

Abstract
Background and purpose:
Traditionally, the stem and root bark of Ulmus davidiana var. japonica (Ulmaceae) have been known to be anti-inflammatory in Korea. Anti-inflammatory effects of torilin, isolated from this plant and the underlying mechanisms were examined by using lipopolysaccharide (LPS)-stimulated microglial BV2 cells.
Experimental approach:
The cells were treated with torilin prior to LPS exposure and the effects on pro-inflammatory enzymes, inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2), and a pro-inflammatory cytokine, interleukin-1β (IL-1β) were analysed by RT-PCR, Western blot or elisa. To reveal the mechanism of action of torilin we investigated the involvement of mitogen-activated protein kinase (MAPK) cascades and their downstream transcription factors, nuclear factor-κB (NF-κB) and cyclic AMP-responsive element (CRE)-binding protein (CREB).
Key results:
Torilin significantly reduced the LPS-induced expression of iNOS, COX-2 and IL-1β, and the subsequent release of NO, prostaglandin E2 and IL-1β into culture medium. LPS stimulation of extracellular signal-regulated kinase 1/2 (ERK1/2) and p38 MAPK was inhibited by torilin. In addition, the inhibitory effect of torilin on NF-κB and CREB was shown by torilin-mediated recovery of LPS-induced degradation of inhibitor κB-α and suppression of LPS-induced phosphorylation of CREB respectively.
Conclusion and implications:
This study indicates that torilin inhibited LPS-induced iNOS, COX-2 and IL-1β via down-regulation of ERK1/2, p38 MAPK, NF-κB and CREB and suggests that torilin has a potential as an anti-inflammatory drug candidate.
Keywords: torilin, lipopolysaccharide, inducible nitric oxide synthase, cyclooxygenase-2, interleukin-1β, extracellular signal-regulated kinase 1/2, p38 MAPK, NF-κB
Introduction
Torilin is a guaiane type sesquiterpene isolated from stem and root bark of Ulmus davidiana var. japonica (Ulmaceae). The stem and root bark of U. davidiana var. japonica have been used in traditional medicine for treatments of inflammation, oedema and gastric cancer (Lee, 1966). We previously reported that three sesquiterpenes from this plant, including torilin, inhibited lipopolysaccharide (LPS)-induced NO production in BV2 cells (Kim et al., 2007). In that study, we used BV2 cells as a screening system for natural products exhibiting neuroprotective activity, based on the role of microglia in neuroinflammation (Minghetti and Levi, 1998; Streit et al., 2004). Thus, the present work was conducted to expand our investigation of torilin to two important pro-inflammatory enzymes, inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) and a pro-inflammatory cytokine, interleukin-1β (IL-1β), and to reveal the underlying anti-inflammatory mechanisms, employing LPS-stimulated microglial BV2 cells as the experimental system.
Two major forms of NOS and COX enzymes have been identified. Under normal conditions, the constitutive isoforms of these enzymes (constitutive NOS and COX-1) are localized in nearly all tissues and are associated with several important physiological events such as anti-platelet activity, vasodilation and cytoprotection (Mollace et al., 2005). The inducible isoforms, iNOS and COX-2, are detected in a variety of cells in inflammatory situations and result in the production of large amounts of pro-inflammatory and cytotoxic NO and prostaglandins (PGs) respectively. The released NO and PGs are involved in a number of pathological processes, including inflammation, pain, neuroinflammatory processes, arthritis, multiple sclerosis, brain ischemia or cancer (Dubois et al., 1998; Kröncke et al., 1998; Bogdan, 2001).
The induction of iNOS and COX-2 is transcriptionally regulated. iNOS and COX-2 genes are found to have binding sites for the same transcription factors, such as CCAAT-enhancer box binding protein (C/EBP), cyclic AMP-responsive element-binding protein (CREB) and nuclear factor-κB (NF-κB), which are known to be involved in the LPS/cytokine-mediated induction of transcription (Appleby et al., 1994; Lin et al., 1996). The pathways regulating iNOS expression are known to vary in different cells or different species. In this regard, activation or inhibition of the Janus kinase/signal transducer and activator of transcription (JAK/STAT) and/or NF-κB pathway are reported to be the central mechanisms explaining the effects of many different regulators (Kleinert et al., 2004). With regard to the activation of the COX-2 promoter, NF-κB, C/EBP and CRE are believed to play a role, and they differently induce COX-2 depending on cell type or stimulator (Appleby et al., 1994; Yamamoto et al., 1995; Caivano and Cohen, 2000). Activation of the mitogen-activated protein kinase (MAPK) signalling cascade has also been linked with the PG biosynthetic pathway (Molina-Holgado et al., 2000).
IL-1β is synthesized in the CNS by tumour-infiltrating immune cells and by endothelial cells (Liu et al., 2003). In the CNS, a number of stimuli, such as LPS, traumatic brain injury and brain viral infection have been shown to produce IL-1β. Under pathological situations involving CNS inflammation, IL-1 is mainly secreted by activated microglia and macrophages (Molina-Holgado et al., 2000; Liu et al., 2003).
Microglia are resident immune cells in the CNS and are also recognized as key cellular mediators of neurodegenerative process (Cuadros and Navascués, 1998; Kaur et al., 2001; Streit et al., 2004; Kim and Joh, 2006). They readily become activated in response to infection or injury. Activated microglia secrete a number of pro-inflammatory and neurotoxic factors such as those mentioned above, which cause neuronal damage (Stoll and Jander, 1999; Liu and Hong, 2003; Lai and Todd, 2006).
In the present study, we attempted to elucidate the anti-inflammatory potential of torilin by investigating the effect of torilin on the inflammatory response induced by LPS in murine microglial BV2 cells. Torilin inhibited the LPS-induced expression of iNOS, COX-2 and IL-1β and subsequent production of NO, PGE2 and IL-1β in BV2 cells. To investigate the underlying mechanisms, the involvement of MAPKs, NF-κB and CREB was also examined. The present study provides information revealing torilin as a potential candidate compound with anti-inflammatory actions and suggests a scientific basis for further investigation of torilin against neuroinflammatory conditions.
Methods
Preparation of torilin
Torilin was isolated from the stem and root bark of U. davidiana var. japonica as previously reported (Kim et al., 2007). For each experiment, torilin was dissolved in dimethylsulphoxide (final culture concentration, 0.05%) and added to the cells at concentrations of 10, 30 and 50 µmol·L−1 for 2 h and then exposed to 0.1 or 1.0 µg·mL−1 LPS (from Escherichia coli; Sigma L4516) for indicated times. Serum-free media were used as a vehicle control for LPS. Preliminary studies indicated that the solvent had no effect on cell viability at the concentration used.
Cell culture
Microglial BV2 cells, originally developed by Dr V Bocchini at University of Perugia (Perugia, Italy; Blasi et al., 1990), were generously provided by Dr Sun Yeou Kim at Kyunghee University (Suwon, Korea). The cells were maintained in Dulbecco's modified Eagle's medium (DMEM; Invitrogen Corporation, Carlsbad, CA, USA) containing 10% fetal bovine serum (FBS; Invitrogen Corporation), 100 unit·mL−1 penicillin G, 100 µg·mL−1 streptomycin and 0.25 µg·mL−1 amphotericin B at 37°C in a humidified atmosphere of 95% O2–5% CO2.
Reverse transcription-polymerase chain reaction (RT-PCR) analysis
BV2 cells were plated overnight in 12-well plates at a density of 5 × 105 cells per plate, then the cells were further incubated in the medium without 10% FBS for at least 4 h before treatments. Total RNA was prepared from BV2 cells by using the TRIzol® reagent (Invitrogen Corporation) according to the manufacturer's protocol. Total RNA was reverse-transcribed by using the AccuPower® RT Premix (Bioneer Corporation, Daejeon, Korea) with 2 µg total RNA and oligo dT. Primer sequences were as follows: iNOS, sense: 5′-GACAAGCTGCATGTGACATC-3′, antisense: 5′-GCTGGTAGGTTCCTGTTGTT-3′; COX-2, sense: 5′-TTGAAGACCAGGAGTACAGC-3′, antisense: 5′-GGTACAGTTCCATGACATCG-3′; IL-1β, sense: 5′-GCTGCTTCCAAACCTT-3′, antisense: 5′-AGGCCACAGGTATTTT-3′; GAPDH, sense: 5′-TCGTGGAGTCTACTGGCGT-3′, antisense: 5′-GCCTGCTTCACCACCTTCT-3′. PCR amplification of the resulting cDNA template was conducted by using the following conditions for 36 (COX-2 and GAPDH), 40 (IL-1β) or 46 (iNOS) cycles; denaturation at 94°C for 30 s, annealing at 48°C (IL-1β), 55°C (iNOS), 57°C (COX-2) or 60°C (GAPDH) for 45 s, and extension at 72°C for 30 s. The amplified DNA products were visualized on 1.2% agarose gels and photographed under ultraviolet light.
Western blot analysis
BV2 cells were plated overnight in 6- or 12-well plates at a density of 5 × 105 cells per plate, then the cells were further incubated in the medium without 10% FBS for at least 4 h before treatments. Cells were harvested with ice-cold PBS and centrifuged at 110×g for 5 min at 4°C. The pellet was lysed in 20–60 µL of PRO-PREP™ protein extraction solution (iNtRON Biotechnology, Seongnam, Korea) containing 4 mmol·L−1 sodium orthovanadate and incubated at −20°C for 20 min. Cell lysates were centrifuged at 20 000×g for 5 min at 4°C, then the supernatants were collected. Protein content was determined by using the BCA protein assay (Pierce, Rockford, IL, USA). Equal amounts of protein (50 µg) were loaded per lane onto 10% SDS-PAGE gel. Proteins were transferred to nitrocellulose membranes (Invitrogen Corporation) and subsequently blocked in 4% bovine serum albumin (BSA)-TBST (100 mmol·L−1 Tris, pH 8.0, 150 mmol·L−1 NaCl and 0.1% Tween 20) for 2 h at room temperature. Anti-iNOS (1:5000 dilution; BD Biosciences, San Jose, CA, USA), anti-phospho p38, anti-phospho extracellular signal-regulated kinase 1/2 (ERK1/2), anti-inhibitor κB-α (IκB-α) (1:1000 dilution; Cell Signaling Technology, Danvers, MA, USA), anti-phospho CREB (1:500 dilution; Cell Signaling Technology) or anti-actin (1:1000 dilution; Santa Cruz Biotechnology, Santa Cruz, CA, USA) antibodies were employed in 4% BSA-TBST. The membranes were incubated with the primary antibody at 4°C overnight. After washing three times with TBST, the immunoreactive bands were visualized by using secondary antibodies conjugated to horseradish peroxidase (1:10 000 dilution; Santa Cruz Biotechnology) and ECL chemiluminescence detection kit (Amersham Biosciences, Piscataway, NJ, USA). The membrane was stripped with Restore™ Western Blot Stripping Buffer (Pierce) for 30–40 min at room temperature and reprobed with anti-p38 or anti-ERK1/2 (1:1000 dilution in 4% BSA-TBST, overnight, 4°C; Cell Signaling Technology) antibodies.
Assay of NO production
BV2 cells were plated overnight in 48-well plates at a density of 3 × 105 cells per plate. NO production was monitored by measuring the nitrite content in culture medium as previously described (Green et al., 1982). The isolated supernatant was mixed with an equal volume of Griess reagent (0.1% N-1-naphthylethylenediamine dihydrochloride and 1% sulphanilamide in 5% phosphoric acid) and incubated at room temperature for 10 min. Absorbance was measured at 550 nm in a microplate reader. Sodium nitrite was used as a standard.
Assay of IL-1β production
BV2 cells were plated overnight in 24-well plates at a density of 3 × 105 cells per plate. The supernatants were collected and stored at −70°C until measurement of IL-1β. The IL-1β concentration in the culture medium was determined by a mouse IL-1β enzyme-linked immunosorbent assay (elisa) system (R&D Systems, Minneapolis, MN, USA).
Assay of PGE2 production
BV2 cells were plated overnight in 6-well plates at a density of 5 × 105 cells per plate. U0126, a selective MEK1/2 inhibitor (Cell Signaling Technology) or SB203580, a selective p38 inhibitor (Calbiochem, Darmstadt, Germany) was added 30 min before treatment with LPS, dissolved in dimethylsulphoxide. The supernatants were collected, and the PGE2 concentration in the culture medium was determined by a PGE2elisa system (R&D Systems). The supernatants were used without dilution, and the assay was performed according to the high sensitivity procedure.
Statistical analysis
Each experiment was performed at least in triplicate. All data are presented as the mean ± standard deviation (SD). Statistical analyses were performed by using SigmaStat® 3.1 (Systat Software, Point Richmond, CA, USA). The differences among groups were analysed by one way anova (analysis of variance) followed by Dunnett's test for comparing a control group with all other groups. Values of P < 0.05 were considered to be statistically significant.
All drug/molecular target nomenclature conform to the Guide to Receptors and Channels (Alexander et al., 2008).
Results
Torilin inhibits LPS-induced iNOS and NO production in BV2 cells
Treatment of BV2 microglia with LPS resulted in increased iNOS expression (Figure 1A,B) and subsequent NO production (Figure 1C). Pre-treatment with torilin significantly inhibited iNOS mRNA levels (Figure 1A) and iNOS protein levels (Figure 1B), compared with LPS-treated control.
Figure 1.
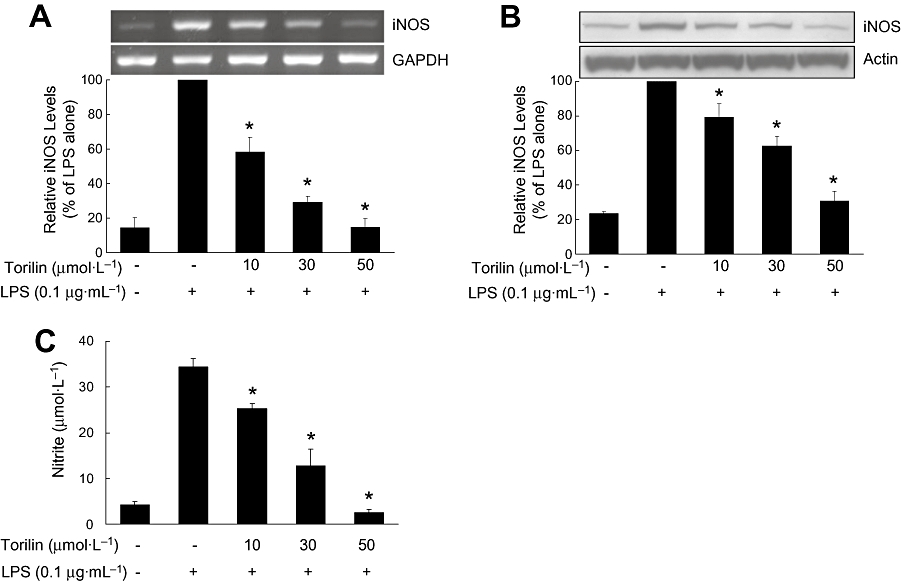
Inhibitory effects of torilin on the LPS-induced expression of iNOS and NO production in BV2 cells. BV2 cells were pre-treated with torilin for 2 h, then exposed to LPS for 6 h (A), 12 h (B) or 24 h (C). (A) Total RNA was extracted from cells, and the level of mRNA encoding iNOS protein was determined by RT-PCR as described in the Methods. (B) Cell lysates (50 µg protein) were prepared from the cells and subjected to Western blot analysis by using an antibody specific for iNOS as described in the Methods. (C) The concentration of nitrite in culture medium was monitored as described in the Methods. The relative mRNA or protein levels were quantified by scanning densitometry and normalized to GAPDH mRNA or actin protein respectively. The values shown are mean ± SD of data from three independent experiments. *Significant compared with LPS alone, P < 0.05.
Levels of LPS-induced NO were measured in BV2 cells pre-treated with torilin by the Griess assay (Figure 1C). LPS increased the nitrite concentration in the medium by eightfold as compared with control. Consistent with the results of iNOS expression, LPS-induced NO production was significantly decreased by torilin in a concentration-dependent manner. Cell viability was not significantly altered by torilin at the concentrations used (data not shown).
Torilin inhibits LPS-induced COX-2 and PGE2 in BV2 cells
Effect of torilin on another pro-inflammatory enzyme, COX-2 was examined by RT-PCR analysis. Stimulation of BV2 cells with LPS led to increased expression of mRNA for COX-2 (Figure 2A). Torilin at 30 and 50 µmol·L−1 significantly inhibited the increase of COX-2 mRNA stimulated by LPS (Figure 2A).
Figure 2.
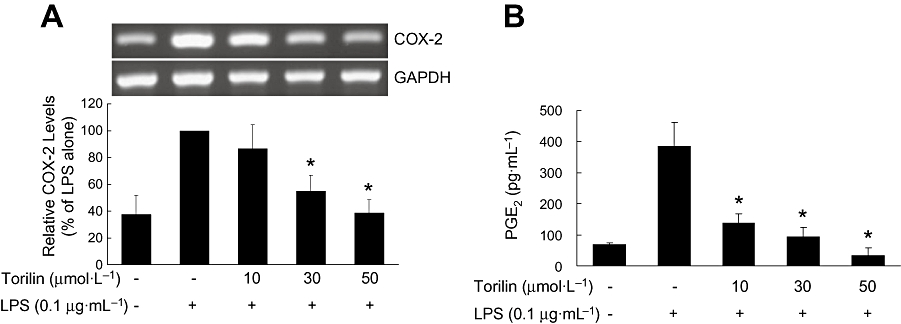
Inhibitory effects of torilin on the LPS-induced expression of COX-2 and PGE2 production in BV2 cells. BV2 cells were pre-treated with torilin for 2 h, then exposed to LPS for 6 h (A) or 17 h (B). (A) Total RNA was extracted from cells, and the level of mRNA encoding COX-2 protein was determined by RT-PCR as described in the Methods. (B) Levels of PGE2 in the culture medium were determined by elisa as described in the Methods. The relative mRNA levels were quantified by scanning densitometry and normalized to GAPDH mRNA. The values shown are mean ± SD of data from three independent experiments. *Significant compared with LPS alone, P < 0.05.
Treatment of BV2 cells with LPS also caused a nearly sixfold increase in PGE2 release in comparison with untreated controls, after 17 h exposure to LPS (Figure 2B). Torilin concentration-dependently inhibited the LPS-induced production of PGE2 over the concentration range used here (Figure 2B).
Torilin inhibits LPS-induced IL-1β in BV2 cells
Effect of torilin on the pro-inflammatory cytokine, IL-1β was examined by RT-PCR analysis. Exposure of BV2 cells to LPS strongly induced the expression of mRNA for IL-1β. Torilin at 30 and 50 µmol·L−1 significantly inhibited the induced IL-1β mRNA, compared with the LPS-treated control (Figure 3A).
Figure 3.
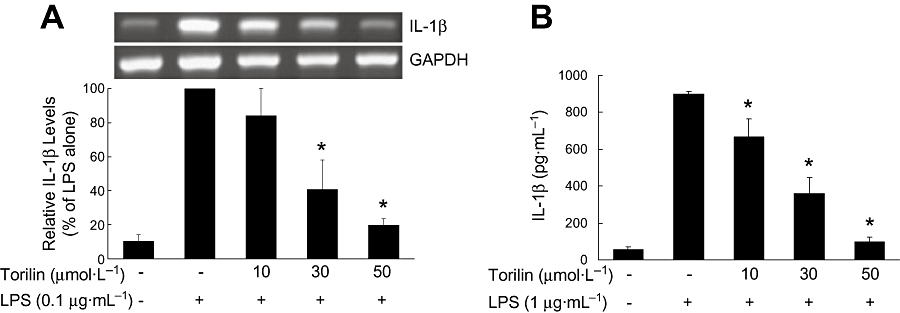
Inhibitory effects of torilin on the LPS-induced IL-1β gene expression and IL-1β release in BV2 cells. BV2 cells were pre-treated with torilin for 2 h, then exposed to LPS for 6 h. (A) Total RNA was extracted from cells, and the level of mRNA encoding IL-1β protein was determined by RT-PCR as described in the Methods. (B) The levels of immunodetectable IL-1β in the culture medium were determined by elisa as described in the Methods. The relative mRNA levels were quantified by scanning densitometry and normalized to GAPDH mRNA. The values shown are mean ± SD of data from three independent experiments. *Significant compared with LPS alone, P < 0.05.
elisa was used to measure immunoreactive IL-1β released into the culture medium from BV2 cells. When BV2 cells were treated with LPS for 24 h, a large increase in IL-1β, 16-fold of control, in the culture medium, was observed. Torilin significantly attenuated LPS-induced IL-1β release, consistent with the result of IL-1β mRNA expression (Figure 3B).
Torilin inhibits LPS-induced degradation of IκB-α and LPS-induced activation of MAPKs and CREB in BV2 cells
We evaluated the involvement of NF-κB in the inhibitory effect of torilin on LPS-induced inflammatory factors. BV2 cells were assayed for the degradation of IκB-α by using Western blotting. As shown in Figure 4A, LPS markedly decreased levels of IκB-α by 51% of those in untreated control BV2 cells. This LPS-induced IκB-α degradation was significantly reversed by torilin, and IκB-α levels were restored to normal by 30 and 50 µmol·L−1 torilin. The data indicate that the anti-inflammatory effect of torilin in LPS-stimulated BV2 cells involved the NF-κB pathway.
Figure 4.
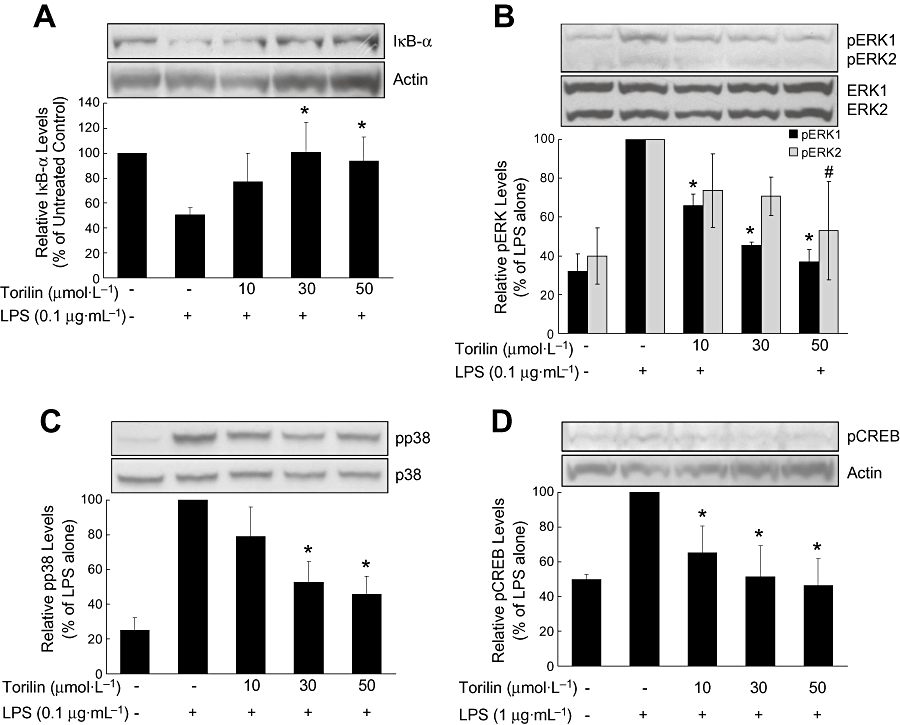
Inhibitory effects of torilin on the LPS-induced degradation of IκB-α and the activation of ERK1/2, p38 MAPK and CREB in BV2 cells. BV2 cells were pre-treated with torilin for 2 h, then exposed to LPS for 1 h (A–C) or 30 min (D). Cell lysates (50 µg protein) were prepared and subjected to Western blot analysis by using antibodies specific for IκB-α or the phosphorylated forms of ERK1/2, p38 MAPK and CREB (shown as pERK, etc.) as described in the Methods. Equivalent loading of cell lysates was determined by reprobing the blots with anti-total ERK1/2, anti-total p38 or anti-actin antibodies. The relative protein levels were quantified by scanning densitometry and normalized to total ERK1/2, total p38 or actin. The values shown are mean ± SD of data from three independent experiments. Significant compared with LPS alone: IκB-α, pp38, pCREB, pERK1, *P < 0.05; pERK2, #P < 0.05.
Then, we investigated the involvement of MAPKs in the torilin-mediated inhibition of inflammatory mediators. To assess activation of these signalling kinases, levels of phosphorylated ERK1/2 and p38 MAPK were determined in LPS-stimulated BV2 cells. ERK2 activation (shown as its phosphorylated form, pERK2) was weakly stimulated by LPS, while ERK1 and p38 MAPK activation were strongly stimulated by LPS (Figure 4B,C). Torilin had significant inhibitory effects on LPS-stimulated ERK1 activation, at 10 µmol·L−1, 30 µmol·L−1 and 50 µmol·L−1 (Figure 4B) and on p38 MAPK activation, at 30 µmol·L−1 and 50 µmol·L−1, compared with the CPS-treated control (Figure 4C). However the LPS-induced increase in pERK2 was inhibited only by the highest concentration of torilin (50 µmol·L−1).
CREB is the physiological substrate for mitogen- and stress-activated protein kinases-1 (MSK1), which is activated by ERK and p38 MAPK-mediated signalling in response to LPS. CREB activation was weakly stimulated by LPS, and this activation was significantly inhibited by pre-treatment with torilin, at all three concentrations (Figure 4D).
MAPKs are involved in LPS-induced PGE2 production in BV2 cells
We used SB203580, a selective p38 inhibitor, and U0126, a selective MEK1/2 inhibitor, to investigate whether p38 MAPK or ERK are involved in the LPS-mediated COX-2 expression, by measuring the release of the COX product PGE2, from BV2 cells. Both SB203580 (50 µmol·L−1) and U0126 (50 µmol·L−1), used separately, blocked the LPS-induced PGE2 production (Figure 5). SB203580 almost completely abolished PGE2 production. These results suggest that one of the possible mechanisms of inhibitory effects of torilin on LPS-stimulated COX-2 is through negative regulation of p38 MAPK and ERK1/2.
Figure 5.
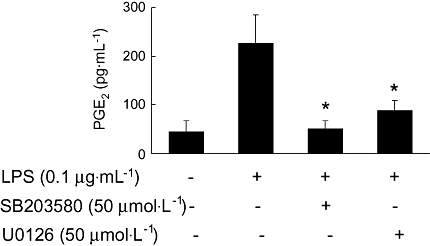
Involvement of ERK1/2 and p38 MAPK in LPS-induced PGE2 production in BV2 cells. BV2 cells were pre-treated with U0126 or SB203580 for 30 min, and then the cells were exposed to LPS for 17 h, followed by elisa for PGE2, as described in the Methods. The values shown are mean ± SD of data from three independent experiments. *Significant compared with LPS alone, P < 0.05.
Discussion
In the present study, torilin exhibited significant inhibitory effects on pro-inflammatory mediators, iNOS, NO, COX-2, PGE2 and IL-1β in LPS-stimulated BV2 cells, and these effects were associated with its negative regulation of LPS-induced phosphorylation of ERK1/2 (mainly ERK1) and p38 MAPK.
In our previous study, ERK and p38 MAPK were found to be positively related to LPS signalling in BV2 cells because inhibitors of MEK1/2 (U0126) and p38 MAPK (SB203580) reduced LPS-induced iNOS protein (Choi et al., 2008). There are also some studies suggesting that MAPKs are critical for the LPS-induced expression of iNOS and release of NO in microglia (Bhat et al., 1998; Pyo et al., 1998; Lu et al., 2008). These results might indicate that torilin-mediated inhibition of ERK or p38 activation is one of the possible mechanisms underlying its inhibitory action on iNOS expression and NO production in LPS-stimulated BV2 cells.
In this study, we examined the involvement of MAPKs in the induction of COX-2 by LPS in BV2 cells. Experiments by using the selective kinase inhibitors, U0126 and SB203580, confirmed that ERK and p38 MAPK were also positively associated with COX-2 regulation in LPS-stimulated BV2 cells. COX-2 is a key enzyme for the biosynthesis of PGE2, and the release of this mediator was decreased by U0126 or SB203580 in LPS-treated BV2 cells, in our results. This observation suggests that the inhibitory effect of torilin on COX-2 induction was regulated via an ERK- or p38-dependent pathway.
These effects of torilin on the activation of ERK and p38 MAPK by LPS may also affect the promoter activity of NF-κB. This is supported by the reversal by torilin of the LPS-induced degradation of IκB-α. Phosphorylation-dependent proteolysis of IκB-α leads to the activation of NF-κB, and MAPKs are known to be involved in NF-κB activation (Aga et al., 2004). As a result, it is suggested that torilin-induced down-regulation of ERK and p38 MAPK phosphorylation negatively affects LPS-induced increase in free NF-κB and thus results in suppression of iNOS and COX-2, which are transcriptionally regulated by NF-κB (Lowenstein et al., 1993; Baldwin, 1996; Eliopoulos et al., 2002).
Another transcription factor, CREB, is essential for inducible expression of COX-2 by LPS (Eliopoulos et al., 2002). ERK1/2 and p38 MAPK are known to be responsible for the LPS-induced activation of the transcription factors CREB and activating transcription factor-1 (ATF1) and subsequent downstream transcription of the COX-2 and IL-1β genes in macrophages (Caivano and Cohen, 2000). On this basis, we examined the effect of torilin on CREB activation in LPS-stimulated BV2 cells. As expected, phosphorylation of CREB was increased in response to LPS and torilin attenuated the CREB activation. These results indicate that torilin-mediated down-regulation of MAPKs leads to the inhibition of CREB, which may contribute to the inhibition of COX-2 expression.
There is evidence supporting the involvement of the MAPKs signalling pathways in LPS-induced IL-1β transcription and production in microglia and other cells (Finco and Baldwin, 1993; Nakajima et al., 1993; Baldassare et al., 1999; Caivano and Cohen, 2000; Kim et al., 2004; Clark et al., 2006). In addition, COX-2 is up-regulated by IL-1β via MAPKs signalling pathways in various cell types, such as astrocytes, airway smooth muscle cells, synovial fibroblasts or endothelial cells (Crofford et al., 1994; Laporte et al., 2000; Molina-Holgado et al., 2000; Liu et al., 2003; Zhai et al., 2004). Further, IL-1β induces iNOS and NO production in C6 astrocytoma cells (Kim et al., 2006). The gene encoding IL-1β also contains a CRE in the promoter region (Sung and Walters, 1991; Tsukada et al., 1994; Caivano and Cohen, 2000). These observations, combined with our results of torilin-mediated inhibition of IL-1β, suggest that torilin-mediated inhibition of the phosphorylation of ERK, p38 and CREB may contribute to the inhibition of IL-1β production and, in turn, affect IL-1β-dependent COX-2 or iNOS induction. Moreover, Faour et al. (2001) showed that the magnitude and duration of the induction of COX-2 mRNA, protein and PGE2 release by IL-1β is primarily the result of PGE2-dependent stabilization of COX-2 mRNA through activation of p38 MAPK in human synovial fibroblasts. In this regard, torilin-mediated inhibition of PGE2 release could affect this PGE2-dependent positive feedback of COX-2 mRNA expression in our cell system.
The mRNA expression of another important pro-inflammatory cytokine, tumour necrosis factor α (TNFα) was also induced by LPS, whereas this was not significantly affected by torilin in our experiments (data not shown). TNFα is one of potent neurotoxins produced by microglia during CNS inflammation, and it is known to be involved in neurodegeneration (Loddick and Rothwell, 1999; Sriram and O'Callaghan, 2007). We paid attention to torilin as a potential neuroprotective agent based on its inhibitory effect on NO production in microglial cells in our previous study (Kim et al., 2007). Thus, anti-inflammatory effects of torilin on microglial cells might be correlated with its protective effects against neurodegenerative diseases due to prevention of neuroinflammation. However it is difficult to discuss our findings in relation to the role of torilin in neurodegenerative diseases at this stage. No significant effect on TNFα could be one of the reasons for them. In this context, we focused our discussion on inflammation rather than neuroinflammation in the present study.
Taken together, the results of our study show that torilin prevented LPS-induced activation of ERK, p38 MAPK, CREB and NF-κB, which induced inhibition of iNOS, COX-2 and IL-1β in BV2 cells. In these processes, the crosstalk between these mediators, such as IL-1β and COX-2/PGE2, IL-1β and iNOS/NO or PGE2 and COX-2, appears to potentiate the effect of torilin, suggesting the possibility of strong anti-inflammatory actions to be exerted by this compound.
Acknowledgments
This research was supported by a grant (M103KV010027-08K2201-02710) from Brain Research Center of the 21st Century Frontier Research Program funded by the Ministry of Science and Technology and by a grant (10524) from Seoul R&BD Program, Korea.
Glossary
Abbreviations
- COX-2
cyclooxygenase-2
- CRE
cyclic AMP-responsive element
- CREB
CRE-binding protein
- ERK1/2
extracellular signal-regulated kinase 1/2
- IκB-α
inhibitor κB-α
- IL-1β
interleukin-1β
- iNOS
inducible nitric oxide synthase
- LPS
lipopolysaccharide
- MAPK
mitogen-activated protein kinase
- NF-κB
nuclear factor-κB
- PGE2
prostaglandin E2
Conflict of interest
The authors state no conflict of interest.
References
- Aga M, Watters JJ, Pfeiffer ZA, Wiepz GJ, Sommer JA, Bertics PJ. Evidence for nucleotide receptor modulation of cross talk between MAP kinase and NF-kappa B signaling pathways in murine RAW 264.7 macrophages. Am J Physiol Cell Physiol. 2004;286:C923–C930. doi: 10.1152/ajpcell.00417.2003. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edn. Br J Pharmacol. 2008;153(Suppl. 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appleby SB, Ristimäki A, Neilson K, Narko K, Hla T. Structure of the human cyclo-oxygenase-2 gene. Biochem J. 1994;302:723–727. doi: 10.1042/bj3020723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldassare JJ, Bi Y, Bellone CJ. The role of p38 mitogen-activated protein kinase in IL-1β transcription. J Immunol. 1999;162:5367–5373. [PubMed] [Google Scholar]
- Baldwin AS. The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- Bhat NR, Zhang P, Lee JC, Hogan EL. Extracellular signal-regulated kinase and p38 subgroups of mitogen-activated protein kinases regulate inducible nitric oxide synthase and tumor necrosis factor-alpha gene expression in endotoxin-stimulated primary glial cultures. J Neurosci. 1998;18:1633–1641. doi: 10.1523/JNEUROSCI.18-05-01633.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasi E, Barluzzi R, Bocchini V, Mazzolla R, Bistoni F. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J Neuroimmunol. 1990;27:229–237. doi: 10.1016/0165-5728(90)90073-v. [DOI] [PubMed] [Google Scholar]
- Bogdan C. Nitric oxide and the immune response. Nat Immunol. 2001;2:907–916. doi: 10.1038/ni1001-907. [DOI] [PubMed] [Google Scholar]
- Caivano M, Cohen P. Role of mitogen-activated protein kinase cascades in mediating lipopolysaccharide-stimulated induction of cyclooxygenase-2 and IL-1β in RAW264 macrophages. J Immunol. 2000;164:3018–3025. doi: 10.4049/jimmunol.164.6.3018. [DOI] [PubMed] [Google Scholar]
- Choi Y, Moon A, Kim YC. A pinusolide derivative, 15-methoxypinusolidic acid from Biota orientalis inhibits inducible nitric oxide synthase in microglial cells: implication for a potential anti-inflammatory effect. Int Immunopharmacol. 2008;8:548–555. doi: 10.1016/j.intimp.2007.12.010. [DOI] [PubMed] [Google Scholar]
- Clark AK, D'Aquisto F, Gentry C, Marchand F, McMahon SB, Malcangio M. Rapid co-release of interleukin 1β and caspase 1 in spinal cord inflammation. J Neurochem. 2006;99:868–880. doi: 10.1111/j.1471-4159.2006.04126.x. [DOI] [PubMed] [Google Scholar]
- Crofford LJ, Wilder RL, Ristimäki AP, Sano H, Remmers EF, Epps HR, et al. Cyclooxygenase-1 and -2 expression in rheumatoid synovial tissues. Effects of interleukin-1β, phorbol ester, and corticosteroids. J Clin Invest. 1994;93:1095–1101. doi: 10.1172/JCI117060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuadros MA, Navascués J. The origin and differentiation of microglial cells during development. Prog Neurobiol. 1998;56:173–189. doi: 10.1016/s0301-0082(98)00035-5. [DOI] [PubMed] [Google Scholar]
- Dubois RN, Abramson SB, Crofford L, Gupta RA, Simon LS, Van De Putte LB, et al. Cyclooxygenase in biology and disease. FASEB J. 1998;12:1063–1073. [PubMed] [Google Scholar]
- Eliopoulos AG, Dumitru CD, Wang CC, Cho J, Tsichlis PN. Induction of COX-2 by LPS in macrophages is regulated by Tpl2-dependent CREB activation signals. EMBO J. 2002;21:4831–4840. doi: 10.1093/emboj/cdf478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faour WH, He Y, He QW, de Ladurantaye M, Quintero M, Mancini A, et al. Prostaglandin E2 regulates the level and stability of cyclooxygenase-2 mRNA through activation of p38 mitogen-activated protein kinase in interleukin-1β-treated human synovial fibroblasts. J Biol Chem. 2001;276:31720–31731. doi: 10.1074/jbc.M104036200. [DOI] [PubMed] [Google Scholar]
- Finco TS, Baldwin AS., Jr κB site-dependent induction of gene expression by diverse inducers of nuclear factor κB requires Raf-1. J Biol Chem. 1993;268:17676–17679. [PubMed] [Google Scholar]
- Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR. Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal Biochem. 1982;126:131–138. doi: 10.1016/0003-2697(82)90118-x. [DOI] [PubMed] [Google Scholar]
- Kaur C, Hao AJ, Wu CH, Ling EA. Origin of microglia. Microsc Res Tech. 2001;54:2–9. doi: 10.1002/jemt.1114. [DOI] [PubMed] [Google Scholar]
- Kim SH, Smith CJ, Van Eldik LJ. Importance of MAPK pathways for microglial pro-inflammatory cytokine IL-1β production. Neurobiol Aging. 2004;25:431–439. doi: 10.1016/S0197-4580(03)00126-X. [DOI] [PubMed] [Google Scholar]
- Kim YC, Lee MK, Sung SH, Kim SH. Sesquiterpenes from Ulmus davidiana var. japonica with the inhibitory effects on lipopolysaccharide-induced nitric oxide production. Fitoterapia. 2007;78:196–199. doi: 10.1016/j.fitote.2006.11.013. [DOI] [PubMed] [Google Scholar]
- Kim YJ, Hwang SY, Oh ES, Oh S, Han IO. IL-1beta, an immediate early protein secreted by activated microglia, induces iNOS/NO in C6 astrocytoma cells through p38 MAPK and NF-kappaB pathways. J Neurosci Res. 2006;84:1037–1046. doi: 10.1002/jnr.21011. [DOI] [PubMed] [Google Scholar]
- Kim YS, Joh TH. Microglia, major player in the brain inflammation: their roles in the pathogenesis of Parkinson's disease. Exp Mol Med. 2006;38:333–347. doi: 10.1038/emm.2006.40. [DOI] [PubMed] [Google Scholar]
- Kleinert H, Pautz A, Linker K, Schwarz PM. Regulation of the expression of inducible nitric oxide synthase. Eur J Pharmacol. 2004;500:255–266. doi: 10.1016/j.ejphar.2004.07.030. [DOI] [PubMed] [Google Scholar]
- Kröncke KD, Fehsel K, Kolb-Bachofen V. Inducible nitric oxide synthase in human diseases. Clin Exp Immunol. 1998;113:147–156. doi: 10.1046/j.1365-2249.1998.00648.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai AY, Todd KG. Microglia in cerebral ischemia: molecular actions and interactions. Can J Physiol Pharmacol. 2006;84:49–59. doi: 10.1139/Y05-143. [DOI] [PubMed] [Google Scholar]
- Laporte JD, Moore PE, Lahiri T, Schwartzman IN, Panettieri RA, Jr, Shore SA. p38 MAP kinase regulates IL-1β responses in cultured airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2000;279:L932–L941. doi: 10.1152/ajplung.2000.279.5.L932. [DOI] [PubMed] [Google Scholar]
- Lee SJ. Korean Folk Medicine. Seoul: Seoul National University Press; 1966. [Google Scholar]
- Lin AW, Chang CC, McCormick CC. Molecular cloning and expression of an avian macrophage nitric-oxide synthase cDNA and the analysis of the genomic 5′-flanking region. J Biol Chem. 1996;271:11911–11919. doi: 10.1074/jbc.271.20.11911. [DOI] [PubMed] [Google Scholar]
- Liu B, Hong JS. Role of microglia in inflammation-mediated neurodegenerative diseases: mechanisms and strategies for therapeutic intervention. J Pharmacol Exp Ther. 2003;304:1–7. doi: 10.1124/jpet.102.035048. [DOI] [PubMed] [Google Scholar]
- Liu W, Reinmuth N, Stoeltzing O, Parikh AA, Tellez C, Williams S, et al. Cyclooxygenase-2 is up-regulated by interleukin-1β in human colorectal cancer cells via multiple signaling pathways. Cancer Res. 2003;63:3632–3636. [PubMed] [Google Scholar]
- Loddick SA, Rothwell NJ. Mechanisms of tumor necrosis factor alpha action on neurodegeneration: interaction with insulin-like growth factor-1. Proc Natl Acad Sci USA. 1999;96:9449–9451. doi: 10.1073/pnas.96.17.9449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowenstein CJ, Alley EW, Raval P, Snowman AM, Snyder SH, Russell SW, et al. Macrophage nitric oxide synthase gene: two upstream regions mediate induction by interferon gamma and lipopolysaccharide. Proc Natl Acad Sci USA. 1993;90:9730–9734. doi: 10.1073/pnas.90.20.9730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine. 2008;42:145–151. doi: 10.1016/j.cyto.2008.01.006. [DOI] [PubMed] [Google Scholar]
- Minghetti L, Levi G. Microglia as effector cells in brain damage and repair: focus on prostanoids and nitric oxide. Prog Neurobiol. 1998;54:99–125. doi: 10.1016/s0301-0082(97)00052-x. [DOI] [PubMed] [Google Scholar]
- Molina-Holgado E, Ortiz S, Molina-Holgado F, Guaza C. Induction of COX-2 and PGE2 biosynthesis by IL-1β is mediated by PKC and mitogen-activated protein kinases in murine astrocytes. Br J Pharmacol. 2000;131:152–159. doi: 10.1038/sj.bjp.0703557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollace V, Muscoli C, Masini E, Cuzzocrea S, Salvemini D. Modulation of prostaglandin biosynthesis by nitric oxide and nitric oxide donors. Pharmacol Rev. 2005;57:217–252. doi: 10.1124/pr.57.2.1. [DOI] [PubMed] [Google Scholar]
- Nakajima T, Kinoshita S, Sasagawa T, Sasaki K, Naruto M, Kishimoto T, et al. Phosphorylation at threonine-235 by a ras-dependent mitogen-activated protein kinase cascade is essential for transcription factor NF-IL6. Proc Natl Acad Sci USA. 1993;90:2207–2211. doi: 10.1073/pnas.90.6.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyo H, Jou I, Jung S, Hong S, Joe EH. Mitogen-activated protein kinases activated by lipopolysaccharide and beta-amyloid in cultured rat microglia. Neuroreport. 1998;9:871–874. doi: 10.1097/00001756-199803300-00020. [DOI] [PubMed] [Google Scholar]
- Sriram K, O'Callaghan JP. Divergent roles for tumor necrosis factor-alpha in the brain. J Neuroimmune Pharmacol. 2007;2:140–153. doi: 10.1007/s11481-007-9070-6. [DOI] [PubMed] [Google Scholar]
- Stoll G, Jander S. The role of microglia and macrophages in the pathophysiology of the CNS. Prog Neurobiol. 1999;58:233–247. doi: 10.1016/s0301-0082(98)00083-5. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Mrak RE, Griffin WS. Microglia and neuroinflammation: a pathological perspective. J Neuroinflammation. 2004;1:14. doi: 10.1186/1742-2094-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung SS, Walters JA. Increased cyclic AMP levels enhance IL-1α and IL-1β mRNA expression and protein production in human myelomonocytic cell lines and monocytes. J Clin Invest. 1991;88:1915–1923. doi: 10.1172/JCI115515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada J, Saito K, Waterman WR, Webb AC, Auron PE. Transcription factors NF-IL6 and CREB recognize a common essential site in the human prointerleukin 1β gene. Mol Cell Biol. 1994;14:7285–7297. doi: 10.1128/mcb.14.11.7285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Arakawa T, Ueda N, Yamamoto S. Transcriptional roles of nuclear factor kappa B and nuclear factor-interleukin-6 in the tumor necrosis factor alpha-dependent induction of cyclooxygenase-2 in MC3T3-E1 cells. J Biol Chem. 1995;270:31315–31320. doi: 10.1074/jbc.270.52.31315. [DOI] [PubMed] [Google Scholar]
- Zhai W, Eynott PR, Oltmanns U, Leung SY, Chung KF. Mitogen-activated protein kinase signalling pathways in IL-1β-dependent rat airway smooth muscle proliferation. Br J Pharmacol. 2004;143:1042–1049. doi: 10.1038/sj.bjp.0705971. [DOI] [PMC free article] [PubMed] [Google Scholar]