

Abstract
Background and purpose:
Ischaemic preconditioning (IPC) and ischaemic post-conditioning (IPoC) activate signal transduction pathways that are also involved in receptor de- and re-sensitization such as phosphatidylinositol (PI) 3-kinase. Therefore, IPC and IPoC may affect post-infarct receptor coupling.
Experimental approach:
Rat isolated hearts (Langendorff mode, constant flow) were exposed to 45 min flow arrest followed by 120 min reperfusion, including IPC or IPoC. Control hearts were perfused without a 45 min flow arrest. Left ventricular developed pressure (LVdevP) was determined. Thirty min after reperfusion, hearts were exposed to parathyroid hormone-related peptide (PTHrP) or isoprenaline for 10 min to monitor receptor responsiveness. Reperfusion injury was quantified by enzyme release.
Key results:
IPC and IPoC significantly reduced enzyme release compared with ischaemia and reperfusion alone by 75% and 62% respectively. Wortmannin or chelerythrine inhibiting either PI 3-kinase or protein kinase C, respectively, attenuated protection. Application of PTHrP 30 min after reperfusion did not change LVdevP in hearts exposed to ischaemia (+1 ± 11%), but IPoC restored the normal and non-ischaemic response to PTHrP characterized by a negative inotropism (−8.3 ± 3.9% and −12.9 ± 6.1%). IPC restored a small negative inotropic effect (−4.4 ± 4.7%). Application of a PTHrP receptor antagonist during the 45 min flow arrest attenuated receptor desensitization (ΔLVdevP: −6.1 ± 1.7%). Wortmannin but not chelerythrine attenuated the re-sensitizing effect of IPoC on post-ischaemic receptor coupling (ΔLVdevP: +6.2 ± 10.5 and −15.0 ± 7.7%). As observed with PTHrP receptors, IPoC restored β-adrenoceptors (ΔLVdevP: +9.3 ± 11.8% vs. 62.3 ± 15.8%).
Conclusions and implications:
IPoC restores PTHrP receptor coupling in a PI 3-kinase-dependent way. A similar mechanism may allow β-adrenoceptor re-sensitization.
Keywords: β-adrenoceptors, parathyroid hormone-related peptide, receptor re-sensitization
Introduction
Reperfusion therapy in acute myocardial infarction has significantly improved patient survival (van Domburg et al., 2005). However, reperfusion alone has its limitations due to the induction of reperfusion injury and the mechanism(s) underlying reperfusion injury are matters for debate. However, it is generally accepted that several procedures reduce infarct size, when they are evoked at the onset of reperfusion (Garcia-Dorado et al., 2006; Gross and Auchampach, 2007). One of the most prominent procedures is the so-called ischaemic post-conditioning (IPoC) that mimics some aspects of ischaemic preconditioning (IPC), and IPC and IPoC are safe when applied clinically (Staat et al., 2005; Ramzy et al., 2006). Nevertheless, many experimental studies have convincingly shown that there is negligible functional improvement of the heart, despite significant reduction of infarct size (Zhao et al., 2003). Moreover, even the effect on infarct size seems to be a delay, rather than an attenuation (Watanabe et al., 2006). On the other hand, this delay in the progressive loss of viable myocardium may provide a therapeutic opportunity if it offers more time in which to start post-infarct therapy that is aimed at attenuating the subsequent loss of function. In order to develop such treatment regimes, it is necessary to know more about receptor function in the post-infarcted myocardium.
We have recently identified an endothelium-derived peptide, parathyroid hormone-related peptide (PTHrP), as an endogenous cardiac protective factor that improves post-ischaemic function (Jansen et al., 2003; Grohéet al., 2004; Lüttecke et al., 2005). Ischaemia-dependent release of PTHrP from endothelial cells is sufficient to improve post-ischaemic heart function after brief periods of ischaemia (Jansen et al., 2003; Grohéet al., 2004). However, this effect is attenuated in prolonged ischaemia due to receptor desensitization because endogenously released PTHrP desensitizes PTHrP receptors (Ross et al., 2007). Therefore, hearts do not benefit either from endogenously released PTHrP or from PTHrP added to the perfusate after prolonged ischaemia. In the present study we hypothesized that IPC or IPoC may re-sensitize PTHrP receptors of the post-ischaemic heart. Although PTHrP is able to improve the function of post-ischaemic hearts it acts as a negative inotropic peptide in non-ischaemic hearts. Thus, even if our hypothesis is right, that PTHrP responsiveness can be reactivated by IPC or IPoC, it is an open question whether this would have beneficial effects for the functional recovery of post-ischaemic hearts.
Ischaemic preconditioning and IPoC activate multiple signal transduction pathways, called the reperfusion injury salvage kinase (RISK) pathway. Among signal transduction processes caused by IPC and IPoC, phosphatidylinositol (PI) 3-kinase activation has been identified leading to a subsequent activation of Akt and p70rsk (Hausenloy and Yellon, 2006). However, PI 3-kinase activation is also important for receptor trafficking and therefore may be involved in receptor re-sensitization (Drake et al., 2006). In the present study we hypothesized therefore, that IPoC may restore receptor coupling to PTHrP after long-term ischaemia. IPC activates similar kinases but at different time points, namely prior to a so-called triggering period and at reperfusion. As PI 3-kinase and other parts of the RISK pathway are involved in both receptor internalization and receptor recovery, one may assume that IPC and IPoC modify receptor trafficking. Indeed, IPC is able to induce receptor internalization and desensitization (Tong et al., 2004). On the other hand, IPC and IPoC also sensitize receptors as has been shown previously for the adenosine A2b receptor (Philipp et al., 2006; Kuno et al., 2007). Whether IPC or IPoC modify PTHrP receptor responsiveness is not known at present and that possibility was analysed in this study. Finally, we addressed the question whether the proposed effect on post-infarct receptor coupling was more generally applicable, that is, to other receptors. For that purpose we chose to study β-adrenoceptor stimulation. As an experimental model we used a constant flow, non-working heart preparation in order to uncouple vascular resistance from perfusion-dependent oxygen supply allowing the study of myocardial function independent of corresponding vascular effects. In these non-working hearts left ventricular developed pressure (LVdevP) recovers well, in the first 30 min of reperfusion, but then falls (Grohéet al., 2004; Ross and Schlüter, 2005; Ross et al., 2007). Therefore, all hearts were treated at this time point, to provide a similar base-line function.
Methods
Animals and analysis of left ventricular developed pressure
All animal procedures in this investigation conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institute of Health (NIH Publication No. 85-23, revised 1996). Adult (5–6 months, 190–250 g) female Wistar rats were used to investigate the impact of ischaemia, IPC or IPoC on post-ischaemic receptor responsiveness. Experiments were performed on isolated hearts from these rats. Hearts were rapidly excised, and the aorta was cannulated for retrograde perfusion with a 16-gauge needle connected to a Langendorff perfusion system. A polyvinyl chloride balloon was inserted into the left ventricle through the mitral valve and held in place by a suture tied around the left atrium. The other end of the tubing was connected to a pressure transducer for continuous measurement of left ventricular pressure. A second transducer connected to the perfusion line just before the heart was used to measure coronary perfusion pressure. The perfusion system consisted of a warmed storage container for perfusate solutions, a rotary pump and a temperature-controlled chamber. Hearts were perfused with an oxygenated saline medium [composition of the perfusate (in mmol·L−1): 140.0 NaCl, 24.0 NaHCO3, 2.7 KCl, 0.4 NaH2PO4, 1.0 MgCl2, 1.8 CaCl2 and 5.0 glucose, gassed with 95% O2-5% CO2, pH 7.4]. After attachment to the Langendorff system, hearts were allowed to stabilize for at least 20 min. The intraventricular balloon was inflated to give a diastolic pressure of 10 mmHg, and the balloon volume was held constant thereafter. The flow was adjusted to give a perfusion pressure of 50 mmHg. Parameters determined were left ventricular diastolic pressure and left ventricular systolic pressure. The developed pressure (LVdevP) was calculated as LVdevP = peak systolic pressure − diastolic pressure. To avoid differences in oxygen supply due to changes in the coronary resistance, hearts were perfused at a constant flow. Hearts were allowed to beat freely. Temperature was held constant at 37°C throughout the whole experiment.
Perfusion protocols
The study consisted of three parts. The different protocols are summarized in Figure 1. In the first part of the study the following protocols were used to investigate the effect of ischaemia and reperfusion (I/R), IPC and IPoC on post-infarct receptor coupling: I/R: 45 min flow arrest and 120 min reperfusion; IPC: three cycles of 3 min flow arrest followed by 5 min reperfusion prior to ischaemia and subsequent flow arrest for 45 min followed by reperfusion for 120 min; IPoC: 45 min flow arrest followed by three cycles of 30 s perfusion and 30 s flow arrest and reperfusion for 117 min; control (C): control experiments were performed on perfused hearts without a 45 min flow arrest as time-matched normoxic controls.
Figure 1.
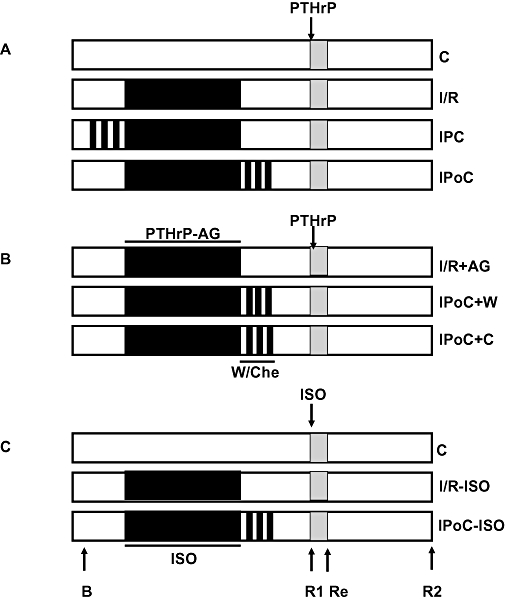
Schematic overview of the experimental design. The study was performed in three steps (A–C). Flow was stopped during the ischaemic periods, which are indicated in black. Functional data were recorded before any intervention (basal, B), 30 min after the onset of reperfusion (R1), 10 min after the addition of PTHrP (100 nmol·L−1) or isoprenaline (ISO, 100 nmol·L−1) (Response, Re) or at the end of the experiments (R2). Where indicated wortmannin (W, 100 nmol·L−1) or chelerythrine (Che, 1 µmol·L−1) were added. When isoprenaline was given during ischaemia, its concentration was 10 nmol·L−1. When the activity of PTHrP released during ischaemia was inhibited, a PTHrP antagonist [PTHrP-AG = PTHrP(7–34), 100 nmol·L−1] was given. I/R, ischaemia and reperfusion; IPC, ischaemic preconditioning; IPoC, ischaemic post-conditioning; PTHrP, parathyroid hormone-related peptide.
Each of the four groups consisted of 16 hearts. Eight of them were exposed to PTHrP(1–36) (100 nmol·L−1) 30 min after reperfusion for 10 min, and the other eight hearts per group received vehicle only (Figure 1A). LVdevP was measured at the end of the stabilization period (Figure 1, arrow ‘B’), after 30 min of reperfusion (Figure 1, arrow ‘R1’), 10 min after the subsequent addition of PTHrP or vehicle (Figure 1, arrow ‘Re’) or at the end of the reperfusion period (Figure 1, arrow ‘R2’). PTHrP receptor responsiveness was defined as the change in LVdevP between ‘R1’ and ‘Re’.
In the second part of the study we investigated (i) whether ischaemia-dependent endogenously released PTHrP desensitized the PTHrP receptors; and (ii) whether the effect of IPoC on PTHrP receptor responsiveness depends on PI 3-kinase and protein kinase C (PKC) activation (Figure 1B). For this purpose, hearts received a PTHrP receptor antagonist [PTHrP(7–34), 100 nmol·L−1)] prior to the 45 min flow arrest. The antagonist was washed out with reperfusion. In order to show that the effect of IPoC on PTHrP receptor coupling depends on PI 3-kinase activation, the IPoC was performed in the presence of wortmannin (100 nmol·L−1) or chelerythrine (1 µmol·L−1) (each n = 8). Parameters under investigation in this part of the study were basal function (Figure 1, arrow ‘B’), 30 min recovery (Figure 1, arrow ‘R1’) and PTHrP responsiveness (Figure 1, ‘Re’).
In the third part of the study we investigated whether IPoC restored β-adrenoceptor coupling (Figure 1C). For this purpose, hearts were exposed to isoprenaline (10 nmol·L−1) prior to the flow arrest, and isoprenaline was washed out during reperfusion. Isoprenaline (100 nmol·L−1) was reperfused, 30 min after starting reperfusion. This part of the study contained another three groups of eight hearts: control (C), which represents a normoxic control perfusion, I/R or IPoC in which isoprenaline was present during 45 min no-flow arrest. Parameters under investigation in this part of the study were basal function (Figure 1, arrow ‘B’), 30 min recovery (Figure 1, arrow ‘R1’) and isoprenaline responsiveness (Figure 1, ‘Re’).
Finally, another 12 hearts were used to monitor activation of the kinase, p70rsk. For that, hearts were exposed to the I/R protocol, IPoC protocol and IPoC protocols with wortmannin or chelerythrine added at reperfusion and keeping the antagonists in the perfusate during the first 5 min of reperfusion (IPoC + W and IPoC + C, Figure 1). In these experiments, reperfusion was stopped after 15 min, and the left ventricles were excised and freeze-clamped in liquid nitrogen. Tissue samples were dissolved in lysis buffer as described previously (Ruf et al., 2002). Levels of p70rsk activity were measured with a commercially available p70 S6K activity assay (Assay Designs, Inc., Ann Arbor, USA) according to the manufacturer's instructions. Briefly, tissue samples were normalized to their total protein levels and incubated with a substrate peptide of the kinase. Phosphorylation of this peptide was determined by application of an antibody directed against the phosphorylated form of the substrate peptide.
Determination of cardiac protection
In order to address the question whether the IPC and IPoC protocols described above, did protect the myocardium, we analysed perfusate samples during the first 15 min of reperfusion every 5 min. The total amount of lactate dehydrogenase (LDH) was measured, as a biochemical correlate of infarct size. The procedure to quantify LDH activity has been published before (Schlüter et al., 1991). From the three different values a curve of LDH release was plotted, and the results are expressed as area under curve (AUC). Perfusate samples at later time points (30, 40, 60 and 90 after the onset of reperfusion) were also assayed for LDH activity but these values did not exceed the basal values of normoxic control hearts. Therefore, they were not included in the calculation of enzyme release.
Statistics
Quantitative results were expressed as means ± SEM as indicated. In experiments in which more than two groups were compared with each other, one-way analysis of variance was used, with Student-Newman-Keuls test for post hoc analysis. P < 0.05 was used as a level of significance.
Materials
Synthetic PTHrP(1–36) was used in this study in order to stimulate PTHrP receptors, and synthetic PTHrP(7–34) was used as an antagonist. These peptides were obtained from Bachem. They were dissolved in double distilled water with addition of 1% (v/v) acetic acid according to the manufacturer's suggestions. The concentration used for PTHrP was obtained from the previously established concentration–response relationship in the same model (Grohéet al., 2004). Isoprenaline was obtained from Sigma/RBI and dissolved in double distilled water without further additions. Wortmannin and chelerythrine were obtained from Calbiochem, dissolved in dimethylsulphoxide (DMSO) and stored as stock solutions (100 µmol·L−1).
Results
Cardiac protective effects of IPC and IPoC
Firstly, we showed that IPC and IPoC procedures used in this study, were cardio-protective. LDH release, taken as a biochemical indicator of infarct size, was increased in I/R compared with normoxic controls, but significantly reduced in IPC and IPoC (Figure 2). These data demonstrate the well-known cardio-protective effects of IPC and IPoC on the myocardial structure in the Langendorff model used for this study. The cardio-protective effect of IPoC was attenuated if PI 3-kinase or PKC activation during IPoC was attenuated by application of wortmannin or chelerythrine respectively (Figure 2). The data confirmed the contribution of both kinases in the salvage pathway activated to reduce infarct sizes. As PI 3-kinase activation is part of a salvage pathway involving PI 3-kinase, Akt and p70rsk, we confirmed the suitability of wortmannin to block this pathway on the biochemical levels as well. IPoC increased the p70rsk activity by 69.0 ± 8.8% (Figure 3, n = 5). When the PI 3-kinase inhibitor wortmannin was given during reperfusion the IPoC-induced activation of p70rsk was abolished. However, the PKC inhibitor chelerythrine did not attenuate p70rsk activation in this model (Figure 3).
Figure 2.
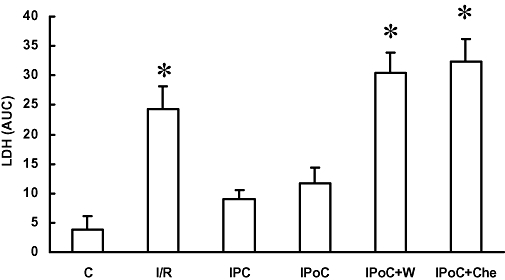
Effects on reperfusion injury, of ischaemia and reperfusion (I/R), ischaemic preconditioning (IPC), ischaemic post-conditioning (IPoC), IPoC with wortmannin (IPoC + W) and IPoC with chelerythrine (IPoC + Che). Reperfusion injury was determined by lactate dehydrogenase (LDH) release into the perfusate. Samples were collected after 5, 10 and 15 min and the area under curve (AUC) was calculated. Data are shown in comparison with normoxic time controls (C). Data are expressed as means ± SEM, n = 8 in each group. *P < 0.05 vs. C.
Figure 3.
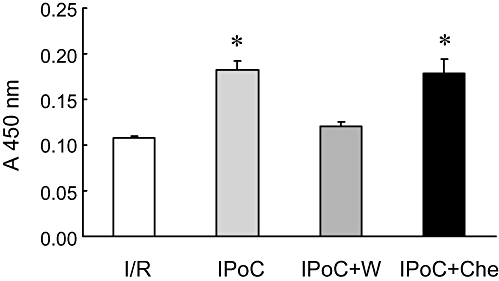
Effect of ischaemic post-conditioning (IPoC) and its inhibition by wortmannin (W) and chelerythrine (Che) on the activation of the reperfusion injury salvage kinase pathway as monitored by p70rsk activity, measured by phosphorylation of a p70rsk substrate peptide and quantified on a microplate reader. Data are expressed as absorbance at 450 nm and are means ± SEM (n = 5). *P < 0.05 vs. ischaemia and reperfusion (I/R).
Effect of IPC and IPoC on PTHrP receptor responsiveness
In order to test our hypothesis that IPoC re-sensitizes PTHrP receptors, we performed experiments in an I/R model, an IPC model and an IPoC model. These results were compared with those obtained for time-matched non-ischaemic controls (C). Hearts were either treated with PTHrP or vehicle after 30 min of reperfusion for the next 10 min. In additional experiments ischaemia was performed in the presence of a PTHrP antagonist, or IPoC was performed in the presence of either wortmannin or chelerythrine. There were no differences on baseline characteristics between the rat hearts from the 11 different groups. LVdevP ranged from 135 to 152 mmHg between these groups (data not shown). Within the first 30 min of reperfusion, the initial depression of cardiac function was improved to nearly normoxic control values in all groups undergoing 45 min flow arrest, irrespective of the treatment regime. Based on the nearly identical 30 min recovery values we decided to test receptor responsiveness at that time point. In Figure 4, these values are given for the seven groups exposed to PTHrP thereafter. When PTHrP was applied 30 min after reperfusion the peptide significantly reduced LVdevP in the IPoC group as it did in normoxic control hearts (Figure 5). However, in the I/R group, PTHrP was unable to change the LVdevP. In the IPC group, a moderate decrease of LVdevP was observed (Figure 5). These data suggested that the endogenous PTHrP, released during ischaemia, desensitized PTHrP receptors and that IPoC re-sensitized the receptor responsiveness. In order to prove this assumption directly, we performed additional experiments in which the endogenously released PTHrP was antagonized by application of a PTHrP receptor antagonist during ischaemia. As expected, PTHrP responsiveness was not attenuated in this group (Figure 5; I/R + AG). In order to test directly whether PI 3-kinase activation during IPoC is responsible for the re-sensitization of PTHrP receptors in this group, we added either wortmannin or chelerythrine at the onset of reperfusion. The data show that wortmannin but not chelerythrine attenuated the recovery of receptor responsiveness induced by IPoC (Figure 5; IPoC + W and IPoC + C).
Figure 4.
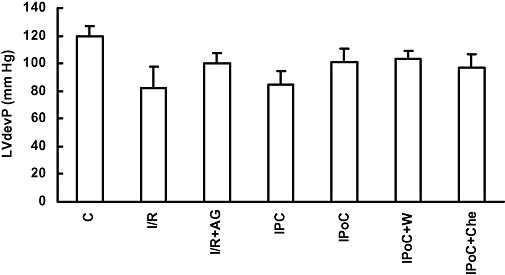
Functional recovery during reperfusion in groups with ischaemia and reperfusion (I/R), ischaemic preconditioning (IPC), ischaemic post-conditioning (IPoC), IPoC and wortmannin (W), IPoC and chelerythrine (Che) and normoxic controls (C). Left ventricular developed pressure (LVdevP) was measured during the first 30 min of reperfusion. Data are means ± SEM; n = 8–16 in each group. P < 0.05 n.s. vs. C. AG, antagonist.
Figure 5.
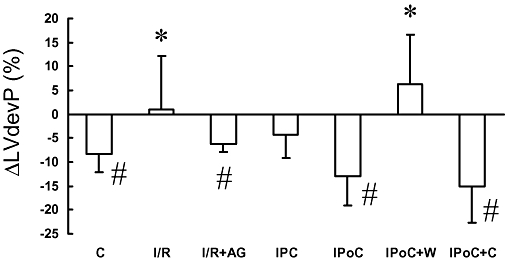
Acute effects of parathyroid hormone-related peptide (PTHrP) (100 nmol·L−1) on functional parameters for the ischaemia and reperfusion (I/R), ischaemic preconditioning (IPC), ischaemic post-conditioning (IPoC), normoxic control (C) group, IPoC with wortmannin (W) and IPoC with chelerythrine (Che). PTHrP was perfused, 30 min after the onset of reperfusion, for 10 min. Data shown in this figure are the changes, 10 min after drug application. Data are given for left ventricular developed pressure (LVdevP) and represent means ± SEM; n = 8 in each group. *P < 0.05 vs. C; #P < 0.05 vs. baseline before application. For the absolute values before application of PTHrP or vehicle see Figure 4. AG, antagonist.
Effect of late PTHrP receptor stimulation of functional recovery
Continuation of reperfusion to 120 min revealed small differences in functional recovery between I/R, IPC and IPoC (Figure 6). In all three groups the mean LVdevP was lower than in the normoxic control group. However, in the IPoC group the mean LVdevP at the end of the reperfusion was not significantly different from normoxic controls. In normoxic controls, transient stimulation of PTHrP receptors had no sustained effect on LVdevP. In the I/R group, PTHrP receptor stimulation did not evoke an acute effect and, in line with this, it did not change LVdevP throughout the reperfusion (Figure 6). In the IPC group, PTHrP receptors were slightly responsive and these hearts showed a slight decrease in LVdevP that did not reach the level of significance. However, in the IPoC group PTHrP receptors showed a negative inotropic effect and, if IPoC was combined with PTHrP receptor stimulation, the mean LVdevP was slightly lower at the end of the experiments compared with IPoC alone (Figure 6).
Figure 6.
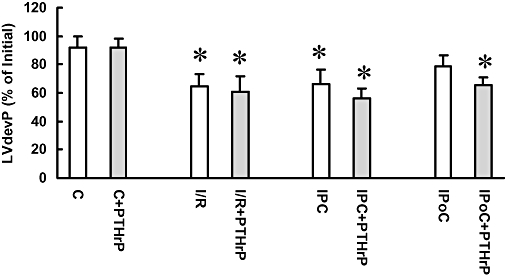
Effect of cardiac protection and parathyroid hormone-related peptide (PTHrP) receptor stimulation on recovery over 120 min. Left ventricular developed pressure (LVdevP) was measured for the ischaemia and reperfusion (I/R), ischaemic preconditioning (IPC), ischaemic post-conditioning (IPoC), normoxic control (C) group and expressed as percentage of initial values (mean: 145 ± 8 mmHg). In the PTHrP groups, the peptide was perfused at 30 min of reperfusion and maintained for 10 min, before the peptide was washed out again. Data are means ± SEM; n = 8 in each group, *P < 0.05 vs. C or C + PTHrP.
Effect of IPoC on β-adrenoceptor responsiveness
To determine if the observed effect of IPoC was specific for the PTHrP receptor or if it might apply to receptors in general, we tested the hypothesis that IPoC re-sensitizes β-adrenoceptors as well. In contrast to PTHrP receptors, that rapidly desensitize by endogenously released PTHrP during 45 min flow arrest, β-adrenoceptors did not desensitize by flow arrest, per se. Subsequent addition of isoprenaline (10 nmol·L−1) to the perfusate at 30 min after reperfusion caused an increase in LVdevP of 7.3 ± 1.6% in control hearts, 12.7 ± 3.4% in hearts exposed to I/R, 7.2 ± 4.2% in hearts exposed to IPC and 9.1 ± 3.7% in hearts exposed to IPoC (each n = 6). However, when isoprenaline (10 nmol·L−1) was added prior to the 45 min flow arrest and maintained during ischaemia, a loss of post-ischaemic receptor coupling was observed for β-adrenoceptors as well (Figure 7, I/R). As expected, non-ischaemic hearts responded to isoprenaline (100 nmol·L−1) with a significant increase in LVdevP. Isoprenaline responsiveness was restored in hearts pre-exposed to isoprenaline (10 nmol·L−1) during the 45 min flow arrest, if an IPoC procedure was performed (Figure 7).
Figure 7.
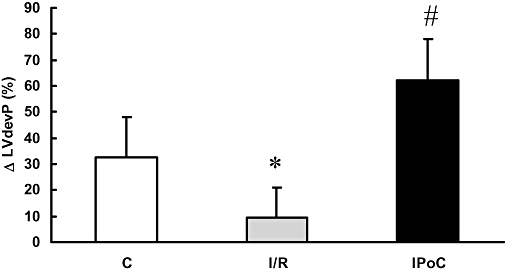
Effect of ischaemic post-conditioning (IPoC) on β-adrenoceptor coupling in hearts with desensitized β-adrenoceptors. Normoxic control hearts were compared with hearts undergoing 45 min flow arrest in the presence of isoprenaline (10 nmol·L−1) subsequently followed by reperfusion (I/R) or IPoC and reperfusion. Isoprenaline (100 nmol·L−1) was perfused 30 min after the onset of reperfusion, and changes in heart function were determined. Data shown in this figure are the changes 5 min after drug application. Data are given for left ventricular developed pressure (LVdevP) and represent means ± SEM; n = 8 in each group; *P < 0.05 vs. C; #P < 0.05 vs. I/R.
Discussion
The present study investigated the hypothesis that IPoC influences receptor coupling in the post-ischaemic heart. The study focused on the responsiveness of the PTHrP receptor, for the following reasons. We had previously shown that an ischaemia-dependent release of PTHrP contributed to the functional recovery of hearts after a moderate period of ischaemia in vitro and in vivo (Jansen et al., 2003; Grohéet al., 2004). However prolonged ischaemia leads to receptor desensitization and thereby to a loss of the cardiac protective effects of PTHrP (Ross et al., 2007). IPoC activates PI 3-kinase pathways that are involved in receptor trafficking in cells. Therefore, IPoC may modify normal PTHrP receptor coupling in post-ischaemic hearts. We found that IPoC restored anormoxic-like receptor coupling in these hearts. Moreover, we demonstrated that IPoC restored β-adrenoceptor responsiveness, too, as long as these receptors were desensitized during ischaemia. IPoC in the presence of wortmannin did not restore receptor coupling. We conclude that PI 3-kinase activation was causally involved in the restoration of receptor coupling.
The role of PTHrP in the response of the heart to I/R has been characterized in greater detail before. While PTHrP exerts a small negative contractile effect in normoxic hearts and isolated cardiomyocytes, it improves contractile performance in the post-ischaemic heart and cardiomyocytes (Jansen et al., 2003; Grohéet al., 2004; Lüttecke et al., 2005). However, this response is lost during prolonged ischaemia, as shown before (Ross et al., 2007). This was reconfirmed in our new study. In a previous study it was found that desensitization of PTHrP receptors (also known as PTH type 1 receptors) enables PTH type 2 receptors to dilate cardiac vessels. This was shown by application of the specific PTH type 2 receptor agonist, the tuberoinfundibular peptide of 39 residues (TIP39) (Ross et al., 2007). However, whether TIP39 is released in the post-ischaemic heart is not known. The lack of effectiveness on cardiac contractility further reduces the ability of TIP39 to improve cardiac function in the reperfused rat heart. We therefore investigated whether we could re-sensitize PTHrP receptors in hearts after prolonged ischaemia and whether this was accompanied by a normoxic-like negative inotropic effect or with a post-ischaemia-like positive contractile effect, as found in post-ischaemic hearts with brief ischaemia. The outcome of these experiments would indicate whether or not PTHrP is a candidate to be used as a positive inotropic mimetic in post-infarcted hearts. In this study, however, a normoxic-like negative contractile effect of PTHrP receptor stimulation was restored by IPoC and no benefit in either sustained improvement or reduction of contractile function was observed, at least under these conditions.
Receptor internalization, but also receptor recovery, requires PI 3-kinase-dependent steps (Tong et al., 2004; Drake et al., 2006). IPoC activates PI 3-kinase, and PI 3-kinase activation induces downstream events that have been summarized as the RISK pathway and include a PI 3-kinase-dependent activation of Akt and p70rsk (Hausenloy and Yellon, 2006). Therefore, we tested the hypothesis that IPoC re-sensitizes receptors via PI 3-kinase activation. We found that IPoC activates p70rsk by approximately 15% above the value found in I/R and that this was indeed antagonized by wortmannin, a PI 3-kinase inhibitor. Another kinase that also leads to cardiac protection and is part of the RISK pathways is PKC. However, although PKC inhibition by chelerythrine significantly attenuated IPoC-induced cell protection it did not modify receptor recoupling. Thus, PI 3-kinase, but not PKC-dependent, steps are part of the receptor recoupling process. Moreover, this experiment shows that the observed effect of IPoC on re-sensitization of PTHrP receptors is not simply the effect of infarct reduction but really depends on an altered functional responsiveness of the surviving cardiomyocytes. The cardio-protective effects evoked by IPoC are elicited during the first minutes of reperfusion but are also responsible for receptor responsiveness. However, receptor re-sensitization was observed 30 min after reperfusion. This is not necessarily a paradox because receptor re-sensitization requires re-expression of receptors to the sarcolemmal membrane and this requires more time than the attenuation of hypercontraction.
The observed effect of IPoC on re-sensitization of PTHrP receptors led to an acute reduction of LVdevP. However, as we have shown previously, receptor-dependent effects evoked by PTHrP, in chronic pressure-overloaded hearts, are switched into a paradoxical positive contractile effect (Ross and Schlüter, 2005). If this holds, re-establishing PTHrP receptor responsiveness in pressure-overloaded hearts may be of greater functional relevance than in hearts from normotensive rats.
In our model, the β-adrenoceptors were affected similarly to the PTHrP receptors. However, this observation may be of academic interest but not necessarily of clinical or therapeutic impact, because the β-adrenoceptors are not desensitized in the post-ischaemic heart. In this study we added isoprenaline during ischaemia and added an excess of isoprenaline to the post-ischaemic hearts in order to demonstrate such an effect of IPoC on β-adrenoceptor re-sensitization. The concentration required to induce a positive contractile effect by isoprenaline was 10-fold higher than that used in normoxic hearts. Nevertheless, although the kinetics of receptor desensitization and recovery are low for the β-adrenoceptors compared with the PTHrP receptors, these experiments indicated that IPoC restores receptor re-sensitization of another G-protein coupled receptor as well. Without any doubt this has consequences for the functional recovery of the post-ischaemic heart and in addition to the post-infarct remodelling that is caused by receptor-dependent processes.
Like IPoC, IPC activates PI 3-kinase-dependent pathways that are involved in receptor internalization and thus in receptor desensitization. Following this suggestion, it is likely hat IPC per se would desensitize PTHrP receptors. As a consequence of this, endogenously released PTHrP should not be able to improve the functional recovery in the IPC hearts. In the IPoC hearts, however receptor re-sensitization occurs, and this may improve the functional recovery. Indeed, the LVdevP was highest in the IPoC group and lowest in the IPC group at 30 min of reperfusion. However, although the different levels of recovery support this hypothesis, such differences did not reach the level of significance.
This IPC- or IPoC-dependent receptor re-sensitization has previously been observed for a subtype of adenosine receptors (A2b) (Philipp et al., 2006; Kuno et al., 2007). However, there is a marked difference between the observations made in our study and those found for adenosine A2b receptors. The adenosine receptors were not desensitized by I/R, but a hypersensitization was found with both IPC and IPoC. In contrast, the PTHrP receptors in this study were completely desensitized, and we observed a re-sensitization together with a restoration of a normoxic-like behaviour. Therefore, the mechanism may be different and the fact that sensitization of adenosine A2b receptors was found with IPC and IPoC, but re-sensitization of PTHrP receptors only with IPoC may already indicate a different mechanism.
The fact that all hearts recovered initially quite well during reperfusion is explained by the experimental model. In non-working, Langendorff-perfused hearts, recovery is not impaired by post-ischaemic vascular resistance because the hearts were perfused under constant flow conditions. Furthermore, we used a lower perfusion pressure (50 mmHg) than those reported in some other papers, for the following reason. In these saline perfused rat hearts, a perfusion pressure of 50 mmHg was sufficient to maintain heart function in normoxic hearts for 3 h. If we increased the perfusion pressure to 80 mmHg, the functional recovery was impaired but IPoC resulted in a functional improvement, relative to the effects of I/R alone (data not shown). However, under these conditions there is a high risk for oedema formation. Thus, at such high perfusion pressures of saline-perfused hearts, it is difficult to distinguish between functional recovery due to improved cardiomyocyte function and that due to improving vascular defects.
In conclusion, this is the first study reporting the influence of IPoC on PTHrP receptor coupling in the post-ischaemic heart. Further studies are required to investigate the relevance of these findings on post-infarct remodelling, that is, altered growth responses to agonists that contribute to reactive myocardial hypertrophy. Our study shows that the effect of IPC and IPoC on infarct size is independent of effects on restoration of receptor recoupling. We need also to identify the signalling steps involved in this process, and finally these results need to be transferred from the rat heart model used in this study to models that are more closely linked to clinical practice, that is, hearts from hypertensive rats and/or aged animals.
Acknowledgments
This study was supported by the Deutsche Forschungsgemeinschaft, SFB 547, project A1.
Glossary
Abbreviations:
- AUC
area under curve
- I/R
ischaemia and reperfusion
- IPC
ischaemic preconditioning
- IPoC
ischaemic post-conditioning
- LVdevP
left ventricular developed pressure
- PI 3-kinase
phosphatidylinositol 3-kinase
- PKC
protein kinase C
- PTHrP
parathyroid hormone-related peptide
- RISK
reperfusion injury salvage kinase
Conflict of interest
None.
References
- Drake MT, Shenoy SK, Lefkowitz RJ. Trafficking of G protein-coupled receptors. Circ Res. 2006;99:570–582. doi: 10.1161/01.RES.0000242563.47507.ce. [DOI] [PubMed] [Google Scholar]
- Garcia-Dorado D, Vinten-Johansen J, Piper HM. Bringing preconditioning and postconditioning into focus. Cardiovasc Res. 2006;70:167–169. doi: 10.1016/j.cardiores.2006.03.009. [DOI] [PubMed] [Google Scholar]
- Grohé C, van Eickels M, Wenzel S, Meyer R, Degenhardt H, Doevendans PA, et al. Sex-specific differences in ventricular expression and function of parathyroid hormone-related peptide. Cardiovasc Res. 2004;61:307–316. doi: 10.1016/j.cardiores.2003.11.025. [DOI] [PubMed] [Google Scholar]
- Gross GJ, Auchampach JA. Reperfusion injury: does it exist? J Mol Cell Cardiol. 2007;42:12–18. doi: 10.1016/j.yjmcc.2006.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausenloy DJ, Yellon DM. Survival kinases in ischemic preconditioning and postconditioning. Cardiovasc Res. 2006;70:240–253. doi: 10.1016/j.cardiores.2006.01.017. [DOI] [PubMed] [Google Scholar]
- Jansen J, Gres P, Umschlag C, Heinzel FR, Degenhardt H, Schlüter K-D, et al. Parathyroid hormone-related peptide improves contractile function of stunned myocardium in rats and pigs. Am J Physiol Heart Circ Physiol. 2003;284:H49–H55. doi: 10.1152/ajpheart.01037.2001. [DOI] [PubMed] [Google Scholar]
- Kuno A, Critz SD, Cui L, Solodushko V, Yang X-M, Krahn T, et al. Protein kinase C protects preconditioning rabbit hearts by increasing sensititvity of adenosine A2b-dependent signaling during early reperfusion. J Mol Cell Cardiol. 2007;43:262–271. doi: 10.1016/j.yjmcc.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüttecke D, Ross G, Abdallah Y, Schäfer C, Piper HM, Schlüter K-D. Parathyroid hormone-related peptide improves contractile responsiveness of adult rat cardiomyocytes with depressed cell function irrespectively of oxidative inhibition. Basic Res Cardiol. 2005;100:320–327. doi: 10.1007/s00395-005-0532-9. [DOI] [PubMed] [Google Scholar]
- Philipp S, Yang X-M, Cui L, Davis AM, Downey JM, Cohen MV. Preconditioning protects rabbit hearts through a protein kinase C-adenosine A2b receptor cascade. Cardiovasc Res. 2006;70:308–314. doi: 10.1016/j.cardiores.2006.02.014. [DOI] [PubMed] [Google Scholar]
- Ramzy D, Rao V, Weisel RD. Clinical applicability of preconditioning and postconditioning: the cardiothoracic surgeons's. view. Cardivasc Res. 2006;70:174–180. doi: 10.1016/j.cardiores.2006.01.020. [DOI] [PubMed] [Google Scholar]
- Ross G, Schlüter K-D. Cardiac-specific effects of parathyroid hormone-related peptide: modification by aging and hypertension. Cardiovasc Res. 2005;66:334–344. doi: 10.1016/j.cardiores.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Ross G, Heinemann MP, Schlüter K-D. Vasodilatory effect of tuberoinfundibular peptide (TIP39): requirement of receptor desensitization and its beneficial effect in the post-ischemic heart. Peptides. 2007;28:878–886. doi: 10.1016/j.peptides.2006.12.010. [DOI] [PubMed] [Google Scholar]
- Ruf S, Piper HM, Schlüter K-D. Specific role for the extracellular signal-regulated kinase pathway in angiotensin II- but not phenylephrine-induced cardiac hypertrophy in vitro. Pflügers Arch – Eur J Physiol. 2002;443:483–490. doi: 10.1007/s004240100710. [DOI] [PubMed] [Google Scholar]
- Schlüter K-D, Schwartz P, Siegmund B, Piper HM. Prevention of the oxygen-paradox in hypoxic-reoxygenated hearts. Am J Physiol Heart Circ Physiol. 1991;261:H416–H423. doi: 10.1152/ajpheart.1991.261.2.H416. [DOI] [PubMed] [Google Scholar]
- Staat P, Rioufol G, Piot C, Cottin Y, Cung TT, L’Huillier I, et al. Postconditioning the human heart. Circulation. 2005;112:143–148. doi: 10.1161/CIRCULATIONAHA.105.558122. [DOI] [PubMed] [Google Scholar]
- Tong H, Rockman HA, Koch WJ, Steenbergen C, Murphy E. G protein-coupled receptor internalization signaling is required for cardioprotection in ischemic preconditioning. Circ Res. 2004;94:1133–1141. doi: 10.1161/01.RES.0000126048.32383.6B. [DOI] [PubMed] [Google Scholar]
- Van Domburg RT, Sonnenschein K, Nieuwlaat R, Kamp O, Storm CJ, Bax JJ, et al. Sustained benefit 20 years after reperfusion therapy in acute myocardial infarction. J Am Coll Cardiol. 2005;46:15–20. doi: 10.1016/j.jacc.2005.03.047. [DOI] [PubMed] [Google Scholar]
- Watanabe K, Yaoita H, Ogawa K, Oikawa M, Maehara K, Maruyama Y. Attenuated cardioprotection by ischemic preconditioning in coronary stenosed heart and its restoration by carvedilol. Cardiovasc Res. 2006;71:537–547. doi: 10.1016/j.cardiores.2006.05.011. [DOI] [PubMed] [Google Scholar]
- Zhao Z-Q, Corvera JS, Halkos ME, Kerendi F, Wang N-P, Guyton RA, et al. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic precondititioning. Am J Physiol Heart Circ Physiol. 2003;285:H579–H788. doi: 10.1152/ajpheart.01064.2002. [DOI] [PubMed] [Google Scholar]