (Silve et al., 2005). So far, no molecular evidence has emerged to support this view, but recently the involvement of T1R3 in calcium-magnesium taste has been implied based on taste studies in vivo (Tordoff et al., 2008), also proposing that T1R3 could have a dimerization partner with Ca2+-sensing properties such as CaR. As delineated above, another potential partner for dimerization is GPRC6A, which intriguingly is also expressed in taste buds (Wellendorph et al., 2007).
Conclusion and outlook
As we have outlined in the present review, the last decade has increased our knowledge of how amino acids bind to the VFTs of family C receptors dramatically. These insights have provided a basis to understand subtype selectivity of mGlu receptors and the initial events in receptor activation. Interestingly, whereas mGlu and GABAB receptors are exclusively activated by one endogenous agonist, CaR, T1R1 and GPRC6A are promiscuously activated/modulated by a range of L-α-amino acids (Figure 4). The molecular mechanism of this promiscuity and the physiological importance of the individual amino acids still remains to be shown. Also the molecular mechanism of cation activation/modulation of family C remains to be fully uncovered. Structural and biophysical studies have unravelled the initial events in activation of the dimeric receptor by closing of the VFT around the agonist, but how this is translated into activation of the 7TM domains is still a mystery. Is it contraction of the two 7TM domains in the dimer, a conformational change in the individual 7TM domains induced via the rigid CRD, or a third mechanism? These and other important unsolved questions ensure that the next decade of family C receptor research will be exciting to follow.
Acknowledgments
We would like to thank all present and past members of our group working on family C receptors for their contributions. Currently, our work on family C receptors is supported by the Danish Medical Research Council, the Drug Research Academy, the Villum Kann Rasmussen Foundation, Simon Foughner Hartmanns Familiefond, the Aase and Ejner Danielsen Foundation and the Alfred Benzon Foundation.
Glossary
Abbreviations:
- 7TM
seven transmembrane
- CaR
calcium-sensing receptor
- CRD
cysteine-rich domain
- DCG-IV
(2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine
- FRET
fluorescence resonance energy transfer
- GABAB
γ-aminobutyric acid type B
- GPCR
G-protein-coupled receptor
- GPRC6A
G-protein-coupled receptor family C, group 6, subtype A
- IMP
5′-inosine monophospate, L-CCG-I, (2S,1'S,2'S)-2-(carboxycyclopropyl)glycine
- L-Glu
(S)-glutamic acid; mGlu, metabotropic glutamate; (S)-MCPG, (S)-(α)-methyl-4-carboxyphenylglycine
- T1R
taste1 receptor
- VFT
Venus flytrap
Conflict of interest
None.
References
- Abe H, Tateyama M, Kubo Y. Functional identification of Gd3+ binding site of metabotropic glutamate receptor 1a. FEBS Lett. 2003;545:233–238. doi: 10.1016/s0014-5793(03)00569-6. [DOI] [PubMed] [Google Scholar]
- Acher FC, Bertrand H-O. Amino acid recognition by Venus flytrap domains is encoded in an 8-residue motif. Biopolymers. 2005;80:357–366. doi: 10.1002/bip.20229. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. (3rd edn) 2008;153(Suppl. 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai M, Quinn S, Trivedi S, Kifor O, Pearce SHS, Pollak MR, et al. Expression and characterization of inactivating and activating mutations in the human Ca2+o-sensing receptor. J Biol Chem. 1996;271:19537–19545. doi: 10.1074/jbc.271.32.19537. [DOI] [PubMed] [Google Scholar]
- Bai M, Trivedi S, Kifor O, Quinn SJ, Brown EM. Intermolecular interactions between dimeric calcium-sensing receptor monomers are important for its normal function. Proc Natl Acad Sci USA. 1999;96:2834–2839. doi: 10.1073/pnas.96.6.2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessis AS, Rondard P, Gaven F, Brabet I, Triballeau N, Prézeau L, et al. Closure of the Venus flytrap module of mGlu8 receptor and the activation process: Insights from mutations converting antagonists into agonists. Proc Natl Acad Sci USA. 2002;99:11097–11102. doi: 10.1073/pnas.162138699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettler B, Kaupmann K, Mosbacher J, Gassmann M. Molecular structure and physiological functions of GABAB receptors. Physiol Rev. 2004;84:835–867. doi: 10.1152/physrev.00036.2003. [DOI] [PubMed] [Google Scholar]
- Bhave G, Nadin BM, Brasier DJ, Glauner KS, Shah RD, Heinemann SF, et al. Membrane topology of a metabotropic glutamate receptor. J Biol Chem. 2003;278:30294–30301. doi: 10.1074/jbc.M303258200. [DOI] [PubMed] [Google Scholar]
- Bjarnadóttir TK, Fredriksson R, Schiöth HB. The gene repertoire and the common evolutionary history of glutamate, pheromone (V2R), taste(1) and other related G protein-coupled receptors. Gene. 2005;362:70–84. doi: 10.1016/j.gene.2005.07.029. [DOI] [PubMed] [Google Scholar]
- Bjarnadóttir TK, Gloriam DE, Hellstrand SH, Kristiansson H, Fredriksson R, Schiöth HB. Comprehensive repertoire and phylogenetic analysis of the G protein-coupled receptors in human and mouse. Genomics. 2006;88:263–273. doi: 10.1016/j.ygeno.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Bowery NG, Hill DR, Hudson AL, Doble A, Middlemiss DN, Shaw J, et al. Baclofen decreases neurotransmitter release in the mammalian CNS by an action at a novel GABA receptor. Nature. 1980;283:92–94. doi: 10.1038/283092a0. [DOI] [PubMed] [Google Scholar]
- Bowery NG, Bettler B, Froestl W, Gallagher JP, Marshall F, Raiteri M, et al. International Union of Pharmacology. XXXIII. Mammalian γ-aminobutyric acidB receptors: structure and function. Pharmacol Rev. 2002;54:247–264. doi: 10.1124/pr.54.2.247. [DOI] [PubMed] [Google Scholar]
- Bräuner-Osborne H, Krogsgaard-Larsen P. Functional pharmacology of cloned heterodimeric GABAB receptors expressed in mammalian cells. Br J Pharmacol. 1999;128:1370–1374. doi: 10.1038/sj.bjp.0702914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bräuner-Osborne H, Krogsgaard-Larsen P. Sequence and expression pattern of a novel human orphan G-protein coupled receptor GPRC5B, a family C receptor with a short amino terminal domain. Genomics. 2000;65:121–128. doi: 10.1006/geno.2000.6164. [DOI] [PubMed] [Google Scholar]
- Bräuner-Osborne H, Jensen AA, Sheppard PO, O’Hara P, Krogsgaard-Larsen P. The agonist-binding domain of the calcium-sensing receptor is located at the amino-terminal domain. J Biol Chem. 1999;274:18382–18386. doi: 10.1074/jbc.274.26.18382. [DOI] [PubMed] [Google Scholar]
- Bräuner-Osborne H, Egebjerg J, Nielsen E, Madsen U, Krogsgaard-Larsen P. Ligands for glutmate receptors: Design and therapeutic prospects. J Med Chem. 2000;43:2609–2645. doi: 10.1021/jm000007r. [DOI] [PubMed] [Google Scholar]
- Bräuner-Osborne H, Jensen AA, Sheppard PO, Brodin B, Krogsgaard-Larsen P, O’Hara P. Cloning and characterization of a human orphan family C G-protein coupled receptor GPRC5D. Biochim Biophys Acta. 2001;1518:237–248. doi: 10.1016/s0167-4781(01)00197-x. [DOI] [PubMed] [Google Scholar]
- Bräuner-Osborne H, Wellendorph P, Jensen AA. Structure, pharmacology and therapeutic prospects of family C G-protein coupled receptors. Curr Drug Targets. 2007;8:169–184. doi: 10.2174/138945007779315614. [DOI] [PubMed] [Google Scholar]
- Brock C, Oueslati N, Soler S, Boudier L, Rondard P, Pin J-P. Activation of a dimeric metabotropic glutamate receptor by intersubunit rearrangement. J Biol Chem. 2007;282:33000–33008. doi: 10.1074/jbc.M702542200. [DOI] [PubMed] [Google Scholar]
- Brown EM. Clinical lessons from the calcium-sensing receptor. Nat Clin Pract Endocrinol Metab. 2007;3:122–133. doi: 10.1038/ncpendmet0388. [DOI] [PubMed] [Google Scholar]
- Brown EM, MacLeod RJ. Extracellular calcium sensing and extracellular calcium signaling. Physiol Rev. 2001;81:239–297. doi: 10.1152/physrev.2001.81.1.239. [DOI] [PubMed] [Google Scholar]
- Brown EM, Gamba G, Riccardi D, Lombardi M, Butters R, Kifor O, et al. Cloning and characterization of an extracellular Ca2+-sensing receptor from bovine parathyroid. Nature. 1993;366:575–580. doi: 10.1038/366575a0. [DOI] [PubMed] [Google Scholar]
- Busque SM, Kerstetter JE, Geibel JP, Insogna K. L-type amino acids stimulate gastric acid secretion by activation of the calcium-sensing receptor in parietal cells. Am J Physiol. 2005;289:G664–G669. doi: 10.1152/ajpgi.00096.2005. [DOI] [PubMed] [Google Scholar]
- Calver AR, Michalovich D, Testa TT, Robbins MJ, Jaillard C, Hill J, et al. Molecular cloning and characterisation of a novel GABAB-related G-protein coupled receptor. Mol Brain Res. 2003;110:305–317. doi: 10.1016/s0169-328x(02)00662-9. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Lotan R. Molecular cloning and characterization of a novel retinoic acid-inducible gene that encodes a putative G protein-coupled receptor. J Biol Chem. 1998;273:35008–35015. doi: 10.1074/jbc.273.52.35008. [DOI] [PubMed] [Google Scholar]
- Christiansen B, Wellendorph P, Bräuner-Osborne H. Activity of L-α-amino acids at the promiscuous goldfish odorant receptor 5.24. Eur J Pharmacol. 2006a;536:98–101. doi: 10.1016/j.ejphar.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Christiansen B, Wellendorph P, Bräuner-Osborne H. Known regulators of nitric oxide synthase and arginase are agonists at the human G-protein-coupled receptor GPRC6A. Br J Pharmacol. 2006b;147:855–863. doi: 10.1038/sj.bjp.0706682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen B, Hansen KB, Wellendorph P, Bräuner-Osborne H. Pharmacological characterization of mouse GPRC6A, an L-α-amino acid receptor with ability to sense divalent cations. Br J Pharmacol. 2007;150:798–807. doi: 10.1038/sj.bjp.0707121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conigrave AD, Brown EM. Taste receptors in the gastrointestinal tract. II. L-amino acid sensing by calcium-sensing receptors: implications for GI physiology. Am J Physiol. 2006;291:G753–761. doi: 10.1152/ajpgi.00189.2006. [DOI] [PubMed] [Google Scholar]
- Conigrave AD, Quinn SJ, Brown EM. L-amino acid sensing by the extracellular Ca2+-sensing receptor. Proc Natl Acad Sci USA. 2000;97:4814–4819. doi: 10.1073/pnas.97.9.4814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conigrave AD, Mun HC, Delbridge L, Quinn SJ, Wilkinson M, Brown EM. L-amino acids regulate parathyroid hormone secretion. J Biol Chem. 2004;279:38151–38159. doi: 10.1074/jbc.M406373200. [DOI] [PubMed] [Google Scholar]
- Conigrave AD, Mun HC, Brennan SC. Physiological significance of L-amino acid sensing by extracellular Ca2+-sensing receptors. Biochem Soc Trans. 2007;35:1195–1198. doi: 10.1042/BST0351195. [DOI] [PubMed] [Google Scholar]
- Conigrave AD, Brown EM, Rizzoli R. Dietary protein and bone health: Roles of amino acid-sensing receptors in the control of calcium metabolism and bone homeostasis. Annu Rev Nutr. 2008;28:131–155. doi: 10.1146/annurev.nutr.28.061807.155328. [DOI] [PubMed] [Google Scholar]
- Costantino G, Macchiarulo A, Guadix AE, Pellicciari R. QSAR and molecular modeling studies of baclofen analogues as GABAB agonists. Insights into the role of the aromatic moiety in GABAB binding and activation. J Med Chem. 2001;44:1827–1832. doi: 10.1021/jm0100133. [DOI] [PubMed] [Google Scholar]
- Coulombe J, Faure H, Robin B, Ruat M. In vitro effects of strontium ranelate on the extracellular calcium-sensing receptor. Biochem Biophys Res Commun. 2004;323:1184–1190. doi: 10.1016/j.bbrc.2004.08.209. [DOI] [PubMed] [Google Scholar]
- Cui M, Jiang P, Maillet E, Max M, Margolskee RF, Osman R. The heterodimeric sweet taste receptor has multiple potential ligand binding sites. Curr Pharm Des. 2006;12:4591–4600. doi: 10.2174/138161206779010350. [DOI] [PubMed] [Google Scholar]
- Fan GF, Ray K, Zhao XM, Goldsmith PK, Spiegel AM. Mutational analysis of the cysteines in the extracellular domain of the human Ca2+ receptor: effects on cell surface expression, dimerization and signal transduction. FEBS Lett. 1998;436:353–356. doi: 10.1016/s0014-5793(98)01165-x. [DOI] [PubMed] [Google Scholar]
- Frauli M, Hubert N, Schann S, Triballeau N, Bertrand H-O, Acher F, Neuville P, Pin J-P, Prézeau L. Amino-pyrrolidine tricarboxylic acids give new insight into group III metabotropic glutamate receptor activation mechanism. Molecular Pharmacology. 2007;71:704–712. doi: 10.1124/mol.106.030254. [DOI] [PubMed] [Google Scholar]
- Frauli M, Neuville P, Vol C, Pin J-P, Prezéau L. Among the twenty classical L-amino acids, only glutamate directly activates metabotropic glutamate receptors. Neuropharmacology. 2006;50:245–253. doi: 10.1016/j.neuropharm.2005.09.016. [DOI] [PubMed] [Google Scholar]
- Galvez T, Parmentier ML, Joly C, Malitschek B, Kaupmann K, Kuhn R, et al. Mutagenesis and modeling of the GABAB receptor extracellular domain support a Venus flytrap mechanism for ligand binding. J Biol Chem. 1999;274:13362–13369. doi: 10.1074/jbc.274.19.13362. [DOI] [PubMed] [Google Scholar]
- Galvez T, Prézeau L, Milioti G, Franek M, Joly C, Froestl W, et al. Mapping the agonist-binding site of GABAB type 1 subunit sheds light on the activation process of GABAB receptors. J Biol Chem. 2000a;275:41166–41174. doi: 10.1074/jbc.M007848200. [DOI] [PubMed] [Google Scholar]
- Galvez T, Urwyler S, Prézeau L, Mosbacher J, Joly C, Malitschek B, et al. Ca2+ requirement for high-affinity γ-aminobutyric acid (GABA) binding at GABAB receptors: involvement of serine 269 of the GABABR1 subunit. Mol Pharmacol. 2000b;57:419–426. doi: 10.1124/mol.57.3.419. [DOI] [PubMed] [Google Scholar]
- Gasparini F, Spooren W. Allosteric modulators for mGlu receptors. Curr Neuropharmacology. 2007;5:187–194. doi: 10.2174/157015907781695900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorodkin J, Heyer LJ, Brunak S, Stormo GD. Displaying the information contents of structural RNA alignments: the structure logos. Comput Appl Biosci. 1997;13:583–586. doi: 10.1093/bioinformatics/13.6.583. [DOI] [PubMed] [Google Scholar]
- Hammerland LG, Krapcho KJ, Garrett JE, Alasti N, Hung BC, Simin RT, et al. Domains determining ligand specificity for Ca2+ receptors. Mol Pharmacol. 1999;55:642–648. [PubMed] [Google Scholar]
- Hampson DR, Huang X-P, Pekhletski R, Peltekova V, Hornby G, Thomsen C, Thøgersen H. Probing the ligand-binding domain of the mGluR4 subtype of metabotropic glutamate receptor. J Biol Chem. 1999;274:33488–33495. doi: 10.1074/jbc.274.47.33488. [DOI] [PubMed] [Google Scholar]
- Hauache OM, Hu J, Ray K, Spiegel AM. Functional interactions between the extracellular domain and the seven-transmembrane domain in Ca2+ receptor activation. Endocrine. 2000;13:63–70. doi: 10.1385/ENDO:13:1:63. [DOI] [PubMed] [Google Scholar]
- Hermit MB, Greenwood JR, Nielsen B, Bunch L, Jørgensen CG, Vestergaard HT, et al. Ibotenic acid and thioibotenic acid: a remarkable difference in activity at group III metabotropic glutamate receptors. Eur J Pharmacol. 2004;486:241–250. doi: 10.1016/j.ejphar.2003.12.033. [DOI] [PubMed] [Google Scholar]
- Hill DR, Bowery NG. 3H-baclofen and 3H-GABA bind to bicuculline-insensitive GABAB sites in rat brain. Nature. 1981;290:149–152. doi: 10.1038/290149a0. [DOI] [PubMed] [Google Scholar]
- Hira T, Nakajima S, Eto Y, Hara H. Calcium-sensing receptor mediates phenylalanine-induced cholecystokinin secretion in enteroendocrine STC-1 cells. FEBS J. 2008. [DOI] [PubMed]
- Hoon MA, Adler E, Lindemeier J, Battey JF, Ryba NJ, Zuker CS. Putative mammalian taste receptors: a class of taste-specific GPCRs with distinct topographic selectivity. Cell. 1999;96:541–551. doi: 10.1016/s0092-8674(00)80658-3. [DOI] [PubMed] [Google Scholar]
- Hrabák A. Common ligands of G-protein-coupled receptors and arginine-utilizing enzymes. Br J Pharmacol. 2006;147:835–837. doi: 10.1038/sj.bjp.0706683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Spiegel AM. Structure and function of the human calcium-sensing receptor: insights from natural and engineered mutations and allosteric modulators. J Cell Mol Med. 2007;11:908–922. doi: 10.1111/j.1582-4934.2007.00096.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Reyes-Cruz G, Chen W, Jacobson KA, Spiegel AM. Identification of acidic residues in the extracellular loops of the seven-transmembrane domain of the human Ca2+ receptor critical for response to Ca2+ and a positive allosteric modulator. J Biol Chem. 2002;277:46622–46631. doi: 10.1074/jbc.M207100200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, McLarnon SJ, Mora S, Jiang J, Thomas C, Jacobson KA, et al. A region in the seven-transmembrane domain of the human Ca2+ receptor critical for response to Ca2+ J Biol Chem. 2005;280:5113–5120. doi: 10.1074/jbc.M413403200. [DOI] [PubMed] [Google Scholar]
- Huang Y, Zhou Y, Yang W, Butters R, Lee HW, Li S, et al. Identification and dissection of Ca2+-binding sites in the extracellular domain of Ca2+-sensing receptor. J Biol Chem. 2007;282:19000–19010. doi: 10.1074/jbc.M701096200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen AA, Bräuner-Osborne H. Allosteric modulation of the calcium-sensing receptor. Curr Neuropharmacology. 2007;5:180–186. doi: 10.2174/157015907781695982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen AA, Spalding TA, Burstein ES, Sheppard PO, O’Hara PJ, Brann MR, et al. Functional importance of the Ala116-Pro136 region in the calcium-sensing receptor. Constitutive activity and inverse agonism in a family C G-protein coupled receptor. J Biol Chem. 2000;275:29547–29555. doi: 10.1074/jbc.M910023199. [DOI] [PubMed] [Google Scholar]
- Jensen AA, Madsen BE, Krogsgaard-Larsen P, Bräuner-Osborne H. Pharmacological characterization of homobaclofen on wild type and mutant GABAB1b receptors coexpressed with the GABAB2 receptor. Eur J Pharmacol. 2001a;417:177–180. doi: 10.1016/s0014-2999(01)00918-9. [DOI] [PubMed] [Google Scholar]
- Jensen AA, Sheppard PO, Jensen LB, O’Hara PJ, Bräuner-Osborne H. Construction of a high affinity zinc binding site in the metabotropic glutamate receptor mGluR1. Non-competitive antagonism originating from the amino terminal domain of a family C G-protein-coupled receptor. J Biol Chem. 2001b;276:10110–10118. doi: 10.1074/jbc.M007220200. [DOI] [PubMed] [Google Scholar]
- Jones KA, Borowsky B, Tamm JA, Craig DA, Durkin MM, Dai M, et al. GABAB receptors function as a heteromeric assembly of the subunits GABABR1 and GABABR2. Nature. 1998;396:674–679. doi: 10.1038/25348. [DOI] [PubMed] [Google Scholar]
- Kammermeier PJ, Yun J. Activation of metabotropic glutamate receptor 1 dimers requires glutamate binding in both subunits. J Pharmacol Exp Ther. 2005;312:502–508. doi: 10.1124/jpet.104.073155. [DOI] [PubMed] [Google Scholar]
- Kang CH, Shin WC, Yamagata Y, Gokcen S, Ames GF, Kim SH. Crystal structure of the lysine-, arginine-, ornithine-binding protein (LAO) from Salmonella typhimurium at 2.7-Å resolution. J Biol Chem. 1991;266:23893–23899. [PubMed] [Google Scholar]
- Kaupmann K, Huggel K, Heid J, Flor PJ, Bischoff S, Mickel SJ, et al. Expression cloning of GABAB receptors uncovers similarity to metabotropic glutamate receptors. Nature. 1997;386:239–246. doi: 10.1038/386239a0. [DOI] [PubMed] [Google Scholar]
- Kaupmann K, Malitschek B, Schuler V, Heid J, Fröstl W, Beck P, et al. GABAB-receptor subtypes assemble into functional heteromeric complexes. Nature. 1998;396:683–687. doi: 10.1038/25360. [DOI] [PubMed] [Google Scholar]
- Kniazeff J, Galvez T, Labesse G, Pin JP. No ligand binding in the GB2 subunit of the GABAB receptor is required for activation and allosteric interaction between the subunits. J Neurosci. 2002;22:7352–7361. doi: 10.1523/JNEUROSCI.22-17-07352.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kniazeff J, Bessis AS, Maurel D, Ansanay H, Prézeau L, Pin JP. Closed state of both binding domains of homodimeric mGlu receptors is required for full activity. Nat Struct Mol Biol. 2004a;11:706–713. doi: 10.1038/nsmb794. [DOI] [PubMed] [Google Scholar]
- Kniazeff J, Saintot PP, Goudet C, Liu J, Charnet A, Guillon G, et al. Locking the dimeric GABAB G-protein-coupled receptor in its active state. J Neurosci. 2004b;24:370–377. doi: 10.1523/JNEUROSCI.3141-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolakowski LF., Jr. GCRDb: a G-protein-coupled receptor database. Receptors Channels. 1994;2:1–7. [PubMed] [Google Scholar]
- Kuang D, Yao Y, Lam J, Tsushima RG, Hampson DR. Cloning and characterization of a family C orphan G-protein coupled receptor. J Neurochem. 2005;93:383–391. doi: 10.1111/j.1471-4159.2005.03025.x. [DOI] [PubMed] [Google Scholar]
- Kuang D, Yao Y, Wang M, Pattabiraman N, Kotra LP, Hampson DR. Molecular similarities in the ligand binding pockets of an odorant receptor and the metabotropic glutamate receptors. J Biol Chem. 2003;278:42551–42559. doi: 10.1074/jbc.M307120200. [DOI] [PubMed] [Google Scholar]
- Kubo Y, Miyashita T, Murata Y. Structural Basis for a Ca2+-Sensing Function of the Metabotropic Glutamate Receptors. Science. 1998;279:1722–1725. doi: 10.1126/science.279.5357.1722. [DOI] [PubMed] [Google Scholar]
- Kuner R, Köhr G, Grünewald S, Eisenhardt G, Bach A, Kornau H-C. Role of heteromer formation in GABAB receptor function. Science. 1999;283:74–77. doi: 10.1126/science.283.5398.74. [DOI] [PubMed] [Google Scholar]
- Kunishima N, Shimada Y, Tsuji Y, Sato T, Yamamoto M, Kumasaka T, et al. Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor. Nature. 2000;407:971–977. doi: 10.1038/35039564. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Mun HC, Lewis NC, Crouch MF, Culverston EL, Mason RS, et al. Allosteric activation of the extracellular Ca2+-sensing receptor by L-amino acids enhances ERK1/2 phosphorylation. Biochem J. 2007;404:141–149. doi: 10.1042/BJ20061826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Staszewski L, Xu H, Durick K, Zoller M, Adler E. Human receptors for sweet and umami taste. Proc Natl Acad Sci USA. 2002;99:4692–4696. doi: 10.1073/pnas.072090199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundstrom K. The future of G protein-coupled receptors as targets in drug discovery. IDrugs. 2005;8:909–913. [PubMed] [Google Scholar]
- Luu P, Acher F, Bertrand HO, Fan J, Ngai J. Molecular determinants of ligand selectivity in a vertebrate odorant receptor. J Neurosci. 2004;24:10128–10137. doi: 10.1523/JNEUROSCI.3117-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen U, Bräuner-Osborne H, Greenwood JR, Johansen TN, Krogsgaard-Larsen P, Liljefors T, et al. Gad SC. Drug Discovery Handbook. New York: Wiley; 2005. GABA and glutamate receptor ligands and their therapeutic potential in CNS disorders; pp. 797–907. [Google Scholar]
- Malherbe P, Knoflach F, Broger C, Ohresser S, Kratzeisen C, Adam G, Stadler H, Kemp JA, Mutel V. Identification of essential residues involved in the glutamate binding pocket of the group II metabotropic glutamate receptor. Mol Pharmacol. 2001;60:944–954. doi: 10.1124/mol.60.5.944. [DOI] [PubMed] [Google Scholar]
- Malitschek B, Ruegg D, Heid J, Kaupmann K, Bittiger H, Fröstl W, et al. Developmental changes of agonist affinity at GABABR1 receptor variants in rat brain. Mol Cell Neurosci. 1998;12:56–64. doi: 10.1006/mcne.1998.0698. [DOI] [PubMed] [Google Scholar]
- Mun HC, Culverston EL, Franks AH, Collyer CA, Clifton-Bligh R, Conigrave AD. A double mutation in the extracellular Ca2+-sensing receptor's Venus fly trap domain that selectively disables L-amino acid sensing. J Biol Chem. 2005;280:29067–29072. doi: 10.1074/jbc.M500002200. [DOI] [PubMed] [Google Scholar]
- Mun HC, Franks AH, Culverston EL, Krapcho K, Nemeth EF, Conigrave AD. The Venus Fly Trap domain of the extracellular Ca2+-sensing receptor is required for L-amino acid sensing. J Biol Chem. 2004;279:51739–51744. doi: 10.1074/jbc.M406164/200. [DOI] [PubMed] [Google Scholar]
- Muto T, Tsuchiya D, Morikawa K, Jingami H. Structures of the extracellular regions of the group II/III metabotropic glutamate receptors. Proc Natl Acad Sci USA. 2007;104:3759–3764. doi: 10.1073/pnas.0611577104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson G, Hoon MA, Chandrashekar J, Zhang Y, Ryba NJ, Zuker CS. Mammalian sweet taste receptors. Cell. 2001;106:381–390. doi: 10.1016/s0092-8674(01)00451-2. [DOI] [PubMed] [Google Scholar]
- Nelson G, Chandrashekar J, Hoon MA, Feng L, Zhao G, Ryba NJP, et al. An amino-acid taste receptor. Nature. 2002;416:199–202. doi: 10.1038/nature726. [DOI] [PubMed] [Google Scholar]
- Nemeth EF, Steffey ME, Hammerland LG, Hung BCP, Van Wagenen BC, DelMar EG, et al. Calcimimetics with potent and selective activity on the parathyroid calcium receptor. Proc Natl Acad Sci USA. 1998;95:4040–4045. doi: 10.1073/pnas.95.7.4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeth EF, DelMar EG, Heaton WL, Miller MA, Lambert LD, Conklin RL, et al. Calcilytic compounds: potent and selective Ca2+ receptor antagonists that stimulate secretion of parathyroid hormone. J Pharmacol Exp Ther. 2001;299:323–331. [PubMed] [Google Scholar]
- Nomura R, Suzuki Y, Kakizuka A, Jingami H. Direct detection of the interaction between recombinant soluble extracellular regions in the heterodimeric metabotropic γ-aminobutyric acid receptor. J Biol Chem. 2008;283:4665–4673. doi: 10.1074/jbc.M705202200. [DOI] [PubMed] [Google Scholar]
- O’Hara PJ, Sheppard PO, Thøgersen H, Venezia D, Haldeman BA, McGrane V, Houamed KM, Thomsen C, Gilbert TL, Mulvihill ER. The ligand-binding domain in metabotropic glutamate receptors is related to bacterial periplasmic binding proteins. Neuron. 1993;11:41–52. doi: 10.1016/0896-6273(93)90269-w. [DOI] [PubMed] [Google Scholar]
- Oh BH, Pandit J, Kang CH, Nikaido K, Gokcen S, Ames GF, et al. Three-dimensional structures of the periplasmic lysine/arginine/ornithine-binding protein with and without a ligand. J Biol Chem. 1993;268:11348–11355. [PubMed] [Google Scholar]
- Pace AJ, Gama L, Breitwieser GE. Dimerization of the calcium-sensing receptor occurs within the extracellular domain and is eliminated by Cys -> Ser mutations at Cys101 and Cys236. J Biol Chem. 1999;274:11629–11634. doi: 10.1074/jbc.274.17.11629. [DOI] [PubMed] [Google Scholar]
- Pearce SH, Bai M, Quinn SJ, Kifor O, Brown EM, Thakker RV. Functional characterization of calcium-sensing receptor mutations expressed in human embryonic kidney cells. J Clin Invest. 1996;98:1860–1866. doi: 10.1172/JCI118987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Garci E, Gassmann M, Bettler B, Larkum ME. The GABAB1b isoform mediates long-lasting inhibition of dendritic Ca2+ spikes in layer 5 somatosensory pyramidal neurons. Neuron. 2006;50:603–616. doi: 10.1016/j.neuron.2006.04.019. [DOI] [PubMed] [Google Scholar]
- Pi M, Faber P, Ekema G, Jackson PD, Ting A, Wang N, et al. Identification of a novel extracellular cation-sensing G-protein-coupled receptor. J Biol Chem. 2005;280:40201–40209. doi: 10.1074/jbc.M505186200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pidasheva S, Grant M, Canaff L, Ercan O, Kumar U, Hendy GN. Calcium-sensing receptor dimerizes in the endoplasmic reticulum: biochemical and biophysical characterization of CASR mutants retained intracellularly. Hum Mol Genet. 2006;15:2200–2209. doi: 10.1093/hmg/ddl145. [DOI] [PubMed] [Google Scholar]
- Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. 2002;3:639–650. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- Pin J-P, Prézeau L. Allosteric modulators of GABAB receptors: Mechanism of action and therapeutic perspective. Curr Neuropharmacol. 2007;5:195–201. doi: 10.2174/157015907781695919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pin JP, Kniazeff J, Binet V, Liu J, Maurel D, Galvez T, et al. Activation mechanism of the heterodimeric GABAB receptor. Biochem Pharmacol. 2004;68:1565–1572. doi: 10.1016/j.bcp.2004.06.035. [DOI] [PubMed] [Google Scholar]
- Quinn SJ, Bai M, Brown EM. pH Sensing by the calcium-sensing receptor. J Biol Chem. 2004;279:37241–37249. doi: 10.1074/jbc.M404520200. [DOI] [PubMed] [Google Scholar]
- Quinn SJ, Kifor O, Trivedi S, Diaz R, Vassilev P, Brown E. Sodium and ionic strength sensing by the calcium receptor. J Biol Chem. 1998;273:19579–19586. doi: 10.1074/jbc.273.31.19579. [DOI] [PubMed] [Google Scholar]
- Quinn SJ, Ye CP, Diaz R, Kifor O, Bai M, Vassilev P, et al. The Ca2+-sensing receptor: A target for polyamines. Am J Physiol. 1997;273:C1315–C1323. doi: 10.1152/ajpcell.1997.273.4.C1315. [DOI] [PubMed] [Google Scholar]
- Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, et al. Crystal structure of the human β2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- Ray K, Northup J. Evidence for distinct cation and calcimimetic compound (NPS 568) recognition domains in the transmembrane regions of the human Ca2+ receptor. J Biol Chem. 2002;277:18908–18913. doi: 10.1074/jbc.M202113200. [DOI] [PubMed] [Google Scholar]
- Ritzén A, Mathiesen JM, Thomsen C. Molecular pharmacology and therapeutic prospects of metabotropic glutamate receptor allosteric modulators. Basic Clin Pharmacol Toxicol. 2005;9:202–213. doi: 10.1111/j.1742-7843.2005.pto_156.x. [DOI] [PubMed] [Google Scholar]
- Robbins MJ, Milchalovich D, Hill J, Calver AR, Medhurst AD, Gloger I, et al. Molecular cloning and characterization of two novel retinoic acid-inducible orphan G-protein-coupled receptors (GPRC5B and GPRC5C) Genomics. 2000;67:8–18. doi: 10.1006/geno.2000.6226. [DOI] [PubMed] [Google Scholar]
- Romano C, Miller JK, Hyrc K, Dikranian S, Mennerick S, Takeuchi Y, et al. Covalent and noncovalent interactions mediate metabotropic glutamate receptor mGlu5 dimerization. Mol Pharmacol. 2001;59:46–53. [PubMed] [Google Scholar]
- Rondard P, Huang S, Monnier C, Tu H, Blanchard B, Oueslati N, et al. Functioning of the dimeric GABAB receptor extracellular domain revealed by glycan wedge scanning. EMBO J. 2008;27:1321–1332. doi: 10.1038/emboj.2008.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondard P, Liu J, Huang S, Malhaire F, Vol C, Pinault A, et al. Coupling of agonist binding to effector domain activation in metabotropic glutamate-like receptors. J Biol Chem. 2006;281:24653–24661. doi: 10.1074/jbc.M602277200. [DOI] [PubMed] [Google Scholar]
- Rosemond E, Peltekova V, Naples M, Thøgersen H, Hampson DR. Molecular determinants of high affinity binding to group III metabotropic glutamate receptors. J Biol Chem. 2002;277:7333–7340. doi: 10.1074/jbc.M110476200. [DOI] [PubMed] [Google Scholar]
- Rosemond E, Wang M, Yao Y, Storjohann L, Stormann T, Johnson EC, et al. Molecular basis for the differential agonist affinities of group III metabotropic glutamate receptors. Mol Pharmacol. 2004;66:834–842. doi: 10.1124/mol.104.002956. [DOI] [PubMed] [Google Scholar]
- Ruat M, Snowman AM, Hester LD, Snyder SH. Cloned and expressed rat Ca2+-sensing receptor. J Biol Chem. 1996;271:5972–5975. doi: 10.1074/jbc.271.11.5972. [DOI] [PubMed] [Google Scholar]
- Sato T, Shimada Y, Nagasawa N, Nakanishi S, Jingami H. Amino acid mutagenesis of the ligand binding site and the dimmer interface of the metabotropic glutamate receptor 1. Identificaiton of crucial residuces for setting the activated state. J Biol Chem. 2003;278:4314–4321. doi: 10.1074/jbc.M210278200. [DOI] [PubMed] [Google Scholar]
- Schneider TD, Stephens RM. Sequence logos: a new way to display consensus sequences. Nucl Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoepp DD, Jane DE, Monn JA. Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology. 1999;38:1431–1476. doi: 10.1016/s0028-3908(99)00092-1. [DOI] [PubMed] [Google Scholar]
- Silve C, Petrel C, Leroy C, Bruel H, Mallet E, Rognan D, et al. Delineating a Ca2+ binding pocket within the venus flytrap module of the human calcium-sensing receptor. J Biol Chem. 2005;280:37917–37923. doi: 10.1074/jbc.M506263200. [DOI] [PubMed] [Google Scholar]
- Speca DJ, Lin DM, Sorensen PW, Isacoff EY, Ngai J, Dittman AH. Functional identification of a goldfish odorant receptor. Neuron. 1999;23:487–498. doi: 10.1016/s0896-6273(00)80802-8. [DOI] [PubMed] [Google Scholar]
- Tateyama M, Abe H, Nakata H, Saito O, Kubo Y. Ligand-induced rearrangement of the dimeric metabotropic glutamate receptor 1α. Nat Struct Mol Biol. 2004;11:637–642. doi: 10.1038/nsmb770. [DOI] [PubMed] [Google Scholar]
- Tfelt-Hansen J, Brown EM. The calcium-sensing receptor in normal physiology and pathophysiology: a review. Crit Rev Clin Lab Sci. 2005;42:35–70. doi: 10.1080/10408360590886606. [DOI] [PubMed] [Google Scholar]
- Tordoff MG, Shao H, Alarcon LK, Margolskee RF, Mosinger B, Bachmanov AA, et al. Involvement of T1R3 in calcium-magnesium taste. Physiol Genomics. 2008;34:338–348. doi: 10.1152/physiolgenomics.90200.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi R, Mithal A, Chattopadhyay N. Recent updates on the calcium-sensing receptor as a drug target. Curr Med Chem. 2008;15:178–186. doi: 10.2174/092986708783330601. [DOI] [PubMed] [Google Scholar]
- Tsuchiya D, Kunishima N, Kamiya N, Jingami H, Morikawa K. Structural views of the ligand-binding cores of a metabotropic glutamate receptor complexed with an antagonist and both glutamate and Gd3+ Proc Natl Acad Sci USA. 2002;99:2660–2665. doi: 10.1073/pnas.052708599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji Y, Shimada Y, Takeshita T, Kajimura N, Nomura S, Sekiyama N, et al. Cryptic dimer interface and domain organization of the extracellular region of metabotropic glutamate receptor subtype 1. J Biol Chem. 2000;275:28144–28151. doi: 10.1074/jbc.M003226200. [DOI] [PubMed] [Google Scholar]
- Vigot R, Barbieri S, Bräuner-Osborne H, Turecek R, Shigemoto R, Zhang YP, et al. Differential compartmentalization and distinct functions of GABAB receptor variants. Neuron. 2006;50:589–601. doi: 10.1016/j.neuron.2006.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Yao Y, Kuang D, Hampson DR. Activation of family C G-protein-coupled receptors by the tripeptide glutathione. J Biol Chem. 2006;281:8864–8870. doi: 10.1074/jbc.M512865200. [DOI] [PubMed] [Google Scholar]
- Wellendorph P, Bräuner-Osborne H. Molecular cloning, expression, and sequence analysis of GPRC6A, a novel family C G-protein-coupled receptor. Gene. 2004;335:37–46. doi: 10.1016/j.gene.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Wellendorph P, Hansen KB, Balsgaard A, Greenwood JR, Egebjerg J, Bräuner-Osborne H. Deorphanization of GPRC6A: a promiscuous L-α-amino acid receptor with preference for basic amino acids. Mol Pharmacol. 2005;67:589–597. doi: 10.1124/mol.104.007559. [DOI] [PubMed] [Google Scholar]
- Wellendorph P, Burhenne N, Christiansen B, Walter B, Schmale H, Bräuner-Osborne H. The rat GPRC6A: cloning and characterization. Gene. 2007;396:257–267. doi: 10.1016/j.gene.2007.03.008. [DOI] [PubMed] [Google Scholar]
- White JH, Wise A, Main MJ, Green A, Fraser NJ, Disney GH, et al. Heterodimerization is required for the formation of a functional GABAB receptor. Nature. 1998;396:679–682. doi: 10.1038/25354. [DOI] [PubMed] [Google Scholar]
- Xu H, Staszewski L, Tang H, Adler E, Zoller M, Li X. Different functional roles of T1R subunits in the heteromeric taste receptors. Proc Natl Acad Sci USA. 2004;101:14258–14263. doi: 10.1073/pnas.0404384101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi S. Basic properties of umami and effects on humans. Physiol Behav. 1991;49:833–841. doi: 10.1016/0031-9384(91)90192-q. [DOI] [PubMed] [Google Scholar]
- Yao Y, Pattabiraman N, Michne WF, Huang XP, Hampson DR. Molecular modeling and mutagenesis of the ligand-binding pocket of the mGlu3 subtype of metabotropic glutamate receptor. J Neurochem. 2003;86:947–957. doi: 10.1046/j.1471-4159.2003.01906.x. [DOI] [PubMed] [Google Scholar]
- Ye C, Ho-Pao CL, Kanazirska M, Quinn S, Rogers K, Seidman CE, et al. Amyloid-beta proteins activate Ca2+-permeable channels through calcium-sensing receptors. J Neurosci Res. 1997;47:547–554. doi: 10.1002/(sici)1097-4547(19970301)47:5<547::aid-jnr10>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Qiu W, Quinn SJ, Conigrave AD, Brown EM, Bai M. Three adjacent serines in the extracellular domains of the CaR are required for L-amino acid-mediated potentiation of receptor function. J Biol Chem. 2002;277:33727–33735. doi: 10.1074/jbc.M200976200. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Sun S, Quinn SJ, Brown EM, Bai M. The extracellular calcium-sensing receptor dimerizes through multiple types of intermolecular interactions. J Biol Chem. 2001;276:5316–5322. doi: 10.1074/jbc.M005958200. [DOI] [PubMed] [Google Scholar]
- Zhao GQ, Zhang Y, Hoon MA, Chandrashekar J, Erlenbach I, Ryba NJ, et al. The receptors for mammalian sweet and umami taste. Cell. 2003;115:255–266. doi: 10.1016/s0092-8674(03)00844-4. [DOI] [PubMed] [Google Scholar]