

Abstract
Background and purpose:
The Na+/Ca2+ exchanger (NCX) may contribute to triggered activity and transmural dispersion of repolarization, which are substrates of torsades de pointes (TdP) type arrhythmias. This study examined the effects of selective inhibition of the NCX by SEA0400 on the occurrence of dofetilide-induced TdP.
Experimental approach:
Effects of SEA0400 (1 µmol·L−1) on dofetilide-induced TdP was studied in isolated, Langendorff-perfused, atrioventricular (AV)-blocked rabbit hearts. To verify the relevance of the model, lidocaine (30 µmol·L−1) and verapamil (750 nmol·L−1) were also tested against dofetilide-induced TdP.
Key results:
Acute AV block caused a chaotic idioventricular rhythm and strikingly increased beat-to-beat variability of the RR and QT intervals. SEA0400 exaggerated the dofetilide-induced increase in the heart rate-corrected QT interval (QTc) and did not reduce the incidence of dofetilide-induced TdP [100% in the SEA0400 + dofetilide group vs. 75% in the dofetilide (100 nmol·L−1) control]. In the second set of experiments, verapamil further increased the dofetilide-induced QTc prolongation and neither verapamil nor lidocaine reduced the dofetilide-induced increase in the beat-to-beat variability of the QT interval. However, lidocaine decreased and verapamil prevented the development of dofetilide-induced TdP as compared with the dofetilide control (TdP incidence: 13%, 0% and 88% respectively).
Conclusions and implications:
Na+/Ca2+ exchanger does not contribute to dofetilide-induced TdP, whereas Na+ and Ca2+ channel activity is involved in TdP genesis in isolated, AV-blocked rabbit hearts. Neither QTc prolongation nor an increase in the beat-to-beat variability of the QT interval is a sufficient prerequisite of TdP genesis in rabbit hearts.
Keywords: NCX, SEA0400, torsades de pointes, QT variability, isolated heart, rabbit, lidocaine, verapamil
Introduction
Torsades de pointes (TdP) is a life-threatening form of polymorphic ventricular tachycardia, which can be evoked by several cardiac or non-cardiac drugs that disturb the process of repolarization of the myocytes. According to the most widely accepted theory, the mechanism of TdP initiation (trigger) involves an early (EAD) or delayed (DAD) afterdepolarization-induced ectopic beat, while the mechanism of the arrhythmia maintenance (substrate) involves re-entry circuits produced by an increase in spatial dispersion of the repolarization of the ventricular wall (Antzelevitch and Oliva, 2006). Dofetilide, an inhibitor of the rapid component of the delayed rectifier potassium current (IKr), prolongs repolarization, induces EAD, increases dispersion of repolarization and can evoke TdP (Belardinelli et al., 2003).
The Na+/Ca2+ exchanger (NCX) is considered to play an important role in Ca2+ handling of cardiac myocytes. In forward mode, the NCX extrudes Ca2+ from the cell and brings Na+ into the cytosol with a ratio of 1 : 3 respectively; in reverse mode, it operates in the opposite direction (Bers, 2001). This exchange of three positive charges for two positive charges makes the exchanger electrogenic and this may cause substantial depolarization in forward mode, leading to EAD- and DAD-induced triggered activity (extrasystoles) (Sipido et al., 2007). Selective inhibition of the NCX by SEA0400 reduced the amplitude of both dofetilide-induced EADs and the strophanthidin-induced DADs in isolated canine ventricular papillary and Purkinje fibres (Nagy et al., 2004). This implies that NCX activity may play a role in the generation of the trigger of drug-induced TdP.
The intrinsic transmural dispersion of repolarization is amplified by any compound that reduces net repolarizing currents, such as dofetilide, by preferential prolongation of the repolarization of the midmyocardial cells (M) cells, which provide the substrate for circus movement re-entrant arrhythmias (Belardinelli et al., 2003). The NCX may contribute to the prolonged action potential of the canine M cells, which produces transmural electrical heterogeneity and re-entry circuits (Zygmunt et al., 2000). This implies that NCX activity may play a role in the generation or the maintenance mechanism of TdP.
To date, the role of NCX in the genesis of TdP has not been examined. As earlier investigations implied that the NCX may contribute to the genesis of drug-induced TdP, the present study examined the anti-arrhythmic effect of the inhibition of the NCX by a selective NCX inhibitor, SEA0400, on the occurrence of dofetilide-induced TdP in isolated, Langendorff-perfused, atrioventricular (AV) nodal-ablated rabbit hearts.
The in vitro, isolated, AV-ablated rabbit heart model is frequently used for testing the proarrhythmic liability of drugs (Thomsen et al., 2006). Interestingly, the ability of pharmacological intervention to reduce the incidence of TdP induced by K+ channel blocking drugs has not been examined in this model. Our results show that NCX inhibition with SEA0400 did not reduce the incidence of dofetilide-induced TdP. As this result questions either the role of NCX in TdP or the validity of this model, we conducted a second series of experiments aimed at validating the model with drugs known to alleviate TdP in other experimental models and in man. Thus, the anti-arrhythmic effect of the inhibition of the inward L-type Ca2+ current (ICaL) by verapamil and the inhibition of the inward Na+ current (INa) by lidocaine was tested against dofetilide-induced TdP in isolated, Langendorff-perfused, AV-ablated rabbit hearts. Also, an attempt was made to determine whether the occurrence of dofetilide-induced TdP was related to the prolongation of the heart rate-corrected QT (QTc) interval or the beat-to-beat variability of the QT interval in the applied model.
Methods
Animals and general methods
Animals were handled in accordance with the European Community guidelines for the use of experimental animals, and the protocol was reviewed and approved by the Ethical Committee for the Protection of Animals in Research at the University of Szeged, Hungary.
Female New Zealand white rabbits weighing 2.5 ± 0.24 kg were used for the experiments. The animals were anticoagulated with sodium heparin (1000 IU) injected into the marginal ear vein and stunned by a blow to the neck. The heart was rapidly removed via thoracotomy and rinsed in ice-cold modified Krebs–Henseleit buffer solution containing (in mmol·L−1): NaCl 118.5, CaCl2 1.8, glucose 11.1, MgSO4 0.5, NaH2PO4 1.2, NaHCO3 25 and KCl 3. A K+ concentration of 3 mmol·L−1 was chosen, as the proarrhythmic action of dofetilide tended to be exacerbated by perfusion with 3 mmol·L−1 K+ versus 4 mmol·L−1 K+ in isolated, AV-blocked rabbit hearts (Barrett et al., 2001). The aorta was cannulated and the heart placed in a Langendorff apparatus and hearts were retrogradely perfused with the modified Krebs–Henseleit buffer solution at a constant temperature of 37°C as described above. A mixture of 95% O2 and 5% CO2 was bubbled through the buffer, which was equilibrated to pH 7.4. All solutions were filtered (5 µm pore size filter) before use. The perfusion pressure was maintained constant at 70 mm Hg. Volume conducted electrocardiogram (ECG) was recorded by using National Instruments data acquisition hardware (PC card, National Instruments, Austin, TX, USA) and SPEL Advanced Haemosys software (version 2.76, Experimetria Ltd. and Logirex Software Laboratory, Budapest, Hungary). Coronary flow was measured with a glass flowmeter (Cole-Parmer Instrument Company, Vernon Hills, IL, USA) positioned immediately above the retrogradely perfused aorta and was later corrected for the weight of each heart to give values in mL·min−1·g−1. An incision was made in the right atrium and the AV node was ablated using forceps 2 min after mounting the heart. AV ablation was regarded successful when the P wave was dissociated from the QRS complex on real-time ECG recording. After AV ablation, the hearts were allowed to beat in their own spontaneous rhythm. As a single ventricular pacing stimulus can initiate TdP in the presence of a Class III antiarrhythmic drug (e.g. E-4031) in isolated, AV-blocked rabbit hearts (Asano et al., 1997), ventricular pacing was not applied in the present experiments in order to avoid the occurrence of electrical pacing-induced arrhythmias. Hearts were equilibrated for 15 min before starting the experimental protocol. At the end of each experiment, the atria were removed from the heart and the ventricles were weighed.
Experimental protocol
In the first set of experiments, four groups (n= 8 hearts in each group) [dofetilide 100 nmol·L−1 (DOF1), SEA0400 1.0 µmol·L−1+ dofetilide 100 nmol·L−1 (SEA + DOF), and control groups distilled water (H2O) and dimethyl sulphoxide (DMSO), solvent of dofetilide and SEA0400 (DMSO)] were compared. A H2O control group was used as DMSO may affect the repolarization (Pinney et al., 1995; Himmel, 2007). The administration of the NCX inhibitor SEA0400 or the solvent of the SEA0400 was started at the beginning of a 20 min ‘pretreatment’ period, after 15 min of initial perfusion with modified Krebs–Henseleit solution. Dofetilide was added to the perfusion solution at the beginning of a 30 min ‘treatment’ period. In the H2O and the DMSO control groups, the hearts were perfused with the equivalent volume of the solvent water or DMSO throughout the whole experiment (Table 1). The concentration of SEA0400 (1 µmol·L−1) was chosen in order to achieve maximal NCX inhibition without significantly affecting any other ion current (Tanaka et al., 2002; Birinyi et al., 2005; Acsai et al., 2007; Farkas et al., 2008).
Table 1.
The experimental protocol applied in isolated, Langendorff-perfused, AV-blocked rabbit hearts
| Group | n | Pretreatment (20 min) | Treatment (30 min) |
|---|---|---|---|
| First set of experiments | |||
| H2O | 8 | H2O | H2O |
| DMSO | 8 | DMSO | DMSO |
| Dofetilide 100 nmol·L−1 | 8 | DMSO | DMSO + dofetilide |
| SEA0400 1.0 µmol·L−1+ dofetilide 100 nmol·L−1 | 8 | DMSO + SEA0400 | DMSO + SEA0400 + dofetilide |
| Second set of experiments | |||
| Dofetilide 100 nmol·L−1 | 8 | DMSO | DMSO + dofetilide |
| Lidocaine 30 µmol·L−1+ dofetilide 100 nmol·L−1 | 8 | DMSO + lidocaine | DMSO + lidocaine + dofetilide |
| Verapamil 750 nmol·L−1+ dofetilide 100 nmol·L−1 | 8 | DMSO + verapamil | DMSO + verapamil + dofetilide |
The groups of hearts, the periods of the experiments and the applied drugs are summarized. In the baseline period of the experiments, all hearts were perfused with Krebs buffer; the perfusion solution was then switched to the test drug in the second period of the experiment (pretreatment); in the last period dofetilide was added to the perfusion (treatment). Group size is indicated by n.
AV, atrioventricular; DMSO, dimethyl sulphoxide.
As SEA0400 administration did not decrease the incidence of dofetilide-induced TdP ventricular tachycardia, a second set of experiments was designed to examine whether it is possible to reduce dofetilide-induced TdP with other drugs in this model. Lidocaine and verapamil were chosen as test drugs as both can successfully reduce the incidence of drug-induced TdP in other experimental models (Carlsson et al., 1993; Milberg et al., 2005). The second set of experiments comprised three groups of hearts: (i) 100 nmol·L−1 dofetilide (DOF2), (ii) 30 µmol·L−1 lidocaine + 100 nmol·L−1 dofetilide (LID + DOF); and (iii) 750 nmol·L−1 verapamil + 100 nmol·L−1 dofetilide (VER + DOF). Each group contained n= 8 hearts. The administration of lidocaine or verapamil or the solvent DMSO was started at the beginning of the ‘pretreatment’ period, whereas dofetilide was added to the perfusion solution at the beginning of the ‘treatment’ period (Table 1). In either set of experiments, the administration of the drug or the solvent was time-matched (Table 1), and the choice of drug solution was made by reference to a randomization table. Randomization was achieved by coding each group with a letter whose meaning was unknown to the operator. Unbiased analysis was achieved by using stock solutions prepared by a second operator, who did not participate in the heart perfusion or data analysis.
Coronary flow measurement and ECG analysis
Coronary flow and ECG intervals were measured at predetermined time points. Coronary flow values were read directly from the flowmeter every 5 min during the experiment and 1 min before and after switching to a different perfusion solution and normalized to heart weight. After completion of the experiments, the data were replayed and the QT and RR intervals were measured by manual positioning of on-screen markers. The QT interval was defined as the time between the first deviation from the isoelectric line of QRS complex until the end of the TU wave. In the rabbit hearts, where the T or U wave overlapped the following QRS complex of the subsequent ventricular beat, the extrapolation method was used to measure the length of the QT (or QU) interval (Farkas et al., 2004).
As QT interval is influenced by the heart rate, baseline data for ventricular heart rates and QT intervals were used to determine the relationship between the RR interval and the QT interval in sinus rhythm before AV ablation and in idioventricular escape rhythm after AV ablation according to Batey and Coker (2002) and Farkas and Curtis (2003). These data were obtained from 56 isolated, Langendorff-perfused rabbit hearts prepared as described above and used here in the present study. Forty consecutive QT intervals were measured together with the corresponding RR intervals in each hearts immediately before and 10 min after the mechanical AV ablation. Simple linear regression revealed a positive correlation between QT and RR intervals in sinus rhythm (QTSinus= 0.326RR + 103.4) as well as in idioventricular escape rhythm (QTAV-block= 0.081RR + 186.8), although the regression coefficient (R) and the slope of the regression line were greater during sinus rhythm as compared with the values calculated in idioventricular escape rhythm (Figure 1A,B). As there was a difference between the slope of the regression line of the sinus and the idioventricular rhythm, a QT correction was calculated for either rhythm in a manner similar to that described by Batey and Coker (2002). The equations were rearranged to allow the calculation of the rate-corrected QT interval in sinus rhythm (QTc-Sinus) at an RR interval of 300 ms (i.e. a ventricular rate of 200 beats·min−1) using the formula QTc-Sinus = QTSinus− 0.326(RR-300) and in idioventricular escape rhythm (QTc-AV) at an RR interval of 500 ms (i.e. a ventricular rate of 120 beats·min−1) using the formula QTc-AV = QTAV-block− 0.081(RR-500). With these equations, plotting QTc-Sinus and QTc-AV against the corresponding RR interval produces a regression line with a slope of zero (Figure 1C,D), indicating that these corrections remove the influence of heart rate. As we examined drug effects after AV ablation in idioventricular escape rhythm, all QTc intervals in this study were corrected according to the QTc-AV.
Figure 1.
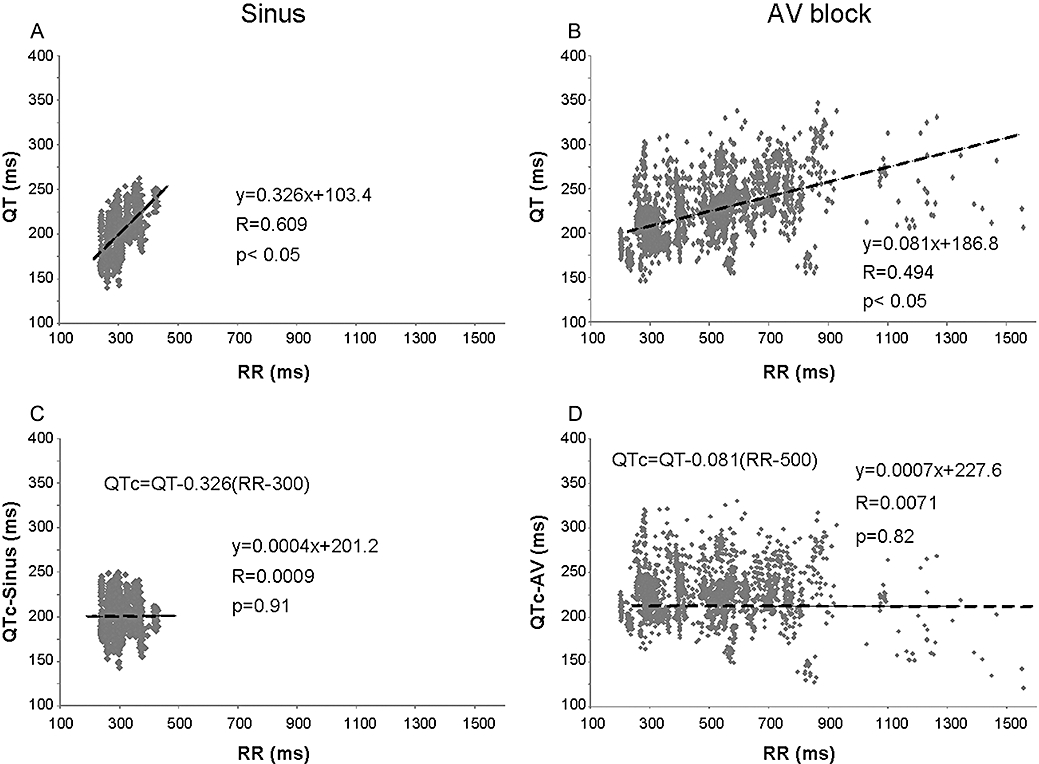
Correlation between individual values of the QT and RR intervals in sinus rhythm (A) and after AV ablation (B) in isolated Langendorff-perfused rabbit hearts. Correlation between individual values of the ‘rate-corrected’ QT intervals and the RR intervals in sinus rhythm (C) and after AV ablation (D) in isolated Langendorff-perfused rabbit hearts. All panels contain 2240 baseline data points obtained from 56 isolated rabbit hearts. QTc-Sinus, the heart rate-corrected QT interval during sinus rhythm; QTc-AV, the ventricular heart rate-corrected QT interval after AV nodal ablation. AV, atrioventricular; QTc, heart rate-corrected QT interval.
From the ECG, the incidence and the time to onset of TdP arrhythmias were obtained. TdP was defined as a polymorphic ventricular tachycardia with runs of four or more ventricular premature beats, where clear twisting of the QRS complexes around the isoelectric axis could be seen in at least one ECG lead.
Measurement of the beat-to-beat variability of the QT intervals and the RR intervals
The beat-to-beat variability of the QT and the RR intervals was determined from the manual measurement data on 40 consecutive QT intervals and the corresponding RR intervals at predetermined time points, that is, in the last minute before AV ablation, in the last minute of drug-free state after AV ablation, in the last minute of the ‘pretreatment’ period before switching to the dofetilide perfusion and in the fifth minute of the dofetilide infusion or before the onset of TdP. A computer program was developed in a .NET environment to obtain the following parameters of the beat-to-beat variability of the QT and RR intervals: for a sequence of RR or QT interval durations, the ‘root mean square’ (RMS) was calculated according to the following definition:  , where di represents the sequence of RR or QT interval durations and N is the total number of intervals. Another approach for characterizing the beat-to-beat variability of RR and QT intervals is to take their successive differences (Δdj=dj+1−dj; 0 ≤j≤N− 2, where dj represents the RR or QT interval durations and N is the total number of intervals) and calculate the RMS of these differences:
, where di represents the sequence of RR or QT interval durations and N is the total number of intervals. Another approach for characterizing the beat-to-beat variability of RR and QT intervals is to take their successive differences (Δdj=dj+1−dj; 0 ≤j≤N− 2, where dj represents the RR or QT interval durations and N is the total number of intervals) and calculate the RMS of these differences: 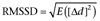 , where E denotes the mean value. The percentage of successive QT intervals that differ by more than 8 ms (PNN8), the ‘instability’ of the RR and QT intervals, the ‘short-term variability’ and the ‘long-term variability’ of the RR and QT intervals were calculated as described earlier (Vincze et al., 2008).
, where E denotes the mean value. The percentage of successive QT intervals that differ by more than 8 ms (PNN8), the ‘instability’ of the RR and QT intervals, the ‘short-term variability’ and the ‘long-term variability’ of the RR and QT intervals were calculated as described earlier (Vincze et al., 2008).
Exclusion criteria
Any heart with a coronary flow <3 mL·min−1·g−1 or asystole longer than 20 s during the whole experimental protocol was excluded. Longer than 20 s asystole was found after the start of dofetilide perfusion in six hearts pretreated with verapamil and in three hearts pretreated with lidocaine, these hearts were excluded. Additional experiments were performed in order to maintain equal group sizes.
Solutions
Perfusion solutions were prepared fresh each day. Dofetilide and SEA0400 were dissolved in DMSO. All final test solutions for heart perfusion contained 0.08 mL DMSO in 1 L of modified Krebs–Henseleit solution. Likewise, the DMSO control solution contained 0.08 mL DMSO in 1 L of modified Krebs–Henseleit solution. The H2O control solution contained 0.08 mL water in 1 L of modified Krebs–Henseleit solution. In the second set of experiments, lidocaine and verapamil were dissolved in water, and all final test solution for heart perfusion contained 0.1 mL water in 1 L of modified Krebs–Henseleit solution.
Statistics
Continuous data were expressed as mean ± standard error of the mean (SEM). All data from independent samples, except the incidences of TdP, were compared with Kruskal–Wallis tests. Within-group comparisons of the variability parameters were performed by Wilcoxon tests. The incidences of TdP were compared by using Fisher's exact probability test with the Bonferoni correction, that is, the P values of Fisher's exact probability test were multiplied by 4 in the first set of experiments or 2 in the second set of experiments (the numbers of comparisons) to allow multiple comparisons (Altman, 1991). P < 0.05 was taken as indicative of a statistically significant difference between values.
Drugs and materials
CaCl2, DMSO, lidocaine and verapamil were purchased from Sigma-Aldrich, Inc., St. Louis, MO, USA. All other salts were purchased from Molar Chemical Ltd., Budapest, Hungary. The dofetilide was a generous gift from Gedeon Richter Ltd. (Budapest, Hungary). The synthesis of 2-[4-[(2,5-difluorophenyl)methoxy]phenoxy]-5-ethoxy-aniline (SEA0400) was performed by Ferenc Fülöp (Department of Pharmaceutical Chemistry, University of Szeged, Szeged, Hungary) according to a method described earlier (Farkas et al., 2008). Water for the preparation of perfusion solution was obtained from a reverse osmosis system (Milli-Q RG, Millipore Ltd., Billerca, MA, USA) fed by distilled water, and had a specific resistivity of >18 MΩ.
Results
The effect of AV block on the ventricular rhythm and repolarization
As expected, the applied mechanical AV block decreased the average ventricular heart rate (Figure 2). Interestingly, AV ablation also led to a chaotic and irregular spontaneous ventricular rhythm (Figure 2B), and it strikingly increased all parameters of the beat-to-beat variability of the RR intervals (Figure 2D). The measurement of the RR variability parameters over a longer period (over 3 min) provided results similar to those of the measurement of the RR variability parameters over a shorter (40-consecutive-beat-long) period (data not shown). In addition, the AV ablation increased all QT variability parameters, which reflected a tentative beat-to-beat repolarization (Figure 2C).
Figure 2.

Representative ECG recordings during sinus rhythm (A) and after AV ablation (B). Beat-to-beat variability parameters of the QT (C) and the RR (D) intervals during sinus rhythm and after AV ablation. Vertical arrows indicate P waves. All values shown as means ± SEM. *P < 0.05 versus sinus rhythm. ECG I-III, electrocardiographic leads similar to standard limb leads I, II and III; sinus, sinus rhythm; AV, atrioventricular; AV block, chaotic idioventricular escape rhythm after AV ablation; mean, the mean of the measured 40 consecutive QT or RR intervals (ms); min, the minimum of the measured 40 consecutive QT or RR intervals (ms); max, the maximum of the measured 40 consecutive QT or RR intervals (ms); SD, the standard deviation of the measured 40 consecutive QT or RR intervals (ms); RMSSD, the root mean square of the successive difference of the QT or RR intervals (ms); instability, QT or RR instability (ms); LTV, long-term variability of the QT or RR intervals (ms); STV, short-term variability of the QT or RR intervals (ms).
TdP incidences and onset times
In the first set of experiments, TdP occurred in the majority of the dofetilide-perfused hearts (Figure 3A). NCX inhibition with SEA0400 did not reduce the incidence of dofetilide-induced TdP (Figure 3B) and did not affect the onset time of this arrhythmia (325 ± 30 s and 409 ± 97 s after switching to dofetilide perfusion in the DOF1 and SEA + DOF group respectively). Interestingly, TdP occurred in one heart in the DMSO-perfused control group 1174 s after switching to DMSO perfusion from Krebs solution.
Figure 3.
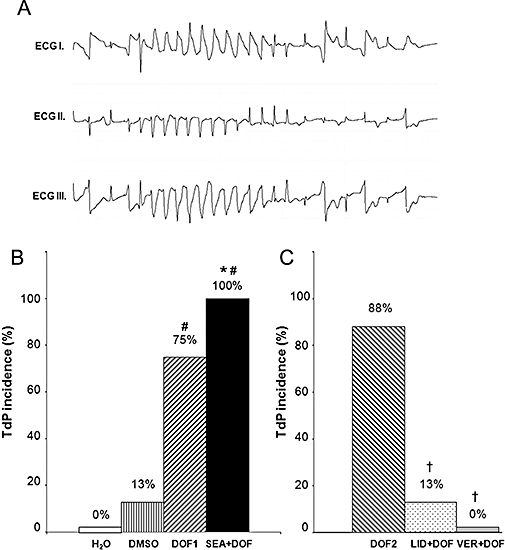
Dofetilide-induced torsades de pointes (TdP) ventricular tachycardia (A). Incidence of TdP in the first set of experiments (B) and in the second set of experiments (C). The hearts were perfused with distilled water (H2O), solvent of dofetilide and SEA0400 (DMSO), 100 nmol·L−1 dofetilide (DOF1) or 1.0 µmol·L−1 SEA0400 and 100 nmol·L−1 dofetilide (SEA + DOF) in the first set of experiments. Hearts were perfused with 100 nmol·L−1 dofetilide (DOF2), 30 µmol·L−1 lidocaine and 100 nmol·L−1 dofetilide (LID + DOF) or 750 nmol·L−1 verapamil and 100 nmol·L−1 dofetilide (VER + DOF) in the second set of experiments. Each group contained eight hearts. Values are percentage incidences of arrhythmias. *P < 0.05 versus DMSO, #P < 0.05 versus H2O, †P < 0.05 versus DOF2. DMSO, dimethyl sulphoxide; ECG, electrocardiogram.
In the second set of experiments, dofetilide provoked TdP in the majority of the hearts as seen in the first set of experiments. Lidocaine significantly decreased the incidence of dofetilide-induced TdP, while verapamil completely prevented the development of this arrhythmia (Figure 3C).
The RR and the QTc intervals and the beat-to-beat variability of the RR and QT intervals
Pretreatment period
In the first set of experiments, neither DMSO nor SEA0400 affected the mean RR interval, the mean QTc interval (Figure 4A,C) or any of the QT or RR variability parameters (Tables 2 and 3). After the exclusion of all hearts that experienced asystole longer than 20 s from the second set of experiments (see exclusion criteria in Methods) the mean RR interval, the mean QTc interval (Figure 5A,C) or any of the QT or RR variability parameters (Tables 2 and 3) did not significantly differ between the lidocaine, verapamil and the control groups before dofetilide perfusion.
Figure 4.
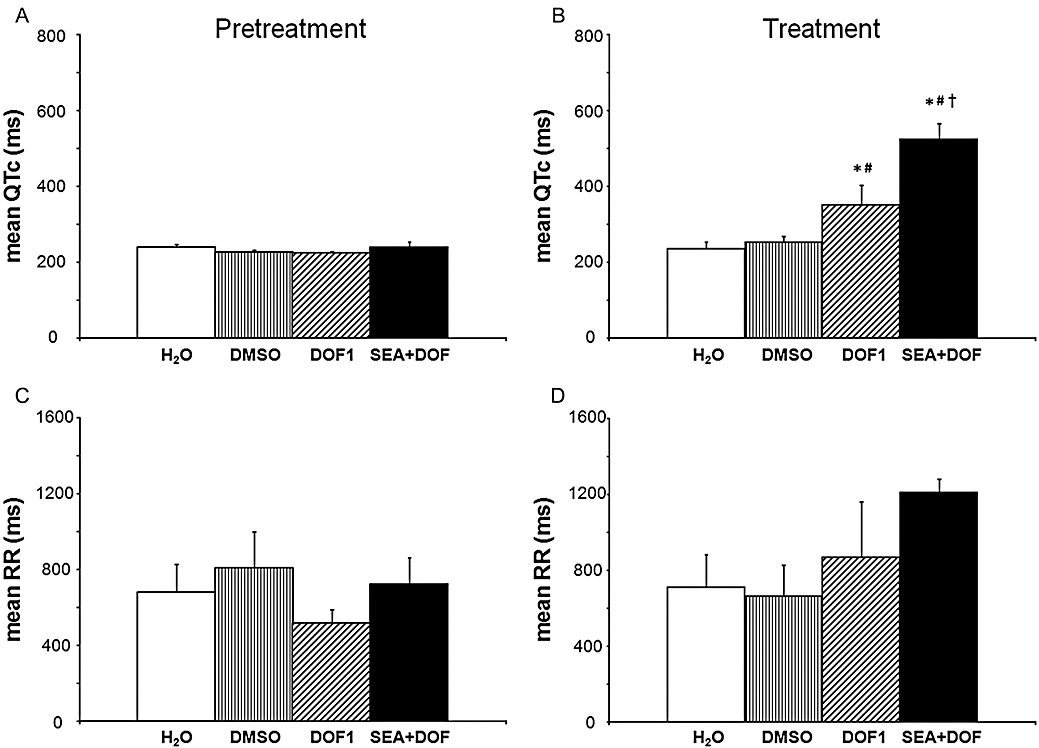
The heart rate-corrected QT (QTc) intervals (A and B) and the RR intervals (C and D) during the pretreatment and the treatment periods in AV-ablated isolated rabbit hearts in the first set of experiments. Values are mean ± SEM. *P < 0.05 versus DMSO; #P < 0.05 versus H2O; †P < 0.05 versus DOF1. For further details, see Figure 3. DMSO, dimethyl sulphoxide.
Table 2.
The beat-to-beat variability of the QT interval in isolated, Langendorff-perfused, AV-blocked rabbit hearts
| n | SD | RMS | RMSSD | PNN8 | Instability | LTV | STV | |
|---|---|---|---|---|---|---|---|---|
| First set of experiments | ||||||||
| H2O | ||||||||
| Baseline | 8 | 11 ± 2 | 225 ± 13 | 15 ± 2 | 25 ± 4 | 12 ± 2 | 8 ± 1 | 8 ± 1 |
| Pretreat | 8 | 11 ± 2 | 254 ± 6 | 11 ± 2 | 23 ± 4 | 14 ± 3 | 10 ± 3 | 6 ± 1 |
| Treat | 8 | 14 ± 2 | 253 ± 19 | 18 ± 3 | 25 ± 3 | 14 ± 2*; | 11 ± 2 | 9 ± 2 |
| DMSO | ||||||||
| Baseline | 8 | 17 ± 4 | 233 ± 12 | 24 ± 7 | 30 ± 4 | 20 ± 5 | 11 ± 3 | 13 ± 4 |
| Pretreat | 8 | 18 ± 4 | 251 ± 13 | 23 ± 5 | 31 ± 3 | 20 ± 3 | 14 ± 3 | 12 ± 2 |
| Treat | 8 | 22 ± 3 | 267 ± 15 | 30 ± 6 | 38 ± 3# | 30 ± 5# | 16 ± 2 | 17 ± 4 |
| DOF1 | ||||||||
| Baseline | 8 | 14 ± 2 | 217 ± 9 | 16 ± 2 | 29 ± 3 | 17 ± 3 | 11 ± 2 | 9 ± 1 |
| Pretreat | 8 | 16 ± 5 | 226 ± 6 | 20 ± 5 | 26 ± 5 | 19 ± 4 | 13 ± 3 | 10 ± 2 |
| Treat | 8 | 73 ± 15*# | 389 ± 55*# | 87 ± 17*# | 41 ± 3# | 65 ± 19# | 63 ± 16*# | 40 ± 8*# |
| SEA + DOF | ||||||||
| Baseline | 8 | 14 ± 4 | 225 ± 12 | 18 ± 6 | 27 ± 3 | 21 ± 7 | 12 ± 3 | 10 ± 4 |
| Pretreat | 8 | 14 ± 2 | 258 ± 20 | 18 ± 3 | 27 ± 4 | 17 ± 3 | 12 ± 2 | 9 ± 2 |
| Treat | 8 | 101 ± 27*# | 589 ± 45*#† | 91 ± 25*# | 40 ± 3# | 145 ± 53*# | 102 ± 32*# | 42 ± 12*# |
| Second set of experiments | ||||||||
| DOF2 | ||||||||
| Baseline | 8 | 15 ± 3 | 242 ± 10 | 23 ± 5 | 30 ± 4 | 21 ± 5 | 9 ± 1 | 13 ± 4 |
| Pretreat | 8 | 12 ± 2 | 245 ± 9 | 17 ± 4 | 27 ± 3 | 14 ± 3 | 9 ± 2 | 9 ± 2 |
| Treat | 8 | 42 ± 12 | 308 ± 17 | 49 ± 14 | 39 ± 4 | 60 ± 16 | 35 ± 12 | 27 ± 8 |
| DOF + LID | ||||||||
| Baseline | 8 | 18 ± 8 | 228 ± 15 | 26 ± 18 | 24 ± 4 | 11 ± 2 | 10 ± 3 | 9 ± 2 |
| Pretreat | 8 | 21 ± 12 | 256 ± 14 | 31 ± 18 | 25 ± 4 | 17 ± 5 | 17 ± 10 | 15 ± 9 |
| Treat | 8 | 31 ± 9 | 459 ± 97‡ | 47 ± 14 | 38 ± 5 | 45 ± 15 | 20 ± 6 | 27 ± 8 |
| DOF + VER | ||||||||
| Baseline | 8 | 12 ± 2 | 225 ± 8 | 16 ± 3 | 27 ± 5 | 12 ± 2 | 8 ± 1 | 9 ± 2 |
| Pretreat | 8 | 19 ± 6 | 293 ± 17 | 22 ± 5 | 29 ± 2 | 17 ± 3 | 13 ± 3 | 10 ± 1 |
| Treat | 8 | 101 ± 36 | 588 ± 115‡ | 135 ± 61 | 38 ± 6 | 161 ± 62 | 72 ± 23 | 78 ± 39 |
The beat-to-beat QT variability parameters in the first set of experiments and in the second set of experiments.
Values shown are mean ± SEM.
P < 0.05 versus DMSO;
P < 0.05 versus H2O;
P < 0.05 versus DOF1;
AV, atrioventricular; DMSO, dimethyl sulphoxide; pretreat, pretreatment period; treat, treatment period; group size is indicated by n. SD, the standard deviation of the measured 40 consecutive QT intervals (ms); RMS, the root mean square of the measured 40 consecutive QT intervals (ms); RMSSD, the root mean square of the successive difference of the QT intervals (ms); PNN8, percentage of QT intervals differing from the preceding one by more than 8 ms (%); instability, QT instability (ms); LTV, long-term variability of the QT intervals (ms); STV, short-term variability of the QT intervals (ms); DOF, dofetilide; LID, lidocaine; SEA, SEA 0400; VER, verapamil.
Table 3.
The beat-to-beat variability of the RR interval in isolated, Langendorff-perfused, AV-blocked rabbit hearts
| n | SD | RMS | RMSSD | Instability | LTV | STV | |
|---|---|---|---|---|---|---|---|
| First set of experiments | |||||||
| H2O | |||||||
| Baseline | 8 | 53 ± 20 | 447 ± 65 | 92 ± 37 | 71 ± 35 | 21 ± 9 | 55 ± 24 |
| Pretreat | 8 | 36 ± 14 | 681 ± 147 | 44 ± 19 | 45 ± 21 | 26 ± 11 | 24 ± 12 |
| Treat | 8 | 174 ± 145 | 783 ± 197 | 191 ± 162 | 44 ± 21 | 105 ± 80 | 48 ± 32 |
| DMSO | |||||||
| Baseline | 8 | 63 ± 35 | 482 ± 67 | 81 ± 56 | 116 ± 68 | 37 ± 30 | 50 ± 38 |
| Pretreat | 8 | 385 ± 211 | 944 ± 258 | 492 ± 254 | 208 ± 66 | 260 ± 157 | 168 ± 69 |
| Treat | 8 | 135 ± 57 | 680 ± 174 | 143 ± 41 | 223 ± 91 | 119 ± 65 | 77 ± 23 |
| DOF1 | |||||||
| Baseline | 8 | 42 ± 20 | 443 ± 24 | 72 ± 38 | 57 ± 36 | 17 ± 9 | 44 ± 25 |
| Pretreat | 8 | 117 ± 93 | 566 ± 88 | 171 ± 140 | 51 ± 35 | 52 ± 37 | 58 ± 46 |
| Treat | 8 | 398 ± 216 | 1057 ± 318 | 535 ± 313 | 166 ± 90 | 231 ± 94 | 149 ± 62 |
| SEA + DOF | |||||||
| Baseline | 8 | 151 ± 48 | 575 ± 29 | 251 ± 91 | 281 ± 85 | 68 ± 30 | 151 ± 56 |
| Pretreat | 8 | 52 ± 26 | 729 ± 137 | 34 ± 18 | 48 ± 21 | 45 ± 21 | 14 ± 7 |
| Treat | 8 | 429 ± 109*# | 1251 ± 81 | 534 ± 152 | 350 ± 82#† | 341 ± 90 | 244 ± 70 |
| Second set of experiments | |||||||
| DOF2 | |||||||
| Baseline | 8 | 141 ± 54 | 541 ± 38 | 262 ± 99 | 232 ± 87 | 39 ± 34 | 165 ± 62 |
| Pretreat | 8 | 75 ± 43 | 590 ± 63 | 113 ± 79 | 102 ± 83 | 37 ± 18 | 65 ± 53 |
| Treat | 8 | 183 ± 49 | 706 ± 75 | 267 ± 76 | 202 ± 72 | 120 ± 33 | 125 ± 34 |
| DOF + LID | |||||||
| Baseline | 8 | 108 ± 40 | 574 ± 66 | 180 ± 81 | 169 ± 82 | 36 ± 11 | 112 ± 57 |
| Pretreat | 8 | 155 ± 107 | 892 ± 122 | 268 ± 208 | 279 ± 202 | 37 ± 30 | 171 ± 140 |
| Treat | 8 | 649 ± 321 | 1660 ± 716 | 1043 ± 505 | 588 ± 375 | 333 ± 175 | 519 ± 267 |
| DOF + VER | |||||||
| Baseline | 8 | 89 ± 54 | 530 ± 61 | 141 ± 85 | 112 ± 54 | 56 ± 44 | 77 ± 42 |
| Pretreat | 8 | 33 ± 16 | 970 ± 182 | 36 ± 17 | 35 ± 15 | 27 ± 13 | 12 ± 5 |
| Treat | 8 | 854 ± 3 | 1920 ± 342‡ | 1301 ± 540 | 11169 ± 495 | 540 ± 211 | 709 ± 304 |
The beat-to-beat RR variability parameters in the first set of experiments and in the second set of experiments. Group size is indicated by n.
Values shown are mean ± SEM.
P < 0.05 versus DMSO;
P < 0.05 versus H2O;
P < 0.05 versus DOF1;
AV, atrioventricular; DMSO, dimethyl sulphoxide; SD, the standard deviation of the measured 40 consecutive RR intervals (ms); RMS, the root mean square of the measured 40 consecutive RR intervals (ms). RMSSD, the root mean square of the successive difference of the RR intervals (ms); Instability, QT instability (ms); LTV, long-term variability of the QT intervals (ms); STV, short-term variability of the QT intervals (ms). DOF, dofetilide: LID, lidocaine; SEA, SEA 0400; VER, verapamil.
Figure 5.
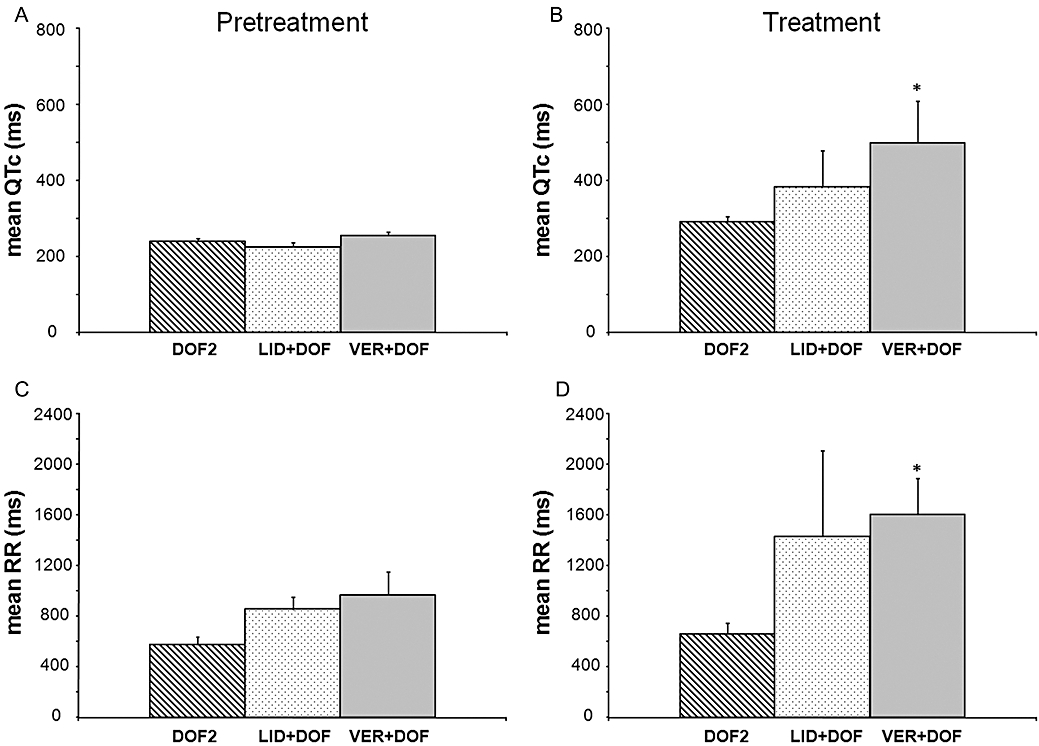
The heart rate-corrected QT (QTc) intervals (A and B) and the RR intervals (C and D) during the pretreatment and the treatment periods in AV-ablated isolated rabbit hearts in the second set of experiments. Values are mean ± SEM. *P < 0.05 versus DOF2. For further details, see Figure 3. AV, atrioventricular.
Treatment period
In the first set of experiments, dofetilide perfusion significantly increased the mean QTc intervals (Figure 4B) and the beat-to-beat variability of the QT interval (Table 2) without affecting the mean RR interval (Figure 4D) and the beat-to-beat variability of the RR interval (Table 3). NCX inhibition with SEA0400 exaggerated the dofetilide-induced increase in the mean QTc interval (Figure 4B) and further increased some of the QT variability parameters (Table 2). Further, SEA0400 upon dofetilide perfusion increased the beat-to-beat variability of the RR interval (Table 3) without affecting the mean RR interval (Figure 4D).
In the second set of experiments, verapamil further increased the dofetilide-induced QTc prolongation whereas lidocaine did not affect the mean QTc interval significantly when compared with that of the DOF2 group (Figure 5B). Verapamil tended to increase the beat-to-beat variability of the QT interval but only the QT RMS parameter reached statistical significance. Similarly, lidocaine added to dofetilide perfusion significantly elevated QT RMS as compared with that of the DOF2 group (Table 2).
Verapamil added to dofetilide perfusion significantly increased the mean RR interval, that is, decreased the ventricular heart rate as compared with that of the DOF2 group (Figure 5D). Verapamil and lidocaine tended to increase the beat-to-beat variability of the RR interval but only the RR RMS parameter of the VER + DOF group reached statistical significance as compared with the DOF2 group (Table 3).
Coronary flow
In the first set of experiments, group mean baseline coronary flows 1 min before the ‘pretreatment’ period ranged from 7.0 ± 0.5 to 11.2 ± 0.4 mL·min−1·g−1 (no significant difference between the groups). There was a small time-dependent fall (range 6–16%) in flow in all groups, which is typical of the model. As the treatment groups were time-matched, this did not influence the interpretation of the results. There was no significant difference between the various groups at any time point of the experiments (data not shown).
In the second set of experiments, group mean baseline coronary flows 1 min before the ‘pretreatment’ period ranged from 7.9 ± 0.5 to 10.4 ± 0.5 mL·min−1·g−1 (no significant difference between the groups). Perfusion with verapamil significantly increased the coronary flow as compared with the DOF2 group (the maximum coronary flow values were 13.3 ± 1.3 and 6.7 ± 0.2 mL·min−1·g−1 in the VER + DOF and the DOF2 group respectively). Lidocaine did not influence the coronary flow as compared with the DOF2 group (data not shown).
Discussion
The present study is the first to examine the role of NCX in the genesis of drug-induced TdP ventricular tachycardia. Selective inhibition of NCX by SEA0400 neither decreased the incidence of dofetilide-induced TdP nor influenced the onset time of this arrhythmia in isolated, Langendorff-perfused, AV-ablated rabbit hearts. On the other hand, SEA0400 significantly prolonged the mean QTc interval in the presence of dofetilide. The inhibition of the Na+ channels by lidocaine as well as the block of the L-type Ca2+ channels by verapamil significantly antagonized the genesis of dofetilide-induced TdP. However, verapamil further increased the dofetilide-induced QTc prolongation and neither verapamil nor lidocaine reduced the dofetilide-induced increase in the beat-to-beat variability of the QT interval.
Selective and effective inhibition of NCX in the isolated rabbit heart: no effect on TdP
SEA0400 is probably the most effective inhibitor of the NCX (Matsuda et al., 2001), with an inhibitory potency 10- to 100-fold higher than that of KB-R7943 (Takahashi et al., 2003). In isolated rabbit myocardial cells, SEA0400 equally suppressed the inward and the outward NCX current (INa/Ca), IC50 values of the inward (forward) and the outward (reverse) currents were 243 and 309 nmol·L−1 respectively (Farkas et al., 2008). SEA0400 at a concentration of 1.0 µmol·L−1 inhibits 70–80% of the NCX function (Matsuda et al., 2001; Tanaka et al., 2002; Birinyi et al., 2005; Farkas et al., 2008). SEA0400 is also considered to be the most selective NCX inhibitor, as it is considered to be selective for the NCX up to a concentration of 1.0 µmol·L−1 without markedly influencing any other ion transport mechanisms (Tanaka et al., 2002; Nagy et al., 2004).
In a recent study, which was performed with the same Langendorff perfusion preparation that we used in the present study, SEA0400 at a concentration of 1.0 µmol·L−1 increased significantly the left ventricular contractility in isolated rat hearts, whereas the drug (0.1–1.0 µmol·L−1) exerted only an insignificant, but concentration-dependent positive inotropic effect in isolated rabbit hearts (Farkas et al., 2008). Likewise, Szentandrássy et al. found that 0.3 and 1.0 µmol·L−1 SEA0400 increased the developed pressure and the amplitude of the Ca2+ transient in isolated, Langendorff-perfused rat hearts (Szentandrássy et al., 2008). These earlier results suggest indirectly that SEA0400 at a concentration of 1.0 µmol·L−1 effectively blocks the NCX in isolated hearts. The effectiveness of verapamil and lidocaine against dofetilide-induced TdP validates our model, as it responded to intervention known to reduce the incidence of drug-induced TdP. This observation suggests that effective inhibition of NCX by SEA0400 did not exert an anti-arrhythmic effect against dofetilide-induced TdP because NCX does not play a role in the genesis of TdP in isolated, AV-blocked rabbit hearts.
NCX does not provide the trigger for TdP
According to the most widely accepted theory, TdP is initiated by an EAD-induced ectopic beat (Antzelevitch and Oliva, 2006). EAD is generated in phase 2 or 3 of the prolonged repolarization as a result of increased depolarizing currents such as ICaL and INa, but also inward NCX current (Sipido et al., 2006). Stimulation of the β-adrenoceptors increases ICaL and may result in an overload of the Ca2+ stores of the sarcoplasmic reticulum (SR), and a subsequent spontaneous SR Ca2+ release can appear as an early Ca2+ aftertransient (Sipido et al., 2006). This activates the forward mode of the NCX leading to EAD generation in guinea pig, canine and human myocytes (Tweedie et al., 1997; Volders et al., 1997). This effect can be evoked with isoprenaline and suppressed by a non-selective NCX blocker Ni2+ (Volders et al., 1997). It was also observed that the Class III antiarrhythmic clofilium and d,l-sotalol prolonged repolarization, but evoked EADs only in the presence of adrenaline, which could be suppressed by reducing inward NCX in canine Purkinje fibre and epicardial myocytes (Patterson et al., 1997). While these results emphasize the role of sympathetic stimulation, NCX-generated EAD as a result of intracellular Ca2+ overload is not always restricted to sympathetic activity. Inward NCX current can generate EAD even in the absence of β- adrenoceptor-stimulated, spontaneous SR Ca2+ release when K+ currents are decreased or inhibited (Volders et al., 2000). Accordingly, the selective inhibition of NCX with SEA0400 effectively decreased the amplitude of EADs evoked by the co-perfusion of BaCl2 and dofetilide in canine ventricular papillary muscles and Purkinje fibres (Nagy et al., 2004). Moreover, acceleration of the pacing rate or a single premature beat could induce EAD activity and prolongation of action potential duration (APD) in canine ventricular M cell preparations pretreated with the IKr blocker E4031 via a mechanism linked to an intracellular calcium-loading-induced electrogenic NCX current (Burashnikov and Antzelevitch, 1998).
Some authors claim that DAD-induced triggered activity (similarly to EAD) can also initiate TdP (Antzelevitch and Oliva, 2006). DADs can be produced by Ca2+ wave-activated, inward currents resulted from the activation of the NCX (Sipido et al., 2006). Accordingly, NCX inhibition antagonized DAD-dependent, digitalis-induced arrhythmias in several in vitro and in vivo experimental models (Watano et al., 1999; Nagy et al., 2004; Nagasawa et al., 2005; Shpak et al., 2006).
Although many results suggest a role of NCX in the genesis of drug-induced TdP, selective inhibition of the NCX did not prevent dofetilide-induced TdP in isolated rabbit hearts in the present study. The initiating mechanism of TdP in isolated, Langendorff-perfused, AV-blocked, non-paced rabbit hearts is unknown. However, in an earlier study with AV-blocked rabbit hearts, an EAD was associated with the initiating beat of d-sotalol-induced TdP (Zabel et al., 1997). If EADs (or DADs) serve as the trigger for TdP in the rabbit hearts in the present study, then the genesis of these triggered activities was not related to the activity of the NCX. Our results are in agreement with earlier findings showing that selective inhibition of the NCX with SEA0400 did not affect the inward rectifier potassium current (Ik1) blocker Ba2+-induced triggered activity and DAD in guinea pig myocytes (Homma et al., 2006) and SEA0400 also did not suppress aconitine-induced, EAD- and DAD-dependent arrhythmias in an in vivo guinea pig model of type-3 long QT syndrome (Amran et al., 2004). Alternatively, it is possible that the trigger of TdP did not originate from EADs or DADs in the present study. It is possible that any beats of the irregular idioventricular escape rhythm that occurred in the vulnerable period of the dofetilide-produced re-entry circuits could serve for a trigger for TdP. These beats do not depend on NCX activity, which may explain the ineffectiveness of NCX inhibition.
NCX does not contribute to the maintenance of TdP
It is widely accepted that the mechanism of maintenance (substrate) of TdP arrhythmia involves re-entry circuits produced by an increase in spatial dispersion of the repolarization of the ventricular wall. The transmural dispersion has been identified as the principal arrhythmogenic substrate in both acquired and congenital long QT syndrome (LQTS) (Antzelevitch and Oliva, 2006). In dogs and humans, the transmural dispersion of repolarization is the consequence of longer APD in M cells, compared with those in epicardial and endocardial cells (Antzelevitch and Oliva, 2006). Zygmunt et al. (2000) showed that the longer APD is due to the smaller slow component of the delayed rectifier potassium current (IKs) and larger late INa and NCX currents in the healthy canine ventricle. The IKr inhibitor d,l-sotalol increased transmural dispersion of repolarization and induced TdP in isolated, Langendorff-perfused, AV-blocked rabbit hearts (Eckardt et al., 2002). Further, verapamil prevented veratridine-induced TdP via reduction of left ventricular transmural dispersion of repolarization (and suppression of EADs) in isolated, Langendorff-perfused, AV-blocked rabbit hearts (Milberg et al., 2005). These earlier observations suggest that transmural dispersion of repolarization may also play a role in the maintenance of TdP in AV-ablated rabbit hearts. Although NCX activity contributes to the generation of transmural dispersion of repolarization in the healthy dog heart (Zygmunt et al., 2000), our results indicate that NCX activity does not increase the transmural dispersion of repolarization and, therefore, does not contribute to the maintenance mechanism of TdP in the rabbit heart.
The effect of SEA0400 on the duration of repolarization
SEA0400 is a highly selective and effective inhibitor of the NCX at a concentration of 1.0 µmol·L−1 (Matsuda et al., 2001; Takahashi et al., 2003; Farkas et al., 2008). To date, no experimental data have been reported on an appreciable effect of this concentration of SEA0400 on the repolarization. Our study did not involve a separate group of hearts with SEA0400 perfusion alone during the 20 min ‘pretreatment’ and the subsequent 30 min ‘treatment’ period; thus, we cannot exclude the possibility that the drug affects repolarization and causes arrhythmias after prolonged exposure. However, 30 min perfusion with SEA0400 at a concentration of 1.0 µmol·L−1, which is the same concentration we used in the present study, did not affect ECG parameters and did not induce any arrhythmias in isolated rat and rabbit hearts in a recent study from our laboratory (Farkas et al., 2008). Similarly, 20 min SEA0400 pretreatment did not affect the control QTc in the present study. In contrast, when the subsequent treatment period started and the IKr inhibitor dofetilide was added to the SEA0400 perfusion, repolarization prolonged quickly and markedly in 5 min, and a significantly longer QTc was measured than that in the group of hearts perfused with dofetilide on its own. These observations suggest that SEA0400 prolongs repolarization only when the repolarization reserve is markedly reduced by the co-administration of another drug. Nevertheless, further investigations would help to characterize the effect of SEA0400 on repolarization after long exposure.
In the present study, SEA0400 pretreatment did not affect the control QTc, but the drug prolonged significantly the QTc in the presence of the IKr inhibitor dofetilide. This observation may have two possible explanations. (i) NCX contributes a small outward current at the end of the action potential plateau that, in the absence of repolarization reserve (i.e. in dofetilide) delays repolarization; and/or (ii) SEA0400 is non-selective for NCX and affects other repolarizing currents.
With regard to the former explanation, Sher et al. (2007) have reported computer simulations in which NCX may generate an outward current at the end of the action potential plateau. Blocking this current, in the absence of repolarization reserve, may thus prolong APD. However, other simulations by Bers et al. (Bers, 2001) have suggested that, apart from a very brief period at the start of the action potential, NCX remains inward throughout the cardiac cycle. The second possibility is that SEA0400 is not selective for NCX and blocks other repolarizing currents. However, in guinea pig myocytes, SEA0400 at a concentration of 1.0 µmol·L−1 caused only 4% and 2% inhibition on Ik (at 37°C) and Ik1 currents respectively (Tanaka et al., 2002). SEA0400 may also inhibit smaller repolarizing currents, for example IKs, but this has never been examined. Either explanation requires that the potential repolarization-influencing effect of SEA0400 was minimal when administered alone, but was revealed when the repolarization reserve was reduced by the potent IKr blocker, dofetilide. Our findings on the effect of SEA0400 on the QTc interval propose that inhibition of the NCX by SEA0400 may exacerbate the proarrhythmic effect of dofetilide. However, this requires further investigation in a setting where control incidence of dofetilide-induced TdP is low.
Neither the duration of the QT interval nor the beat-to-beat variability of the QT interval correlated with the occurrence of TdP
A substantial amount of experimental and clinical data suggest that QT prolongation alone is an unreliable predictor of drug-induced TdP (Thomsen et al., 2006). In our study, verapamil exaggerated the dofetilide-induced QTc prolongation while the drug prevented TdP. Likewise, lidocaine decreased significantly the incidence of TdP but did so without affecting the QTc interval. These results corroborate earlier reports showing that QT prolongation did not correlate with the proarrhythmic liability of repolarization-prolonging drugs in isolated, AV-blocked rabbit hearts (Eckardt et al., 2002; Milberg et al., 2002).
The temporal dispersion of repolarization defined as the beat-to-beat variability of the monophasic APD has been shown to predict the proarrhythmic activity of repolarization-prolonging drugs in isolated rabbit hearts (Hondeghem et al., 2001) and in chronic AV-blocked dogs in vivo (Thomsen et al., 2004). In accordance with this, Lengyel et al. (2007) found that the beat-to-beat variability of the QT interval predicted the occurrence of drug-induced TdP in anaesthetized rabbits. In contrast, the beat-to-beat variability of the QT interval failed to predict repolarization-prolonging-drug-induced TdP in two recent investigations with adrenergically stimulated, anaesthetized rabbits (Michael et al., 2007; Vincze et al., 2008). Thus, the predictive power of an increase in the beat-to-beat variability of the QT interval is debatable. In our isolated heart study, 100 nmol·L−1 dofetilide perfusion increased each QT variability parameter, and the drug induced TdP reproducibly in nearly all hearts. On the other hand, neither verapamil nor lidocaine decreased any of the QT variability parameters; nevertheless, they successfully block the development of TdP. Thus, the predictive value of an increase in the beat-to-beat variability of the QT interval in isolated, AV-ablated, non-paced rabbit hearts is questionable. Further investigation is needed to clarify whether it is because an increase in the beat-to-beat variability of the action potential is not a contributor to TdP or rather it is because an increase in the beat-to-beat variability of the QT interval is only one out of several contributing factors that need to be present at the same time to allow generation of TdP in the model.
AV block-induced electrical instability
In our study, acute AV block was applied in order to slow down the ventricular heart rate as bradycardia has been reported to increase the proarrhythmic activity of IKr blockers in isolated rabbit hearts (Eckardt et al., 1998). Surprisingly, AV block resulted in a chaotic idioventricular rhythm in all hearts during control conditions. This chaotic idioventricular rhythm is characterized by an elevated beat-to-beat variability of the RR and QT intervals in our study. Similar electrical instability was reported in isolated mouse hearts after AV ablation, and the level of instability (i.e. the incidence of baseline arrhythmias after AV block) inversely correlated with the K+ concentration of the perfusion solution (Fabritz et al., 2003). Accordingly, perfusion with 3 mmol·L−1 K+ induced afterdepolarizations in nearly all mouse hearts and evoked polymorphic ventricular tachycardia in one mouse heart during control conditions, after AV ablation (Fabritz et al., 2003). This may explain why the evaluation of our experiments with 3 mmol·L−1 K+ in the perfusion solution found a run of ‘TdP’ in a heart in the DMSO control group. Further, the baseline electrical instability in the AV-blocked rabbit hearts perfused with 3 mmol·L−1 K+ in the present study might contribute to the high sensitivity of these hearts to the proarrhythmic activity of dofetilide.
The antiarrhythmic effect of verapamil and lidocaine against TdP
The ICaL channel blocker verapamil significantly prolonged the QT interval in the presence of dofetilde and prevented the development of TdP. Verapamil (which has never been reported to cause TdP and indeed has been proposed as a therapy for long QT-related arrhythmias) inhibited IKr in the same concentration range as quinidine and amiodarone (Yang et al., 2001). This may explain why the drug prolonged further the QT interval, when the repolarization reserve was very small as a result of dofetilide perfusion. Verapamil also prolonged the mean RR interval, that is, reduced the number of idioventricular beats mostly when it was co-perfused with dofetilide. This effect might reduce the number of trigger beats for the initiation of TdP irrespective of the fact that the trigger was either the idioventricular beat itself or the corresponding afterdepolarization. As lidocaine tended to have the same effect on the ventricular rate, this may also explain its antiarrhythmic effect in our investigations. Shimizu et al. (1995) reported that verapamil suppressed spontaneous or adrenaline-induced EADs and TdP in patients with congenital LQTS. Milberg et al. (2005) found that the same concentration of verapamil we used (750 nmol·L−1) prevented TdP via the reduction of EAD and ventricular transmural dispersion of repolarization in an isolated rabbit heart model of the acquired LQT3 syndrome. Likewise, lidocaine suppressed the IKr blocker almokalant-induced dispersion of repolarization and the development of EADs in rabbit Purkinje fibres in vitro (Abrahamsson et al., 1996). Thus, the antiarrhythmic effect of verapamil and lidocaine in our experiments may also be related their direct effect on the development of EAD and/or dispersion of the repolarization.
Conclusions
Na+/Ca2+ exchanger neither contributed to the initiation nor assisted the maintenance of dofetilide-induced TdP in our in vitro experimental model, in which inhibition of the INa and ICaL currents successfully antagonized the genesis of this arrhythmia. Neither QTc prolongation nor an increase in the beat-to-beat variability of the QT interval is a sufficient prerequisite of TdP genesis in this model. AV ablation resulted in a chaotic idioventricular rhythm, which might make the hearts more sensitive to the proarrhythmic activity of dofetilide in the applied isolated rabbit heart model.
Acknowledgments
Eleonora Grandi and Donald M. Bers (Department of Pharmacology, University of California, Davis, CA, USA) are thanked for their useful comments on the results of the present study. This work was supported by the Hungarian Ministry of Health (ETT 203/2003 and 353/2006), the Hungarian National Research Fund (OTKA F-046776, OTKA K-69018 and NI-61902), the National Research and Development Programmes (NKFP 1 A/046/2004), the European Community (EU FP7 grant ICT-2008-224381, preDiCT) and the Hungarian Academy of Sciences.
Glossary
Abbreviations:
- APD
action potential duration
- AV
atrioventricular
- EAD
early afterdepolarization
- ECG
electrocardiogram
- DAD
delayed afterdepolarization
- DMSO
dimethyl sulphoxide
- ICaL
inward L-type calcium current
- IKr
the rapid component of the delayed rectifier potassium current
- IKs
the slow component of the delayed rectifier potassium current
- IK1
inward rectifier potassium current
- INa
inward sodium current
- M cells
midmyocardial cells
- NCX
Na+/Ca2+ exchanger
- QTc
heart rate-corrected QT interval
- TdP
torsades de pointes
Conflicts of interest
None.
References
- Abrahamsson C, Carlsson L, Duker G. Lidocaine and nisoldipine attenuate almokalant-induced dispersion of repolarization and early afterdepolarizations in vitro. J Cardiovasc Electrophysiol. 1996;7:1074–1081. doi: 10.1111/j.1540-8167.1996.tb00483.x. [DOI] [PubMed] [Google Scholar]
- Acsai K, Kun A, Farkas AS, Fülöp F, Nagy N, Balázs M, et al. Effect of partial blockade of the Na(+)/Ca(2+)-exchanger on Ca(2+) handling in isolated rat ventricular myocytes. Eur J Pharmacol. 2007;576:1–6. doi: 10.1016/j.ejphar.2007.07.047. [DOI] [PubMed] [Google Scholar]
- Altman DG. Practical Statistics for Medical Research. 1st edn. London: Chapman & Hall; 1991. [Google Scholar]
- Amran MS, Hashimoto K, Homma N. Effects of sodium-calcium exchange inhibitors, KB-R7943 and SEA0400, on aconitine-induced arrhythmias in guinea pigs in vivo, in vitro, and in computer simulation studies. J Pharmacol Exp Ther. 2004;310:83–89. doi: 10.1124/jpet.104.066951. [DOI] [PubMed] [Google Scholar]
- Antzelevitch C, Oliva A. Amplification of spatial dispersion of repolarization underlies sudden cardiac death associated with catecholaminergic polymorphic VT, long QT, short QT and Brugada syndromes. J Intern Med. 2006;259:48–58. doi: 10.1111/j.1365-2796.2005.01587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano Y, Davidenko JM, Baxter WT, Gray RA, Jalife J. Optical mapping of drug-induced polymorphic arrhythmias and torsade de pointes in the isolated rabbit heart. J Am Coll Cardiol. 1997;29:831–842. doi: 10.1016/s0735-1097(96)00588-8. [DOI] [PubMed] [Google Scholar]
- Barrett TD, Hennan JK, Fischbach PS, O’Neill BP, Driscoll EM, Jr, Lucchesi BR. Tedisamil and dofetilide-induced torsades de pointes, rate and potassium dependence. Br J Pharmacol. 2001;132:1493–1500. doi: 10.1038/sj.bjp.0703967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batey AJ, Coker SJ. Proarrhythmic potential of halofantrine, terfenadine and clofilium in a modified in vivo model of torsade de pointes. Br J Pharmacol. 2002;135:1003–1012. doi: 10.1038/sj.bjp.0704550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belardinelli L, Antzelevitch C, Vos MA. Assessing predictors of drug-induced torsade de pointes. Trends Pharmacol Sci. 2003;24:619–625. doi: 10.1016/j.tips.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Bers DM. Excitation-contraction Coupling and Cardiac Contractile Force. 2nd edn. Dordrecht: Kluwer Academic Publishers; 2001. [Google Scholar]
- Birinyi P, Acsai K, Bányász T, Tóth A, Horváth B, Virág L, et al. Effects of SEA0400 and KB-R7943 on Na(+)/Ca(2+) exchange current and L-type Ca(2+) current in canine ventricular cardiomyocytes. Naunyn Schmiedebergs Arch Pharmacol. 2005;372:63–70. doi: 10.1007/s00210-005-1079-x. [DOI] [PubMed] [Google Scholar]
- Burashnikov A, Antzelevitch C. Acceleration-induced action potential prolongation and early afterdepolarizations. J Cardiovasc Electrophysiol. 1998;9:934–948. doi: 10.1111/j.1540-8167.1998.tb00134.x. [DOI] [PubMed] [Google Scholar]
- Carlsson L, Drews L, Duker G, Schiller-Linhardt G. Attenuation of proarrhythmias related to delayed repolarization by low-dose lidocaine in the anesthetized rabbit. J Pharmacol Exp Ther. 1993;267:1076–1080. [PubMed] [Google Scholar]
- Eckardt L, Haverkamp W, Mertens H, Johna R, Clague JR, Borggrefe M, et al. Drug-related torsades de pointes in the isolated rabbit heart: comparison of clofilium, d,l-sotalol, and erythromycin. J Cardiovasc Pharmacol. 1998;32:425–434. doi: 10.1097/00005344-199809000-00013. [DOI] [PubMed] [Google Scholar]
- Eckardt L, Breithardt G, Haverkamp W. Electrophysiologic characterization of the antipsychotic drug sertindole in a rabbit heart model of torsade de pointes: low torsadogenic potential despite QT prolongation. J Pharmacol Exp Ther. 2002;300:64–71. doi: 10.1124/jpet.300.1.64. [DOI] [PubMed] [Google Scholar]
- Fabritz L, Kirchhof P, Franz MR, Eckardt L, Monnig G, Milberg P, et al. Prolonged action potential durations, increased dispersion of repolarization, and polymorphic ventricular tachycardia in a mouse model of proarrhythmia. Basic Res Cardiol. 2003;98:25–32. doi: 10.1007/s00395-003-0386-y. [DOI] [PubMed] [Google Scholar]
- Farkas A, Curtis MJ. Does QT widening in the Langendorff-perfused rat heart represent the effect of repolarization delay or conduction slowing? J Cardiovasc Pharmacol. 2003;42:612–621. doi: 10.1097/00005344-200311000-00006. [DOI] [PubMed] [Google Scholar]
- Farkas A, Batey AJ, Coker SJ. How to measure electrocardiographic QT interval in the anaesthetized rabbit. J Pharmacol Toxicol Methods. 2004;50:175–185. doi: 10.1016/j.vascn.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Farkas AS, Acsai K, Nagy N, Tóth A, Fülöp F, Seprényi G, et al. Na(+)/Ca(2+) exchanger inhibition exerts a positive inotropic effect in the rat heart, but fails to influence the contractility of the rabbit heart. Br J Pharmacol. 2008;154:93–104. doi: 10.1038/bjp.2008.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himmel HM. Suitability of commonly used excipients for electrophysiological in-vitro safety pharmacology assessment of effects on hERG potassium current and on rabbit Purkinje fiber action potential. J Pharmacol Toxicol Methods. 2007;56:145–158. doi: 10.1016/j.vascn.2007.04.004. [DOI] [PubMed] [Google Scholar]
- Homma N, Amran MS, Nagasawa Y, Hashimoto K. Topics on the Na+/Ca2+ exchanger: involvement of Na+/Ca2+ exchange system in cardiac triggered activity. J Pharmacol Sci. 2006;102:17–21. doi: 10.1254/jphs.fmj06002x3. [DOI] [PubMed] [Google Scholar]
- Hondeghem LM, Carlsson L, Duker G. Instability and triangulation of the action potential predict serious proarrhythmia, but action potential duration prolongation is antiarrhythmic. Circulation. 2001;103:2004–2013. doi: 10.1161/01.cir.103.15.2004. [DOI] [PubMed] [Google Scholar]
- Lengyel C, Varró A, Tábori K, Papp JG, Baczkó I. Combined pharmacological block of I(Kr) and I(Ks) increases short-term QT interval variability and provokes torsades de pointes. Br J Pharmacol. 2007;151:941–951. doi: 10.1038/sj.bjp.0707297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda T, Arakawa N, Takuma K, Kishida Y, Kawasaki Y, Sakaue M, et al. SEA0400, a novel and selective inhibitor of the Na+-Ca2+ exchanger, attenuates reperfusion injury in the in vitro and in vivo cerebral ischemic models. J Pharmacol Exp Ther. 2001;298:249–256. [PubMed] [Google Scholar]
- Michael G, Dempster J, Kane KA, Coker SJ. Potentiation of E-4031-induced torsade de pointes by HMR1556 or ATX-II is not predicted by action potential short-term variability or triangulation. Br J Pharmacol. 2007;152:1215–1227. doi: 10.1038/sj.bjp.0707513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milberg P, Eckardt L, Bruns HJ, Biertz J, Ramtin S, Reinsch N, et al. Divergent proarrhythmic potential of macrolide antibiotics despite similar QT prolongation: fast phase 3 repolarization prevents early afterdepolarizations and torsade de pointes. J Pharmacol Exp Ther. 2002;303:218–225. doi: 10.1124/jpet.102.037911. [DOI] [PubMed] [Google Scholar]
- Milberg P, Reinsch N, Osada N, Wasmer K, Monnig G, Stypmann J, et al. Verapamil prevents torsade de pointes by reduction of transmural dispersion of repolarization and suppression of early afterdepolarizations in an intact heart model of LQT3. Basic Res Cardiol. 2005;100:365–371. doi: 10.1007/s00395-005-0533-8. [DOI] [PubMed] [Google Scholar]
- Nagasawa Y, Zhu BM, Chen J, Kamiya K, Miyamoto S, Hashimoto K. Effects of SEA0400, a Na+/Ca2+ exchange inhibitor, on ventricular arrhythmias in the in vivo dogs. Eur J Pharmacol. 2005;506:249–255. doi: 10.1016/j.ejphar.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Nagy ZA, Virág L, Tóth A, Biliczki P, Acsai K, Bányász T, et al. Selective inhibition of sodium-calcium exchanger by SEA-0400 decreases early and delayed after depolarization in canine heart. Br J Pharmacol. 2004;143:827–831. doi: 10.1038/sj.bjp.0706026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson E, Scherlag BJ, Szabo B, Lazzara R. Facilitation of epinephrine-induced afterdepolarizations by class III antiarrhythmic drugs. J Electrocardiol. 1997;30:217–224. doi: 10.1016/s0022-0736(97)80007-6. [DOI] [PubMed] [Google Scholar]
- Pinney SP, Koller BS, Franz MR, Woosley RL. Terfenadine increases the QT interval in isolated guinea pig heart. J Cardiovasc Pharmacol. 1995;25:30–34. doi: 10.1097/00005344-199501000-00006. [DOI] [PubMed] [Google Scholar]
- Sher AA, Noble PJ, Hinch R, Gavaghan DJ, Noble D. The role of the Na(+)/Ca(2+) exchangers in Ca(2+) dynamics in ventricular myocytes. Prog Biophys Mol Biol. 2007;96:377–398. doi: 10.1016/j.pbiomolbio.2007.07.018. [DOI] [PubMed] [Google Scholar]
- Shimizu W, Ohe T, Kurita T, Kawade M, Arakaki Y, Aihara N, et al. Effects of verapamil and propranolol on early afterdepolarizations and ventricular arrhythmias induced by epinephrine in congenital long QT syndrome. J Am Coll Cardiol. 1995;26:1299–1309. doi: 10.1016/0735-1097(95)00313-4. [DOI] [PubMed] [Google Scholar]
- Shpak B, Gofman Y, Shpak C, Hiller R, Boyman L, Khananshvili D. Effects of purified endogenous inhibitor of the Na+/Ca2+ exchanger on ouabain-induced arrhythmias in the atria and ventricle strips of guinea pig. Eur J Pharmacol. 2006;553:196–204. doi: 10.1016/j.ejphar.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Sipido KR, Varró A, Eisner D. Sodium Calcium Exchange as a Target for Antiarrhythmic Therapy. 2006/04/14 edn. Berlin, Heidelberg: Springer-Verlag; 2006. [DOI] [PubMed] [Google Scholar]
- Sipido KR, Bito V, Antoons G, Volders PG, Vos MA. Na/Ca exchange and cardiac ventricular arrhythmias. Ann N Y Acad Sci. 2007;1099:339–348. doi: 10.1196/annals.1387.066. [DOI] [PubMed] [Google Scholar]
- Szentandrássy N, Birinyi P, Szigeti G, Farkas A, Magyar J, Tóth A, et al. SEA0400 fails to alter the magnitude of intracellular Ca(2+) transients and contractions in Langendorff-perfused guinea pig heart. Naunyn Schmiedebergs Arch Pharmacol. 2008;378:65–71. doi: 10.1007/s00210-008-0296-5. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Takahashi T, Suzuki T, Onishi M, Tanaka Y, Hamano-Takahashi A, et al. Protective effects of SEA0400, a novel and selective inhibitor of the Na+/Ca2+ exchanger, on myocardial ischemia-reperfusion injuries. Eur J Pharmacol. 2003;458:155–162. doi: 10.1016/s0014-2999(02)02732-2. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Nishimaru K, Aikawa T, Hirayama W, Tanaka Y, Shigenobu K. Effect of SEA0400, a novel inhibitor of sodium-calcium exchanger, on myocardial ionic currents. Br J Pharmacol. 2002;135:1096–1100. doi: 10.1038/sj.bjp.0704574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen MB, Verduyn SC, Stengl M, Beekman JD, de Pater G, van Opstal J, et al. Increased short-term variability of repolarization predicts d-sotalol-induced torsades de pointes in dogs. Circulation. 2004;110:2453–2459. doi: 10.1161/01.CIR.0000145162.64183.C8. [DOI] [PubMed] [Google Scholar]
- Thomsen MB, Matz J, Volders PG, Vos MA. Assessing the proarrhythmic potential of drugs: current status of models and surrogate parameters of torsades de pointes arrhythmias. Pharmacol Ther. 2006;112:150–170. doi: 10.1016/j.pharmthera.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Tweedie D, O’Gara P, Harding SE, MacLeod KT. The effect of alterations to action potential duration on beta-adrenoceptor-mediated aftercontractions in human and guinea-pig ventricular myocytes. J Mol Cell Cardiol. 1997;29:1457–1467. doi: 10.1006/jmcc.1997.0385. [DOI] [PubMed] [Google Scholar]
- Vincze D, Farkas AS, Rudas L, Makra P, Csík N, Leprán I, et al. Relevance of anaesthesia for dofetilide-induced torsades de pointes in alpha1-adrenoceptor-stimulated rabbits. Br J Pharmacol. 2008;153:75–89. doi: 10.1038/sj.bjp.0707536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volders PG, Vos MA, Szabo B, Sipido KR, de Groot SH, Gorgels AP, et al. Progress in the understanding of cardiac early afterdepolarizations and torsades de pointes: time to revise current concepts. Cardiovasc Res. 2000;46:376–392. doi: 10.1016/s0008-6363(00)00022-5. [DOI] [PubMed] [Google Scholar]
- Volders PG, Kulcsar A, Vos MA, Sipido KR, Wellens HJ, Lazzara R, et al. Similarities between early and delayed afterdepolarizations induced by isoproterenol in canine ventricular myocytes. Cardiovasc Res. 1997;34:348–359. doi: 10.1016/s0008-6363(96)00270-2. [DOI] [PubMed] [Google Scholar]
- Watano T, Harada Y, Harada K, Nishimura N. Effect of Na+/Ca2+ exchange inhibitor, KB-R7943 on ouabain-induced arrhythmias in guinea-pigs. Br J Pharmacol. 1999;127:1846–1850. doi: 10.1038/sj.bjp.0702740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Snyders D, Roden DM. Drug block of I(kr): model systems and relevance to human arrhythmias. J Cardiovasc Pharmacol. 2001;38:737–744. doi: 10.1097/00005344-200111000-00010. [DOI] [PubMed] [Google Scholar]
- Zabel M, Hohnloser SH, Behrens S, Li YG, Woosley RL, Franz MR. Electrophysiologic features of torsades de pointes: insights from a new isolated rabbit heart model. J Cardiovasc Electrophysiol. 1997;8:1148–1158. doi: 10.1111/j.1540-8167.1997.tb01001.x. [DOI] [PubMed] [Google Scholar]
- Zygmunt AC, Goodrow RJ, Antzelevitch C. I(NaCa) contributes to electrical heterogeneity within the canine ventricle. Am J Physiol Heart Circ Physiol. 2000;278:H1671–H1678. doi: 10.1152/ajpheart.2000.278.5.H1671. [DOI] [PubMed] [Google Scholar]