

Abstract
Background and purpose:
Atherosclerotic plaque rupture and thrombosis are the main cause of acute coronary syndrome. The study was aimed to test the hypothesis that oral administration of rapamycin may attenuate inflammation, inhibit progression and enhance stability of atherosclerotic plaques.
Experimental approach:
Thirty New Zealand rabbits were subjected to balloon-induced endothelial injury of the abdominal aorta and were fed a diet of 1% cholesterol for 20 weeks. From week 9 to week 20, the animals were treated with oral rapamycin (0.5 mg·kg−1·day−1; group A), oral simvastatin (5 mg·kg−1·day−1; group B) and no drugs (group C). At the end of week 20, all rabbits were challenged with injection of Chinese Russell's viper venom and histamine. Serological, ultrasonographic, pathological, immunohistochemical and gene expression studies were performed.
Key results:
Rapamycin significantly increased the thickness of the fibrous caps and decreased plaque vulnerability index in group A rabbits. Serum lipid levels were higher whereas plaque burden was lower in group A than in group B (P < 0.05). The incidence of plaque rupture in group A (0%) and group B (0%) was significantly lower than that in group C (56.0%, P < 0.05).
Conclusions and implications:
Oral administration of rapamycin effectively attenuated inflammation, inhibited progression and enhanced stability of atherosclerotic plaques in rabbits, without altering serum lipid levels. Our findings suggest a novel approach to the treatment of atherosclerosis.
Keywords: vulnerable plaque, inflammation, rapamycin, atherosclerosis
Introduction
Atherosclerotic plaque rupture is the major cause of acute cardiovascular events and treatment aimed at stabilization of vulnerable plaques is of great clinical importance (Naghavi et al., 2003). Although statins have been recognized as the most potent drugs for stabilizing plaques (Aikawa et al., 1998; Libby and Aikawa, 2002), the Prove-It trial found that 22.4% of enrolled patients experienced an acute coronary event despite an intensive statin therapy for 2 years (Ridker et al., 2005). Moreover, liver dysfunction as a side effect of statin administration caused a number of patients to withdraw from statin therapy. Therefore, it is of high priority to develop safer and more effective drugs to stabilize vulnerable plaques.
Previous studies have demonstrated that atherosclerosis is a complex and persistent inflammatory disease involving aorta and its major branches and active inflammation and immunoreactions are important features of vulnerable plaques (Carr et al., 1997; Armstrong et al., 2006). Consequently, drugs targeting inflammation and immunoreactivity may have a considerable potential to stabilize atherosclerotic plaques. Rapamycin, an antifungal antibiotic and a potent immunosuppressant, has been used in patients to prevent rejection of transplanted kidneys and to inhibit coronary restenosis after stenting with a rapamycin-coated stent (Wessely et al., 2007). In experimental studies, Pakala et al. (2005) reported that rapamycin inhibited monocyte chemotaxis and attenuated atherosclerotic plaque progression in apolipoprotein E knockout mice (apoE−/−) whereas Farb et al. (2002) found that oral rapamycin significantly inhibited in-stent neointimal growth in rabbit iliac arteries. Recent studies showed that daily oral administration of rapamycin for 14 days after stenting significantly reduced the incidence of coronary restenosis in patients receiving coronary intervention (Rodriguez et al., 2006). The Oral Rapamune to Inhibit Restenosis study found that oral administration of rapamycin was safe for prevention of restenosis, and associated with a low rate of repeat revascularization (Waksman et al., 2004). However, the effect of rapamycin on plaque stability is unknown. The present study was designed to test the hypothesis that oral administration of rapamycin may attenuate inflammation and enhance stability of atherosclerotic plaques in a rabbit model of atherosclerosis, independent of serum lipid levels.
Methods
Animal model
All animal care and experimental protocols complied with the Animal Management Rules of the Ministry of Health of the People's Republic of China (document No 55, 2001) and were approved by the Animal Care Committee of Shandong University. Thirty New Zealand white rabbits weighing 2.0–3.0 kg were randomly divided into three groups (n= 10 each). All rabbits were subjected to balloon-induced endothelial injury of the abdominal aorta and were fed a diet of 1% cholesterol for 20 weeks. From the end of week 8 to the end of week 20, rabbits in group A were given a daily oral dose of rapamycin (0.5 mg·kg−1), whereas rabbits in group B received a daily oral dose of simvastatin (5 mg·kg−1). These drugs were dissolved in water and administered by oral gavage. Rabbits in group C received no drugs and served as controls. At the end of week 20, all rabbits were exposed to pharmacological triggering as described previously (Chen et al., 2007b). In brief, 0.15 mg·kg−1 of Chinese Russell's viper venom was injected intraperitoneally, followed 30 min later by an intravenous injection of 0.02 mg·kg−1 histamine (Sigma, St. Louis, MO, USA). High-frequency ultrasonography and intravascular ultrasound imaging were performed before and after pharmacological triggering to study the morphological changes of the plaques in the abdominal aorta. Rabbits were killed 24 h later for pathological studies.
Biochemical studies
Blood samples were collected from all rabbits at the beginning of the experiment and before pharmacological triggering. Serum levels of total cholesterol (TC), triglyceride (TGs), high-density lipoprotein cholesterol (HDL-C) and low-density lipoprotein cholesterol (LDL-C) were measured by enzymatic assays. Serum levels of high sensitive C-reactive protein (hs-CRP), interleukin (IL)-8, IL-1, monocyte chemoattracted-1 (MCP-1) matrix metalloproteinases-1 (MMP-1) and P-selectin were assayed by use of ELISA kits (R&D Systems, Chicago, IL, USA).
Ultrasonographic study
High-frequency ultrasonography
A high-frequency duplex ultrasonographic system (HP SONOS 5500, Andover, MA, USA) and a 7.5 MHz transducer were used to detect atherosclerotic plaques in the abdominal aorta before and after pharmacological triggering. After the aortic longitudinal and transversal axis views were scanned, the aortic diameter at end-diastole (Dd) and the maximal intima-media thickness (IMT) were measured by two-dimensional echocardiography, and the aortic peak velocity (Vp), mean velocity (Vm) and velocity-time integral (VTI) were recorded by the pulsed Doppler technique.
Integrated backscatter analysis
Ultrasonic integrated backscatters from the aortic wall and the atherosclerotic plaques were analysed by the acoustic densitometry technique. The ultrasonic intensity (AII) of the aortic intima and adventitia in normal segments and in the atherosclerotic plaques were measured and averaged and the corrected AII (AIIc%) was derived by calculating the ratio of AII of the intima to the AII of the adventitia in both normal segments and plaques.
Intravascular ultrasound studies
Intravascular ultrasound (IVUS) studies were performed before and after pharmacological triggering by use of a 3.2 F catheter containing a single rotating element transducer of 40 MHz connected to an IVUS system (Galaxy, Boston Scientific Corporation, Fremont, CA, USA). The catheter was withdrawn from the aortic arch to the abdominal aorta by a motorized pullback device at a constant speed of 0.5 mm·s−1. The following parameters were measured from the abdominal aortic cross-sectional images: external elastic membrane area (EEMA), lumen area (LA), plaque area (PA = EEMA − LA) and plaque burden (PB% = PA/EEMA × 100%) (Mintz et al., 2001; Chen et al., 2007a).
Histopathology and immunohistochemistry
All rabbits were killed by intravenous overdose of pentobarbital. The abdominal aorta was excised to observe the occurrence of plaque rupture and thrombosis and the thoracic aorta was removed to analyse the systemic effects of rapamycin on atherosclerotic plaques. Tissue samples (2 cm long) were taken from the abdominal aorta and fixed in 4% formaldehyde, with some segments embedded in paraffin and cut into 5 µm sections for staining with haematoxylin and eosin (H&E) and Masson's trichrome.
Tissue samples from the thoracic and abdominal aorta were embedded in paraffin and cut into 5 µm sections. Sections were stained with sirius red and Oil-red-O (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and reacted with mouse anti-rabbit metalloproteinase 3 (MMP-3) monoclonal antibody (Oncogene, USA), mouse anti-rabbit RAM-11 monoclonal antibody (Dako, USA), mouse anti-rabbit α-smooth-muscle-cell (SMC) actin monoclonal antibody (Sigma Chemical, USA), mouse anti-human MMP-2 and MMP-9 monoclonal antibody (Chemicon, Chemicon Internatonal, USA), goat anti-human MMP-1, MMP-12, P-selectin and MCP-1 polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Sections reacted with non-immune mouse or goat IgG, secondary antibody only and no primary and secondary antibodies were used as negative controls.
Histopathological slides were analysed by use of a computer-assisted morphometric analysis system (Image-Pro Plus 5.0, Media Cybernetics, Cambridge, MA, USA). The fibrous cap thickness and IMT of the abdominal aorta were measured at 10 equidistant points around the cap in each slice; three slices per section were measured, values were averaged and the ratio of fibrous cap thickness to IMT was calculated. The area of positive staining of α-actin (SMCs), sirius red (collagens), Oil-red-O (lipids) and RAM-11 (macrophages) was expressed as a percentage of stained area divided by the plaque area of the abdominal aorta in at least 10 high-power fields (×400). The vulnerability index was calculated as: (macrophage staining % + lipid staining %)/(smooth muscle cell % + collagen fibre %) (Torzewski et al., 1998). Plaque rupture was defined as fibrous cap disruption with luminal thrombosis or a buried fibrous cap within a plaque (Williams et al., 2002).
Molecular biological studies
Tissue samples from the abdominal aorta were frozen with liquid nitrogen. Total RNA was extracted and mRNA expression of MCP-1, MMP-1, MMP-2, MMP-3, MMP-9, MMP-12 and P-selectin in plaques was examined by quantitative RT-PCR with use of LightCycler (Roche Applied Science, Indianapolis, IN, USA) following the manufacturer's instruction. The mRNA sequences were obtained from GenBank (Bethesda, MD, USA). The transcript amount of glyceraldehyde 3-phosphate dehydrogenase was quantified as an internal RNA control. Quantitative values were obtained from the threshold cycle value, the point at which a significant increase in fluorescence was first detected (Livak and Schmittgen, 2001). Experiments were performed in triplicate for each data point. The results of RT-PCR were confirmed by gel electrophoresis.
Statistical analysis
All statistical analyses were carried out with spss, v11.0 (SPSS Inc, Chicago, IL, USA). Quantitative variables are expressed as means ± SD. Independent sample t-tests were used to compare continuous data for between-group differences and paired t-tests were applied to within-group comparisons at different time points. Variables with skewed distribution were log-transformed before the t-test. The anova test was adopted to compare the difference among three groups of rabbits. The Kruskal–Wallis test and χ2 analysis were applied to analyse non-parametric and categorical data respectively. Two-tailed P < 0.05 was considered statistically significant.
Materials
The sources of the materials used are as follows: rapamycin, Hebei Huabei Pharmaceutic Co. Ltd., Hebei, China; simavastatin, Merck & Co. Inc, Hangzhou, China; pentobarbital, Sigma, St. Louis, USA; Russell viper venom, Snake Venom Institute of Guangzhou, Guangzhou, China.
Results
Serum lipid profile and inflammatory markers
One rabbit in group A died of myocardial infarction at week 14, one rabbit in group B died of cerebral infarction at week 12 and one rabbit in group C died of ileus at week 17. At the beginning of the experiment, there was no significant difference in serum lipid levels among the three groups of rabbits (Table 1). At the end of week 20, group B showed a significant reduction of serum TC and LDL-C levels in comparison with those in group A and group C (both P < 0.05), but group A and group C did not differ in serum levels of TC, TG, HDL-C and LDL-C (Table 2). The serum levels of MCP-1, hs-CRP, IL-8, IL-18, MMP-1 and P-selectin were significantly lower in groups A and group B than those in group C, with no difference between group A and group B (Table 3).
Table 1.
Serum lipid profile in rabbits at week 8 before intervention
| Groups | TC (mmol·L−1) | TG (mmol·L−1) | HDL-C (mmol·L−1) | LDL-C (mmol·L−1) |
|---|---|---|---|---|
| Group A (n= 10) | 30.84 ± 11.26 | 2.26 ± 1.04 | 1.12 ± 1.04 | 26.35 ± 9.43 |
| Group B (n= 9) | 28.0 ± 8.09 | 3.08 ± 2.47 | 1.01 ± 0.50 | 24.90 ± 7.50* |
| Group C (n= 9) | 28.23 ± 6.09 | 3.09 ± 1.06 | 1.05 ± 0.09 | 26.00 ± 5.12 |
HDL-C, high-density lipoprotein; LDL-C, low-density lipoprotein; TC, total cholesterol; TG, triglyceride.
Table 2.
Serum lipid profile in three groups after 12 week intervention
| Groups | TC (mmol·L−1) | TG (mmol·L−1) | HDL-C (mmol·L−1) | LDL-C (mmol·L−1) |
|---|---|---|---|---|
| Group A (n= 9) | 28.76 ± 5.24# | 2.12 ± 1.91 | 0.94 ± 0.25 | 25.7 ± 3.21# |
| Group B (n= 9) | 3.60 ± 1.42* | 0.94 ± 0.03 | 0.95 ± 0.42 | 2.22 ± 0.32* |
| Group C (n= 9) | 30.12 ± 8.91 | 2.98 ± 2.12 | 1.02 ± 0.23 | 27.75 ± 5.45 |
P < 0.05 versus group C;
P < 0.05 versus group B.
HDL-C, high-density lipoprotein; LDL-C, low-density lipoprotein; TC, total cholesterol; TG, triglyceride.
Table 3.
Serum inflammatory markers in three groups after 12 week intervention
| Groups | hs-CRP (ng·mL−1) | MCP-1 (pg·mL−1) | IL-8 (pg·mL−1) | IL-18 (pg·mL−1) | MMP-1 (ng·mL−1) | P-selectin (ng·mL−1) |
|---|---|---|---|---|---|---|
| Group A (n= 9) | 35.7 ± 9.1* | 24.9 ± 7.2* | 3.5 ± 2.8* | 24.5 ± 16.7* | 16.9 ± 5.4* | 8.7 ± 2.9* |
| Group B (n= 9) | 40.7 ± 8.9* | 26.8 ± 4.6* | 5.2 ± 2.7* | 34.8 ± 12.7* | 11.7 ± 4.2* | 7.4 ± 1.3* |
| Group C (n= 9) | 127.2 ± 44.9 | 86.3 ± 10.4 | 20.1 ± 14.7 | 92.7 ± 40.8 | 59.4 ± 8.9 | 32.1 ± 4.9 |
P < 0.05 versus group C.
hs-CRP, high sensitive C-reactive protein; IL-8, interleukin-8; IL-18, interleukin-18; MCP-1, monocyte chemoattractant protein-1; MMP-1, matrix metalloproteinase-1.
High-frequency ultrasonographic measurements
Measurements of IMT in the abdominal aorta were significantly lower in group A and group B than those in group C (P < 0.01 and P < 0.05 respectively), and also lower in group A than in group B (P < 0.05; Table 4). Similarly, values of Vm in group A were significantly lower than those in groups B and group C, whereas AIIc% in groups A and group B was significantly higher than that in group C (both P < 0.05). However, these groups did not differ in Dd, Vp or VTI (Table 4).
Table 4.
Ultrasonographic measurements in three groups after 12 week intervention
| Groups | IMT (mm) | Dd (mm) | Vp (cm·s−1) | Vm (cm·s−1) | VTI (cm·s−1) | AIIc% |
|---|---|---|---|---|---|---|
| Group A (n= 9) | 0.56 ± 0.12*# | 3.42 ± 0.32 | 79.1 ± 13.7 | 61.2 ± 7.3* | 9.4 ± 2.5 | 81.3 ± 9.8* |
| Group B (n= 9) | 0.79 ± 0.14* | 3.62 ± 0.28 | 89.1 ± 19.7 | 70.2 ± 8.7 | 9.7 ± 2.4 | 79 ± 10.22* |
| Group C (n= 9) | 1.33 ± 0.26 | 3.56 ± 0.28 | 96 ± 25.68 | 73.9 ± 11.37 | 9.9 ± 4.2 | 63.6 ± 12.5 |
P < 0.05 versus group C;
P < 0.05 versus group B.
AIIc%, corrected averaged ultrasonic intensity (AII); Dd, end-diastolic diameters; IMT, intima-media thickness; Vm, mean velocity; Vp, peak velocity; VTI, velocity-time integral.
IVUS measurements
Measurements of EEMA, PA and PB% in the abdominal aorta in group A and group B were significantly lower than that in group C, and the value of PB% was remarkably reduced in group A compared with that in group B. However, the three groups did not differ in LA (Table 5).
Table 5.
Intravascular ultrasonographic measurements in three groups after 12 week intervention
| Groups | LA (mm2) | EEMA (mm2) | PA (mm2) | PB (%) |
|---|---|---|---|---|
| Group A (n= 9) | 5.62 ± 2.47 | 7.28 ± 2.52* | 1.66 ± 1.45* | 17.8 ± 2.3*# |
| Group B (n= 9) | 6.32 ± 2.42 | 8.42 ± 2.12* | 2.10 ± 1.61* | 24.9 ± 2.5* |
| Group C (n= 9) | 8.3 ± 4.24 | 14.27 ± 2.38 | 5.97 ± 3.29 | 41.8 ± 3.6 |
P < 0.05 versus group C;
P < 0.05 versus group B.
EEMA, external elastic membrane area; LA, lumen area; PA, plaque area; PB, plaque burden.
Histopathological and immunohistochemical analysis
Groups A and B showed a significantly thicker fibrous cap of the abdominal aortic plaque than group C (268 ± 57 and 283 ± 72, respectively, vs. 123 ± 52 µm, both P < 0.01) and thinner abdominal aortic IMT (620 ± 189 and 575 ± 196, respectively, vs. 986 ± 278 µm, both P < 0.05). Consequently, the ratio of fibrous cap thickness to IMT of the abdominal aorta was significantly larger in group A and group B (0.43 ± 0.02 and 0.49 ± 0.08 respectively) than in group C (0.15 ± 0.05, both P < 0.05). After pharmacological triggering, no rabbits in group A and group B, but five rabbits in group C (5/9, 56%), showed abdominal aortic plaque rupture, with the occurrence of plaque disruption significantly different in group C as compared with that in group A and group B (both P < 0.05, Figure 1).
Figure 1.
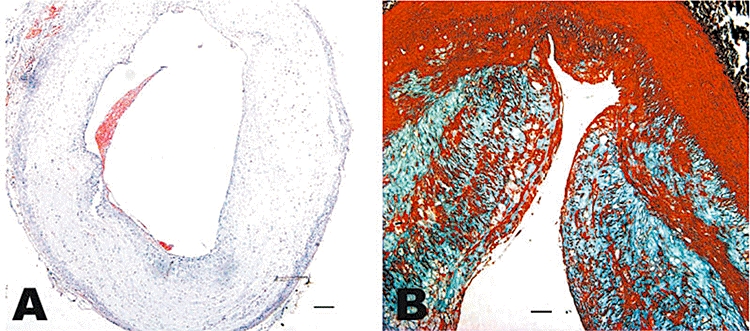
Histopathological assays in group C. Haematoxylin and eosin staining of the cross section of the abdominal aorta in a rabbit of group C showing plaque rupture and thrombosis (A, bars = 200 µm) and Masson staining of the equivalent cross section in a rabbit of group C showing buried fibrous cap (B, bars = 100 µm).
As observed in the abdominal aorta, thoracic aortic plaques in group A and group B showed a significantly thicker fibrous cap than those in group C (216 ± 37 and 207 ± 40, respectively, vs. 142 ± 55 µm, both P < 0.01) and thinner abdominal aortic IMT (584 ± 87 and 578 ± 67, respectively, vs. 676 ± 87 µm, both P < 0.05). Consequently, the ratio of fibrous cap thickness to IMT was significantly larger in group A and group B (0.39 ± 0.12 and 0.36 ± 0.06 respectively) than in group C (0.22 ± 0.10, both P < 0.01).
Compared with group C, group A and group B showed increased positive staining area of α-actin (16 ± 2.8% and 15.5 ± 3.5% vs. 6.8 ± 4.2% respectively, both P < 0.05) and sirius red (14.2 ± 3.6% and 15.2 ± 4.2% vs. 7.1 ± 4.2% respectively, both P < 0.05) in the abdominal aortic segments (Figure 2). Conversely, group A and group B showed decreased positive staining area of RAM-11 (5.6 ± 2.9% and 6.5 ± 3.6% vs. 25.8 ± 9.6% respectively, both P < 0.01) and Oil-red-O (13.3 ± 6.9% and 9.6 ± 7.3% vs. 26. ± 12.6% respectively, both P < 0.01) in the abdominal aortic segments in comparison with group C (Figure 2). As a result, the vulnerability index in group A and group B was significantly lower than that in group C (0.62 ± 0.13% and 0.52 ± 0.10% vs. 3.71 ± 0.21% respectively, both P < 0.01). The percentage of positive-stained cells for MCP-1, MMP-1, MMP-2, MMP-3, MMP-9, MMP-12 and P-selectin of the abdominal aortic sections was noticeably lower in group A and group B than that in group C (all P < 0.05), with no significant difference between group A and group B (Figures 3 and 4).
Figure 2.
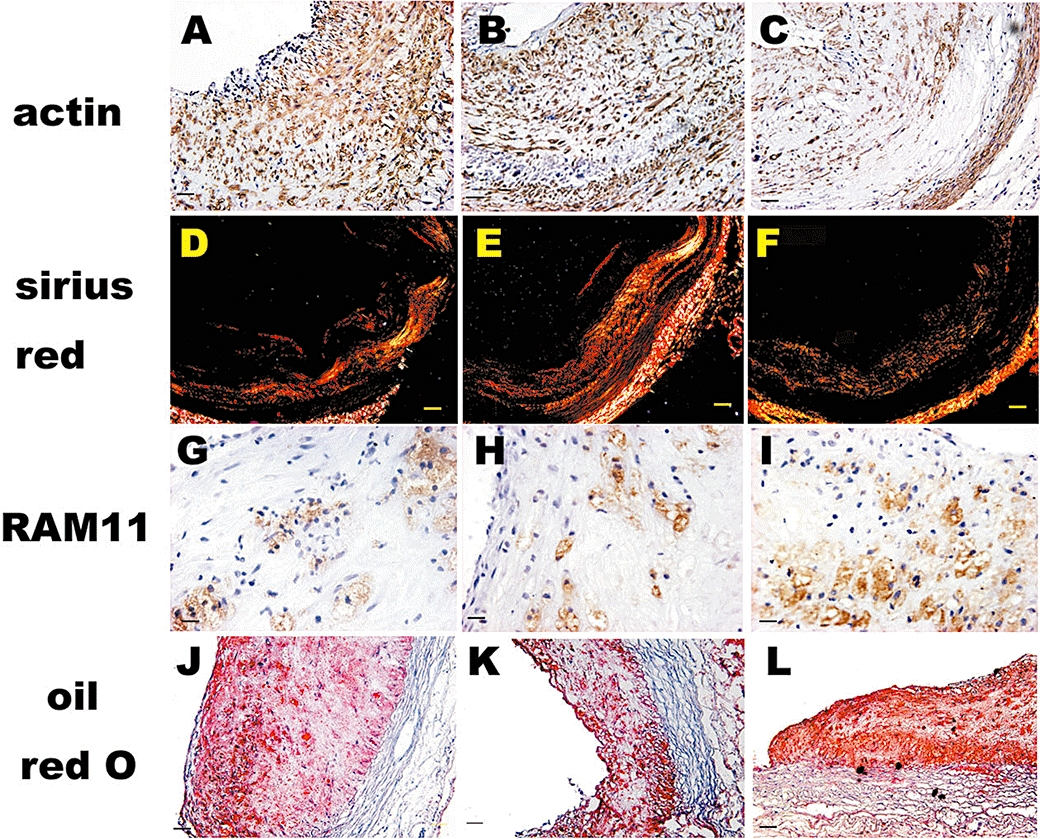
Immunohistochemical, sirius red and oil red O staining in plaques of three groups of rabbits after 12 week intervention. Immunohistological staining of the abdominal aortic cross section showing dense positive staining for α-actin in smooth muscle cells in groups A (A) and B (B) and sparse positive α-actin in smooth muscle cells in group C (C) (bars = 50 µm). Sirius red staining shows abundant collagen in groups A (D) and B (E) and less collagen in group C (F) (bars = 100 µm). Immunohistological staining shows few RAM-11-positive cells in groups A (G) and B (H) and ample RAM-11-positive cells in group C (I) (bars = 50 µm). Oil red O staining shows a small amount of lipids in groups A (J) and B (K) and a large amount of lipids in group C (L) (bars = 100 µm).
Figure 3.
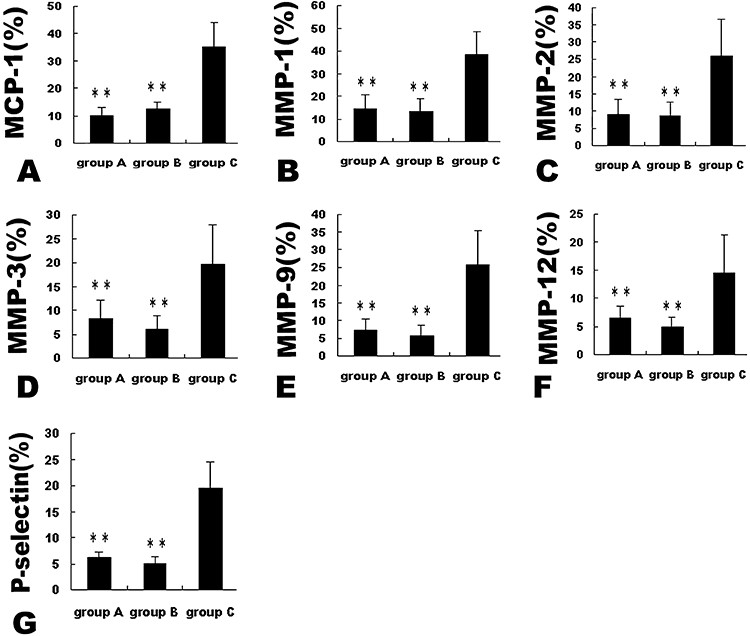
Expression of inflammatory markers by immunohistochemistry in plaques of three groups of rabbits after 12 week intervention. **P < 0.01 versus group C. MCP-1, monocyte chemoattractant protein-1; MMP, matrix metalloproteinases.
Figure 4.
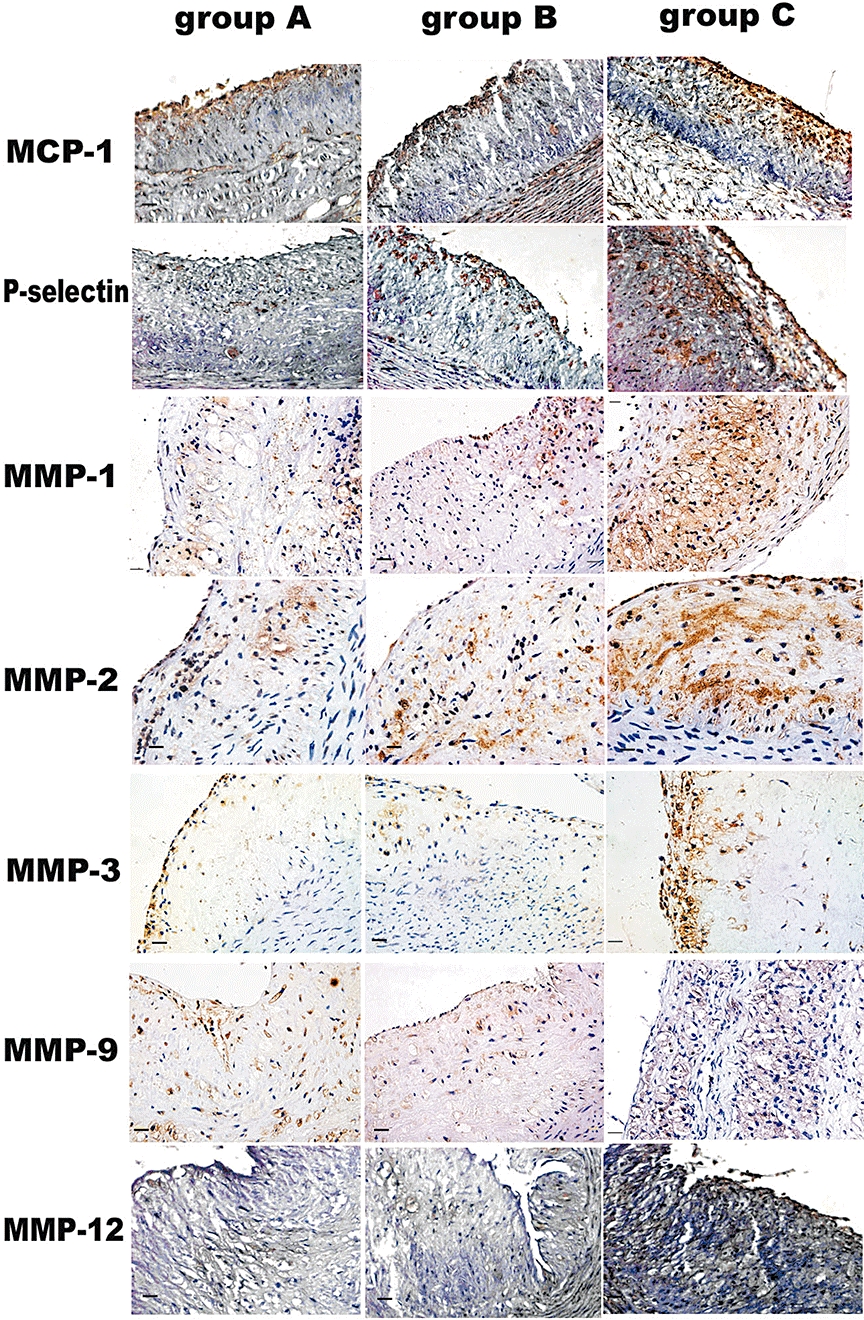
Immunohistochemical staining showing MCP-1, P-selectin, MMP-1, MMP-2, MMP-3, MMP-9 and MMP-12 expression in plaques of three groups of rabbits after 12 week intervention. Bars = 50 µm. MCP-1, monocyte chemoattractant protein-1; MMP, matrix metalloproteinases.
RT-PCR analysis
The expression of mRNAs for MCP-1, MMP-1, MMP-2, MMP-3, MMP-9, MMP-12 and P-selectin in the abdominal aortic plaques was clearly lower in group A and group B than that in group C (all P < 0.05), with no significant difference between group A and group B (Figure 5).
Figure 5.
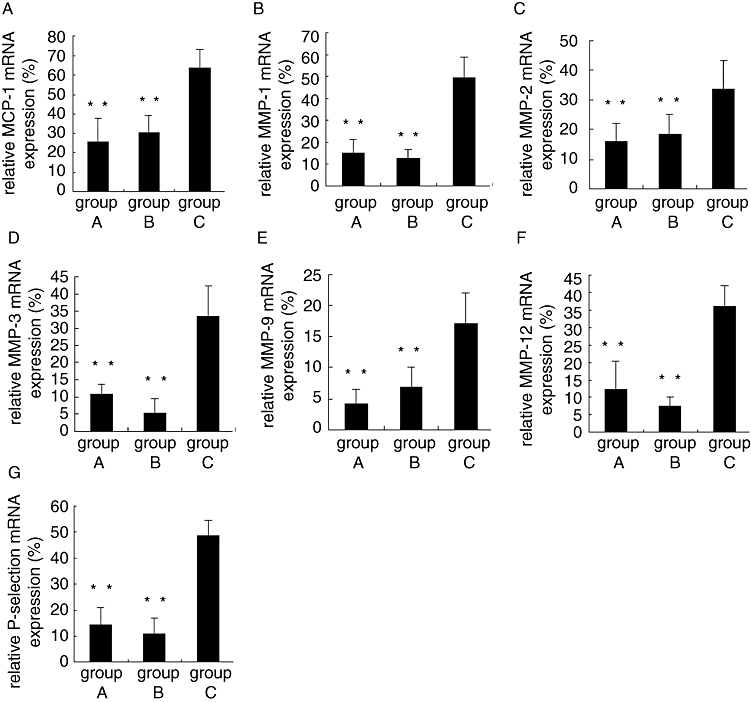
Relative mRNA expression of inflammatory markers in plaques of three groups of rabbits after 12 week intervention. **P < 0.01 versus group C. MCP-1, monocyte chemoattractant protein-1; MMP, matrix metalloproteinases.
Discussion and conclusions
The major finding of the present study is that oral administration of rapamycin resulted in a significant reduction of atherosclerotic plaque inflammation, burden and vulnerability in rabbits, which was independent of serum lipid levels. These molecular and cellular effects translated into a successful prevention of plaque disruption even in the presence of endothelial injury, hyperlipidemia and pharmacological triggering. To the best of our knowledge, this is the first study to show that oral administration of rapamycin had effects comparable to those of simvastatin, in attenuating local and systemic inflammation and enhancing stability of atherosclerotic plaques, although rapamycin had no impact on serum lipid levels.
Recent studies have demonstrated that a large proportion of patients with acute coronary syndrome may have multiple plaque disruption in their coronary arteries and stable plaques may become vulnerable to rupture or erosion once they develop active inflammation. Therefore, long-term administration of oral anti-inflammatory drugs may be more effective than local therapy with a drug-eluting stent for the prevention of plaque rupture, particularly in patients with multiple vulnerable plaques. Although statins have proven effective in stabilizing atherosclerotic plaques, lack of therapeutic effects, liver dysfunction and muscle pathology seen in some patients after statin treatment are the major limitations of statin therapy. Thus, there is a clear need to develop new drugs for stabilizing vulnerable plaques (Kovanen et al., 2005).
Rapamycin, a potent immunosuppressant, has been widely used in drug-eluting stents that have proved effective in reducing the incidence of coronary restenosis after percutaneous coronary intervention. However, acute in-stent thrombosis, probably caused by incomplete and delayed endothelialization of stented plaques, has been found to be a major complication of rapamycin-coated stents. For this reason, some investigators have shifted to the study of the safety and effects of oral rapamycin in preventing coronary restenosis. The Oral Rapamycin in Argentina trial found that oral rapamycin administered after percutaneous coronary intervention was safe and with few minor side effects. High rapamycin blood concentrations were associated with significantly lower late lumen loss and angiographic in-stent restenosis (Rodríguez et al., 2005). In addition, several studies have shown that rapamycin attenuated atherosclerotic plaque progression in apo-E−/− mice (Waksman et al., 2003; Castro et al., 2004; Naoum et al., 2005; Pakala et al., 2005). The mechanisms underlying the therapeutic effects involved alteration in endothelial NO synthase and MCP-1 expression and MMP-2 and MMP-9 activation (Waksman et al., 2003; Castro et al., 2004) and selective deletion of macrophages in plaques (Martinet et al., 2007; Verheye et al., 2007; Danner et al., 2008).
In the present study, a rabbit model of vulnerable plaques was developed by high-fat feeding, endothelial injury and pharmacological triggering, as reported earlier (Chen et al., 2007b). We found that plaques formed in the rabbit abdominal aorta had a large lipid core, a thin cap and abundant macrophages, which are features of vulnerable plaques. Furthermore, the abdominal aorta offers an optimal site for balloon endothelial injury and ultrasonic imaging. For these reasons, we focused on the abdominal aorta for therapeutic interventions. The atherosclerotic burden of the thoracic aorta, without balloon endothelial injury, was less prominent than that of the abdominal aorta, but was reduced in group A and B compared with that in group C, suggesting that rapamycin and simvastatin had the same attenuation effects on both thoracic and abdominal aortic plaques. However, no plaque rupture was detected in the thoracic aorta after pharmacological triggering in all three groups of rabbits. Using this model, we found that oral administration of rapamycin reduced inflammation, inhibited progression and enhanced stability of atherosclerotic plaques as shown by the following evidence: first, after 12 week treatment with rapamycin, ultrasonographic measurements of IMT, EEMA, PA and PB% were greatly decreased and those of AIIc% increased. Pathologically, the fibrous cap became thicker and the IMT thinner; second, serum levels of MCP-1, hs-CRP, IL-8, IL-18, MMP-1 and P-selectin after treatment were significantly reduced; third, the mRNA expression and positive staining for MCP-1, MMP-1, MM-3, MMP-12 and P-selectin in the plaques as well as the vulnerability index were consistently decreased; and finally, no rabbits in group A developed plaque disruption, indicating successful prevention of plaque rupture. A notable finding in this study is that treatment with rapamycin did not affect serum lipid levels and only moderately reduced the lipid contents of plaques. Nonetheless, rapamycin therapy induced a marked anti-inflammatory effect, which led to stabilization of plaques. These results suggest that potent suppression of inflammation alone, without lipid lowering, can inhibit the progression and enhance the stability of atherosclerotic plaques.
The reason why rapamycin treatment induces selective deletion of macrophages in plaques is not completely understood, but may be related to the decreased macrophage viability by inhibiting mammalian target of rapamycin and macrophage autophagy in plaques (Martinet et al., 2007; Verheye et al., 2007; Danner et al., 2008; Mita et al., 2008; Mueller et al., 2008). The fact that serum levels of inflammatory factors were significantly reduced in the rapamycin group suggests that rapamycin not only decreased the number of macrophages, but also inhibited the secretion of inflammatory factors. Although many studies have reported that rapamycin-coated stent or oral rapamycin inhibited proliferation and migration of SMCs after coronary stenting (Burke et al., 1999; Marx and Marks, 2001; Sousa et al., 2001; Farb et al., 2002; Moses et al., 2003), the effect of rapamycin on SMCs in plaques without stenting is in dispute. Recently, two groups of investigators found that the rapamycin derivative, everolimus, selectively deleted macrophages without altering SMC content in atherosclerotic plaques of rabbits (Martinet et al., 2007; Verheye et al., 2007). In our rabbit model of vulnerable plaques, we found that rapamycin treatment significantly increased SMCs in the fibrous cap of plaques, indicating that rapamycin exerts its stabilization effects by decreasing macrophages as well as increasing SMCs in vulnerable plaques.
A major concern with rapamycin therapy is that this drug induces hyperlipidemia in patients receiving at least 12 month treatment (Eisen et al., 2003; Celik et al., 2008). In the present study, however, such an adverse effect was not observed after 12 week treatment with rapamycin. The reason for this discrepancy may be threefold: first, before rapamycin treatment, our rabbits already had high serum lipid levels after being on a high-cholesterol diet for 8 weeks and the adverse effects of rapamycin were thus concealed; second, the 12 week duration of rapamycin intervention was too short to induce further increase in serum lipid levels; and third, the difference among animal species should be also taken into account. Another concern is the effect of rapamycin on the intimal thickness of plaques. Eisen et al. (2003) found that although 12 month everolimus treatment prevented allograft rejection in cardiac-transplant recipients, the intimal thickness of coronary arteries was increased significantly. However, Gregory et al. (1993) found that rapamycin inhibited carotid intimal thickening in rabbits after carotid endothelial balloon injury. Similarly, Mueller et al. (2008) reported that 12 week everolimus treatment decreased atherosclerotic plaque area significantly in LDLR−/− mice. Other animal experiments also found a significant reduction of neointimal formation by rapamycin (Burke et al., 1999; Suzuki et al., 2001; Farb et al., 2002). In our experiments, we found that oral administration of rapamycin for 12 weeks resulted in a marked decrease of IMT in plaques as measured by ultrasonography and pathology, which is consistent with most previous studies.
A head-to-head comparison between simvastatin and rapamycin was carried out in the present study and the major difference between the two treatment groups was found to be a much higher serum level of TC and LDL-C, but a much lower ultrasonic value of IMT and PB% in group A as compared with group B. Although rapamycin had no effect on lipid lowering, the net effects of the two drugs in attenuating inflammation and enhancing stability of atherosclerotic plaques were similar. This was demonstrated by the non-significant difference in local and systemic inflammatory marker levels, vulnerability index and incidence of plaque rupture between group A and group B. The fact that IMT and PB% measured by ultrasonic imaging in group A were significantly lower than those in group B indicates that rapamycin may have a relatively stronger effects on inhibiting plaque progression, which is beneficial for the prevention of plaque disruption, as our previous studies revealed that plaque area (PA) was an independent predictor of plaque rupture. The mechanisms underlying these inhibitory effects are not clear, but may be due to the more potent efficacy of rapamycin in reducing macrophages and attenuating inflammatory reaction in plaques than simvastatin.
Our study contains several limitations. First, the sample size in each of the three animal groups was small, and further studies involving a larger sample size and other types of animals, such as apoE−/− mice, are warranted to confirm our results. Second, the incidence of plaque rupture after pharmacological triggering in group C rabbits was relatively low because of the simplicity of our methods in inducing plaque vulnerability, which may explain in part the zero incidence of plaque rupture in groups A and B rabbits. Therefore, the therapeutic effects of rapamycin in animals with a high incidence of plaque disruption need to be further investigated. Finally, although 12 week rapamycin treatment effectively stabilized vulnerable plaques, the possible changes of plaque vulnerability after rapamycin discontinuation was not addressed in this study and, thus, further studies are required to clarify the necessity and safety of long-term administration of rapamycin.
In conclusion, oral administration of rapamycin provides a new and effective approach to attenuating inflammation, inhibiting progression and enhancing stability of atherosclerotic plaques. These therapeutic effects are independent of serum lipid levels and demonstrate that potent inhibition of plaque inflammation alone, without lipid lowering, can stabilize vulnerable plaques.
Acknowledgments
This work was supported by the National 973 Basic Research Program of China (No.2006CB503803), the National High-tech Research and Development Program of China (No.2006AA02A406), the Program of Introducing Talents of Discipline to Universities (No.B07035) and the State Key Program of National Natural Science of China (No. 60831003).
Glossary
Abbreviations
- AII
ultrasonic intensity
- AIIc%
corrected AII
- Dd
aortic diameter at end-diastole
- EEMA
external elastic membrane area
- HDL-C
high-density lipoprotein cholesterol
- hs-CRP
high sensitive C-reactive protein
- H&E
haematoxylin and eosin
- IL
interleukin
- IMT
maximal intima- media thickness
- IVUS
intravascular ultrasound
- LA
lumen area
- LDL-C
low-density lipoprotein cholesterol
- MCP-1
monocyte chemoattractant protein-1
- MMP
matrix metalloproteinases
- PA
plaque area
- PB
plaque burden
- SMC
smooth muscle cell
- TC
total cholesterol
- TG
triglyceride
- Vm
mean velocity
- Vp
aortic peak velocity
- VTI
velocity-time integral
Conflict of interest
None.
References
- Aikawa M, Rabkin E, Okada Y, Voglic SJ, Clinton SK, Brinckerhoff CE, et al. Lipid lowering by diet reduces matrix metalloproteinase activity and increases collagen content of rabbit atheroma: a potential mechanism of lesion stabilization. Circulation. 1998;97:2433–2444. doi: 10.1161/01.cir.97.24.2433. [DOI] [PubMed] [Google Scholar]
- Armstrong EJ, Morrow DA, Sabatine MS. Inflammatory biomarkers in acute coronary syndromes. Part I: introduction and cytokines. Circulation. 2006;113:e72–e75. doi: 10.1161/CIRCULATIONAHA.105.595520. [DOI] [PubMed] [Google Scholar]
- Burke SE, Lubbers NL, Chen YW, Hsieh GC, Mollison KW, Luly JR, et al. Neointimal formation after balloon-induced vascular injury in Yucatan minipigs is reduced by oral rapamycin. J Cardiovasc Pharmacol. 1999;33:829–835. doi: 10.1097/00005344-199906000-00001. [DOI] [PubMed] [Google Scholar]
- Carr SC, Farb A, Pearce WH, Virmani R, Yao JS. Activated inflammatory cells are associated with plaque rupture in carotid artery stenosis. Surgery. 1997;122:757–763. doi: 10.1016/s0039-6060(97)90084-2. [DOI] [PubMed] [Google Scholar]
- Castro C, Campistol JM, Sancho D, Sanchez-Madrid F, Casals E, Andres V. Rapamycin attenuates atherosclerosis induced by dietary cholesterol in apolipoprotein-deficient mice through a p27 Kip1-independent pathway. Atherosclerosis. 2004;172:31–38. doi: 10.1016/j.atherosclerosis.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Celik S, Doesch A, Erbel C, Blessing E, Ammon K, Koch A, et al. Beneficial effect of omega-3 fatty acids on sirolimus- or everolimus-induced hypertriglyceridemia in heart transplant recipients. Transplantation. 2008;86:245–250. doi: 10.1097/TP.0b013e318177281e. [DOI] [PubMed] [Google Scholar]
- Chen WQ, Zhang M, Ji XP, Ding SF, Zhao YX, Chen YG, et al. Usefulness of high frequency vascular ultrasound imaging and serum inflammatory markers to predict plaque rupture in patients with stable and unstable angina pectoris. Am J Cardiol. 2007a;100:1341–1346. doi: 10.1016/j.amjcard.2007.06.044. [DOI] [PubMed] [Google Scholar]
- Chen WQ, Zhang L, Liu YF, Chen L, Ji XP, Zhang M, et al. Prediction of atherosclerotic plaque ruptures with high frequency ultrasound imaging and serum inflammatory markers. Am J Physiol Heart Circ Physiol. 2007b;293:H2836–H2844. doi: 10.1152/ajpheart.00472.2007. [DOI] [PubMed] [Google Scholar]
- Danner S, Sigrist S, Moreau F, Mandes K, Vodouhé C, Langlois A, et al. Influence of rapamycin on rat macrophage viability and chemotaxis toward allogenic pancreatic islet supernates. Transplant Proc. 2008;40:470–472. doi: 10.1016/j.transproceed.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Eisen HJ, Tuzcu EM, Dorent R, Kobashigawa J, Mancini D, Valantine-von Kaeppler HA, et al. Everolimus for the prevention of allograft rejection and vasculopathy in cardiac-transplant recipients. N Engl J Med. 2003;349:847. doi: 10.1056/NEJMoa022171. [DOI] [PubMed] [Google Scholar]
- Farb A, John M, Acampado E, Kolodgie FD, Prescott MF, Virmani R. Oral everolimus inhibits in-stent neointimal growth. Circulation. 2002;106:2379–2384. doi: 10.1161/01.cir.0000033973.06059.04. [DOI] [PubMed] [Google Scholar]
- Gregory CR, Huie P, Billingham ME, Morris RE. Rapamycin inhibits arterial intimal thickening caused by both alloimmune and mechanical injury: its effect on cellular, growth factor, and cytokine response in injured vessels. Transplantation. 1993;55:1409–1418. doi: 10.1097/00007890-199306000-00037. [DOI] [PubMed] [Google Scholar]
- Kovanen PT, Mayranpaa M, Lindstedt KA. Drug therapies to prevent coronary plaque rupture and erosion: present and future. Handb Exp Pharmacol. 2005;170:745–776. doi: 10.1007/3-540-27661-0_28. [DOI] [PubMed] [Google Scholar]
- Libby P, Aikawa M. Stabilization of atherosclerotic plaques: new mechanisms and clinical targets. Nat Med. 2002;8:1257–1262. doi: 10.1038/nm1102-1257. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Martinet W, Verheye S, De Meyer GR. Everolimus-induced mTOR inhibition selectively depletes macrophages in atherosclerotic plaques by autophagy. Autophagy. 2007;3:241–244. doi: 10.4161/auto.3711. [DOI] [PubMed] [Google Scholar]
- Marx SO, Marks AR. Bench to bedside: the development of rapamycin and its application to stent restenosis. Circulation. 2001;104:852–855. doi: 10.1161/01.cir.104.8.852. [DOI] [PubMed] [Google Scholar]
- Mintz GS, Nissen SE, Anderson WD, Rosenfield K, Bailey S, Siegel R, et al. American college of cardiology clinical expert consensus document on standards for acquisition, measurement and reporting of Intravascular Ultrasound Studies (IVUS) J Am Coll Cardiol. 2001;37:1478–1492. doi: 10.1016/s0735-1097(01)01175-5. [DOI] [PubMed] [Google Scholar]
- Mita A, Ricordi C, Miki A, Barker S, Haertter R, Hashikura Y, et al. Anti-proinflammatory effects of sirolimus on human islet preparations. Transplantation. 2008;86:46–53. doi: 10.1097/TP.0b013e31817c79c0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moses JW, Leon MB, Popma JJ, Fitzgerald PJ, Holmes DR, O'Shaughnessy C, et al. Sirolimus-eluting stents versus standard stents in patients with stenosis in a native coronary artery. N Engl J Med. 2003;349:1315–1323. doi: 10.1056/NEJMoa035071. [DOI] [PubMed] [Google Scholar]
- Mueller MA, Beutner F, Teupser D, Ceglarek U, Thiery J. Prevention of atherosclerosis by the mTOR inhibitor everolimus in LDLR-/- mice despite severe hypercholesterolemia. Atherosclerosis. 2008;198:39–48. doi: 10.1016/j.atherosclerosis.2007.09.019. [DOI] [PubMed] [Google Scholar]
- Naghavi M, Libby P, Falk E, Casscells SW, Litovsky S, Rumberger J, et al. From vulnerable plaque to vulnerable patient. A call for new definition and risk assessment strategies: part I. Circulation. 2003;108:1664–1672. doi: 10.1161/01.CIR.0000087480.94275.97. [DOI] [PubMed] [Google Scholar]
- Naoum JJ, Woodside KJ, Zhang S, Rychahou PG, Hunter GC. Effects of rapamycin on the arterial inflammatory response in atherosclerotic plaques in Apo-E knockout mice. Transplant Proc. 2005;37:1880–1884. doi: 10.1016/j.transproceed.2005.02.080. [DOI] [PubMed] [Google Scholar]
- Pakala R, Stabile E, Jang GJ, Clavijo L, Waksman R. Rapamycin attenuates atherosclerotic plaque progression in apolipoprotein E knockout mice: inhibitory effect on monocyte chemotaxis. J Cardiovasc Pharmacol. 2005;46:481–486. doi: 10.1097/01.fjc.0000177985.14305.15. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Cannon CP, Morrow D, Rifai N, Rose LM, McCabe CH, et al. For the pravastatin or atorvastatin evaluation and infection therapy–thrombolysis in myocardial infarction 22 (PROVE IT-TIMI 22) investigators. C-reactive protein levels and outcomes after statin therapy. N Engl J Med. 2005;352:20–28. doi: 10.1056/NEJMoa042378. [DOI] [PubMed] [Google Scholar]
- Rodríguez AE, Rodríguez Alemparte M, Vigo CF, Fernández Pereira C, Llauradó C, Vetcher D, et al. Role of oral rapamycin to prevent restenosis in patients with de novo lesions undergoing coronary stenting: results of the Argentina single centre study (ORAR trial) Heart. 2005;91:1433–1437. doi: 10.1136/hrt.2004.050617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez AE, Granada JF, Rodriguez-Alemparte M, Vigo CF, Delgado J, Fernandez-Pereira C, et al. ORAR II investigators. Oral rapamycin after coronary bare-metal stent implantation to prevent restenosis: the prospective, randomized Oral Rapamycin in Argentina (ORAR II) Study. J Am Coll Cardiol. 2006;47:1522–1529. doi: 10.1016/j.jacc.2005.12.052. [DOI] [PubMed] [Google Scholar]
- Sousa JE, Costa MA, Abizaid A, Abizaid AS, Feres F, Pinto IM, et al. Lack of neotintimal proliferation after implantation of sirolimus-coated stents in human coronary arteries. Circulation. 2001;103:192–195. doi: 10.1161/01.cir.103.2.192. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Kopia G, Hayashi S, Bailey LR, Llanos G, Wilensky R, et al. Stent-based delivery of sirolimus reduces neointimal formation in a porcine coronary model. Circulation. 2001;104:1188–1193. doi: 10.1161/hc3601.093987. [DOI] [PubMed] [Google Scholar]
- Torzewski M, Klouche M, Hock J, Messner M, Dorweiler B, Torzewski J, et al. Immunohistochemical demonstration of enzymatically modified human LDL and its colocalization with the terminal complement complex in the early atherosclerotic lesion. Arterioscler Thromb Vasc Biol. 1998;18:369–378. doi: 10.1161/01.atv.18.3.369. [DOI] [PubMed] [Google Scholar]
- Verheye S, Martinet W, Kockx MM, Knaapen MW, Salu K, Timmermans JP, et al. Selective clearance of macrophages in atherosclerotic plaques by autophagy. J Am Coll Cardiol. 2007;49:706–715. doi: 10.1016/j.jacc.2006.09.047. [DOI] [PubMed] [Google Scholar]
- Waksman R, Pakala R, Burnett MS, Gulick CP, Leborgne L, Fournadjiev J, et al. Oral rapamycin inhibits growth of atherosclerotic plaque in apoE knock-out mice. Cardiovasc Radiat Med. 2003;4:34–38. doi: 10.1016/s1522-1865(03)00121-5. [DOI] [PubMed] [Google Scholar]
- Waksman R, Ajani AE, Pichard AD, Torguson R, Pinnow E, Canos D, et al. Oral rapamune to inhibit restenos study. Oral rapamycin to inhibit restenosis after stenting of de novo coronary lesions: the Oral Rapamune to Inhibit Restenosis (ORBIT) study. J Am Coll Cardiol. 2004;44:1386–1392. doi: 10.1016/j.jacc.2004.06.069. [DOI] [PubMed] [Google Scholar]
- Wessely R, Kastrati A, Mehilli J, Dibra A, Pache J, Schömig A. Randomized trial of rapamycin- and paclitaxel-eluting stents with identical biodegradable polymeric coating and design. Eur Heart J. 2007;28:2720–2725. doi: 10.1093/eurheartj/ehm425. [DOI] [PubMed] [Google Scholar]
- Williams H, Johnson JL, Carson KG, Jackson CL. Characteristics of intact and ruptured atherosclerotic plaques in brachiocephalic arteries of apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol. 2002;22:788–779. doi: 10.1161/01.atv.0000014587.66321.b4. [DOI] [PubMed] [Google Scholar]