

Abstract
Background and purpose:
The selective inhibition of prostaglandin (PG)E2 formation via interference with microsomal PGE2 synthase (mPGES)-1 could have advantages in the treatment of PGE2-associated diseases, such as inflammation, fever and pain, compared with a general suppression of all PG biosynthesis, provided by inhibition of cyclooxygenase (COX)-1 and 2. Here, we addressed whether the naturally occurring acylphloroglucinol myrtucommulone (MC) from Myrtus communis L. (myrtle) affected mPGES-1.
Experimental approach:
The effect of MC on PGE2 formation was investigated in a cell-free assay by using microsomal preparations of interleukin-1β-stimulated A549 cells as the source of mPGES-1, in intact A549 cells, and in lipopolysaccharide-stimulated human whole blood. Inhibition of COX-1 and COX-2 activity in cellular and cell-free assays was assessed by measuring 12(S)-hydroxy-5-cis-8,10-trans-heptadecatrienoic acid and 6-oxo PGF1α formation.
Key results:
MC concentration-dependently inhibited cell-free mPGES-1-mediated conversion of PGH2 to PGE2 (IC50 = 1 µmol·L−1). PGE2 formation was also diminished in intact A549 cells as well as in human whole blood at low micromolar concentrations. Neither COX-2 activity in A549 cells nor isolated human recombinant COX-2 was significantly affected by MC up to 30 µmol·L−1, and only moderate inhibition of cellular or cell-free COX-1 was evident (IC50 > 15 µmol·L−1).
Conclusions and implications:
MC is the first natural product to inhibit mPGES-1 that efficiently suppresses PGE2 formation without significant inhibition of the COX enzymes. This provides an interesting pharmacological profile suitable for interventions in inflammatory disorders, without the typical side effects of coxibs and non-steroidal anti-inflammatory drugs.
Keywords: microsomal prostaglandin E2 synthase-1, prostaglandin E2, prostanoids, cyclooxygenase, natural products, NSAIDs, inflammation
Introduction
Prostaglandin (PG)E2 is a prominent product of the cyclooxygenase (COX) pathway and has long been considered as a mediator of inflammation, pain, fever and cancer, but it is also known to regulate physiological functions in the gastrointestinal tract, the kidney and in the immune and nervous systems (Smith, 1989; Sugimoto and Narumiya, 2007). Three terminal isoforms of PGE2 synthases (PGES) have been identified so far, and co-transfection of COX-1 and 2 with PGES isoenzymes in mammalian cells suggests that molecular interactions may cause preferential functional coupling (Murakami et al., 2002; Samuelsson et al., 2007). Thus, the constitutively expressed cytosolic PGE2 synthase (cPGES) was found to be coupled to COX-1 being necessary for homeostasis, whereas the microsomal PGE2 synthase (mPGES)-1 is the predominant isoform involved in COX-2-mediated PGE2 production, responsible for distinct pathophysiological effects (Park et al., 2006). mPGES-1 belongs to the membrane-associated proteins involved in eicosanoid and glutathione metabolism (MAPEG) family, and its expression is induced by pro-inflammatory stimuli (such as interleukin-1β) congruent with expression of COX-2 (Jakobsson et al., 1999). Knockout (KO) experiments have revealed that mice deficient in mPGES-1 develop normally but exhibit reduced inflammatory responses (Trebino et al., 2003) and do not show fever in response to lipopolysaccharide (Engblom et al., 2003).
Selective inhibition of inflammatory PGE2 formation is considered superior over inhibition of all COX-derived products (Samuelsson et al., 2007), as other prostanoids such as PGI2 and PGD2 possess important physiological functions (Funk, 2001). Thus, mPGES-1 may constitute a potential target for therapeutic intervention.
Only a few inhibitors of mPGES-1 have been described, including polyunsaturated fatty acids, such as arachidonic acid and 15-deoxy-Δ12,14-PGJ2 (IC50 = 0.3 µmol·L−1, (Quraishi et al., 2002)). Further, mPGES-1 is inhibited by MK-886 [3-[1-(4-chlorobenzyl)-3-t-butyl-thio-5-isopropylindol-2-yl]-2,2-dimethylpropanoic acid, IC50 = 2.4 µmol·L−1, (Claveau et al., 2003)] and its structural derivatives (Riendeau et al., 2005) including licofelone (Koeberle et al., 2008). Synthetic phenanthrene imidazoles constitute another group of potent, selective, and orally active mPGES-1 inhibitors with IC50 values of 0.42 and 1.3 µmol·L−1 in A549 cells and human whole blood respectively (Cote et al., 2007; Xu et al., 2008).
Myrtucommulone (MC, Figure 1A) consists of an isobutyrophenone core (IBP-C), substituted with two syncarpic acid moieties and represents an oligomeric, nonprenylated acylphloroglucinol which is present in the leaves of myrtle (Myrtus communis L., Myrtaceae) (Appendino et al., 2002). Myrtle is used in traditional folk medicine as an antiseptic agent, and extracts of myrtle have been reported to possess antibacterial (Bonjar, 2004), analgesic (Levesque and Lafont, 2000) and antioxidative properties (Rosa et al., 2003; Romani et al., 2004). MC acts as antioxidant (Rosa et al., 2003), shows antibacterial activity against clinically relevant bacteria (Appendino et al., 2002), induces selective apoptosis of cancer cells (Tretiakova et al., 2007) and suppresses inflammatory neutrophil functions (Feisst et al., 2005). Nevertheless, little is known about the molecular targets of MC. Recently, we have shown that 5-lipoxygenase in intact polymorphonuclear leukocytes (IC50 = 2 µmol·L−1) and to a lesser extent also COX-1 in human platelets (IC50 = 17 µmol·L−1) are directly targeted by MC (Feisst et al., 2005). Here we provide strong evidence that MC potently interferes with mPGES-1.
Figure 1.

Effects of myrtucommulone (MC) on the activity of isolated cyclooxygenase (COX)-1 and COX-2. (A) Chemical structures of MC, semi-myrtucommulone (S-MC) and the isobutyrophenone core (IBP-C). (B) Purified ovine COX-1 (50 units) or human recombinant COX-2 (20 units) were added to a COX reaction mix. Vehicle (w/o), MC (0.3 to 33 µmol·L−1), indomethacin (Indo, 10 µmol·L−1) and celecoxib (Cele, 5 µmol·L−1) were added, and after 5 min the reaction was started by addition of arachidonic acid (5 µmol·L−1 for COX-1, 2 µmol·L−1 for COX-2). After another 5 min at 37°C, the formation of 12-HHT (12(S)-hydroxy-5-cis-8,10-trans-heptadecatrienoic acid) was determined by RP-HPLC as described in the Methods section. Data are given as mean ± SE, n = 3, ***P < 0.001 vs. vehicle (0.1% dimethyl sulphoxide) control, anova + Tukey HSD post hoc tests.
Methods
Cells and cell viability assay
A549 cells were cultured in DMEM high glucose (4.5 g·L−1) medium supplemented with heat-inactivated fetal calf serum (FCS) (10%, v,v), penicillin (100 U·mL−1) and streptomycin (100 µg·mL−1) at 37°C in a 5% CO2 incubator. After 3 days, confluent cells were detached by using 1× trypsin-EDTA solution and reseeded at 2 × 106 cells in 20 mL medium in 175 cm2 flasks. Cell viability was measured by using the colorimetric thiazolyl blue tetrazolium (MTT) dye reduction assay. A549 cells (4 × 104 cells per 100 µL medium) were plated into a 96-well microplate and incubated at 37°C and 5% CO2 for 16 h. Then, MC, semi-myrtucommulone (S-MC) or the IBP-C (30 µmol·L−1, each) were added, and the samples were incubated for another 5 h. MTT (20 µL, 5 mg·mL−1) was added, and the incubations were continued for 4 h. The formazan product was solubilized with SDS (10%, m,v in 20 mmol·L−1 HCl), and the absorbance of each sample was measured at 595 nm relative to the absorbance of vehicle (dimethyl sulphoxide, DMSO)-treated control cells by using a multi-well scanning spectrophotometer (Victor3 plate reader, PerkinElmer, Rodgau-Juegesheim, Germany). None of the substances significantly reduced cell viability (data not shown), excluding possible acute cytotoxic effects of the compounds in the cellular assays.
Activity assays of isolated COX-1 and 2
Inhibition of the activities of purified ovine COX-1 and human COX-2 was performed as described (Mitchell et al., 1993; Capdevila et al., 1995). Although the purified COX-1 is not of human origin, ovine COX-1 is generally used for inhibitor studies when examining the effectiveness of compounds on the activity of isolated COX-1 enzyme (Mitchell et al., 1993). Briefly, purified COX-1 (ovine, 50 units) or COX-2 (human recombinant, 20 units) were diluted in 1 mL reaction mixture containing 100 mmol·L−1 Tris buffer pH 8, 5 mmol·L−1 glutathione, 5 µmol·L−1 haemoglobin and 100 µmol·L−1 EDTA at 4°C and pre-incubated with the test compounds for 5 min. Samples were pre-warmed for 60 s at 37°C, and arachidonic acid (5 µmol·L−1 for COX-1, 2 µmol·L−1 for COX-2) was added to start the reaction. After 5 min at 37°C, the COX product 12-HHT (12(S)-hydroxy-5-cis-8,10-trans-heptadecatrienoic acid) was extracted and then analysed by high performance liquid chromatography (HPLC) as described (Guichardant et al., 1989; Albert et al., 2002).
Induction of mPGES-1 in A549 cells and isolation of microsomes
Preparation of A549 cells was performed as described (Koeberle et al., 2008). In brief, cells (2 × 106 cells in 20 mL medium) were plated in 175 cm2 flasks and incubated for 16 h at 37°C and 5% CO2. Subsequently, the culture medium was replaced by fresh DMEM high glucose medium containing FCS (2%, v,v). In order to induce mPGES-1 expression, interleukin-1β (1 ng·mL−1) was added, and cells were incubated for another 48 h. Thereafter, cells were detached with trypsin-EDTA, washed with PBS and frozen in liquid nitrogen. Ice-cold homogenization buffer (0.1 mol·L−1 potassium phosphate buffer pH 7.4, 1 mmol·L−1 PMSF, 60 µg·mL−1 soybean trypsin inhibitor, 1 µg·mL−1 leupeptin, 2.5 mmol·L−1 glutathione and 250 mmol·L−1 sucrose) was added and after 15 min, cells were resuspended and sonicated on ice (3 × 20 s). The homogenate was subjected to differential centrifugation at 10 000×g for 10 min and 174 000×g for 1 h at 4°C. The pellet (microsomal fraction) was resuspended in 1 mL homogenization buffer, and the total protein concentration was determined by Coomassie protein assay (Bradford, 1976). Microsomal membrane fractions were stored at −80°C for several weeks.
Determination of PGE2 synthase activity in microsomes of A549 cells
Prostaglandin E2 synthase activity was determined as previously reported (Thoren and Jakobsson, 2000). Microsomal membranes were diluted in potassium phosphate buffer (0.1 mol·L−1, pH 7.4) containing 2.5 mmol·L−1 glutathione. Test compounds or vehicle were added and after 15 min at 4°C, the reaction (100 µL total volume) was initiated by addition of PGH2 (20 µmol·L−1, final concentration). After 1 min at 4°C, the reaction was terminated by using stop solution (100 µL; 40 mmol·L−1 FeCl2, 80 mmol·L−1 citric acid and 10 µmol·L−1 of 11β-PGE2). PGE2 was separated by solid phase extraction on reversed phase (RP)-C18 material by using acetonitrile (200 µL) as eluent, and analysed by RP-HPLC [30% acetonitrile in water + 0.007% TFA (v,v), Nova-Pak® C18 column, 5 × 100 mm, 4 µm particle size, flow rate 1 mL·min−1] with UV detection at 195 nm. 11β-PGE2 was used as internal standard to quantify PGE2 product formation by integration of the area under the peaks.
Determination of PGE2 and 6-oxo PGF1α formation in intact A549 cells
A549 cells (2 × 106 cells) were plated in a 175 cm2 flask and incubated for 16 h at 37°C and 5% CO2. Then, the medium was changed, and the cells were stimulated with interleukin-1β (1 ng·mL−1) in DMEM high glucose medium containing FCS (2%, v,v) for 48 h. After trypsination, cells were washed with PBS twice. For determination of PGE2, 4 × 106 cells·per·mL PBS containing CaCl2 (1 mmol·L−1) were pre-incubated with the indicated compounds at 37°C for 10 min, and PGE2 formation was started by addition of ionophore A23187 (2.5 µmol·L−1), arachidonic acid (1 µmol·L−1) and [3H]arachidonic acid (18.4 kBq). The reaction was stopped after 15 min at 37°C, and the samples were put on ice. After centrifugation (800×g, 5 min, 4°C), the supernatant was acidified (pH 3) by addition of citric acid (20 µL, 2 mol·L−1), and the internal standard 11β-PGE2 (2 nmol) was added. Radiolabelled PGE2 was separated by RP-18 solid phase extraction and eluted with acetonitrile (200 µL). Solid phase extraction and HPLC analysis were performed as described above. The amount of 11β-PGE2 was quantified by integration of the area under the eluted peaks. For quantification of radiolabelled PGE2, fractions (0.5 mL) were collected and mixed with Ultima Gold™ XR (2 mL) for liquid scintillation counting in a LKB Wallac 1209 Rackbeta Liquid Scintillation Counter.
For determination of 6-oxo PGF1α, 5 × 106 cells resuspended in 1 mL PBS containing CaCl2 (1 mmol·L−1) were pre-incubated with the indicated compounds for 15 min at 37°C, and 6-oxo PGF1α formation was initiated by addition of arachidonic acid (30 µmol·L−1). After 15 min at 37°C, the reaction was stopped by cooling on ice. Cells were centrifuged (300×g, 5 min, 4°C), and the amount of released 6-oxo PGF1α was assessed by enzyme-linked immunosorbent assay (elisa) by using a monoclonal antibody against 6-oxo PGF1α according to the protocol described by Yamamoto et al. (1987). For the elisa, the monoclonal antibody (0.2 µg per 200 µL) was coated on microtiter plates via a goat anti-mouse immunoglobulin G antibody. 6-oxo PGF1α (15 µg) was linked to bacterial β-galactosidase (0.5 mg), and the enzyme activity bound to the antibody was determined in an elisa reader at 550 nm (reference wavelength: 630 nm) by using chlorophenol-red-β-D-galactopyranoside (CPRG, Roche Diagnostic GmbH) as substrate.
Determination of PGE2 and 12-HHT in whole blood
Peripheral blood from healthy adult volunteers, who had not received any medication for at least 2 weeks under informed consent, was obtained by venepuncture and collected in syringes containing heparin (20 U·mL−1). For determination of PGE2, aliquots of whole blood (0.8 mL) were mixed with the thromboxane synthase inhibitor CV4151 (1 µmol·L−1) and with aspirin (50 µmol·L−1). A total volume of 1 mL was adjusted with sample buffer (10 mmol·L−1 potassium phosphate buffer pH 7.4, 3 mmol·L−1 KCl, 140 mmol·L−1 NaCl and 6 mmol·L−1 D-glucose). After pre-incubation with the indicated compounds for 5 min at room temperature, the samples were stimulated with lipopolysaccharide (10 µg·mL−1) for 5 h at 37°C. PGE2 formation was stopped on ice; the samples were centrifuged (2300×g, 10 min, 4°C), and citric acid (30 µL, 2 mol·L−1) was added to the supernatant. After another centrifugation step (2300×g, 10 min, 4°C), solid phase extraction and HPLC analysis of PGE2 was performed as described above. The PGE2 peak (3 mL), identified by co-elution with the authentic standard, was collected, and acetonitrile was removed under a nitrogen stream. The pH was adjusted to 7.2 by addition of 10× PBS buffer pH 7.2 (230 µL) before PGE2 contents were quantified by using a PGE2 High Sensitivity EIA Kit (Assay Designs, Ann Arbor, MI) according to the manufacturer's protocol.
For determination of the COX product 12-HHT, whole blood (2 mL) was pre-incubated with the indicated compounds at 37°C for 10 min, and formation of 12-HHT was initiated by addition of 30 µmol·L−1 Ca2+-ionophore A23187 and 100 µmol·L−1 arachidonic acid. After 10 min at 37°C, the reaction was stopped on ice, and the samples were centrifuged (600×g, 10 min, 4°C). Aliquots of the resulting plasma (500 µL) were then mixed with 2 mL of methanol, and 200 ng of PGB1 was added as internal standard. The samples were held at −20°C for 2 h and centrifuged again (600×g, 15 min, 4°C). The supernatants were collected and diluted with 2.5 mL PBS and 75 µL HCl 1 N. Formed 12-HHT was extracted and analysed by HPLC as described (Albert et al., 2002).
SDS-PAGE and Western blot
Cells (4 × 106 cells·per·mL) were resuspended in 50 µL PBS buffer pH 7.2, mixed with the same volume of 2 × SDS-PAGE sample loading buffer [20 mmol·L−1 Tris–HCl, pH 8, 2 mmol·L−1 EDTA, 5% (m,v) SDS and 10% (v,v) β-mercaptoethanol] and boiled for 5 min at 95°C. Aliquots (20 µL) equivalent to 0.8 × 106 cells were mixed with 4 µL glycerol, and 0.1% bromophenol blue (1:1, v,v) and proteins were separated by SDS-PAGE. After electroblotting to nitrocellulose membrane (GE Healthcare, Munich, Germany) and blocking with 5% BSA for 1 h at room temperature, membranes were washed and incubated with primary antibodies overnight at 4°C. The membranes were washed and incubated with 1:1000 dilution of alkaline phosphatase-conjugated immunoglobulin G for 3 h at room temperature. After washing, proteins were visualized with nitro blue tetrazolium and 5-bromo-4-chloro-3-indolylphosphate.
Statistics
Data are expressed as mean ± SE. IC50 values were calculated by non-linear regression by using SigmaPlot 9.0 (Systat Software Inc., San Jose, USA) one site binding competition. The programme Graphpad Instat (Graphpad Software Inc., San Diego, CA) was used for statistical comparisons. Statistical evaluation of the data was performed by one-way anova for independent or correlated samples followed by Tukey HSD post hoc tests. Where appropriate, Student's t-test for paired and correlated samples was applied. A P-value of <0.05 (*) was considered significant.
Materials
Myrtucommulone and S-MC (Figure 1) were isolated from myrtle leaves as described previously (Appendino et al., 2002). The IBP-C (Figure 1) was synthesized from S-MC (G. Appendino, unpubl. data). The compounds were dissolved in DMSO and kept in the dark at −20°C, and freezing–thawing cycles were kept to a minimum. The thromboxane synthase inhibitor CV4151 (Kato et al., 1985) and celecoxib were generous gifts by Dr S Laufer (University of Tuebingen, Germany), and the anti-6-oxo PGF1α antibody was provided by Dr T Dingermann (University of Frankfurt, Germany). DMEM high glucose (4.5 g·L−1) medium, penicillin, streptomycin, trypsin-EDTA solution, sodium pyruvate were from PAA (Coelbe, Germany); PGH2 from Larodan (Malmö, Sweden); 11β-PGE2, PGB1, MK-886, 6-oxo PGF1α, human recombinant COX-2, isolated ovine COX-1 from Cayman Chemical (Ann Arbor, MI); [5, 6, 8, 9, 11, 12, 14, 15-3H]arachidonic acid ([3H]arachidonic acid) from BioTrend Chemicals GmbH (Cologne, Germany); Ultima Gold™ XR from Perkin Elmer (Boston, MA). All other chemicals were obtained from Sigma-Aldrich (Deisenhofen, Germany) unless stated otherwise.
Results
Effects of myrtucommulone on the activity of isolated COX-1 and 2
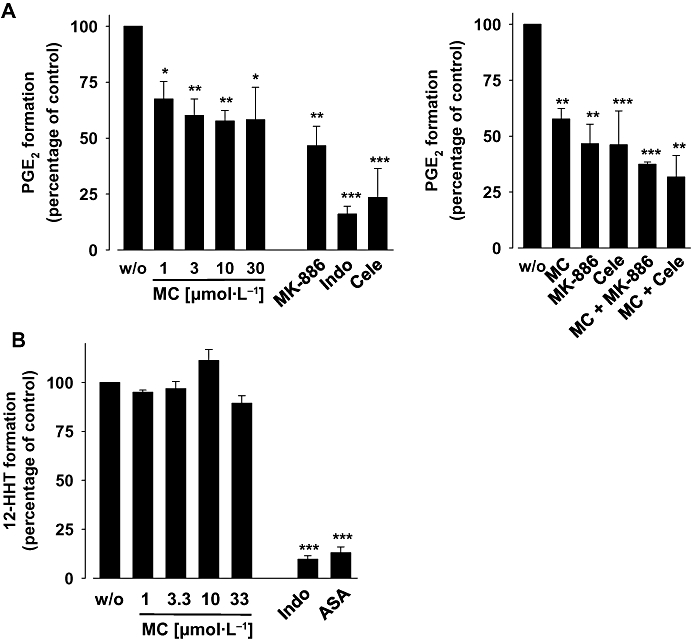
We have previously shown that MC suppresses cellular COX-1 product formation in intact platelets (IC50 = 17 µmol·L−1), whereas MC up to 30 µmol·L−1 did not inhibit COX-2 activity in monocytic Mono Mac 6 cells (Feisst et al., 2005). Here, we analysed the direct effects of MC on isolated COX enzymes. Purified ovine COX-1 and purified human recombinant COX-2 were pre-incubated with MC for 5 min, arachidonic acid (5 µmol·L−1 for COX-1, 2 µmol·L−1 for COX-2) was added, and after 5 min at 37°C the formation of 12-HHT, which is the major COX-1 and 2-derived product under these experimental conditions (Capdevila et al., 1995), was determined. MC up to 33 µmol·L−1 caused no significant inhibition of COX-2 activity and hardly suppressed COX-1 activity (IC50 > 33 µmol·L−1) (Figure 1B), excluding a strong interaction of MC with these enzymes. In control experiments, the COX inhibitor indomethacin [10 µmol·L−1, (Mitchell et al., 1993)] suppressed the activity of COX-1, and the selective COX-2 inhibitor celecoxib (5 µmol·L−1) inhibited the activity of COX-2, as expected (Figure 1B).
Effects of myrtucommulone on the activity of mPGES-1
Next, we investigated if MC interferes with mPGES-1 activity in a cell-free assay. A549 cells treated with 1 ng·mL−1 interleukin-1β expressed mPGES-1 after 48 h, as analysed by SDS-PAGE and Western blotting (Figure 2A). Microsomes were isolated from interleukin-1β-stimulated A549 cells, pre-incubated with the test compounds for 15 min, and PGH2 (20 µmol·L−1) was added as substrate for mPGES-1. The indole derivative MK-886, a well-recognized inhibitor of mPGES-1 (Claveau et al., 2003; Kojima et al., 2004), was used as a reference compound, and this compound concentration-dependently blocked PGE2 formation with an IC50 ≈ 2 µmol·L−1 (Figure 2B). As shown in Figure 2B, MC potently and concentration-dependently suppressed PGE2 formation with an IC50 = 1 µmol·L−1. Variation of the pre-incubation periods (2 to 60 min) did not affect the efficiency of MC (not shown). The MC derivative, S-MC, which has only one syncarpic acid moiety (Figure 1), also inhibited mPGES-1 activity, although less potently (IC50 = 10 µmol·L−1, Figure 2B). As the IBP-C (Figure 1) was hardly active (Figure 2B), the syncarpic acid moieties of MC are seemingly important for inhibition of mPGES-1. The polyphenol resveratrol failed to inhibit mPGES-1 up to 10 µmol·L−1, and neither indomethacin nor celecoxib (10 µmol·L−1, each) caused suppression of PGE2 formation in this assay (data not shown).
Figure 2.
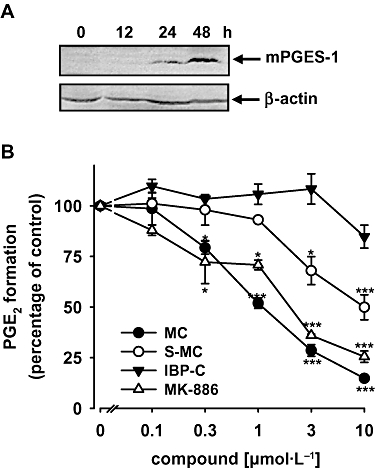
Effects of myrtucommulone (MC) on the activity of microsomal prostaglandin E2 synthase (mPGES)-1 in a cell-free assay. (A) Expression of mPGES-1 in interleukin-1β-stimulated A549 cells. Treatment of A549 cells with interleukin-1β (1 ng·mL−1) in culture medium containing 2% fetal calf serum resulted in a time-dependent increase of mPGES-1 (16 kDa) protein. Proteins were subjected to gel electrophoresis on a 14% SDS-PA gel and analysed by Western blotting. β-Actin (45 kDa) was used as loading control. Data are representative of three independent experiments. (B) Microsomal preparations of interleukin-1β-stimulated A549 cells were pre-incubated with vehicle (dimethyl sulphoxide, DMSO), MK-886, MC, semi-myrtucommulone (S-MC) and the isobutyrophenone-core (IBP-C) for 15 min at 4°C, and the reaction was started by addition of 20 µmol·L−1 PGH2. After 1 min at 4°C the reaction was terminated by using a stop solution containing 11β-PGE2 (1 nmol) as internal standard. PGE2 was quantified by RP-HPLC as described in the Methods section. Data are given as mean ± SE, n = 3, *P < 0.05 or ***P < 0.001 vs. vehicle (0.1% DMSO) control, anova + Tukey HSD post hoc tests.
Myrtucommulone is a reversible and substrate concentration-independent inhibitor of mPGES-1
To investigate if MC blocks mPGES-1 in a reversible manner, microsomal preparations of A549 cells were pre-incubated with MC or MK-886 at 0.3 or 3 µmol·L−1 for 15 min, each. The sample containing 3 µmol·L−1 inhibitor was split: one aliquot was diluted 10-fold with reaction buffer to obtain a final inhibitor concentration of 0.3 µmol·L−1; the other aliquot was not altered. Then, PGH2 was added to each sample to start PGE2 formation. Both MK-886 and MC moderately reduced PGE2 formation at 0.3 µmol·L−1, whereas at 3 µmol·L−1, PGE2 synthesis was potently inhibited (Figure 3A). However, dilution of the latter mixture to yield a final concentration of 0.3 µmol·L−1, prior to addition of substrate abolished the inhibitory effect of MK-886 and of MC, implying that both compounds inhibit mPGES-1 in a reversible fashion.
Figure 3.

Myrtucommulone (MC) inhibits microsomal prostaglandin E2 synthase (mPGES)-1 in a reversible and substrate concentration-independent manner. (A) Microsomal preparations of interleukin-1β-stimulated A549 cells were pre-incubated with 3 µmol·L−1 MK-886 or MC, or with vehicle (dimethyl sulphoxide, DMSO, w/o) for 15 min at 4°C, each. Then, one aliquot of the samples was diluted with assay buffer 10-fold, whereas the other one was not altered, and 20 µmol·L−1 PGH2 was added to start the reaction. For comparison, microsomal preparations were pre-incubated for 15 min at 4°C with 0.3 µmol·L−1 MK-886 or MC, or with vehicle (DMSO), and then 20 µmol·L−1 PGH2 was added (no dilution). Then, all samples were incubated for 1 min on ice, and PGE2 formation was analysed as described. Data are given as mean + SE, n = 3, *P < 0.05, anova + Tukey HSD post hoc tests. (B) The potency of MC for mPGES-1 inhibition was compared at 1 and 20 µmol·L−1 PGH2 as substrate. In a variation of the general procedure, the amount of PGE2 was quantified for 1 µmol·L−1 PGH2 by use of a PGE2 high sensitivity EIA Kit according to the manufacturer's protocol. Data are given as mean ± SE, n = 3–4. (C) The activity of mPGES-1 was determined at different PGH2 and different MC concentrations, as indicated (left panel). In a variation of the general procedure, the amount of PGE2 was quantified by use of a PGE2 high sensitivity EIA Kit according to the manufacturer's protocol. Data are given as mean ± SE, n = 3–4. Eadie-Hofstee analysis (right panel) was performed by using the original data.
Inhibition of mPGES-1 by MC could be influenced by the concentration of PGH2 as substrate. However, decreasing the PGH2 concentration from 20 µmol·L−1 to 1 µmol·L−1 did not affect the potency of MC for inhibition of mPGES-1, and also not of MK-886 (Figure 3B), which may exclude a substrate-competitive inhibitory mechanism. More detailed studies by using different concentrations of MC at varying substrate concentrations were performed to evaluate the kinetic mechanism for inhibition of mPGES-1. Eadie Hofstee plots indicate a non-competitive type of inhibition (Figure 3C).
Effects of myrtucommulone on the formation of PGE2 and 6-oxo PGF1α in intact A549 cells
A549 cells treated with interleukin-1β for 48 h were shown to strongly express COX-2 and mPGES-1 (Jakobsson et al., 1999), whereas COX-1 was not detected (Asano et al., 1996). Hence, in activated A549 cells, PGH2 should derive from COX-2, and the conversion to PGE2 should be catalysed by mPGES-1. However, cPGES and mPGES-2 are still likely to contribute, although to a lesser extent (Park et al., 2006). A549 cells, treated with interleukin-1β for 48 h were pre-incubated with MC, MK-886 or vehicle (DMSO) for 10 min prior to stimulation with 2.5 µmol·L−1 A23187 plus 1 µmol·L−1 arachidonic acid and [3H]arachidonic acid (18.4 kBq). Under these stimulation conditions, cells are activated via a marked elevation of intracellular Ca2+, and the need for the release of endogenous arachidonic acid by cPLA2 as substrate for COX-2 is circumvented, allowing the exclusion of indirect inhibitory effects of MC. The PGE2 formed was separated by RP-HPLC and quantified by liquid scintillation counting.
The formation of PGE2 was concentration-dependently reduced by MC with an apparent IC50 of approximately 30 µmol·L−1 (Figure 4A). Notably, a complete inhibition of PGE2 synthesis could not be achieved, neither by MC nor by MK-886, and compared with the cell-free assay, both compounds were obviously less potent. Thus, at 33 µmol·L−1, MK-886 reduced PGE2 formation only by approximately 60% of control. In contrast to the mPGES-1 inhibitors, indomethacin (10 µmol·L−1) or celecoxib (5 µmol·L−1) essentially abolished PGE2 formation (Figure 4A). In parallel, the effects of MC and MK-886 on the formation of the PGI2 degradation product 6-oxo PGF1α in A549 cells was analysed as a measure of cellular COX-2 activity. In fact, no reduction of 6-oxo PGF1α formation was observed for MC (1 to 33 µmol·L−1) or for MK-886 (30 µmol·L−1) (Figure 4B), whereas indomethacin (10 µmol·L−1) as well as celecoxib (5 µmol·L−1) reduced 6-oxo PGF1α formation under these conditions.
Figure 4.

Effects of myrtucommulone (MC) on the formation of prostaglandin (PG)E2 and 6-oxo PGF1α in intact A549 cells. (A) PGE2 formation. A549 cells (4 × 106 per·mL) were pre-incubated with MK-886, MC or vehicle (dimethyl sulphoxide, DMSO, w/o) for 10 min, then cellular PGE2 synthesis was elicited by addition of 2.5 µmol·L−1 A23187 plus 1 µmol·L−1 arachidonic acid and [3H]arachidonic acid (18.4 kBq). After 15 min at 37°C, formed [3H]PGE2 was analysed by RP-HPLC and liquid scintillation counting as described in the Methods section. (B) 6-oxo PGF1α formation. Interleukin-1β-stimulated A549 cells (5 × 106 per·mL) were pre-incubated with the indicated concentrations of MC, with MK-886 (30 µmol·L−1), indomethacin (Indo, 10 µmol·L−1), celecoxib (Cele, 5 µmol·L−1) or vehicle (DMSO, w/o) for 15 min prior to addition of 30 µmol·L−1 arachidonic acid. After 15 min at 37°C the amount of 6-oxo PGF1α was assessed by enzyme-linked immunosorbent assay as described in the Methods section. Data are given as mean ± SE, n = 3, **P < 0.01 or ***P < 0.001 vs. vehicle (0.1% DMSO) control, anova + Tukey HSD post hoc tests.
Effects of myrtucommulone on the formation of PGE2 and 12-HHT in human whole blood
Heparinized human whole blood was pre-incubated with MC for 5 min, prior to stimulation with lipopolysaccharide (10 µg·mL−1) for 5 h. For measuring PGE2, we established a novel assay system consisting of a combination of RP-HPLC and elisa because separation of PGE2 by HPLC was necessary prior to immunological detection. In fact, when we assessed PGE2 solely by elisa, neither MC nor MK-886 led to reduced PGE2 levels, instead rather increased PGE2 signals were obtained (not shown). Possibly, inhibition of mPGES-1 shunts PGH2 into other metabolic pathways, and these metabolites seemingly interfere with the PGE2 antibody (cross reactivity) in the elisa. Such shunting of PGH2 was recognized also in mPGES-1 KO mice, where enhanced levels of thromboxane (Tx)A2, PGD2, PGF2α and PGI2 were detected (Scholich and Geisslinger, 2006). First, the formed PGE2 was isolated from plasma by solid phase extraction and separated by RP-HPLC. The semi-purified PGE2 was then collected and quantified by elisa. As shown in Figure 5A, at 1 µmol·L−1 MC PGE2 synthesis was reduced by 33 ± 8%, but higher concentrations of MC did not decrease the PGE2 level further (42 ± 5% inhibition at 30 µmol·L−1). Also, inhibition of mPGES-1 by 30 µmol·L−1 MK-886 failed to completely suppress PGE2 synthesis, thus, 61 ± 9% PGE2 still remained, and higher concentrations (up to 100 µmol·L−1) caused no further decrease of PGE2 levels. In contrast, COX inhibition by celecoxib (20 µmol·L−1) or indomethacin (50 µmol·L−1) efficiently reduced PGE2 formation to 24 ± 13% and 16 ± 3% respectively (Figure 5A, left panel). It should be noted that combination of these inhibitors did not increase their effects. Thus, addition of 10 µmol·L−1 MC did not significantly improve the potency of MK-886 (30 µmol·L−1) or of celecoxib (5 µmol·L−1, suboptimal concentration) in PGE2 synthesis (Figure 5A, right panel).
Figure 5.
Effects of myrtucommulone (MC) on the formation of prostaglandin (PG)E2 and 12-HHT (12(S)-hydroxy-5-cis-8,10-trans-heptadecatrienoic acid in human whole blood). (A) PGE2 formation. Heparinized human whole blood, treated with 1 µmol·L−1 thromboxane synthase inhibitor CV4151 and 50 µmol·L−1 aspirin, was pre-incubated with vehicle (dimethyl sulphoxide, DMSO, w/o), MC (at the indicated concentrations), MK-886 (30 µmol·L−1), indomethacin (Indo, 50 µmol·L−1) and celecoxib (Cele, 20 µmol·L−1 left panel, 5 µmol·L−1 right panel), or with combinations of MC (10 µmol·L−1), MK-886 (30 µmol·L−1) and celecoxib (5 µmol·L−1) as indicated. After 5 min at room temperature, PGE2 formation was induced by addition of 10 µg·mL−1 lipopolysaccharide. After 5 h at 37°C, PGE2 was extracted from plasma by solid phase extraction, separated by RP-HPLC and quantified by enzyme-linked immunosorbent assay as described in the Methods section. (B) 12-HHT formation. Heparinized whole blood was pre-incubated with MC or vehicle (DMSO, w/o), at the indicated concentrations for 10 min, and arachidonic acid (100 µmol·L−1) and Ca2+-inophore (30 µmol·L−1) were added to induce cyclooxygenase-product formation. After 10 min at 37°C, 12-HHT was extracted form blood plasma by RP-18 solid phase extraction and analysed by RP-HPLC as described in the Methods section. Indomethacin (Indo, 20 µmol·L−1) and aspirin (ASA, 30 µmol·L−1) were used as controls. Data are given as mean ± SE, n = 3, *P < 0.05, **P < 0.01 or ***P < 0.001 vs. vehicle (0.1% DMSO) control, anova + Tukey HSD post hoc tests.
In order to discriminate between the effects of MC on mPGES-1 and on COX enzymes in whole blood, we also determined the influence of MC on the formation of the COX product 12-HHT. The formation of 12-HHT in whole blood (no pretreatment with aspirin and CV4151) after stimulation with Ca2+-inophore and arachidonic acid, mainly deriving from COX-1, was not inhibited by MC (up to 33 µmol·L−1), whereas aspirin (30 µmol·L−1) or indomethacin (20 µmol·L−1) potently inhibited 12-HHT synthesis (Figure 5B). These findings indicate that suppression of PGE2 production by MC in whole blood was not the result of interference with the COX enzymes.
Discussion
The development of selective mPGES-1 inhibitors is of major interest as an alternative strategy to traditional NSAIDs (non-steroidal anti-inflammatory drugs) and coxibs for the treatment of chronic inflammation (Jachak, 2007). The plant kingdom is a valuable source for anti-inflammatory substances, and several hundred natural compounds of plant origin have been described to interfere with PG biosynthesis. Investigations aiming to reveal the underlying molecular mechanisms of these active compounds have mainly aimed at COX-1 and 2 or phospholipase A2 as relevant targets. However, to our knowledge, no natural compound has been described yet that affects a PGES. Here, we demonstrate that MC from myrtle inhibits mPGES-1, leading to suppression of PGE2 synthesis without significant effects on the COX enzymes.
The functionality of MC as inhibitor of mPGES-1 was demonstrated in this study by applying three different assays. These were: (i) a cell-free assay measuring the direct conversion of PGH2 to PGE2 by mPGES-1; (ii) a cellular assay determining PGE2 formation from exogenously added arachidonic acid; and finally, (iii) a whole blood assay measuring PGE2 after exposure to lipopolysaccharide. MC reduced the formation of PGE2 in all these assays at approximately 1 to 3 µmol·L−1. Nevertheless, PGE2 formation in intact A549 cells or whole blood was not completely suppressed by MC, even not at high concentrations. A similar pattern was found for the mPGES-1 inhibitor MK-886, whereas the COX-2 selective inhibitor, celecoxib, and the non-selective COX inhibitor, indomethacin, efficiently blocked PGE2 synthesis in these assays. The basal PGE2 formation in whole blood, which was not inhibited by MC, is presumably mPGES-1-independent and could also not be reduced by the COX-2-selective celecoxib or by combinations of celecoxib with MC or with MK-886, implying that the remaining PGE2 formation could occur via COX-1 and other PGES (Park et al., 2006), not affected by MC or MK-886. Intriguingly, interleukin-1β-treated mouse embryo fibroblasts of mPGES-1 KO mice still produced about 30% of PGE2 relative to the output in wild type (WT) animals (Kapoor et al., 2006), and the in vivo levels of PGE2 in mPGES-1 KO mice were only reduced by 52%, compared with WT animals (Trebino et al., 2003). Finally, PGE2 was formed from PGH2 in the cell-free assay containing heat-inactivated PGES, suggesting non-enzymatic conversion of PGH2 to PGE2 (Koeberle A, Werz O, unpubl. data). Consequently, a complete suppression of PGE2 formation in vivo may be achieved by blocking PGH2 generation (i.e. by COX-1 and 2 inhibition) but apparently not by blockade of mPGES-1 alone. Although this might be detrimental in respect to the potency of mPGES-1 inhibitors, selective inhibition of mPGES-1 without affecting constitutively expressed PGES isoforms is considered to be an advantage in terms of side effects (Samuelsson et al., 2007).
Interestingly, both MC and MK-886 potently inhibit cellular 5-lipoxygenase product formation (Gillard et al., 1989; Feisst et al., 2005). As a consequence, blockade of leukotriene formation and shunting of arachidonic acid into prostanoid biosynthetic pathways might counteract the inhibitory effect of MC and MK-886 on mPGES-1. However, although shunting effects might impair the efficiency of MC and MK-886 in whole blood compared with cell-free assays, they should be negligible in A549 cells, which do not express significant levels of 5-lipoxygenase, as monitored by Western blotting (Brock et al., 2005).
Inhibition of PGE2 formation was not observed until 3 µmol·L−1 MC in A549 cells, whereas MC was effective at 1 µmol·L−1 in the cell-free test system and in the whole blood assay. Similarly, MK-886 (10 µmol·L−1) only marginally reduced PGE2 formation in intact A549, despite potent mPGES-1 inhibition in the cell-free assay. These discrepancies in potency might be explained by the different assay conditions. Thus, A23187 rapidly induces substantial PGH2 synthesis in A549 cells, compared with the slower effects of lipopolysaccharide in whole blood. Possibly, MC (and also MK-886) inhibits the transfer of PGH2 from COX-2 to mPGES-1 resulting in lower inhibitory efficiency when high amounts of PGH2 are provided by COX-2. On the other hand, A549 cells express high amounts of multidrug resistance-1 (MDR-1) and lung resistance protein (LRP) (Trussardi et al., 1998), resulting potentially in enhanced cellular efflux of MC.
Mechanistic investigations revealed that MC as well as MK-886 inhibited mPGES-1 in a reversible fashion, not dependent on substrate concentration, and our kinetic data indicated a non-competitive mode of inhibition for MC. Both MC and MK-886 represent lipophilic carboxylic acids and based on their fatty acid-like character, they may bind to the same sites at mPGES-1 as polyunsaturated fatty acids, like arachidonic acid and 15-deoxy-Δ12,14-PGJ2 that inhibit mPGES-1 (Quraishi et al., 2002). In fact, MK-886 functions as potent leukotriene (LT) synthesis inhibitor [IC50 = 2.5 nmol·L−1, (Gillard et al., 1989)] by competing with arachidonic acid for binding to 5-lipoxygenase-activating protein (FLAP) (Mancini et al., 1993), and also MC potently blocks cellular LT biosynthesis, partially due to direct inhibition of 5-lipoxygenase (Feisst et al., 2005). On the other hand, MC could simply scavenge PGH2 by a redox mechanism, because PGH2 is sensitive to reducing agents, causing formation of PGF2 (Thoren and Jakobsson, 2000; Mancini et al., 2001). However, MC is a weak antioxidant [IC50 up to 162 µmol·L−1, (Rosa et al., 2003)], and also incubation with MC in the test tube caused no significant enhanced degradation of PGH2 (data not shown). Therefore, a specific interaction between mPGES-1 and MC, depending on defined structural elements, is reasonable.
In addition to the effectiveness of MC to block PGE2 formation, focus has been placed also on its selectivity for mPGES-1 as the point of attack. MC did not inhibit the activity of isolated COX-2 and hardly interfered with isolated COX-1. Also, despite significant suppression of PGE2 formation in cell-based assays, the concomitant generation of 6-oxo PGF1α in A549 cells and 12-HHT in whole blood was not reduced. Hence, interference with COX enzymes is seemingly not the reason for reduced PGE2 formation by MC in the intact cell assays. Moreover, suppression of PLA2 enzymes by MC as possible reason for reduced PGE2 levels can be ruled out, because PGE2 formation was suppressed in A549 cells in the presence of exogenously added arachidonic acid.
In conclusion, MC is the first natural compound of plant origin reported to inhibit mPGES-1. Moreover, MC advantageously blocks LT formation (Feisst et al., 2005), and suppression of both the LT and the PG biosynthetic pathway would be of advantage over inhibition of a single pathway, not only in terms of anti-inflammatory effectiveness, but also due to a lower incidence of (gastrointestinal) side effects (Celotti and Laufer, 2001). MC might exhibit a similar pharmacological profile as MK-886, because both compounds potently inhibit LT and PGE2 formation, without interfering with COX isoforms at low concentrations. Unfortunately, no studies have been performed yet that aimed to assess the bioavailability and achievable plasma concentrations of MC, either in animals or in humans. Detailed analysis of MC or MC-containing extracts of myrtle in appropriate animal models of inflammation (our ongoing studies) will give further insights into the pharmacological potential of MC and of myrtle for therapeutic use. Altogether, in regard of its effectiveness in whole blood and its functionality on mPGES-1, but not on COX enzymes, MC has an interesting pharmacological profile as a dual mPGES-1 and 5-lipoxygenase inhibitor.
Acknowledgments
The authors would like to acknowledge the financial support by the Deutsche Forschungsgemeinschaft. Further, we thank Gertrud Kleefeld for expert technical assistance.
Glossary
Abbreviations:
- 12-HHT
12(S)-hydroxy-5-cis-8, 10-trans-heptadecatrienoic acid
- COX
cyclooxygenase
- cPGES
cytosolic prostaglandin E2 synthase
- cPLA2
cytosolic phospholipase A2
- elisa
enzyme-linked immunosorbent assay
- FCS
fetal calf serum
- MAPEG
membrane-associated proteins involved in eicosanoid and glutathione metabolism
- MC
myrtucommulone
- mPGES
microsomal prostaglandin E2 synthase
- NSAIDs
non-steroidal anti-inflammatory drugs
- PG
prostaglandin
Conflict of interest
None.
References
- Albert D, Zundorf I, Dingermann T, Muller WE, Steinhilber D, Werz O. Hyperforin is a dual inhibitor of cyclooxygenase-1 and 5-lipoxygenase. Biochem Pharmacol. 2002;64:1767–1775. doi: 10.1016/s0006-2952(02)01387-4. [DOI] [PubMed] [Google Scholar]
- Appendino G, Bianchi F, Minassi A, Sterner O, Ballero M, Gibbons S. Oligomeric acylphloroglucinols from myrtle (Myrtus communis) J Nat Prod. 2002;65:334–338. doi: 10.1021/np010441b. [DOI] [PubMed] [Google Scholar]
- Asano K, Lilly CM, Drazen JM. Prostaglandin G/H synthase-2 is the constitutive and dominant isoform in cultured human lung epithelial cells. Am J Physiol. 1996;271:L126–L131. doi: 10.1152/ajplung.1996.271.1.L126. [DOI] [PubMed] [Google Scholar]
- Bonjar GH. Antibacterial screening of plants used in Iranian folkloric medicine. Fitoterapia. 2004;75:231–235. doi: 10.1016/j.fitote.2003.12.013. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Brock TG, Lee YJ, Maydanski E, Marburger TL, Luo M, Paine R, et al. Nuclear localization of leukotriene A4 hydrolase in type II alveolar epithelial cells in normal and fibrotic lung. Am J Physiol Lung Cell Mol Physiol. 2005;289:L224–L232. doi: 10.1152/ajplung.00423.2004. [DOI] [PubMed] [Google Scholar]
- Capdevila JH, Morrow JD, Belosludtsev YY, Beauchamp DR, DuBois RN, Falck JR. The catalytic outcomes of the constitutive and the mitogen inducible isoforms of prostaglandin H2 synthase are markedly affected by glutathione and glutathione peroxidase(s) Biochemistry. 1995;34:3325–3337. doi: 10.1021/bi00010a023. [DOI] [PubMed] [Google Scholar]
- Celotti F, Laufer S. Anti-inflammatory drugs: new multitarget compounds to face an old problem. The dual inhibition concept. Pharmacol Res. 2001;43:429–436. doi: 10.1006/phrs.2000.0784. [DOI] [PubMed] [Google Scholar]
- Claveau D, Sirinyan M, Guay J, Gordon R, Chan CC, Bureau Y, et al. Microsomal prostaglandin E synthase-1 is a major terminal synthase that is selectively up-regulated during cyclooxygenase-2-dependent prostaglandin E2 production in the rat adjuvant-induced arthritis model. J Immunol. 2003;170:4738–4744. doi: 10.4049/jimmunol.170.9.4738. [DOI] [PubMed] [Google Scholar]
- Cote B, Boulet L, Brideau C, Claveau D, Ethier D, Frenette R, et al. Substituted phenanthrene imidazoles as potent, selective, and orally active mPGES-1 inhibitors. Bioorg Med Chem Lett. 2007;17:6816–6820. doi: 10.1016/j.bmcl.2007.10.033. [DOI] [PubMed] [Google Scholar]
- Engblom D, Saha S, Engstrom L, Westman M, Audoly LP, Jakobsson PJ, et al. Microsomal prostaglandin E synthase-1 is the central switch during immune-induced pyresis. Nat Neurosci. 2003;6:1137–1138. doi: 10.1038/nn1137. [DOI] [PubMed] [Google Scholar]
- Feisst C, Franke L, Appendino G, Werz O. Identification of molecular targets of the oligomeric nonprenylated acylphloroglucinols from Myrtus communis and their implication as anti-inflammatory compounds. J Pharmacol Exp Ther. 2005;315:389–396. doi: 10.1124/jpet.105.090720. [DOI] [PubMed] [Google Scholar]
- Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- Gillard J, Ford-Hutchinson AW, Chan C, Charleson S, Denis D, Foster A, et al. L-663,536 (MK-886) (3-(1-(4-chlorobenzyl)-3-t-butyl-thio-5-isopropylindol-2-yl)-2,2-dimethylpropanoic acid), a novel, orally active leukotriene biosynthesis inhibitor. Can J Physiol Pharmacol. 1989;67:456–464. doi: 10.1139/y89-073. [DOI] [PubMed] [Google Scholar]
- Guichardant M, Petit F, Lagarde M. Metabolism of endogenous arachidonic acid in weakly activated platelets. Absence of leukocyte cooperative products in whole blood. Eicosanoids. 1989;2:117–121. [PubMed] [Google Scholar]
- Jachak SM. PGE synthase inhibitors as an alternative to COX-2 inhibitors. Curr Opin Investig Drugs. 2007;8:411–415. [PubMed] [Google Scholar]
- Jakobsson PJ, Thoren S, Morgenstern R, Samuelsson B. Identification of human prostaglandin E synthase: a microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proc Natl Acad Sci USA. 1999;96:7220–7225. doi: 10.1073/pnas.96.13.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor M, Kojima F, Qian M, Yang L, Crofford LJ. Shunting of prostanoid biosynthesis in microsomal prostaglandin E synthase-1 null embryo fibroblasts: regulatory effects on inducible nitric oxide synthase expression and nitrite synthesis. FASEB J. 2006;20:2387–2389. doi: 10.1096/fj.06-6366fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato K, Ohkawa S, Terao S, Terashita Z, Nishikawa K. Thromboxane synthetase inhibitors (TXSI). Design, synthesis, and evaluation of a novel series of omega-pyridylalkenoic acids. J Med Chem. 1985;28:287–294. doi: 10.1021/jm00381a005. [DOI] [PubMed] [Google Scholar]
- Koeberle A, Siemoneit U, Buehring U, Northoff H, Laufer S, Albrecht W, et al. Licofelone suppresses prostaglandin E2 formation by interference with the inducible microsomal prostaglandin E2 synthase-1. J Pharmacol Exp Ther. 2008;326:975–982. doi: 10.1124/jpet.108.139444. [DOI] [PubMed] [Google Scholar]
- Kojima F, Naraba H, Miyamoto S, Beppu M, Aoki H, Kawai S. Membrane-associated prostaglandin E synthase-1 is upregulated by proinflammatory cytokines in chondrocytes from patients with osteoarthritis. Arthritis Res Ther. 2004;6:R355–R365. doi: 10.1186/ar1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levesque H, Lafont O. Aspirin throughout the ages: a historical review. Rev Med Interne. 2000;21(Suppl. 1):8s–17s. doi: 10.1016/s0248-8663(00)88720-2. [DOI] [PubMed] [Google Scholar]
- Mancini JA, Abramovitz M, Cox ME, Wong E, Charleson S, Perrier H, et al. 5-Lipoxygenase-activating protein is an arachidonate binding protein. FEBS Lett. 1993;318:277–281. doi: 10.1016/0014-5793(93)80528-3. [DOI] [PubMed] [Google Scholar]
- Mancini JA, Blood K, Guay J, Gordon R, Claveau D, Chan CC, et al. Cloning, expression, and up-regulation of inducible rat prostaglandin E synthase during lipopolysaccharide-induced pyresis and adjuvant-induced arthritis. J Biol Chem. 2001;276:4469–4475. doi: 10.1074/jbc.M006865200. [DOI] [PubMed] [Google Scholar]
- Mitchell JA, Akarasereenont P, Thiemermann C, Flower RJ, Vane JR. Selectivity of nonsteroidal antiinflammatory drugs as inhibitors of constitutive and inducible cyclooxygenase. Proc Natl Acad Sci USA. 1993;90:11693–11697. doi: 10.1073/pnas.90.24.11693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami M, Nakatani Y, Tanioka T, Kudo I. Prostaglandin E synthase. Prostaglandins Other Lipid Mediat. 2002;68–69:383–399. doi: 10.1016/s0090-6980(02)00043-6. [DOI] [PubMed] [Google Scholar]
- Park JY, Pillinger MH, Abramson SB. Prostaglandin E2 synthesis and secretion: the role of PGE2 synthases. Clin Immunol. 2006;119:229–240. doi: 10.1016/j.clim.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Quraishi O, Mancini JA, Riendeau D. Inhibition of inducible prostaglandin E(2) synthase by 15-deoxy-Delta(12,14)-prostaglandin J(2) and polyunsaturated fatty acids. Biochem Pharmacol. 2002;63:1183–1189. doi: 10.1016/s0006-2952(02)00844-4. [DOI] [PubMed] [Google Scholar]
- Riendeau D, Aspiotis R, Ethier D, Gareau Y, Grimm EL, Guay J, et al. Inhibitors of the inducible microsomal prostaglandin E2 synthase (mPGES-1) derived from MK-886. Bioorg Med Chem Lett. 2005;15:3352–3355. doi: 10.1016/j.bmcl.2005.05.027. [DOI] [PubMed] [Google Scholar]
- Romani A, Coinu R, Carta S, Pinelli P, Galardi C, Vincieri FF, et al. Evaluation of antioxidant effect of different extracts of Myrtus communis L. Free Radic Res. 2004;38:97–103. doi: 10.1080/10715760310001625609. [DOI] [PubMed] [Google Scholar]
- Rosa A, Deiana M, Casu V, Corona G, Appendino G, Bianchi F, et al. Antioxidant activity of oligomeric acylphloroglucinols from Myrtus communis L. Free Radic Res. 2003;37:1013–1019. doi: 10.1080/10715760310001595739. [DOI] [PubMed] [Google Scholar]
- Samuelsson B, Morgenstern R, Jakobsson PJ. Membrane prostaglandin E synthase-1: a novel therapeutic target. Pharmacol Rev. 2007;59:207–224. doi: 10.1124/pr.59.3.1. [DOI] [PubMed] [Google Scholar]
- Scholich K, Geisslinger G. Is mPGES-1 a promising target for pain therapy? Trends Pharmacol Sci. 2006;27:399–401. doi: 10.1016/j.tips.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Smith WL. The eicosanoids and their biochemical mechanisms of action. Biochem J. 1989;259:315–324. doi: 10.1042/bj2590315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto Y, Narumiya S. Prostaglandin E receptors. J Biol Chem. 2007;282:11613–11617. doi: 10.1074/jbc.R600038200. [DOI] [PubMed] [Google Scholar]
- Thoren S, Jakobsson PJ. Coordinate up- and down-regulation of glutathione-dependent prostaglandin E synthase and cyclooxygenase-2 in A549 cells. Inhibition by NS-398 and leukotriene C4. Eur J Biochem. 2000;267:6428–6434. doi: 10.1046/j.1432-1327.2000.01735.x. [DOI] [PubMed] [Google Scholar]
- Trebino CE, Stock JL, Gibbons CP, Naiman BM, Wachtmann TS, Umland JP, et al. Impaired inflammatory and pain responses in mice lacking an inducible prostaglandin E synthase. Proc Natl Acad Sci USA. 2003;100:9044–9049. doi: 10.1073/pnas.1332766100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tretiakova I, Blaesius D, Maxia L, Wesselborg S, Schulze-Osthoff K, Cinatl J, et al. Myrtucommulone from Myrtus communis induces apoptosis in cancer cells via the mitochondrial pathway involving caspase-9. Apoptosis. 2007;13:119–131. doi: 10.1007/s10495-007-0150-0. [DOI] [PubMed] [Google Scholar]
- Trussardi A, Poitevin G, Gorisse MC, Faroux MJ, Bobichon H, Delvincourt C, et al. Sequential overexpression of LRP and MRP but not P-gp 170 in VP16-selected A549 adenocarcinoma cells. Int J Oncol. 1998;13:543–548. doi: 10.3892/ijo.13.3.543. [DOI] [PubMed] [Google Scholar]
- Xu D, Rowland SE, Clark P, Giroux A, Cote B, Guiral S, et al. MF63 {2-(6-chloro-1H-phenanthro[9,10-d]imidazol-2-yl)isophthalonitrile}, a selective microsomal prostaglandin E synthase 1 inhibitor, relieves pyresis and pain in preclinical models of inflammation. J Pharmacol Exp Ther. 2008;326:754–763. doi: 10.1124/jpet.108.138776. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Yokota K, Tonai T, Shono F, Hayashi Y. Enzyme Immunoassay. Prostaglandins and Related Substances – a Practical Approach. Oxford: IRL Press; 1987. [Google Scholar]