

Abstract
Background and purpose:
The incidence of diabetes mellitus is increased in patients with liver cirrhosis. Oltipraz is currently in trials to treat patients with liver fibrosis and cirrhosis induced by chronic hepatitis types B and C and is primarily metabolized via hepatic cytochrome P450 isozymes CYP1A1/2, 2B1/2, 2C11, 2D1 and 3A1/2 in rats. We have studied the influence of diabetes mellitus on pharmacokinetics of oltipraz and on expression of hepatic, CYP1A, 2B1/2, 2C11, 2D and 3A in rats with experimental liver cirrhosis.
Experimental approach:
Oltipraz was given intravenously (10 mg·kg−1) or orally (30 mg·kg−1) to rats with liver cirrhosis induced by N-dimethylnitrosamine (LC rats) or with diabetes, induced by streptozotocin (DM rats) or to rats with both liver cirrhosis and diabetes (LCD rats) and to control rats, and pharmacokinetic variables measured. Protein expression of hepatic CYP1A, 2B1/2, 2C11, 2D and 3A was measured using Western blot analysis.
Key results:
After i.v. or p.o. administration of oltipraz to LC and DM rats, the AUC was significantly greater and smaller, respectively, than that in control rats. In LCD rats, the AUC was that of LC and DM rats (partially restored towards control rats). Compared with control rats, the protein expression of hepatic CYP1A increased, that of CYP2C11 and 3A decreased, but that of CYP2B1/2 and 2D was not altered in LCD rats.
Conclusions and implications:
In rats with diabetes and liver cirrhosis, the AUC of oltipraz was partially restored towards that of control rats.
Keywords: oltipraz; pharmacokinetics; liver cirrhotic rats; diabetes mellitus rats; liver cirrhotic rats with diabetes mellitus; hepatic CYP1A, 2B1/2, 2C11, 2D and 3A
Introduction
Oltipraz [5-(2-pyrazinyl)-4-methyl-1,2-dithiol-3-thione], a synthetic dithiolthione, was developed by Rhône-Poulenc (Vitry-sur-Seine, France) for the treatment of schistosomiasis (Bueding et al., 1982). After single p.o. administration of oltipraz to normal subjects and subjects with increased risk for colorectal carcinoma, the terminal half-lives ranged from 4.16 h to 11.1 h (Gupta et al., 1995) and from 22.7 h to 9.3 h (O'Dwyer et al., 2000) respectively. After 6 months p.o. administration of oltipraz to patients, the side-effect profile was: gastrointestinal problems, photosensitivities/heat intolerance, and neurological problems (Benson et al., 2000). Based on the therapeutic effects of oltipraz on rats with liver cirrhosis (LC rats) (Kang et al., 2002; Bae et al., 2006a), oltipraz is being evaluated in phase II clinical trial in South Korea as an p.o. agent to treat patients with liver fibrosis and cirrhosis induced by chronic hepatitis types B and C.
Bae et al. (2005a) reported that oltipraz is primarily metabolized via hepatic cytochrome P450 (CYP) 1A1/2, 2B1/2, 2C11, 2D1 and 3A1/2, but not via CYP2E1, in male Sprague–Dawley rats. Kim et al. (2005) reported that in rats with diabetes mellitus induced by streptozotocin (DM rats), the protein expression and mRNA levels of hepatic CYP1A2, 2B1/2 and 3A1 increase, whereas those of CYP2C11 decrease. Similar results have also been reported in other studies (Yamazoe et al., 1989; Shimojo et al., 1993; Raza et al., 2000; Sindhu et al., 2006). Sakuma et al. (2001) reported that the protein expression of CYP2D1 is not altered in DM rats. However, to our knowledge, no studies on changes in hepatic CYP isozymes in LC rats, with or without diabetes mellitus, have yet been reported.
Intestinal CYP isozymes have also been reported to be changed in DM rats. For example, activity of intestinal CYP1A and 2B increased (Al-Turk et al., 1981) but that of CYP3A1/2 decreased (Borbás et al., 2006). In rat intestine, mostly CYP1A and 3A are expressed (Kamninsky and Zhang, 2003), whereas CYP2D is little expressed (Aiba et al., 2003) and CYP2B2, 2C11 and 3A2 are not detectable (Kamninsky and Zhang, 2003).
The pharmacokinetics of oltipraz are changed in DM rats (Bae et al., 2006b) and LC rats (Bae et al., 2004; 2006a) but there is no data on pharmacokinetics in LCD rats. Bae et al. (2005b) reported that the hepatic first-pass effect of oltipraz after absorption into the portal vein is 40% and intestinal first-pass effect is 32% in rats. The hepatic first-pass effect of 40% is equivalent to 25% of the oral dose considering the 32% of the intestinal first-pass effect.
The association between liver disease and diabetes mellitus is well known (Vidal et al., 1994; Kwon, 2003; Moscatiello et al., 2007) and the overall prevalence of diabetes mellitus in patients with liver cirrhosis is significantly higher than that expected.
Pharmacokinetic studies on oltipraz in patients with diabetes mellitus and liver cirrhosis, alone or combined, have not been performed. Hence, the objectives of the current studies were to evaluate, using a rat model, the effects of DM and LC, alone and in combination (LCD), on the pharmacokinetics of oltipraz. Changes in the protein expression of hepatic CYP1A, 2B1/2, 2C11, 2D and 3A in rats with LC and DM, alone and in combination (LCD) using Western blot analysis, were also investigated.
Methods
Animals
The protocols for the animal studies were approved by the Animal Center and Use Committee of the College of Pharmacy of Seoul National University, Seoul, South Korea. Male Sprague–Dawley rats (4–5 weeks old, weighing 180–200 g) were purchased from the Charles River Company Korea (Orient, Seoul, South Korea). They were randomly divided into three disease groups (LC, DM and LCD) and a control group. They were maintained in a clean room (Animal Center for Pharmaceutical Research, College of Pharmacy, Seoul National University) at a temperature of 22 ± 2°C with 12 h light (0700–1900 h) and dark (1900–0700 h) cycles and a relative humidity of 55 ± 5%. Rats were housed in metabolic cages (Tecniplast, Varese, Italy) under filtered pathogen-free air and with food (Sam Yang Company, Pyungtaek, South Korea) and water available ad libitum.
Induction of LC by N-dimethylnitrosamine injection
Freshly prepared N-dimethylnitrosamine (diluted to 0.01 mg·mL−1 in 0.9% NaCl-injectable solution) was injected i.p. at a dose of 0.01 mg (1 mL)·kg−1 on three consecutive days per week for 4 weeks (Ohara and Kusano, 2002; Bae et al., 2004; 2006a). On day 29, one dose of citrate buffer (pH 7.4; 1 mL·kg−1) was injected via the tail vein. On day 36, the rats were treated with oltipraz. Laboratory rats with N-dimethylnitrosamine-induced liver cirrhosis have clinical features similar to those of humans with liver cirrhosis such as increased mortality, hepatic parenchymal cell destruction, formation of connective tissue and nodular regeneration (Kang et al., 2002). Liver cirrhosis in LC rats was evident by liver histological analysis, which revealed extensive micronodular cirrhosis with regenerative hepatocellular changes, and bile duct proliferation was also detected (Bae et al., 2004; 2006a). It has also been reported that N-dimethylnitrosamine-induced liver cirrhosis in rats is a reproducible effect (Jenkins et al., 1985; Jezequel et al., 1987; Kang et al., 2002).
Induction of DM by streptozotocin injection
A 0.9% NaCl-injectable solution was injected i.p. (1 mL·kg−1) on three consecutive days per week for 4 weeks. On day 29, one dose [45 mg (1 mL)·kg−1] of freshly prepared streptozotocin [dissolved in citrate buffer (pH 4.5) to 45 mg·mL−1] was administered via the tail vein (Kim et al., 2005). On day 36, the rats were treated with oltipraz.
Induction of LCD with N-dimethylnitrosamine and streptozotocin injections
Liver cirrhosis was induced by intraperitoneal injection of N-dimethylnitrosamine as described above. Then, on day 29, diabetes mellitus was induced by injection of streptozotocin via the tail vein as described above. On day 36, the rats were treated with oltipraz.
Control rats
Control rats were injected intraperitoneally with 0.9% NaCl-injectable solution (1 mL·kg−1) on three consecutive days per week for 4 weeks. On day 29, one dose (1 mL·kg−1) of citrate buffer (pH 4.5) was administered via the tail vein. On day 36, the rats were treated with oltipraz.
During the pretreatment, food and water were available ad libitum to all rats. Immediately before the experiment, blood glucose levels in all rats were measured using the Medisense Optium kit (Abbott Laboratories, Bedford, MA), and rats with blood glucose levels greater than 13.9 mmol·L−1 were chosen as diabetic rats (DM and LCD rats).
Preparation of hepatic and intestinal microsomes
The procedures used for the preparation of hepatic (Bae et al., 2004; 2005a; 2006b) and intestinal (Peng et al., 2004) microsomes from LC, DM, LCD and control rats (n= 4–6, each) were similar to reported methods. Hepatic and intestinal microsomes were stored at −70°C until use. Protein contents in hepatic and intestinal microsomes were measured using the method of Bradford (1976).
Immunoblot analyses of hepatic CYP1A, 2B1/2, 2C11, 2D and 3A
The procedures used were similar to a reported method (Kim et al., 2001). Hepatic microsomes were resolved by sodium dodecyl sulphate (SDS) gel electrophoresis on a 7.5% polyacrylamide gel (10 µg protein per lane; n= 3, each). Proteins were transferred to a nitrocellulose membrane (Bio-Rad Laboratories) that was then blocked for 1 h in 5% milk powder in phosphate-buffered 0.9% NaCl-injectable solution containing 0.05% (v/v) Tween 20 (PBS-T). For immunodetection, blots were incubated overnight at 4°C with rabbit anti-human CYP1A, 2B1/2, 2C11, 2D and 3A antibodies (diluted 1:10 000 in PBS-T containing 5% bovine serum albumin), followed by incubation for 1 h at room temperature with horseradish peroxidase-conjugated goat anti-rabbit secondary antibody (diluted 1 : 10 000 in PBS-T containing 5% milk powder). Hepatic CYP1A, 2B1/2, 2C11, 2D and 3A were detected by enhanced chemiluminescence on Kodak X-OMAT film and quantified by densitometry with a microcomputer imaging device (model M1; Imaging Research, St. Catharines, Ontario, Canada). The β-actin band was used as a loading control. We performed calibration assays in some of the replicate blots to ensure the linearity of band intensity quantification.
Measurement of Vmax, Km and CLint for the disappearance of oltipraz in hepatic and intestinal microsomes
The procedures used were similar to a reported method (Ahn et al., 2008). The Vmax (the maximum velocity) and the Km (the apparent Michaelis–Menten constant; the concentration at which the rate is one-half of the Vmax) for the disappearance of oltipraz in LC, DM, LCD and control rats (n= 4–6, each) were determined after incubating the above microsomes (equivalent to 0.5 and 0.1 mg protein for hepatic and intestinal microsomes respectively), a 5 µL aliquot of dimethylsulphoxide containing final oltipraz concentrations of 2.5, 5, 10, 20, 50 and 100 µmol·L−1 (hepatic microsomes) or 1, 2.5, 5, 7.5, 10 and 20 µmol·L−1 (intestinal microsomes), and 0.1 mol·L−1 phosphate buffer (pH 7.4) containing a 50 and 25 µL aliquot (for hepatic and intestinal microsomes respectively) of 1 mmol·L−1 NADPH in a final volume of 0.5 and 0.25 mL (for hepatic and intestinal microsomes respectively) by adding 0.1 mol·L−1 phosphate buffer (pH 7.4) in a water-bath shaker [kept at 37°C, 50 oscillations·min−1 (opm)]. All of the above microsomal incubation conditions were linear. The reaction was terminated by adding 1 mL of acetonitrile after 5 min incubation for both hepatic and intestinal microsomes. Oltipraz was determined using a reported HPLC method (Bae et al., 2001). The kinetic constants (Km and Vmax) for the disappearance of oltipraz were calculated using a non-linear regression method (Duggleby, 1995). The intrinsic clearance for the disappearance of oltipraz (CLint) was calculated by dividing the Vmax by the Km.
i.v. study
The procedures used for the handling of rats including the cannulation of the carotid artery (for blood sampling) and the jugular vein (for drug administration in the intravenous study) were similar to methods previously reported (Kim et al., 1993).
Oltipraz, suspended in PEG 400: distilled water (40:60, v/v) (Bae et al., 2004; 2005a–c; 2006a,b) at a dose of 10 mg (2 mL)·kg−1 was infused for 1 min via the jugular vein of rats in each group (n= 7, 8, 11 and 8 for the LC, DM, LCD and control rats respectively). A blood sample (approximately 0.12 mL) was collected via the carotid artery at 0 (control), 1 (end of the infusion), 5, 15, 30, 60, 90, 120, 180, 240 and 360 min after the start of the i.v. infusion of oltipraz. Each blood sample was immediately centrifuged and a 50 µL aliquot of each plasma sample was stored at −70°C for later analysis by HPLC. The procedures used for the preparation and handling of the 24 h urine (Ae0–24 h) samples and gastrointestinal tract (including its contents and faeces) samples at 24 h (GI24 h) were similar to reported methods (Bae et al., 2004; 2005a–c; 2006a,b).
p.o. study
Oltipraz (the same suspension used in the i.v. study) at a dose of 30 mg (3 mL)·kg−1 was administered orally using a feeding tube to rats in each group (n= 8, 9, 4 and 8 for the LC, DM, LCD and control rats respectively). A blood sample was collected at 0, 15, 30, 60, 90, 120, 180, 240, 360, 480, 600, 720 and 960 min after the p.o. administration of oltipraz. Other procedures were similar to those described above for the i.v. study.
Measurement of rat plasma protein binding of oltipraz using equilibrium dialysis
Binding of oltipraz to fresh plasma from LC, DM, LCD and control rats (n= 5, each) was measured using equilibrium dialysis (Bu et al., 2001). Plasma (1 mL) was dialysed against 1 mL of isotonic Sørensen phosphate buffer (pH 7.4) containing 3% (w/v) dextran ('the buffer') to minimize volume shift (Boudinot and Jusko, 1984) in a 1 mL dialysis cell (Fisher Scientific, Fair Lawn, NJ) fitted with a Spectra/Por 4 membrane (mol. wt. cutoff 12–14 kDa; Spectrum Medical Industries Inc., Los Angeles, CA). It took 8 h incubation to reach equilibrium between ‘the buffer’ and plasma compartments, and the binding values were not influenced up to 24 h incubation. Thus, 24 h incubation was employed in the present study. The binding of oltipraz to 4% human serum albumin was independent of oltipraz concentrations ranging from 1 to 100 µg·mL−1. Thus, a 10 µg·mL−1 was chosen in the present plasma protein binding study. Total protein contents in plasma from control, LC, DM and LCD rats were 5.5 ± 0.14%, 4.2 ± 0.34%, 5.2 ± 0.53% and 4.6 ± 0.54%, respectively, and the corresponding values for albumin were 3.4 ± 0.12%, 2.5 ± 0.31%, 3.3 ± 0.30% and 2.9 ± 0.14% respectively (Ahn et al., 2008).
HPLC analysis of oltipraz
Concentrations of oltipraz in the samples were determined using an HPLC method (Bae et al., 2001). Briefly, a 100 µL aliquot of acetonitrile was added to deproteinize (Chiou et al., 1978) a 50 µL aliquot of sample. After vortex-mixing and centrifugation (16 000×g for 1 min), a 50 µL aliquot of the supernatant was directly injected onto a reversed-phase (C18) HPLC column. The mobile phase, acetonitrile: 0.5 mmol L−1 ammonium acetate [55:45 (v/v) for both rat plasma and gastrointestinal tract samples, and 45:55 (v/v) for the rat urine samples], was run at a flow rate of 1.5 mL·min−1, and the column eluent was monitored using an ultraviolet detector at 305 nm at room temperature. The retention time of oltipraz was approximately 5.8 min in both rat plasma and gastrointestinal tract samples, and 8.6 min in rat urine samples. The detection limits of oltipraz in rat plasma and urine samples were 20 and 50 ng·mL−1 respectively. The mean within-day coefficients of variation (CV) in rat plasma and urine samples were 2.29% and 1.01%, respectively, and the corresponding between-day CVs of the analysis of the same samples on consecutive 3 days were 3.37% and 1.51% respectively. As oltipraz in solution was reported to be photo-degraded (Christensen and Malone, 1992), all samples in the present study were covered (or wrapped) with aluminum foil or kept in the dark during the experiment or when they are not in use.
Pharmacokinetic analysis
The total area under the plasma concentration-time curve from time zero to time infinity (AUC) was calculated using the trapezoidal rule-extrapolation method (Chiou, 1978). The area from the last datum point to time infinity was estimated by dividing the last measured plasma concentration by the terminal-phase rate constant.
Standard methods (Gibaldi and Perrier, 1982) were used to calculate the following pharmacokinetic parameters using a non-compartmental analysis (WinNonlin®; Pharsight Corporation, Mountain View, CA): the time-averaged total body clearance (CL), the terminal half-life (t1/2), the first moment of AUC (AUMC), the mean residence time (MRT), the apparent volume of distribution at steady state (Vss) and the extent of absolute oral bioavailability (F) (Kim et al., 1993). The peak plasma concentration (Cmax) and time to reach Cmax (Tmax) were directly read from the experimental data.
Statistical analysis
A P-value < 0.05 was deemed to be statistically significant using a Duncan's multiple range test with the Statistical Package for the Social Sciences (spss) posteriori analysis of variance (anova) among the four (or three) means for the unpaired data. All data are expressed as mean ± SD.
Materials
Oltipraz was donated by the R & D Center of Pharmaceuticals, Institute of Science & Technology, CJ Corporation (Ichon, South Korea). NADPH, streptozotocin, β-actin, primary monoclonal antibody for β-actin and Kodak X-OMAT film were purchased from Sigma–Aldrich Corporation (St. Louis, MO). Polyethylene glycol 400 (PEG 400) and N-dimethylnitrosamine were products from Duksan Chemical Company (Seoul, South Korea) and Tokyo Kasei Kogyo Company (Tokyo, Japan) respectively. Polyclonal rabbit anti-human CYP1A, 2B1/2, 2C11, 2D and 3A antibodies and horseradish peroxidase-conjugated goat anti-rabbit secondary antibody were purchased from Detroit R&D (Detroit, MI) and Bio-Rad Laboratories (Hercules, CA) respectively. Enhanced chemiluminescence reagents were products from Amersham Biosciences Corporation (Piscataway, NJ). Other chemicals were of reagent or HPLC grade.
Results
Protein expression of hepatic CYP1A, 2B1/2, 2C11, 2D and 3A
The protein expression of hepatic CYP1A, 2B1/2, 2C11, 2D and 3A in control, DM, LC and LCD rats is shown in Figure 1. In DM rats, the protein expression of CYP1A, 2B1/2, 2D and 3A was significantly increased, but that of CYP2C11 was significantly decreased, compared with control rats. In LC rats, the protein expression of CYP1A, 2B1/2, 2C11, 2D and 3A was significantly decreased, compared with control rats. In LCD rats, the protein expression of CYP1A was significantly increased, that of CYP2C11 and 3A was significantly decreased, and that of CYP2B1/2 and 2D was comparable to control values.
Figure 1.
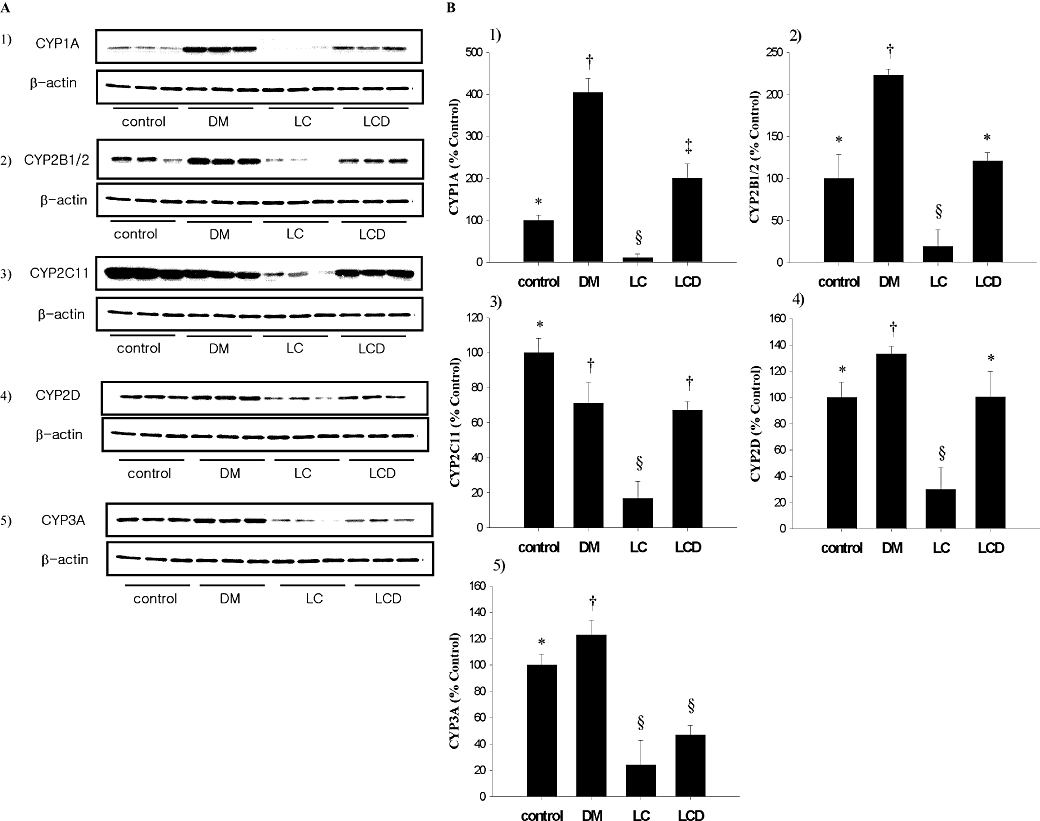
Immunoblotting of hepatic CYP1A, 2B1/2, 2C11, 2D and 3A in LC, DM, LCD and control rats (A). Representative Western blots for each hepatic CYP isoform studied. The β-actin band was used as a loading control. (B). Summary data from the Western blots. The protein expression in LC DM and LCD groups was expressed in terms of that in control rats, set to 100%. Data are presented as mean ± SD and values with different marks (*, †, §, #) are significantly different (P < 0.05). (i) For CYP1A, all three experimental groups were different from control and each group was significantly different; (ii) For CYP2B1/2, only the DM group and the LC group were significantly different from control and the LCD groups; (iii) For CYP2C11, all three experimental groups were different from control and the LC group was different from the DM and LCD groups; (iv) For CYP2D, the DM and the LC groups were significantly different from control and the LCD groups (P < 0.05); and (v) For CYP3A, all three experimental groups were different from control (*P < 0.05) and the DM group was significantly different from the LC and LCD groups.
Vmax, Km and CLint for the disappearance of oltipraz in hepatic and intestinal microsomes
The Vmax, Km and CLint for the disappearance of oltipraz in hepatic microsomes from the four groups of rats are listed in Table 1. The Vmax was significantly different in each group, increasing in the order LC < LCD < control < DM. However, the Km values were not significantly different among the four groups of rats. Thus, the affinity of the hepatic enzyme(s) for oltipraz in the liver is not changed in DM, LC or LCD rats. As a result, the CLint showed the same trends as shown in Vmax, and the overall formation of oltipraz metabolite(s) in the liver (Bieder et al., 1983) was the greatest in DM rats and the lowest in LC rats. Total hepatic protein was significantly different among control rats, DM rats, and LC and LCD rats.
Table 1.
Vmax, Km, and CLint for the disappearance of oltipraz in hepatic and intestinal microsomes from LC, DM, LCD and control rats
| Parameter | Control | LC | DM | LCD |
|---|---|---|---|---|
| Hepatic | ||||
| Vmax (nmol·min−1 mg·protein−1) | 5.38 ± 0.382* | 1.84 ± 0.480* | 9.49 ± 1.22* | 2.91 ± 0.497* |
| Km (µmol·L−1) | 29.3 ± 5.59 | 46.4 ± 34.5 | 38.2 ± 5.88 | 24.8 ± 6.12 |
| CLint (mL·min−1 mg·protein−1) | 0.184 ± 0.0129* | 0.0530 ± 0.0228* | 0.250 ± 0.0125* | 0.119 ± 0.0111* |
| Total protein (mg whole liver−1) | 373 ± 113 | 121 ± 24.7# | 239 ± 13.6 | 143 ± 35.7# |
| Intestinal | ||||
| Vmax (nmol·min−1 mg·protein−1) | 0.523 ± 0.231 | 0.268 ± 0.169 | 0.257 ± 0.137 | 0.212 ± 0.182† |
| Km (µmol·L−1) | 6.41 ± 2.60 | 4.60 ± 2.24 | 2.54 ± 1.76† | 4.28 ± 2.38 |
| CLint (mL·min−1 mg·protein−1) | 0.0807 ± 0.00444 | 0.0556 ± 0.00950# | 0.109 ± 0.0307 | 0.0451 ± 0.0122# |
| Total protein (mg whole intestine−1) | 6.93 ± 1.23 | 5.37 ± 2.99 | 7.13 ± 2.90 | 4.83 ± 3.29 |
Data are expressed as mean ± SD (n= 4–6, each).
Each group was significantly different (P < 0.05).
Control and DM groups were significantly different from LCD and LC groups (P < 0.05).
Significantly different (P < 0.05) from control.
The Vmax, Km and CLint for the disappearance of oltipraz in intestinal microsomes from four groups of rats are also listed in Table 1. Compared with control rats, the Vmax was significantly slower in LCD rats (60% decrease). Compared with control rats, the Km was significantly lower in DM rats (60% decrease). The CLint was significantly slower (31% and 44% decrease respectively) in LC and LCD rats (not significantly different between LC and LCD rats), but was significantly faster (35% increase) in DM rats, all relative to control values. This suggests that the formation of oltipraz metabolite(s) in the intestine increased in DM rats, but decreased in LC and LCD rats. The total intestinal protein was comparable among the four groups of rats.
Rat plasma protein binding of oltipraz
The binding values of oltipraz to fresh plasma from four groups of rats (n= 5, each) are 88 ± 2% (control rats), 70 ± 5% (LC rats), 83 ± 3% (DM rats) and 80 ± 3% (LCD rats). Only the value in LC rats was significantly different from those in the other groups and this could be at least partly due to significantly lower albumin concentrations in plasma from LC rats (Ahn et al., 2008).
Pharmacokinetics of oltipraz after i.v. administration
The mean arterial plasma oltipraz concentration-time profiles for the i.v. administration of oltipraz (10 mg·kg−1) to LC, DM, LCD and control rats are shown in Figure 2. The relevant pharmacokinetic parameters are listed in Table 2. Compared with control rats, the AUC was significantly greater, CL was significantly slower, terminal t1/2 and MRT were significantly longer, and Vss was significantly smaller in LC rats. The CL was significantly faster in DM rats than that in control rats. Interestingly, CL, terminal t1/2 and MRT were similar for LCD and control rats, but Vss in LCD rats was significantly smaller than that in control rats. Oltipraz was below the detection limit in the 24 h urine samples (Ae0–24 h) and gastrointestinal tract (including its contents and faeces) samples at 24 h (GI24 h) for all rats.
Figure 2.
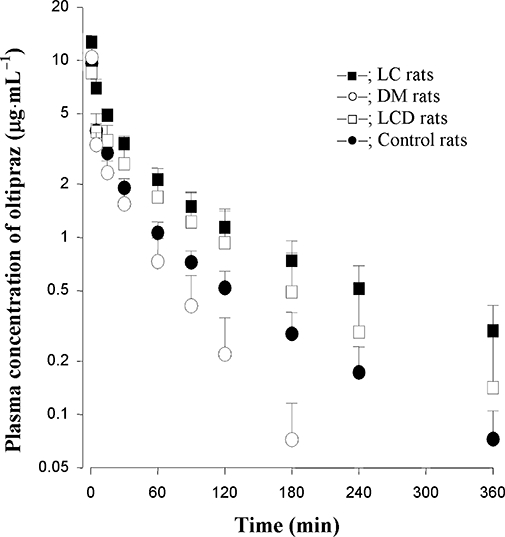
Mean arterial plasma concentration-time profiles of oltipraz after i.v. infusion at a dose of 10 mg·kg−1 to LC (n= 9), DM (n= 7), LCD (n= 7) and control (n= 8) rats. Data are presented as mean ± SD.
Table 2.
Pharmacokinetic parameters of oltipraz after i.v. administration at a dose of 10 mg·kg−1 to LC, DM, LCD and control rats
| Parameter | Control | LC | DM | LCD |
|---|---|---|---|---|
| Initial body weight (g) | 188 ± 10.3 | 198 ± 9.94 | 199 ± 12.5 | 194 ± 8.50 |
| Final body weight (g) | 376 ± 24.7† | 299 ± 9.45* | 282 ± 13.2* | 250 ± 26.9# |
| Blood glucose (mmol·L−1) | 5.90 ± 0.314† | 5.80 ± 0.751† | 20.7 ± 4.44* | 19.1 ± 4.62* |
| AUC (µg·min−1·mL−1) | 249 ± 32.9† | 531 ± 103* | 157 ± 34.2† | 350 ± 132# |
| CL (mL·min−1·kg−1) | 40.8 ± 6.03† | 19.3 ± 3.06* | 65.3 ± 15.5 | 34.1 ± 18.6† |
| Terminal t1/2 (min) | 80.5 ± 11.9† | 116 ± 20.0* | (33.2 ± 5.65)a | 68.9 ± 27.4† |
| MRT (min) | 84.1 ± 17.1† | 132 ± 25.8* | (39.9 ± 9.04) | 90.0 ± 37.2† |
| Vss (mL·kg−1) | 3350 ± 351† | 2500 ± 391* | (2570 ± 278) | 2700 ± 901 |
| Ae0–24 h (% of dose) | BD | BD | BD | BD |
| GI24 h (% of dose) | BD | BD | BD | BD |
Data are expressed as mean ± SD (control and DM, n= 8; LC, n= 7; LCD, n= 11).
The numbers in parentheses were not included in statistical analysis, because the detection of plasma concentrations of oltipraz in DM rats was shorter than other rats.
Values of each parameter with different marks (†, *, #) are significantly different; P < 0.05.
BD, below the detection limit.
Pharmacokinetics of oltipraz after p.o. administration
The mean arterial plasma oltipraz concentration-time profiles for oltipraz following oral administration of 30 mg·kg−1 to LC, DM, LCD and control rats are shown in Figure 3. The relevant pharmacokinetic parameters are listed in Table 3. Compared with control rats, the AUC was significantly greater, terminal t1/2 was significantly longer, and Cmax was significantly lower in LC rats. Compared with control rats, the AUC was significantly smaller and Cmax was significantly lower in DM rats. Interestingly, the terminal t1/2 in LCD rats was similar to that in control rats and AUC in LCD rats was in between LC and DM rats. The F values were in the order, LC > LCD, control > DM groups.
Figure 3.
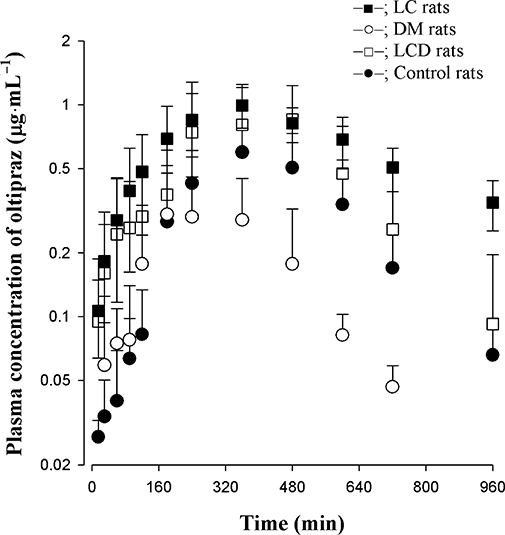
Mean arterial plasma concentration-time profiles of oltipraz after p.o. administration at a dose of 30 mg·kg−1 to LC (n= 7), DM (n= 8), LCD (n= 8) and control (n= 7) rats. Data are presented as mean ± SD.
Table 3.
Pharmacokinetic parameters of oltipraz after p.o. administration at a dose of 30 mg·kg−1 to LC, DM, LCD and control rats
| Parameter | Control | LC | DM | LCD |
|---|---|---|---|---|
| Initial body weight (g) | 214 ± 16.9 | 223 ± 9.64 | 201 ± 7.66 | 216 ± 13.6 |
| Final body weight (g) | 334 ± 10.3† | 246 ± 16.2* | 285 ± 13.2# | 221 ± 22.0§ |
| Blood glucose (mmol·L−1) | 5.42 ± 0.531† | 5.61 ± 0.583† | 16.3 ± 2.59* | 16.9 ± 3.50* |
| AUC (µg·min−1·mL−1) | 286 ± 78.7† | 812 ± 188* | 134 ± 29.1# | 438 ± 164§ |
| Terminal t1/2 (min) | 168 ± 46.6† | 423 ± 111* | 157 ± 58.8† | 126 ± 29.8† |
| Cmax (µg·mL−1) | 0.689 ± 0.166† | 1.06 ± 0.227* | 0.440 ± 0.148# | 1.16 ± 0.322* |
| Tmax (min) | 368 ± 113 | 308 ± 74.8 | 267 ± 124 | 370 ± 134 |
| GI24 h (% of dose) | 3.68 ± 3.85 | 4.26 ± 6.17 | 3.12 ± 4.46 | 5.63 ± 4.63 |
| F (%) | 38.3 | 51.0 | 28.5 | 41.7 |
Data are expressed as mean ± SD (control and LC, n= 8; DM, n= 9; LCD, n= 6). Values of each parameter with different marks (†, *, #, §) are significantly different; P < 0.05.
Discussion
Bae et al. (2005c) reported that the pharmacokinetic parameters of oltipraz (such as AUC, terminal half-life, MRT, Vss, CL and/or CLR) were dose-independent after i.v. (at doses of 5–20 mg·kg−1) and p.o. (at doses of 25–100 mg·kg−1) administration to rats. Thus, the 10 mg (for intravenous study) and 30 mg (for oral study) of oltipraz were chosen for this study.
Bae et al. (2005b) also reported that the non-renal clearance of oltipraz could represent the metabolic clearance of the drug in rats. In the present study, oltipraz was below the detection limit in the 24 h urine samples (Table 2). Thus, the CL of oltipraz listed in Table 2 could represent the metabolic clearance of the drug in rats. Additionally, changes in the CL of oltipraz could be due to the changes in the metabolism of the drug.
In LC rats, the AUC of i.v. oltipraz was significantly greater (113% increase) than that in control rats, possibly as a result of the significantly slower CL of oltipraz (52.7% decrease) (Table 2), as reported earlier (Bae et al., 2004). The slower CL could be supported by significantly slower (71% decrease) hepatic CLint for the disappearance of oltipraz (Table 1) and slower hepatic blood flow rate (Goeting et al., 1986) than those in control rats (Wilkinson and Shand, 1975), because oltipraz has an intermediate hepatic extraction ratio in rats (Bae et al., 2005b). Goeting et al. (1986) reported that the hepatic blood flow was lower in rats with liver cirrhosis induced by carbon tetrachloride. The slower hepatic CLint was attributable to a decreased protein expression of hepatic CYP1A, 2B1/2, 2C11, 2D and 3A in LC rats (Figure 1). Moreover, the hepatic protein content in LC rats was significantly lower (68% decrease) than in control rats (Table 1). However, in LC rats, the free fraction of oltipraz in plasma was clearly greater (150% increase) than that in control rats.
In LC rats, the AUC of p.o. oltipraz was also significantly greater (184% increase) than that in control rats (Table 3), as reported in other studies (Bae et al., 2004; 2006a). However, this was not likely due to the increased gastrointestinal absorption of oltipraz in LC rats. The ‘true’ fractions of the dose unabsorbed (‘Funabs’) after p.o. administration of oltipraz were estimated using the equation of Lee and Chiou (1983). The ‘Funabs’ values thus estimated were 3.68% and 4.26% for control and LC rats respectively. The 184% increase in p.o. AUC (Table 3) was considerably greater than 113% increase in i.v. AUC (Table 2). This could have been due to inhibited intestinal metabolism of oltipraz in addition to inhibited hepatic metabolism of oltipraz, compared with control rats. The decreased intestinal metabolism of oltipraz in LC rats could be supported by significantly slower (31% decrease) intestinal CLint than that in control rats (Table 1), because oltipraz is close to a low intestinal clearance drug in rats (Bae et al., 2005b). The above results could explain somewhat greater F value in LC rats than that in control rats (Table 3).
In DM rats, the AUC of i.v. oltipraz was significantly smaller (36.9% decrease) than that in control rats, possibly as a result of the significantly faster CL (60.0% increase) in DM rats (Table 2), as reported in other studies (Bae et al., 2006b). The faster CL of oltipraz could be supported by significantly faster (35.9% increase) hepatic CLint for the disappearance of oltipraz (Table 1) and higher hepatic blood flow (Sato et al., 1991) than those in control rats. The faster hepatic CLint was attributable to an increased protein expression of hepatic CYP1A, 2B1/2, 2D and 3A, relative to that in control rats (Figure 1). Note that CYP2C11 decreased in DM rats (Figure 1), as has been found earlier (Yamazoe et al., 1989; Shimojo et al., 1993; Kim et al., 2005; Sindhu et al., 2006). The free fraction of oltipraz in plasma was comparable between control and DM rats.
In DM rats, the AUC of p.o. oltipraz was also significantly smaller (53.1% decrease) than that in control rats (Table 3), as has been reported (Bae et al., 2006b). Again, this was not likely to be due to the decreased gastrointestinal absorption of oltipraz in DM rats. The estimated ‘Funabs’ values were 3.68% and 3.12% for control and DM rats respectively. In DM rats, the 53% decrease in p.o. AUC (Table 3) was somewhat greater than 37% decrease in i.v. AUC (Table 2). This could have been due to an increased intestinal metabolism of oltipraz in addition to increased hepatic metabolism of oltipraz in DM rats. The increased intestinal metabolism of oltipraz in DM rats could be supported by the significantly faster (35.1% increase) intestinal CLint than that in control rats. The faster intestinal CLint of oltipraz in DM rats could have been due to increased intestinal CYP1A (Al-Turk et al., 1981), because CYP3A is decreased in DM rats (Borbás et al., 2006). The above results could explain why the F value was lower in DM rats, than that in control rats (Table 3).
In LCD rats, the AUC of i.v. oltipraz was significantly greater (40.6% increase) than that in control rats, possibly as a result of the significantly slower CL (16.4% decrease) in LCD rats (Table 2). The slower CL of oltipraz in LCD rats could be supported by significantly slower (35.3% decrease) hepatic CLint for the disappearance of oltipraz (Table 2) than that in control rats. The slower hepatic CLint could be attributed to a significantly decreased protein expression of CYP2C11 and 3A compared with that in control rats, because expression of CYP1A protein increased, but that of CYP2B1/2 and 2D was comparable to that in control rats (Figure 1). The free fractions of oltipraz in plasma were comparable between control and LCD rats. No studies on changes in hepatic blood flow rate in LCD rats have yet been reported.
In LCD rats, the AUC of p.o. oltipraz was also significantly greater (53.1% increase) than that in control rats (Table 3). Again, this was not likely due to an increased gastrointestinal absorption of oltipraz in LCD rats. The estimated ‘Funabs’ values were 3.68% and 5.63% for control and LCD rats respectively. In LCD rats, the 53% increase in p.o. AUC (Table 3) was somewhat greater than 41% increase in i.v. AUC (Table 2). This could have been due to decreased intestinal metabolism of oltipraz in addition to decreased hepatic metabolism of oltipraz in LCD rats. The decreased intestinal metabolism of oltipraz in LCD rats could be supported by significantly slower (44% decrease) intestinal CLint than that in control rats (Table 1).
After i.v. administration of oltipraz to LC and LCD rats, the Vss was significantly smaller than that in control rats (25% and 19% decrease respectively; Table 2). Although the exact reason is not clear, this was not likely due to a decrease in free fractions of oltipraz in plasma from LC and LCD rats; the free fractions were rather greater than that in control rats (the free fractions of 12%, 30% and 21% for the control, LC and LCD rats respectively).
Changes in the CYP isozymes in patients with liver cirrhosis are somewhat different from those reported here in LC rats. For example, Elbekai et al. (2004) reported that in patients with liver cirrhosis, CYP1A and 3A levels and related enzyme activities are usually reduced and CYP2B and 2C are mostly unaltered. Yang et al. (2003) reported that in patients with liver cirrhosis, the enzyme activity, protein expression and mRNA level of CYP3A was reduced. Frye et al. (2006) reported that in patients with liver disease, CYP2C11, 1A2, 2D6 and 2E1 decreased. Only antipyrine metabolism was reported in patients with type I diabetes mellitus. For example, Sotaniemi et al. (2001) reported that in patients with insulin-responsive, untreated type I diabetes, antipyrine metabolism (markers of CYP1A2, 2B6, 2C and 3A) was clearly increased. However, changes in CYP isozymes in patients with liver cirrhosis and type I diabetes mellitus did not seem to be reported. Moreover, Wiwi and Waxman (2004) reported that endogenous regulation of CYPs showed pronounced species differences, particularly of CYP2C isoforms. Thus, the present experimental data in rats should be extrapolated with care to the human situation.
In conclusion, compared with control rats, the protein expression of CYP1A and 3A increased and decreased, respectively, and that of CYP2B1/2 and 2D was comparable in LCD rats (Figure 1). Thus, it could be expected that the pharmacokinetic parameters of oltipraz in LCD rats would be fully or partially (in between DM and LC rats) restored to the values in control rats. This could be supported by the following; in LCD rats, the CL, terminal t1/2 and MRT of oltipraz after i.v. administration (Table 2), and terminal t1/2 and F of oltipraz after p.o. administration (Table 3) were fully restored to those in the control rats. In LCD rats, the in vitro Vmax and CLint for the disappearance of oltipraz (Table 1), and AUC of oltipraz after i.v. (Table 2) and p.o. administration (Table 3) were in between LC and DM rats (partially restored to those in control rats).
Acknowledgments
This study was supported in part by a grant from the 2007 BK21 Project for Applied Pharmaceutical Life Sciences.
Glossary
Abbreviations:
- Ae0–24 h
percentage of the dose excreted in the 24-h urine
- AUC
total area under the plasma concentration–time curve from time zero to time infinity
- CL
time-averaged total body clearance
- CLint
intrinsic clearance
- CLR
time-averaged renal clearance
- CLNR
time-averaged non-renal clearance
- Cmax
peak plasma concentration
- CYP
hepatic cytochrome P450
- DM
diabetes mellitus
- F
extent of absolute oral bioavailability
- GI24 h
percentage of the dose recovered from the entire gastrointestinal tract (including its contents and feces) at 24 h
- HPLC
high-performance liquid chromatography
- Km
apparent Michaelis–Menten constant
- LC
liver cirrhosis
- LCD
liver cirrhosis with diabetes mellitus
- MRT
mean residence time
- NADPH
reduced form of β-nicotinamide adenine dinucleotide phosphate
- Tmax
time to reach Cmax
- Vmax
maximum velocity
- Vss
apparent volume of distribution at steady state
Conflict of interest
The authors state no conflict of interest.
References
- Ahn CY, Bae SK, Jung YS, Lee I, Kim YC, Lee MG, et al. Pharmacokinetic parameters of chlorzoxazone and its main metabolite, 6-hydroxychlorzoxazone, after intravenous and oral administration of chlorzoxazone to liver cirrhotic rats with diabetes mellitus. Drug Metab Dispos. 2008;36:1233–1241. doi: 10.1124/dmd.107.017442. [DOI] [PubMed] [Google Scholar]
- Aiba T, Takehara Y, Okuno M, Hashimoto Y. Poor correlation between intestinal and hepatic metabolic rates of CYP3A4 substrates in rats. Pharm Res. 2003;20:745–748. doi: 10.1023/a:1023429401738. [DOI] [PubMed] [Google Scholar]
- Al-Turk WA, Stohs SJ, Roche EB. Activities of hepatic and extrahepatic microsomal mixed function oxidase enzymes in diabetic and gonadectomized-diabetic rats. Gen Pharmacol. 1981;12:345–350. doi: 10.1016/0306-3623(81)90088-4. [DOI] [PubMed] [Google Scholar]
- Bae SK, Bu SC, Kim EJ, Kim SH, Kim SG, Lee MG. Determination of chemopreventive agent, oltipraz, in rat plasma and urine by high-performance liquid chromatography. Res Commun Mol Pathol Pharmacol. 2001;110:133–138. [PubMed] [Google Scholar]
- Bae SK, Lee SJ, Lee JY, Lee YS, Lee I, Kim SG, et al. Pharmacokinetic changes of oltipraz after intravenous and oral administration to rats with liver cirrhosis induced by dimethylnitrosamine. Int J Pharm. 2004;275:227–238. doi: 10.1016/j.ijpharm.2004.02.004. [DOI] [PubMed] [Google Scholar]
- Bae SK, Lee SJ, Kim YH, Kim T, Lee MG. Effects of enzyme inducers and inhibitors on the pharmacokinetics of oltipraz in rats. J Pharm Pharmacol. 2005a;26:129–134. doi: 10.1211/0022357055704. [DOI] [PubMed] [Google Scholar]
- Bae SK, Kim JW, Kim YH, Kim YG, Kim SG, Lee MG. Hepatic and intestinal first-pass effects of oltipraz in rats. Biopharm Drug Dispos. 2005b;26:129–134. doi: 10.1002/bdd.439. [DOI] [PubMed] [Google Scholar]
- Bae SK, Lee SJ, Kim YG, Kim SH, Kim JW, Kim T, et al. Interspecies pharmacokinetic scaling of oltipraz in mice, rats, rabbits, and dogs, and prediction of human pharmacokinetics. Biopharm Drug Dispos. 2005c;26:99–115. doi: 10.1002/bdd.437. [DOI] [PubMed] [Google Scholar]
- Bae SK, Lee SJ, Kim T, Lee I, Kim SG. Pharmacokinetics and therapeutic effects of oltipraz after consecutive or intermittent oral administration in rats with liver cirrhosis induced by dimethylnitrosamine. J Pharm Sci. 2006a;95:985–997. doi: 10.1002/jps.20597. [DOI] [PubMed] [Google Scholar]
- Bae SK, Kim JY, Yang SH, Kim JW, Kim T, Lee MG. Pharmacokinetics of oltipraz in rat models of diabetes mellitus induced by alloxan or streptozotocin. Life Sci. 2006b;78:2287–2294. doi: 10.1016/j.lfs.2005.09.031. [DOI] [PubMed] [Google Scholar]
- Benson AB, Olopade OI, Ratain MJ, Rademaker A, Mobarhan S, Stucky-Marshall L, et al. Chronic daily low dose of 4-methyl-5(2-pyrazinyl)-1,2-dithiole-3-thione (Oltipraz) in patients with previously resected colon polyps and first-degree female relatives of breast cancer patients. Clin Cancer Res. 2000;6:3870–3877. [PubMed] [Google Scholar]
- Bieder A, Decouvelaere B, Gaillard G, Depaire H, Heusse D, Leudoux C, et al. Comparison of the metabolism of oltipraz in the mouse, rat and monkey and in man. Distribution of the metabolites in each species. Arzneim-Forsch/Drug Res. 1983;33:1289–1297. [PubMed] [Google Scholar]
- Borbás T, Benkö B, Dalmadi B, Szabó I, Tihanyi K. Insulin in flavin-containing monooxygenase regulation. Flavin-containing monooxygenase and cytochrome P450 activities in experimental diabetes. Eur J Pharmacol Sci. 2006;28:51–58. doi: 10.1016/j.ejps.2005.12.011. [DOI] [PubMed] [Google Scholar]
- Boudinot FD, Jusko WJ. Fluid shifts and other factors affecting plasma protein binding of prednisolone by equilibrium dialysis. J Pharm Sci. 1984;73:774–780. doi: 10.1002/jps.2600730617. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Bu SC, Kim EJ, Kim SH, Kim SG, Lee MG. Stability, blood partition and protein binding of an antifibrotic agent, oltipraz. Res Commun Mol Pathol Pharmacol. 2001;109:333–344. [PubMed] [Google Scholar]
- Bueding E, Dolan P, Leroy JP. The antischistosomal activity of oltipraz. Res Commun Chem Pathol Pharmacol. 1982;37:293–303. [PubMed] [Google Scholar]
- Chiou WL. Critical evaluation of potential error in pharmacokinetic studies using the linear trapezoidal rule method for the calculation of the area under the plasma level–time curve. J Pharmacokinet Biopharm. 1978;6:539–546. doi: 10.1007/BF01062108. [DOI] [PubMed] [Google Scholar]
- Chiou WL, Nation RL, Peng GW, Huang SM. Improved micro-scale high-pressure liquid-chromatographic assay of gentamicin in plasma. Clin Chem. 1978;24:1846–1847. [PubMed] [Google Scholar]
- Christensen RG, Malone W. Determination of oltipraz in serum by high-performance liquid chromatography with optical absorbance and mass spectrometric detection. J Chromatogr. 1992;584:207–212. doi: 10.1016/0378-4347(92)80577-d. [DOI] [PubMed] [Google Scholar]
- Duggleby RG. Analysis of enzyme progress curves by nonlinear regression. Methods Enzymol. 1995;249:61–90. doi: 10.1016/0076-6879(95)49031-0. [DOI] [PubMed] [Google Scholar]
- Elbekai RH, Korashy HM, El-kadi AOS. The effect of liver cirrhosis on the regulation and expression of drug metabolizing enzymes. Curr Drug Metab. 2004;5:157–167. doi: 10.2174/1389200043489054. [DOI] [PubMed] [Google Scholar]
- Frye RF, Zgheib NK, Matzke GR, Chaves-Gnecco D, Rabinovits M, Shaikh OS, et al. Liver disease selectively modulates cytochrome P450-mediated metabolism. Clin Pharmacol Ther. 2006;80:235–245. doi: 10.1016/j.clpt.2006.05.006. [DOI] [PubMed] [Google Scholar]
- Gibaldi M, Perrier D. Pharmacokinetics. 2nd. New York: Marcel–Dekker; 1982. [Google Scholar]
- Goeting NL, Fleming JS, Gallagher P, Walmsely BH, Karran SJ. Alterations in liver blood flow and reticuloendothelial function in progressive cirrhosis in the rat. J Nucl Med. 1986;27:1751–1754. [PubMed] [Google Scholar]
- Gupta E, Olopade OI, Ratain MJ, Mick R, Baker TM, Berezin FK, et al. Pharmacokinetics and pharmacodynamics of oltipraz as a chemoprevetive agent. Clin Cancer Res. 1995;1:1133–1138. [PubMed] [Google Scholar]
- Jenkins SA, Grandison A, Baxter JN, Day DW, Taylor I, Shields R. A dimethylnitrosamine-induced model of cirrhosis and portal hypertension in the rat. J Hepatol. 1985;1:489–499. doi: 10.1016/s0168-8278(85)80747-9. [DOI] [PubMed] [Google Scholar]
- Jezequel AM, Mancini R, Rinaldesi ML, Macarri G, Venturini C, Orlandi F. A morphological study of the early stages of hepatic fibrosis induced by low doses of dimethylnitrosamine in the rat. J Hepatol. 1987;6:174–181. doi: 10.1016/s0168-8278(87)80570-6. [DOI] [PubMed] [Google Scholar]
- Kamninsky LS, Zhang QY. The small intestine as a xenobiotic-metabolizing organ. Drug Metab Dispos. 2003;31:1510–1525. doi: 10.1124/dmd.31.12.1520. [DOI] [PubMed] [Google Scholar]
- Kang KW, Kim YG, Cho MK, Bae SK, Kim CW, Lee MG, et al. Oltipraz regenerates cirrhotic liver through CCAAT/enhancer binding protein-mediated stellate cell inactivation. FASEB J. 2002;16:1988–1990. doi: 10.1096/fj.02-0406fje. [DOI] [PubMed] [Google Scholar]
- Kim SG, Kim EJ, Kim YG, Lee MG. Expression of cytochrome P-450s and glutathione S-transferases in the rat liver during water deprivation: effects of glucose supplementation. J Appl Toxicol. 2001;21:123–129. doi: 10.1002/jat.734. [DOI] [PubMed] [Google Scholar]
- Kim SH, Choi YM, Lee MG. Pharmacokinetics and pharmacodynamics of furosemide in protein–calorie malnutrition. J Pharmacokinet Biopharm. 1993;21:1–17. doi: 10.1007/BF01061772. [DOI] [PubMed] [Google Scholar]
- Kim YC, Lee AK, Lee JH, Lee I, Lee DC, Kim SH, et al. Pharmacokinetics of theophylline in diabetes mellitus rats: induction of CYP2E1 on 1,3-dimethyluric acid formation. Eur J Pharmacol Sci. 2005;26:114–123. doi: 10.1016/j.ejps.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Kwon SY. Prevalence and clinical significance of diabetes mellitus in patients with liver cirrhosis. Taehan Kan Hakhoe Chi. 2003;9:205–211. [PubMed] [Google Scholar]
- Lee MG, Chiou WL. Evaluation of potential causes for the incomplete bioavailability of furosemide: gastric first-pass metabolism. J Pharmacokinetic Biopharm. 1983;11:623–640. doi: 10.1007/BF01059061. [DOI] [PubMed] [Google Scholar]
- Moscatiello S, Manini R, Marchesini G. Diabetes and liver disease: an ominous association. Nutr Metab Cardiovasc Dis. 2007;17:63–70. doi: 10.1016/j.numecd.2006.08.004. [DOI] [PubMed] [Google Scholar]
- O'Dwyer PJ, Szarka C, Brennan JM, Laub PB, Gallo JM. Pharmacokinetics of the chemopreventive agent oltipraz and of its metabolite M3 in human subjects after a single oral dose. Clin Cancer Res. 2000;6:4692–4696. [PubMed] [Google Scholar]
- Ohara K, Kusano M. Anti-transforming growth factor-β1 antibody improves survival rate following partial hepatectomy in cirrhotic rats. Hepatol Res. 2002;24:174–183. doi: 10.1016/s1386-6346(02)00031-1. [DOI] [PubMed] [Google Scholar]
- Peng JZ, Remmel RP, Sawchuk RK. Inhibition of murine cytochrome P4501A by tacrine: in vitro studies. Drug Metab Dispos. 2004;32:805–812. doi: 10.1124/dmd.32.8.805. [DOI] [PubMed] [Google Scholar]
- Raza H, Ahmed I, John A, Sharma AK. Modulation of xenobiotic metabolism and oxidative stress in chronic streptozotocin-induced diabetic rats fed with Momprdica charantia fruit extract. J Biochem Mol Toxicol. 2000;14:131–139. doi: 10.1002/(sici)1099-0461(2000)14:3<131::aid-jbt2>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Sakuma T, Honma R, Maguchi S, Tamaki H, Nemoto N. Different expression of hepatic and renal cytochrome P450s between the streptozotocin-induced diabetic mouse and rat. Xenobiotica. 2001;31:223–237. doi: 10.1080/00498250110046451. [DOI] [PubMed] [Google Scholar]
- Sato H, Terasaki T, Okumura K, Tsuji A. Effect of receptor up-regulation on insulin pharmacokinetics in streptozotocin-treated diabetic rats. Pharm Res. 1991;8:563–569. doi: 10.1023/a:1015888203572. [DOI] [PubMed] [Google Scholar]
- Shimojo N, Ishizaki T, Imaoka S, Funae Y, Fujii S, Okuda K. Changes in amounts of cytochrome P450 isozymes and levels of catalytic activities in hepatic and renal microsomes of rats with streptozocin-induced diabetes. Biochem Pharmacol. 1993;46:621–627. doi: 10.1016/0006-2952(93)90547-a. [DOI] [PubMed] [Google Scholar]
- Sindhu RK, Koo JR, Sindhu KK, Ehdaie A, Farmand F, Robert CK. Differential regulation of hepatic cytochrome P450 monooxygenase in streptozotocin-induced diabetic rats. Free Radic Res. 2006;40:921–928. doi: 10.1080/10715760600801272. [DOI] [PubMed] [Google Scholar]
- Sotaniemi EA, Pelkonen O, Arranto AJ, Tapanainen P, Rautio A, Pasanen M. Diabetes and elimination of antipyrine in man: an analysis of 298 patients classified by type of diabetes, age, sex, duration of disease and liver involvement. Pharmacol Toxicol. 2001;90:155–160. doi: 10.1034/j.1600-0773.2002.900308.x. [DOI] [PubMed] [Google Scholar]
- Vidal J, Ferrer JP, Esmatjes E, Salmeron JM, Gonzalez-Clemente JM, Gomis R, et al. Diabetes mellitus in patients with liver cirrhosis. Diabetes Res Clin Pract. 1994;25:19–25. doi: 10.1016/0168-8227(94)90157-0. [DOI] [PubMed] [Google Scholar]
- Wilkinson GR, Shand DG. A physiological approach to hepatic drug clearance. Clin Pharmacol Ther. 1975;18:377–390. doi: 10.1002/cpt1975184377. [DOI] [PubMed] [Google Scholar]
- Wiwi CA, Waxman DJ. Role of hepatocyte nuclear factors in growth hormone-regulated, sexually dimorphic expression of liver cytochrome P450. Growth Factors. 2004;22:79–88. doi: 10.1080/08977190410001715172. [DOI] [PubMed] [Google Scholar]
- Yamazoe Y, Murayama N, Shimada M, Yamauchi K, Kato R. Cytochrome P450 in livers of diabetic rats: regulation by growth hormone and insulin. Arch Biochem Biophys. 1989;268:567–575. doi: 10.1016/0003-9861(89)90324-x. [DOI] [PubMed] [Google Scholar]
- Yang LQ, Li SJ, Cao YF, Man XB, Yu WF, Wang HY, et al. Different alterations of cytochrome P450 3A4 isoform and its gene expression in livers of patients with chronic liver disease. World J Gastroenterol. 2003;9:359–363. doi: 10.3748/wjg.v9.i2.359. [DOI] [PMC free article] [PubMed] [Google Scholar]