

Abstract
Glycogen synthase kinase 3 (GSK3), a constitutively acting multi-functional serine threonine kinase is involved in diverse physiological pathways ranging from metabolism, cell cycle, gene expression, development and oncogenesis to neuroprotection. These diverse multiple functions attributed to GSK3 can be explained by variety of substrates like glycogen synthase, τ protein and β catenin that are phosphorylated leading to their inactivation. GSK3 has been implicated in various diseases such as diabetes, inflammation, cancer, Alzheimer's and bipolar disorder. GSK3 negatively regulates insulin-mediated glycogen synthesis and glucose homeostasis, and increased expression and activity of GSK3 has been reported in type II diabetics and obese animal models. Consequently, inhibitors of GSK3 have been demonstrated to have anti-diabetic effects in vitro and in animal models. However, inhibition of GSK3 poses a challenge as achieving selectivity of an over achieving kinase involved in various pathways with multiple substrates may lead to side effects and toxicity. The primary concern is developing inhibitors of GSK3 that are anti-diabetic but do not lead to up-regulation of oncogenes. The focus of this review is the recent advances and the challenges surrounding GSK3 as an anti-diabetic therapeutic target.
British Journal of Pharmacology (2009) doi:10.1111/j.1476-5381.2008.00085.x
Keywords: diabetes, pancreatic β cells, β catenin, Wnt signalling, inhibitors
Introduction
Glycogen synthase kinase 3 (GSK3), a Ser/Thr kinase derives its name from its substrate glycogen synthase (GS) a key enzyme involved in the glycogen synthesis (Embi et al., 1980; Frame and Cohen, 2001). The name glycogen synthase kinase does not adequately describe the multitude of diverse substrates and functions attributed to GSK3. It is involved in a variety of cellular processes ranging from glycogen metabolism, insulin signalling, cell proliferation, neuronal function, oncogenesis to embryonic development. Of the many diverse substrates, some of the key molecules mediating GSK3 functions in regulation of several cellular processes are glycogen synthase (GS), tau protein and beta catenin proteins (Eldar-Finkelman, 2002; Jope and Johnson, 2004). Given the multiple processes regulated by GSK3, it is not surprising that GSK3 has been implicated in various diseases such as diabetes, cancer, bipolar disorder and Alzheimer's to name a few, and GSK inhibitors are being actively developed as drugs for the treatment of the various disorders.
Glycogen synthase kinase 3 is ubiquitously expressed with high levels of expression in brain and is associated with a variety of neurological disorders like Alzheimer's, bipolar disorder, Huntington disease and other neurodegenerative disorders (Jope and Roh, 2006; Mazanetz and Fischer, 2007). GSK3-mediated hyper phosphorylation of τ protein has been implicated in Alzheimer's disorder, and currently there are several GSK3 inhibitors being developed for Alzheimer's and bipolar disorder (Jope and Roh, 2006; Mazanetz and Fischer, 2007). This area is beyond the scope of this article, and readers are referred to other reviews in this area (Mazanetz and Fischer, 2007; Rowe et al., 2007).
The focus of the present review is regulation of GSK3, its function in insulin signalling and its role in maintenance of glucose homeostasis. Further, the involvement of GSK3 in Wnt signalling and its implication for oncogenesis (Jope and Johnson, 2004; Patel et al., 2004) and the impact of this on the development of GSK3 inhibitors for treatment of diabetes mellitus will be discussed.
GSK3 a unique multi-tasking kinase
Apart from its diverse substrates and several patho-physiological roles, GSK3 is a unique kinase for several other reasons. First, unlike other kinases the enzyme is constitutively active and is inactivated in response to cellular signals (Harwood, 2001; Doble and Woodgett, 2003). Second, phosphorylation of its substrates generally leads to their inactivation. Further, it has unique substrate specificity wherein many of its substrates need a ‘priming phosphate’ (which is a phosphorylated Ser/Thr residue) located at four amino acids C terminal to the site of phosphorylation (Frame and Cohen, 2001; Harwood, 2001; Doble and Woodgett 2003). Several protein kinases have been identified, which can phosphorylate the ‘priming site’ with their identity depending on the substrate in question. The consensus recognition sequence is Ser/Thr-(X-X-X)-pSer/pThr with X being any amino acid and pSer/pThr being the phosphorylated serine/threonine, which serves as a priming phosphate (Frame and Cohen, 2001; Harwood, 2001; Doble and Woodgett 2003). Multiple phosphorylations by GSK3, is carried out in a ‘relay’ fashion with the ‘primed’ phosphate serving to initiate the process of phosphorylation. Subsequently, the phosphorylated residue itself serves as a ‘priming’ phosphate to the subsequent residue to be phosphorylated. This relay mechanism may continue further ranging from two (transcription factor c-Jun) to five residues (human GS) (Frame and Cohen, 2001; Frame et al., 2001).The mechanism underlying the requirement of ‘priming phosphate’ was revealed subsequently, with the identification of a specific binding site for the priming phosphate on GSK3 (Frame and Cohen, 2001; Frame et al., 2001). Interestingly, GSK3 itself is subject to regulation by phosphorylation and is phosphorylated at Ser/Thr and also at Try residues at N and C terminus respectively (Wang et al., 1994).
Glycogen synthase kinase 3 is involved in several diverse pathways, and several substrates have been identified, which are phosphorylated in vitro. However, the physiological relevance and function of many of the substrates has not been determined so far. Of the several pathways in which GSK3 is implicated, its role in insulin signalling and Wnt signalling has been studied in detail (Figure 1). GSK3 regulates insulin signalling (Figure 1) via inhibition of GS leading to decreased glycogen synthesis and hence inhibition of GSK3 leads to increase in glycogen synthesis and increase in insulin sensitivity (Henriksen and Dokken, 2006; Lee and Kim, 2007). In the Wnt pathway (Figure 2), GSK3 exists in a multimeric complex comprising of adenomatous polyposis coli (APC) protein, scaffolding protein axin and β catenin (Figure 2) (Rubinfeld et al., 1996; Seidensticker and Behrens, 2000). Activation of Wnt signalling pathway through its receptors Frizzled and LDL receptor related protein (LRP)5/6 co-receptors and intracellular protein Dishevelled, leads to inactivation of GSK3 (Cadigan and Liu, 2006; Zeng et al., 2008). GSK3-mediated phosphorylation of the N terminal Ser/Thr of β catenin leads to its degradation and regulates the levels, cellular localization and function of β catenin (Figure 2) (Cadigan and Liu, 2006).
Figure 1.
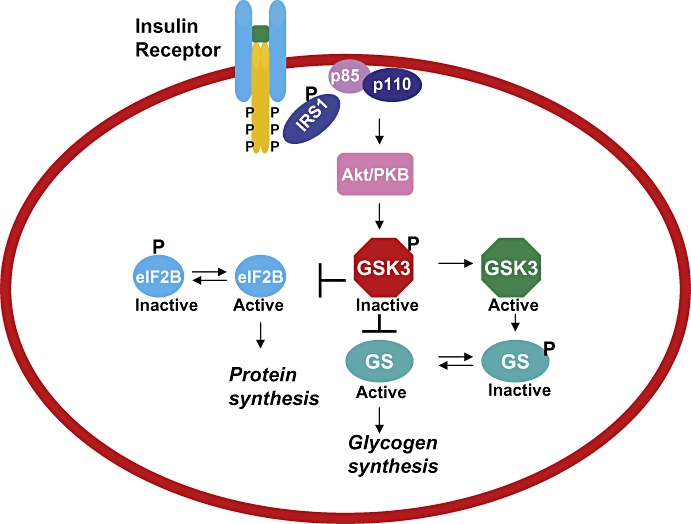
Schematic representation of insulin-signalling events and role of GSK3. Binding of insulin to its receptor leads to autophosphorylation, followed by binding of IRS that is phosphorylated leading to activation of PI3 kinase and Akt/PKB that phosphorylates GSK3. Inactive phosphorylated GSK3 relieves glycogen synthase inhibition and eIF2B phosphorylation, leading to active form of GS and eIF2B, which leads to increase in glycogen and protein synthesis. eIF2B, eukaryotic initiation factor 2B; GS, glycogen synthase; GSK3, glycogen synthase kinase 3; IRS, insulin receptor substrates; PKB, protein kinase B.
Figure 2.
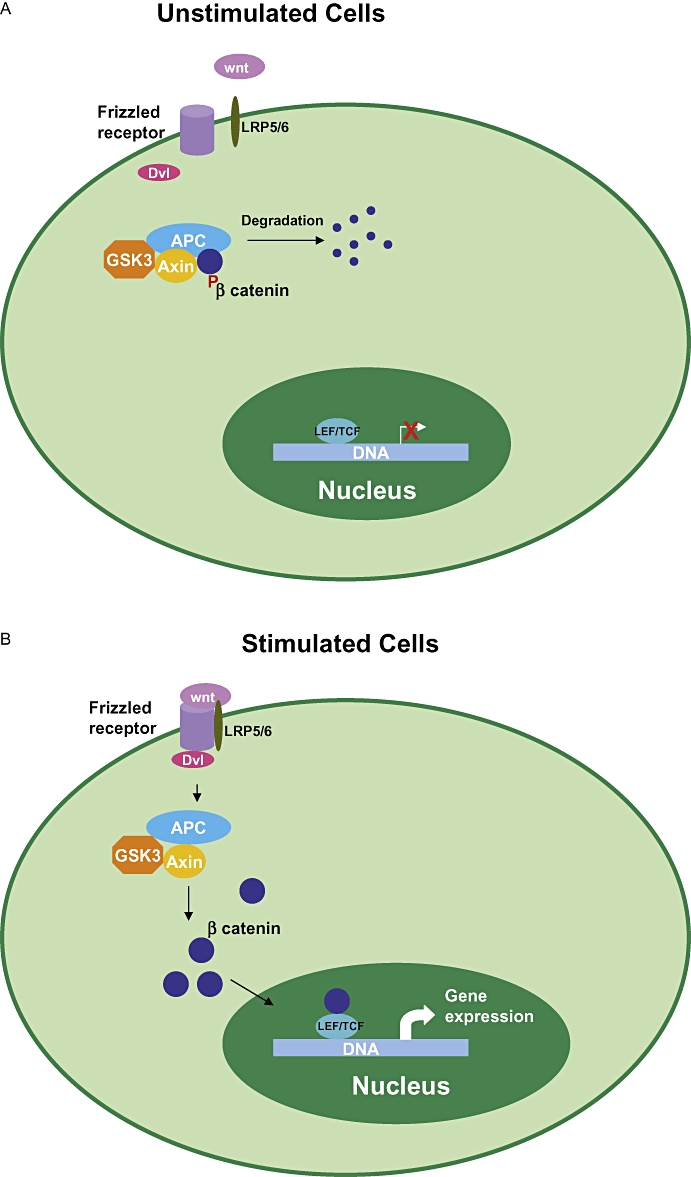
Schematic illustration of role of GSK3 in Wnt signalling. In the unstimulated cells β catenin is present in a multi-protein complex with GSK3, Axin and APC. Phosphorylation of β catenin by GSK3 targets β catenin to proteosome-mediated degradation. In stimulated cells binding of Wnt to its receptor Frizzled and its co-receptor LRP5/6 leads to activation of Dishevelled (Dvl) leading to inhibition of phosphorylation of β catenin by the multimeric complex and its stabilization and accumulation in the cytosol. Stabilized β catenin translocates to the nucleus and in association with transcription factors LEF/TCF leads to increase in gene expression of target genes such as c-myc. APC, adenomatous polyposis coli; GSK3, glycogen synthase kinase 3; LEF/TCF, lymphoid enhancer factor/T cell factor; LRP, LDL receptor related protein.
GSK3α/β
Glycogen synthase kinase 3 has two isoforms encoded by two different genes with GSK3α having a higher molecular weight than GSK3β owing to an extension at the N terminus (Woodgett, 1990;1991). The two isoforms share extensive homology, especially at the kinase domain with 97% homology between the two. Perhaps, not surprisingly, they both have similar functions. However, despite this the two are not functionally redundant as demonstrated by the gene knockout studies. Deletion of GSK3β leads to embryonic lethality at E16 due to hepatocyte apoptosis thought to be mediated by regulation of nuclear factor κB by GSK3β (Hoeflich et al., 2000). This suggests that the function of GSK3β is not compensated by GSK3α, which is intriguing as both the isoforms display similar substrate specificity and functions. In this context knockout of shaggy, a Drosophila GSK3 homolog is rescued by over-expression of human GSK3β but not GSK3α suggesting that they might have different physiological functions (Ruel et al., 1993). It has been demonstrated that GSK3α and GSK3β have distinct role during zebrafish cardiogenesis with GSK3α being necessary for cardiomyocyte survival and GSK3β modulating left right symmetry and affecting heart positioning (Lee et al., 2007; Markou et al., 2008). However, in certain pathways both the isoforms are implicated, as in axon formation in hippocampal neurons (Garrido et al., 2007). Although most of the research so far has primarily been on GSK3β, recent advances on the understanding of the function of GSK3α in Alzheimer's disease (Phiel et al., 2003) and glucose metabolism (MacAulay et al., 2007) may change the course.
Another variant of GSK3β called as GSK3β2 that contains a 13-amino acid residue insert within the kinase domain has recently been discovered (Mukai et al., 2002). Although its physiological functions are not well characterized, GSK3β2 has been recently shown to be localized in neuronal cell bodies and not in neuronal processes too unlike GSK3β, which is localized in both. Further, GSK3β2 has reduced activity towards τ protein as compared with GSK3β whose significance yet needs to be understood.
Regulation of GSK3
As GSK3 is involved in diverse processes, regulation of its activity is critical to ensure that the pathways are insulated and yet could be appropriately coordinated. To achieve this GSK3 is subject to multiple levels of regulation mediated by phosphorylation, cellular localization and protein–protein interactions. GSK3β can phosphorylate at both primed substrates like GS, eukaryotic initiation factor 2B (eIF2B), β catenin and unprimed substrates like cyclin D3, axin and APC although it phosphorylates the unprimed substrates less efficiently (Frame and Cohen, 2001; Frame and Zheleva, 2006).The mechanistic details for the requirement of the priming site have been well understood, and the phosphorylated priming site of the substrate interacts with three positive residues in GSK3β, Arg 96, Arg 180 and Lys 205, which form a binding pocket for the priming phosphate (Dajani et al., 2001; Frame et al., 2001; ter Haar, 2001). Mutation of Arg 96 to Ala prevents the phosphorylation of primed substrates by GSK3 (Frame et al., 2001). Apart from the requirement of priming phosphate, substrate recognition requires GSK3β residues, Phe 67, Gln 89 and Asn 95, which facilitates precise positioning of the substrate within the substrate binding pocket and provides an insight into the substrate binding and specificity (Ilouz et al., 2006). GSK3 protein itself undergoes multiple phosphorylation events, which impact its activity depending on the amino acid being modified (Hughes et al., 1993; Wang et al., 1994). Phosphorylation of the N terminal Ser 21 for GSK3α and Ser 9 for GSK3β has an inhibitory effect and plays an important role in regulation of GSK3 function during insulin signalling (Stambolic and Woodgett, 1994; Cross et al., 1995). The Ser/Thr-phosphorylated N terminus serves as a pseudo substrate and inhibits its interaction with the binding of the priming phosphate resulting in inactivation of GSK3. These Ser residues are phosphorylated by Akt/PKB (protein kinase B) during insulin signalling leading to the inactivation of the enzyme and resulting in activation of GS and increased glycogen synthesis. Ser 21/9 could also be phosphorylated by several other kinases like AGC kinase, p70 ribosomal S6 kinase, p90 ribosomal S6 kinase (Frame and Cohen, 2001). Phosphorylation at Tyr in the C terminus at 279 for α and 216 for β activates the enzyme, and its mutation decreases the activity of the enzyme. Kinases phosphorylating GSK3 at this site are not well characterized (Hughes et al., 1993; Wang et al., 1994). In a recent development, phosphorylation at C terminus of GSK3 by p38 mitogen-activated protein kinase (MAPK) leading to its inactivation has been reported (Thornton et al., 2008). Significantly, p38 MAPK phosphorylates only GSK3β on Thr 390 and not GSK3α residue leading to an increased accumulation of β catenin (Thornton et al., 2008). In addition, Thr 390 residue is not conserved between the two isoforms (Thornton et al., 2008). This suggests that apart from AKT/PKB-mediated inactivation of GSK3, p38 MAPK could be an alternative mechanism for regulation of GSK3 activity especially in brain (Thornton et al., 2008).
Within the Wnt pathway, it is intriguing that GSK3 activates/inactivates Wnt signalling and functions as an on/off switch of Wnt signalling pathway by its ability to phosphorylate primed and unprimed substrates. Unlike most of the phosphorylation events mediated by GSK3β that leads to inactivation, phosphorylation of LRP6 by GSK3 leads to activation and promotes the engagement of LRP6 with scaffolding protein axin (Zeng et al., 2005). LRP6 is an unprimed substrate, and phosphorylation of LRP6 promotes axin recruitment and Wnt activation. On the other hand phosphorylation of β catenin a primed substrate by GSK3 leads to inactivation of Wnt signalling (Zeng et al., 2005). It is also likely that different pools of GSK3 have different functions where cytosolic GSK3 phosphorylates β catenin and antagonizes Wnt signalling and the plasma membrane bound GSK3 phosphorylates LRP6 and activates Wnt/β catenin signalling (Zeng et al., 2005).
Glycogen synthase kinase 3β can phosphorylate τ protein, implicated in Alzheimer's disease, at both primed and unprimed substrates with phosphorylation at primed sites (pThr 231) being important for the function of τ than phosphorylation at unprimed substrates (pSer 396/404) (Cho and Johnson, 2004). Presenilin-1 is another unprimed substrate of GSK3 involved in Alzheimer's disease (Twomey and McCarthy, 2006). Significantly, role of different GSK3 substrates in regulating nervous system development and specially axon morphology has been examined (Kim et al., 2006). By using GSK3 R96A mutant that inhibits GSK3 activity towards primed substrates, it was demonstrated that differential regulation of primed substrates is associated with defects in axon branching (Kim et al., 2006). It has been suggested that partial inhibition due to low concentrations of inhibitors or inhibition of primed substrates produces an intermediate condition and suggests the importance of dosage effects and also spatial regulation of GSK3 substrates. Another new form of regulation of GSK3 involving N terminal cleavage by calpain, which removes the N terminal inhibitory domain and activates its kinase activity has also been recently described (Goñi-Oliver et al., 2007).
Role of GSK3 in insulin resistance in type II diabetes mellitus
Homeostasis of blood glucose levels is controlled by efficient absorption of glucose and storage of excess glucose as glycogen. Insulin resistance is one of the main causes underlying the initiation and progression of type II diabetes mellitus (T2DM) (Bouchéet al., 2004). Insulin resistance is caused by the inability of the insulin-sensitive tissues to respond to insulin and efficiently clear the blood glucose by decreasing the hepatic glucose output and reduced glucose uptake by peripheral tissues (Bouchéet al., 2004). The various steps involved in insulin signal transduction have been well elucidated, and a general mechanism has emerged. Stimulation of insulin receptor (IR) upon binding of insulin leads to autophosphorylation of several tyrosine residues on the IR. This phosphorylation event triggers a cascade of phosphorylation events, where the insulin receptor substrates 1 and 2 (IRS1, IRS2) recognize and bind to the phosphorylated IR and are themselves-phosphorylated. Subsequently, the activation of phosphoinositol 3 kinase converts phosphatidylinositol bisphosphate to phosphatidylinositol triphosphate, which in turn stimulates and activates PKB. The next step in the signalling event involves the activated PKB to phosphorylate GSK3 leading to its inactivation (Figure 1) (Cross et al., 1995; Eldar-Finkelman and Ilouz, 2003). This inactivation leads to a decrease in phosphorylation of GS resulting in its activation. Apart from GS, eIF2B is also activated in a similar fashion leading to an increase in glycogen synthesis and protein synthesis. Increased expression and activity of GSK3 has been reported in the skeletal muscle of T2DM patients (Nikoulina et al., 2000) and in mice having diabetes (Eldar-Finkelman et al., 1999) suggesting that dysregulation of GSK3β might result in impaired insulin signalling and hence diabetes. Evidence for this has been obtained by using pharmacological inhibitors of GSK3, which leads to improved insulin signalling and glucose levels in animal models of diabetes (Meijer et al., 2004).
GSK3 as a regulator of glycogen synthase activity
Glycogen synthase a key enzyme mediating the conversion of glucose to glycogen is kept in an inactive state by phosphorylation of a cluster of three serine residues at the C terminus termed site 3. The kinase chiefly responsible for phosphorylating the site 3 has been identified as GSK3. Insulin activates GS by inactivation of GSK3 and hence promoting dephosphorylation of serine residues Ser 641, Ser 645, Ser 549 and Ser 653 at C terminus of GS. Regulation of GS by insulin is multifactorial, and insulin activates GS by inducing glycogen-associated form of protein phosphatase 1 (PP1) that dephosphorylates the residues phosphorylated by GSK3 (Suzuki, et al., 2001, Delibegovic et al., 2003). Apart from site 3, site 2 in GS has also been implicated in regulation of GS activity, and PP1 and PP2 can dephosphorylate site 2 and site 3. Also, non-GSK3-mediated insulin-dependent GS activation has also been proposed (Semiz and McNeill, 2002). Glycogen synthesis is also stimulated in an insulin-independent manner after exercise when glycogen levels are depleted (Halse et al., 2001; Nielsen and Wojtaszewski, 2004). However, data suggest that primarily the activation of GS by insulin is mediated by inactivation of GSK3. It has been proposed that hepatic GS activation by glucose is through stimulation of a protein phosphatase rather than Ser 21/Ser 9-mediated inactivation of GSK3 isoforms (McManus et al., 2005). However, GSK3 inhibitors stimulate liver GS and are responsible for the blood glucose lowering effects of GSK3 inhibition observed in animal models of T2DM (Cline et al., 2002).
The activity of GS is regulated by GSK3α and β and insulin in both the skeletal muscle and liver. GS is a primed substrate, and casein kinase 2 (CK2) has been proposed to be the priming kinase that phosphorylates Ser 657 (Hanashiro and Roach, 2002; Semiz and McNeill, 2002). By using pharmacological inhibitors of GSK3, it has been demonstrated that inactivation of GS in muscle is mediated by GSK3, although the relative importance of the two isoforms to this process is not known. By using selective cell permeable inhibitors of GSK3, CT98014 and CHIR98023, acute treatment of human skeletal muscle cells, activation of GS was observed with an increase in glycogen synthesis. Significantly, increase in glucose uptake was observed only upon prolonged or chronic exposure of the inhibitors, and an increase in IRS1 levels was also observed (Nikoulina et al., 2002).
Role for GSK3 in regulation of GS in muscle has been proposed (MacAulay et al., 2005), and over-expression of GSK3β in muscle results in reduced muscle glycogen deposition, which suggests that GSK3β is the primary kinase regulating GS in muscle (Pearce et al., 2004). By using GSK3β9A/9A knock in mice, GSK3β has been proposed to be the principal isoform regulating GS in muscle (McManus et al., 2005), and about threefold to fourfold higher level of expression of GSK3β has been reported in humans and mice, as compared with GSK3α. However, down-regulation of GSK3α by using antisense oligonucleotides in muscle cells leads to an increase in insulin-stimulated GS and glucose uptake in the muscle cells (Ciaraldi et al., 2007). Further, GSK3α over-expression in muscle cells impaired insulin-stimulated glycogen synthesis and glucose uptake but not basal glycogen synthesis and glucose uptake levels, implying that GSK3α also may have a functional role in insulin action in muscle (Ciaraldi et al., 2007). In contrast to this, recently MacAulay et al. (2007) demonstrated that GSK3α regulates glycogen metabolism in liver and not in muscle using GSK3α knockout mice (MacAulay et al., 2007). Knockout animals are viable and show increased glucose tolerance and insulin sensitivity with a significant reduction in total body fat mass as compared with wild type. In the knockout mice, liver GS was more active, accompanied by increased levels of glycogen and IRS1 in contrast to the GS and IRS1 of muscle from knockout mice. No significant increase in muscle or liver glucose transport was detected in the knockout animals (MacAulay et al., 2007). Insulin-stimulated glycogen synthesis has been demonstrated in skeletal muscle in absence of GSK3 phosphorylation of GS by using GSK3 knockin mice with wild-type GSK3α and GSK3β replaced with insulin-non-responsive mutant forms (S21A/S21A/S9A/S9A) suggesting that other mechanisms like allosteric regulation of GS by glucose-6-phosphate may be involved (Bouskila et al., 2008).
Glycogen synthase kinase 3 also plays an important role in glucose metabolism in adipocytes apart from muscle and contributes to insulin regulation of glycogen synthesis but not in glucose transport and glucose transporter 4 (GLUT4) translocation (Summers et al., 1999). However, some reports suggest that in 3T3L-1 adipocytes, insulin can stimulate GS despite low or lack of expression of GSK3β (Ueki et al., 1998). Also, decrease in GSK3β expression has been reported during adipocyte differentiation (Brady et al., 1998). Obese type II diabetic patients treated with troglitazone for 3–4 months displayed increased glucose disposal accompanied by decreased GSK3α and GSK3β levels in skeletal muscle but with no changes in adipocytes (Ciaraldi et al., 2006). This suggests that in humans GSK3 plays a role in skeletal muscle, while its role in adipose tissue is not clear (Ciaraldi et al., 2006).
GSK3 and glucose homeostasis
Apart from its role in regulation of GS activity that is chiefly responsible for its function in glucose homeostasis it has also been implicated in glucose uptake. GSK3 has been proposed to phosphorylate light chains of kinesin motor proteins, which mediate the translocation of the GLUT4 glucose transporters (Morfini et al., 2002). This phosphorylation of kinesin results in its inhibition, and inactivation of GSK3 by insulin may increase the glucose uptake. However, the data from knock in studies suggest no change in the insulin-stimulated glucose uptake in muscle (McManus et al., 2005; MacAulay et al., 2007). GSK3 can also phosphorylate the IRS (IRS1) and influence insulin action (Eldar-Finkelman and Krebs, 1997). Insulin mediates most of its actions through IRS1 and IRS2 that recruit src homology 2 proteins such as p85 regulatory subunit of phosphoinositol 3 kinase, src homology protein tyrosine phosphatase 2, growth factor receptor bound protein 2 (Eldar-Finkelman and Krebs, 1997). In unstimulated cells IRS1 is phosphorylated on Ser/Thr residues, and upon stimulation of cells with insulin, IRS1 is phosphorylated on Tyr residues (Greene and Garofalo, 2002). Hyper phosphorylation of IRS1 on Ser/Thr residues leads to a reduction of the ability of the IR to phosphorylate IRS1, and this has been implicated in insulin resistance (Hotamisligil et al., 1996). GSK3-mediated phosphorylation of IRS1 on Ser residues has been reported (Eldar-Finkelman and Krebs, 1997), and over-expression of GSK3 leads to an increase in serine phosphorylation of IRS1 and decreased tyrosine phosphorylation of IRS1 and IR (Aguirre et al., 2002; Liberman and Eldar-Finkelman, 2005). However, apart from GSK3 other kinases like CK2 have been implicated in IRS1 phosphorylation, and several other factors like tumour necrosis factor α that induce insulin resistance also lead to serine phosphorylation of IRS1 by c-Jun N terminal kinase (JNK) and inhibitor κB kinase (IKKβ) (Eldar-Finkelman and Krebs, 1997; Liberman and Eldar-Finkelman, 2005). The serine residues phosphorylated by GSK3 have been identified as Ser 332 with Ser 336 serving as the ‘priming’ phosphate required for phosphorylation. Mutation of these sites to Ala increased the tyrosine phosphorylation of IR and GSK3 inhibitor lithium reduced Ser 322 phosphorylation and over-expression of GSK3 increased its phosphorylation (Liberman and Eldar-Finkelman, 2005). Hyper phosphorylation of IRS1 on serine residues induces IRS1 degradation in proteosomes, and hence inhibition of GSK3 could reduce its phosphorylation and its degradation. Increase in IRS1 protein expression in skeletal muscle could augment insulin action. GSK3 has been implicated in repression of gluconeogenic enzymes such as phosphoenolpuruvate carboxy kinase and glucose 6 phosphatase that reduces hepatic glucose production and helps in modulating glucose homeostasis (Lochhead et al., 2001). However, there are contradicting reports regarding the regulation of gluconeogenic enzymes by GSK3. While some reports suggest that inhibition of GSK3 leads to the repression of the gluconeogenic enzymes, other reports suggest that the gluconeogenic enzymes can be repressed without inhibiting GSK3 (Lipina et al., 2005).
The data so far suggest that apart from its primary effect on GS, GSK3 is also involved in upstream and downstream insulin-signalling events. GSK3 can suppress GS activity in short term and in long term negatively regulate glucose uptake and insulin signalling. Chronic effects of GSK3 inhibitors could also be mediated by regulation of protein expression and inactivation of eIF2B, can lead to up-regulation of protein translation.
Role of GSK3 in pancreas
Imbalanced glucose homeostasis due to the combined effect of insulin resistance and β cell failure is a hallmark of progression of T2DM (Donath et al., 2005; Bouchéet al., 2004). Progressive insulin resistance leads to increased insulin secretion by pancreas finally resulting in the failure of pancreatic β cells (Donath et al., 2005; Bouchéet al., 2004). Hence, as the disease progresses, there is a reduction in β cells of pancreas and desired goal of anti-diabetic agents is to preserve and increase the β cell mass and restore their function. Recently developed glucagon like peptide 1 analogs and dipeptidly peptidase IV inhibitors have been demonstrated to improve β cell function in preclinical models (Baggio and Drucker, 2006). The function of GSK3 in T2DM has been primarily focused on the insulin signal transduction in liver and skeletal muscle. Recent studies have provided novel insights into the function of GSK3 in pancreas.
It is likely that GSK3 has a function in pancreas as it is a substrate of PI3 kinase/Akt pathway, which previously has been shown to be a central regulator of β cell growth. GSK3 has also been implicated in endoplasmic reticulum stress in pancreatic β cells, and down-regulation of GSK3 can protect the cells from death (Srinivasan et al., 2005). Further, GSK3 has been reported to be inactivated by several ligands like glucagon like peptide 1, glucose-dependent insulinotropic peptide, insulin-like growth factor-1, which promote β cell preservation and proliferation (Mussmann et al., 2007).
Transcription factor IPF1/PDX1 (insulin promoter factor or pancreatic/duodenal homeobox 1 transcription factor 1) plays a crucial role in pancreas development and maintenance of β cell function as its deletion leads to diabetes in mice and reduced expression affects insulin expression and secretion (Boucher et al., 2006). IPF1/PDX1 has been shown to be phosphorylated by GSK3 on Ser 61 and/or Ser 66 in β cells, which targets it for degradation by proteasomes (Boucher et al., 2006). However, it is not clear if IPF1/PDX1 is indeed substrate of GSK3 or an indirect effect of oxidative stress observed in diabetic state, which increases this phosphorylation event and decreases the levels of IPF1/PDX1.
Apart from PDX1, it has been recently demonstrated that Maf A, a β cell transcription factor is constitutively phosphorylated by GSK3 at its amino terminus leading to its rapid degradation under low-glucose conditions and not in high-glucose conditions (Han et al., 2007). Surprisingly, although the GSK3-mediated phosphorylation status of Maf A is not altered by glucose concentration it is required for its degradation (Han et al., 2007).
Mussman et al., have demonstrated that inhibition of GSK3β promotes replication and survival of pancreatic β cells against death induced by high glucose and fatty acid palmitate (Mussmann et al., 2007). Interestingly, experiments by using siRNA demonstrate that inhibition of GSK3α and GSK3β is required for stimulation of replication as inhibition of either of them alone did not stimulate the proliferation (Mussmann et al., 2007). Transgenic mice over-expressing constitutively active form GSK3β (S9A) in β cells showed impaired glucose tolerance after 5 months of age. The β cell mass and proliferation were decreased along with a 50% decrease in the PDX1 levels (Liu et al., 2008).
Recently, role of GSK3β in regulation of β cell mass (Liu et al., 2008) has been examined in mice lacking one subunit of IR (Ir+/−) and in mice lacking two alleles of IRS (Irs2−/−) (Tanabe et al., 2008). Although both the genotypes exhibit insulin resistance Ir+/− mice exhibit doubling of β cell mass, and Irs2−/− mice develop β cell loss and severe diabetes. Significantly, increased levels of GSK3β activity were reported in β cells of Irs2−/− mice, and these mice heterozygous for GSK3β have reduced insulin resistance and preserved β cell mass due to increase in proliferation and decreased apoptosis. Ir+/−Gsk3β+/− mice exhibit improved insulin sensitivity, reduced hyperinsulinemia and reduced β cell mass. GSK3β has been proposed to phosphorylate and stabilize cyclin-dependent kinase (CDK) inhibitor p27 kip1 levels and destabilize PDX1 levels. Although insulin resistance was reduced in Irs2−/−Gsk3β+/− mice, it was still insulin-resistant as compared with wild-type, suggesting that the beneficial effects of GSK3β deficiency on restoration of glucose homeostasis is not solely due to altered insulin sensitivity. Contribution of increased insulin sensitivity in tissues like muscle to the preservation of β cell mass has been ruled out by using β cell specific knockout of GSK3 mice, which also prevented diabetes of Irs2−/− mice. These results demonstrate that GSK3β plays an important role in β cell regulation and that inhibitors of GSK3 have a good therapeutic potential to preserve β cell function and delay or prevent diabetes progression (Tanabe et al., 2008). GSK3β has also been implicated in improved β cell mass observed by inhibition of JNK kinase (Fornoni et al., 2008).
Using small molecule GSK3 inhibitors like 1-azapaullones derived from 1-paullone, stimulation of β cell replication and also protection from cell death induced by glucolipotoxicity has been demonstrated in insulinoma cell line (Stukenbrock, et al., 2008). In addition, Cazpaullone (9-cyano-1-azapaullone) stimulated replication of primary β cells and increased expression of β cell transcription factor Pax4 involved in β cell growth and development (Stukenbrock, et al., 2008). The evidence so far suggests that GSK3 has an important function in β cells of pancreas, which is mediated by modulating key factors like PDX1, Maf A and Pax4 that adds yet another feature to this multifaceted protein.
GSK3 in Wnt signalling and oncogenesis
Apart from regulation of GS and insulin signalling, GSK3 also plays an important role in canonical Wnt signalling pathway, which is critical for embryonic development (Logan and Nusse, 2004). As discussed earlier, GSK3 exists in a multimeric complex with APC, axin and β catenin, where GSK3 phosphorylates the N terminal Ser/Thr of β catenin leading to its degradation mediated by ubiquitin/proteasomes (Figure 2) (Rubinfeld et al., 1996; Chen et al., 2000). Activation of Wnt signalling leads to GSK inactivation resulting in dephosphorylation and stabilization of β catenin protein in the cytosol. The increased levels of β catenin lead to its nuclear translocation, and its interaction with transcription factors LEF/TCF (lymphoid enhancer factor/T cell factor) activate the expression of target genes like c-myc and cyclin D1, leading to an increase in cell proliferation (Jope and Johnson, 2004; Patel et al., 2004). Abnormal Wnt signalling (Figure 2) has been associated with several human diseases like cancers (Luo et al., 2007). Role of β catenin was first implicated in colorectal cancers as mutations in either APC or GSK3β sites in β catenin lead to increase in levels of β catenin as observed in colon cancer (Luo et al., 2007). Aberrant Wnt signalling has also been reported in wide range of cancers (Luo et al., 2007). Further, GSK3 has been implicated in accurate chromosome segregation based on studies in cultured cells, suggesting that GSK3 inhibitors may induce chromosome instability (Tighe et al., 2007), which might promote tumorigenesis. However, GSK3 inhibitors-induced chromosome instability needs to be examined in animal models and humans. Recently, GSK3β has also been implicated in skin tumorigenesis (Ma et al., 2007). Apart from GSK3β, GSK3α also regulates β catenin levels and β catenin-mediated transcription through Wnt pathway (Asuni et al., 2006; Doble et al., 2007).
Importantly, as inhibitors of GSK3 can improve insulin resistance and serve as therapeutic agents for diabetes, it is of concern that GSK3 inhibition via Wnt pathway leads to stabilization of β catenin that may promote tumorigenesis (Jope and Johnson, 2004; Patel et al., 2004). Hence, it is important to address these issues, which would be aided by a detailed understanding of the mechanism of regulation of GSK3 in the insulin and Wnt pathway. Significantly, although both Wnt and insulin decrease the GSK3β activity to a similar extent, research demonstrates that the downstream effect is specific to each of the pathways. In the Wnt pathway GSK3 activity is regulated by protein–protein interactions in multi-protein complexes. Insulin signalling does not regulate β catenin levels and regulates only GS suggesting that there are different pools of GSK3β complexes, which are differentially regulated (Patel et al., 2004). Ser 9 of GSK3β is not regulated by Wnt or Dishevelled, and GSK3β in the complex is not phosphorylated on Ser 9 upon insulin stimulation (Ding et al., 2000). Further, in GSK3α/β 21A/21A/9A/9A mice, Wnt induced inhibition of phosphorylation of β catenin on the sites targeted by GSK3, suggesting that Wnt3α-mediated inactivation of GSK3 does not involve phosphorylation of GSK3α and GSK3β at Ser 21 and Ser 9 (McManus et al., 2005). It has been proposed that inactivation of GSK3 isoforms by Ser 21/Ser 9 phosphorylation does not play a major role in regulating development, cell growth and proliferation (McManus et al., 2005). This elaborate mechanism of substrate specificity, different binding partners and compartmentalization ensures that there is little or no crosstalk between the two pathways (Ding et al., 2000). In addition, protein kinase C (PKC) has also been shown to be involved in Wnt-induced inactivation of GSK3β (Chen et al., 2000). It has been suggested that phosphorylation of β catenin by GSK3 does not require priming in vitro, in contrast to the phosphorylation of GS (Hagen et al., 2006). However, recent reports provide evidence that phosphorylation of β catenin also requires priming phosphate (Hagen and Vidal-Puig, 2002), and it has been demonstrated that CK1 is the priming kinase for β catenin (Jope and Johnson 2004, Patel et al., 2004; Bustos et al., 2006). Ser 33, Ser 37, Thr 41 phosphorylation on β catenin requires prior phosphorylation on Ser 45, and the Arg 96 mutant is unable to induced degradation and indeed stabilizes β catenin (Jope and Johnson 2004, Patel et al., 2004).
Another protein of the Wnt pathway regulating GSK3 is intracellular protein FRAT (frequently rearranged in advanced T cell lymphoma) for which two isoforms FRAT1 and FRAT2 have been identified in humans (van Amerongen et al., 2004; Stoothoff et al., 2005; Hagen et al., 2006). FRAT1 and FRAT2 inhibit GSK3 activity towards β catenin by competing with axin for binding to GSK3β (Ferkey and Kimelman, 2002; Stoothoff et al., 2005). Both FRAT1 and 2 have a conserved GSK3β-interacting domain, and it has been proposed that FRAT1 competes with axin for binding GSK3β as the binding sites on GSK3β for these two proteins overlap (Ferkey and Kimelman, 2002; Stoothoff et al., 2005). FRATtide a 39 amino acid peptide derived from FRAT1 is sufficient to bind to GSK3 and inhibit axin association with GSK3 (Thomas et al., 1999; Jope and Johnson 2004, Patel et al., 2004). Significantly, inhibition of GSK3 activity by FRATide is specific to axin and β catenin and does not inhibit its activity towards GS and eIF2B (Thomas et al., 1999; van Amerongen and Berns, 2005). In addition, FRATide inhibits GSK3β-mediated phosphorylation of unprimed sites on τ and not the primed phosphorylation sites (van Amerongen and Berns, 2005; Stoothoff et al., 2005). On the other hand, FRAT2 interacts with GSK3β directly, and it may enhance the GSK3β activity selectively directed towards primed substrates (Stoothoff et al., 2005).
It was earlier proposed that as priming phosphate is not required for phosphorylation of β catenin by GSK3β it is feasible to develop drugs that selectively inhibit the phosphorylation of insulin-signalling proteins without affecting the unprimed substrates like β catenin thereby reducing the potential of the inhibitors being oncogenic (Frame et al., 2001). However, although with the demonstration that β catenin is also a primed substrate, that approach might not be feasible, it is significant note that Gsk3α−/− mice do not show increased stabilization of proto oncogene β catenin, which has been believed to lead to enhanced oncogenesis. Also, the knockout mice show no increase in the propensity for tumorigenesis. In another significant development it has been demonstrated that loss of at least three if not all the four alleles of GSK3α and β is required for significant dysregulation of β catenin levels and its signalling. Deletion of either both the alleles of GSK3α alone or β alone does not disrupt β catenin signalling and there is no compensatory up-regulation of the other isoform that might explain for the lack of tumorigenesis in the GSK3α knockout mice. These findings suggest that despite the concern of GSK3 inhibitors leading to increased β catenin levels and enhanced oncogenesis, this might not hinder the development of GSK3 inhibitors for diabetes.
GSK 3 inhibitors for type II diabetes
One of the best studied inhibitors of GSK3 is lithium, which is ATP-non-competitive inhibitor and competes with magnesium and inhibits GSK3 with a Ki of 2 mmol·L−1 (Ryves and Harwood, 2001). Lithium has been used as a therapeutic agent for treatment of manic depression and bipolar disorders (O'Brien et al., 2004). Lithium inhibition stimulates glucose uptake, glycogen synthesis and normalizes insulin sensitivity in diabetic rats (Rossetti, 1989). However, importantly lithium is not a selective inhibitor of GSK3 as it can inhibit several other enzymes like CK2, p38-regulated/activated kinase and MAPK-activated protein kinase-2 (Phiel and Klein, 2001). Lithium also inhibits polyphosphate 1-phosphatase and inositol monophosphate (Phiel and Klein, 2001). Hence, the phenotypes attributed to inhibition of lithium would be difficult to assign to sole inhibition of GSK3. In this regard it would be important to use specific inhibitors of GSK3.
Inhibitors of GSK3 have enormous potential as therapeutics for diabetes, bipolar disorder and Alzheimer's and several other neurological disorders. The anti-diabetic effect of GSK3 is exerted by enhancing glucose disposal and improving insulin resistance. Apart from this, the recently demonstrated beneficial effects on β cells provide a distinct advantage over other anti-diabetic therapies. GSK3 inhibitors could also have a beneficial effect for type I diabetes and also islet transplantation patients. Several potent GSK3 inhibitors have been identified and characterized in preclinical models for treatment against T2DM.
The inhibitors identified so far belong to diverse chemotypes described following and significantly, most are directed to the ATP-binding site and function as ATP-competitive inhibitors. Of these, maleimides were some of the earliest GSK3 inhibitors with SB-216763 and SB-415286 inhibiting GSK3α and GSK3β with Ki of 9 and 31 nmol·L−1 (Coghlan et al., 2000; MacAulay et al., 2003).These compounds stimulate glycogen synthesis in human liver cells and also induce expression of β catenin – LEF/TCF reporter gene (Coghlan et al., 2000). Arylmaleimide (Smith et al., 2001), pyrazolo pyridines (Witherington et al., 2003; Engler et al., 2005), pyrimidin amines (Tavares et al., 2004), series of pyrazolo arylhydrazones (Peat et al., 2004), thiazolo [5,4-f] quinazolin-9-ones (Testard et al., 2006), 7-hydroxy-1H-benzoimidazole derivatives (Shin et al., 2007), bis(indolyl)maleimide pyridinophanes (Zhang et al., 2007) and pyrimidine derivatives (Miyazaki et al., 2008) have all been reported as potent and selective GSK3 inhibitors with some reported to have cellular efficacy. Of these 2-cyanoethylalsterpaullones are most potent with IC50s in picomolar range (Kunick et al., 2005). Significantly, as discussed above Cazpaullone a GSK3 inhibitor has been reported to stimulate replication and protection of pancreatic β cells (Stukenbrock et al., 2008) extending the role of GSK3 to pancreatic function in T2DM.
Importantly, as most of the inhibitors are targeted to the ATP-binding site, achieving selectivity over other related kinases is challenging and is one of the key issues that need to be addressed. As discussed below, some of the inhibitors have been shown to be selective over a small panel of about 20–50 kinases. Also, importantly it has been difficult to obtain selectivity over CDKs, which are key regulators of cell cycle. Most potent GSK3 inhibitors also inhibit CDKs as the homology between the two ATP-binding sites is 86%. It is desirable to avoid inhibition of CDKs for the treatment of diabetes as it is a chronic disease needing long-term treatment although CDK inhibitors are reported to function as anti-cancer agents (Fischer, 2003). However, for neurodegenerative diseases it might be desirable to have dual GSK3/CDK inhibitors as CDKs especially CDK5 has been shown to phosphorylate similar substrates as GSK3 (Fischer, 2003). In this context several inhibitors have been reported that exhibit moderate to good selectivity over CDKs. SB-216763 and SB-415286 are reported to be selective over 25 Ser/Thr kinases (Coghlan et al., 2000), macrocyclic bisindolylmaleimides that inhibit GSK3β and PKCβ (Zhang et al., 2003); a series of pyrazolo pyridines are selective over the closely related CDK2 (Witherington et al., 2003); azaindolylmaleimides are slightly selective over CDK2 with little or no inhibition over 62 protein kinases (Kuo et al., 2003; Shen et al., 2004). Moderate selectivity over PKC and CDK with good selectivity over 50 kinases has been reported for macrocyclic polyoxygenated bis-7-azaindolylmaleimides (Kuo et al., 2003), and AR-A014418 was selective over CDK2 and CDK5 and 26 other kinases (Bhat et al., 2003).
Preclinical data
In acute experiments several GSK3 inhibitors like CT118637, CT98014, CHIR98023 have been demonstrated to enhance insulin signalling in skeletal muscle from diabetic animals (Nikoulina et al., 2002; Henriksen et al., 2003; Dokken et al., 2005; Henriksen and Teachey, 2007). Acute inhibition with CT118637 in muscles of pre-diabetic obese Zucker rat enhances insulin signalling with increased basal GS, insulin stimulated glucose transport only in insulin-resistant skeletal muscle (Dokken et al., 2005). Up-regulation of IR and IRS1 tyrosine phosphorylation, reduction in the serine phosphorylation of IRS1 (Ser 307) along with increased GSK3β Ser 9 and AKt Ser 473 phosphorylation were reported when muscles from Zucker diabetic fatty rats were treated with CT98014 in vitro (Henriksen and Teachey, 2007). An increase in liver glycogen synthesis with little effect on muscle glycogen synthesis was reported with GSK3 inhibitors CHIR98023 and CHIR99021 in Zucker fa/fa rats (Cline et al., 2002).
Chronic treatment of GSK3 inhibitors like CT118637 (Dokken and Henriksen, 2006), aminopyrimidine derivatives CHIR 98014 and CHIR 99021 (Ring et al., 2003) in Zucker rats enhanced glucose tolerance, activated GS, with improved insulin sensitivity and increased IRS1-dependent insulin signalling (Dokken and Henriksen, 2006). Other inhibitors like bisarylmaleimide that are >160- to >10 000-fold selective over CDK2/4 and PKCβII demonstrated lowering of plasma glucose levels in Zucker diabetic fatty rats (Engler et al., 2004).
Among the non-ATP-competitive GSK inhibitors, thienyl and phenyl alpha-halomethyl ketones (Conde et al., 2003) and thiadiazolidinone derivatives have been reported (Castro et al., 2008). Substrate-competitive peptide inhibitors have also been reported for GSK3 in contrast to most other inhibitors that are ATP-competitive (Plotkin et al., 2003). They are selective for several related kinases like Cdc2 and increase GS activity and glucose uptake in cell-based systems and also improved glucose tolerance in insulin-resistant obese mice (Plotkin et al., 2003). Further, chronic treatment in ob/ob mice, reduced blood glucose levels, improved glucose tolerance, suppressed hepatic phosphoenolpuruvate carboxy kinase, increased hepatic glycogen content and lead to up-regulation of GLUT4 in skeletal muscle (Kaidanovich-Beilin and Eldar-Finkelman, 2006). Also, in high fat fed C57BL/6J mice, it has been shown to improve hepatic and peripheral insulin resistance by increasing liver GS activity and hepatic glycogen synthesis (Rao et al., 2007).
Many GSK3 inhibitors have been reported during the identification of inhibitors for CDKs with anti-tumour properties like paullones (Leost et al., 2000) and indirubins that inhibit CDK5 and GSK3β (Leclerc et al., 2001). Also, pyrazolo [3,4-b] quinoxalines (Ortega et al., 2002) and aloisines (Mettey et al., 2003) were found to inhibit both CDK5 and GSK3. Not, all CDK inhibitors inhibit GSK3 (Leclerc et al., 2001) and 1-azakenpaullone has been reported to be selective for GSK3β over CDK1 (Kunick et al., 2004). 9-oxo-thiazolo [5,4-f] quinazoline-2-carbonitrile derivatives have been reported as dual CDK1 and GSK3 inhibitors with potency in sub-micromolar range (Logéet al., 2007). Significantly, CDK/GSK3 inhibitors have been proposed as therapy for proliferative renal disease, and efficacy has been demonstrated in preclinical models of mesangial proliferative glomerulonephritis (Soos et al., 2006).
Glycogen synthase kinase 3 inhibitors have also been identified from natural sources, like hymenialdisine from marine sponge (Meijer et al., 2000). Bisindole indirubin from a traditional Chinese medicine and other indirubins have been reported to be potent inhibitors of CDKs and GSK3β (Leclerc et al., 2001). Subsequently, 6-bromo indirubin was identified as a potent and selective GSK3 inhibitor from ‘tyrian purple’ dye of mollusks (Meijer et al., 2003). By using a cell permeable derivative 6-bromoindirubin-3′-oxime (BIO) it was shown that it inhibits phosphorylation of GSK3α/β on Tyr276/216 and also reduces β catenin phosphorylation (Meijer et al., 2003). Moreover, BIO was demonstrated to have >16-fold selectivity over CDK2 and CDK5 (Meijer et al., 2003). Novel derivatives of indirubin like 5-substituted indirubins inhibit CDKs and GSK3 (Beauchard et al., 2006) where as 7-bromoindirubin-3′-oxime (7BIO) is less potent for CDK and GSK3 and has anti-tumour function (Ribas et al., 2006). Recently, manzamine A and related derivatives from an Indonesian sponge have been reported as a new class of GSK3 inhibitors (Hamann et al., 2007). Surprisingly, these inhibitors appear to inhibit specifically GSK3β and CDK5 and not CDK1, protein kinase A, MAPK, GSK3α (Hamann et al., 2007). Although several potent inhibitors belonging to different chemical classes have been reported to exhibit efficacy in various animal models of diabetes the critical translation of these results in human diabetic patients is awaited.
Conclusion
Glycogen synthase kinase 3 is undoubtedly, a promising target for diabetes given its role in improving insulin resistance along with the added benefit of protecting pancreatic β cells. However, it has proven to be challenging given the complexity of its regulation, implication in several diverse pathways and the proposed side effect of GSK3 inhibitors leading to cancer. The fact that most of the GSK3 inhibitors are targeted at the ATP-binding site, obtaining selectivity over other kinases is a key issue and is important to achieve good selectivity encompassing a broad range of kinases. As GSK3 has been implicated in tumorigenesis it is important that the safety of GSK3 inhibitors be demonstrated in long-term treatment of chronic diseases like T2DM. Intriguingly, GSK3 has been implicated in cancer therapy, and recently GSK3 inhibitors have been proposed as a novel class of therapeutic agents for colon cancer (Shakoori et al., 2007). Significantly, it has been suggested that inhibition of GSK3 alone in untransformed cells is not a primary event to increase levels of β catenin unless accompanied by other transformation event (Frame and Zheleva, 2006). Further, GSK3β-independent regulation of β catenin by other kinases like CK1, CK2, protein kinase A, IKKα and IKKβ has also been proposed, which might regulate the levels of β catenin. In this context recently, Cyclacel has reported GSK3 inhibitors that do not increase levels of β catenin although the mechanistic basis for this is not clear (Frame and Zheleva, 2006).
Long-term use of lithium the well studied GSK3 inhibitor for bipolar disorder has not been shown to be associated with an increased risk of cancer. Recent research has also demonstrated that inactivation of at least three alleles of GSK3β and GSK3α is required for the increase of β catenin levels. Hence, it is possible that moderate inhibition of GSK3 might result in beneficial effects without up-regulation of β catenin. Further, a modest inhibition of GSK3 has been shown to be sufficient for stimulation of insulin action and hence an optimal level of inhibition of GSK3 can be achieved without affecting the β catenin levels as heterozygous mice are not cancer-prone.
Research so far has demonstrated that GSK3α and β forms are similar yet functionally not redundant. Future research into the isoform specific functions and their implication for various disease in which GSK3 is implicated might facilitate development of isoform specific inhibitors. It would be interesting to identify the regions and interacting partners within GSK3α and β contributing to the differential physiological function. Differential involvement of either isoform in specific pathways would provide a selective tool for drug discovery and enable development of inhibitors, which may not target the active site and achieve isoform selectivity and also selectivity over several other related kinases and minimize the side effects associated with them. Although at present it appears that most substrates of GSK3 are primed, use of inhibitors that might specifically affect primed versus unprimed might also facilitate the development of target specific GSK3 inhibitors. Although GSK3 inhibitors identified so far have demonstrated efficacy in preclinical models, it is important that efficacy and safety of GSK3 inhibitors be demonstrated in humans, which will aid in the development of novel drugs for T2DM and especially with a potential for protective and beneficial effects on pancreatic β cells, which further extends the scope of GSK3 inhibitors to T1DM.
Glossary
Abbreviations:
- APC
adenomatous polyposis coli
- BIO
6-bromoindirubin-3′-oxime
- CDK
cyclin-dependent kinase
- CK
casein kinase
- eIF2B
eukaryotic initiation factor 2B
- GLUT4
glucose transporter 4
- GS
glycogen synthase
- GSK3
glycogen synthase kinase 3
- IKKβ
inhibitor κB kinase
- IPF1/PDX1
insulin promoter factor or pancreatic/duodenal homeobox 1 transcription factor 1
- IR
insulin receptor
- IRS
insulin receptor substrates
- JNK
c-Jun N terminal kinase
- LEF/TCF
lymphoid enhancer factor/T cell factor
- LRP
LDL receptor related protein
- MAPK
mitogen-activated protein kinase
- PKB
protein kinase B
- PKC
protein kinase C
- PP1
protein phosphatase 1
- T2DM
type II diabetes mellitus
Conflict of interest
None.
References
- Aguirre V, Werner ED, Giraud J, Lee YH, Shoelson SE, White MF. Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J Biol Chem. 2002;277:1531–1537. doi: 10.1074/jbc.M101521200. [DOI] [PubMed] [Google Scholar]
- van Amerongen R, Berns A. Re-evaluating the role of Frat in Wnt-signal transduction. Cell Cycle. 2005;4:1065–1072. [PubMed] [Google Scholar]
- van Amerongen R, van der Gulden H, Bleeker F, Jonkers J, Berns A. Characterization and functional analysis of the murine Frat2 gene. J Biol Chem. 2004;279:26967–26974. doi: 10.1074/jbc.M400439200. [DOI] [PubMed] [Google Scholar]
- Asuni AA, Hooper C, Reynolds CH, Lovestone S, Anderton BH, Killick R. GSK3alpha exhibits beta-catenin and tau directed kinase activities that are modulated by Wnt. Eur J Neurosci. 2006;24:3387–3392. doi: 10.1111/j.1460-9568.2006.05243.x. [DOI] [PubMed] [Google Scholar]
- Baggio LL, Drucker DJ. Therapeutic approaches to preserve islet mass in type 2 diabetes. Annu Rev Med. 2006;57:265–281. doi: 10.1146/annurev.med.57.110104.115624. [DOI] [PubMed] [Google Scholar]
- Beauchard A, Ferandin Y, Frère S, Lozach O, Blairvacq M, Meijer L, et al. Synthesis of novel 5-substituted indirubins as protein kinases inhibitors. Bioorg Med Chem. 2006;14:6434–6443. doi: 10.1016/j.bmc.2006.05.036. [DOI] [PubMed] [Google Scholar]
- Bhat R, Xue Y, Berg S, Hellberg S, Ormö M, Nilsson Y, et al. Structural insights and biological effects of glycogen synthase kinase 3-specific inhibitor AR-A014418. J Biol Chem. 2003;278:45937–45945. doi: 10.1074/jbc.M306268200. [DOI] [PubMed] [Google Scholar]
- Bouché C, Serdy S, Kahn CR, Goldfine AB. The cellular fate of glucose and its relevance in type 2 diabetes. Endocr Rev. 2004;25:807–830. doi: 10.1210/er.2003-0026. [DOI] [PubMed] [Google Scholar]
- Boucher MJ, Selander L, Carlsson L, Edlund H. Phosphorylation marks IPF1/PDX1 protein for degradation by glycogen synthase kinase 3-dependent mechanisms. J Biol Chem. 2006;281:6395–6403. doi: 10.1074/jbc.M511597200. [DOI] [PubMed] [Google Scholar]
- Bouskila M, Hirshman MF, Jensen J, Goodyear LJ, Sakamoto K. Insulin promotes glycogen synthesis in the absence of GSK3 phosphorylation in skeletal muscle. Am J Physiol Endocrinol Metab. 2008;294:E28–E35. doi: 10.1152/ajpendo.00481.2007. [DOI] [PubMed] [Google Scholar]
- Brady MJ, Bourbonais FJ, Saltiel AR. The activation of glycogen synthase by insulin switches from kinase inhibition to phosphatase activation during adipogenesis in 3T3-L1 cells. J Biol Chem. 1998;273:14063–14066. doi: 10.1074/jbc.273.23.14063. [DOI] [PubMed] [Google Scholar]
- Bustos VH, Ferrarese A, Venerando A, Marin O, Allende JE, Pinna LA. The first armadillo repeat is involved in the recognition and regulation of beta-catenin phosphorylation by protein kinase CK1. Proc Natl Acad Sci USA. 2006;103:19725–19730. doi: 10.1073/pnas.0609424104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadigan KM, Liu Y. Wnt signaling: complexity at the surface. J Cell Sci. 2006;119:395–402. doi: 10.1242/jcs.02826. [DOI] [PubMed] [Google Scholar]
- Castro A, Encinas A, Gil C, Bräse S, Porcal W, Pérez C, et al. Non-ATP competitive glycogen synthase kinase 3beta (GSK-3beta) inhibitors: study of structural requirements for thiadiazolidinone derivatives. Bioorg Med Chem. 2008;6:495–510. doi: 10.1016/j.bmc.2007.09.016. [DOI] [PubMed] [Google Scholar]
- Chen RH, Ding WV, McCormick F. Wnt signaling to beta-catenin involves two interactive components. Glycogen synthase kinase-3beta inhibition and activation of protein kinase C. J Biol Chem. 2000;275:7894–7899. doi: 10.1074/jbc.M905336199. [DOI] [PubMed] [Google Scholar]
- Cho JH, Johnson GV. Primed phosphorylation of tau at Thr231 by glycogen synthase kinase 3beta (GSK3beta) plays a critical role in regulating tau's ability to bind and stabilize microtubules. J Neurochem. 2004;88:349–358. doi: 10.1111/j.1471-4159.2004.02155.x. [DOI] [PubMed] [Google Scholar]
- Ciaraldi TP, Oh DK, Christiansen L, Nikoulina SE, Kong AP, Baxi S, et al. Tissue-specific expression and regulation of GSK-3 in human skeletal muscle and adipose tissue. Am J Physiol Endocrinol Metab. 2006;291:E891–E898. doi: 10.1152/ajpendo.00176.2006. [DOI] [PubMed] [Google Scholar]
- Ciaraldi TP, Nikoulina SE, Bandukwala RA, Carter L, Henry RR. Role of glycogen synthase kinase-3 alpha in insulin action in cultured human skeletal muscle cells. Endocrinology. 2007;148:4393–4399. doi: 10.1210/en.2006-0932. [DOI] [PubMed] [Google Scholar]
- Cline GW, Johnson K, Regittnig W, Perret P, Tozzo E, Xiao L, et al. Effects of a novel glycogen synthase kinase-3 inhibitor on insulin-stimulated glucose metabolism in Zucker diabetic fatty (fa/fa) rats. Diabetes. 2002;51:2903–2910. doi: 10.2337/diabetes.51.10.2903. [DOI] [PubMed] [Google Scholar]
- Coghlan MP, Culbert AA, Cross DA, Corcoran SL, Yates JW, Pearce NJ, et al. Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem Biol. 2000;7:793–803. doi: 10.1016/s1074-5521(00)00025-9. [DOI] [PubMed] [Google Scholar]
- Conde S, Pérez DI, Martínez A, Perez C, Moreno FJ. Thienyl and phenyl alpha-halomethyl ketones: new inhibitors of glycogen synthase kinase (GSK-3beta) from a library of compound searching. J Med Chem. 2003;46:4631–4633. doi: 10.1021/jm034108b. [DOI] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- Dajani R, Fraser E, Roe SM, Young N, Good V, Dale TC, et al. Crystal structure of glycogen synthase kinase 3 beta: structural basis for phosphate-primed substrate specificity and autoinhibition. Cell. 2001;105:721–732. doi: 10.1016/s0092-8674(01)00374-9. [DOI] [PubMed] [Google Scholar]
- Delibegovic M, Armstrong CG, Dobbie L, Watt PW, Smith AJ, Cohen PT. Disruption of the striated muscle glycogen targeting subunit PPP1R3A of protein phosphatase 1 leads to increased weight gain, fat deposition, and development of insulin resistance. Diabetes. 2003;52:596–604. doi: 10.2337/diabetes.52.3.596. [DOI] [PubMed] [Google Scholar]
- Ding VW, Chen RH, McCormick F. Differential regulation of glycogen synthase kinase 3beta by insulin and Wnt signaling. J Biol Chem. 2000;275:32475. doi: 10.1074/jbc.M005342200. [DOI] [PubMed] [Google Scholar]
- Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doble BW, Patel S, Wood GA, Kockeritz LK, Woodgett JR. Functional redundancy of GSK-3alpha and GSK-3beta in Wnt/beta-catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev Cell. 2007;12:957–971. doi: 10.1016/j.devcel.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokken BB, Henriksen EJ. Chronic selective glycogen synthase kinase-3 inhibition enhances glucose disposal and muscle insulin action in prediabetic obese Zucker rats. Am J Physiol Endocrinol Metab. 2006;291:E207–E213. doi: 10.1152/ajpendo.00628.2005. [DOI] [PubMed] [Google Scholar]
- Dokken BB, Sloniger JA, Henriksen EJ. Acute selective glycogen synthase kinase-3 inhibition enhances insulin signaling in prediabetic insulin-resistant rat skeletal muscle. Am J Physiol Endocrinol Metab. 2005;288:E1188–E1194. doi: 10.1152/ajpendo.00547.2004. [DOI] [PubMed] [Google Scholar]
- Donath MY, Ehses JA, Maedler K, Schumann DM, Ellingsgaard H, Eppler E, et al. Mechanisms of beta-cell death in type 2 diabetes. Diabetes. 2005;54:S108–S113. doi: 10.2337/diabetes.54.suppl_2.s108. [DOI] [PubMed] [Google Scholar]
- Eldar-Finkelman H. Glycogen synthase kinase 3: an emerging therapeutic target. Trends Mol Med. 2002;8:126–132. doi: 10.1016/s1471-4914(01)02266-3. [DOI] [PubMed] [Google Scholar]
- Eldar-Finkelman H, Ilouz R. Challenges and opportunities with glycogen synthase kinase-3 inhibitors for insulin resistance and Type 2 diabetes treatment. Expert Opin Investig Drugs. 2003;12:1511–1519. doi: 10.1517/13543784.12.9.1511. [DOI] [PubMed] [Google Scholar]
- Eldar-Finkelman H, Krebs EG. Phosphorylation of insulin receptor substrate 1 by glycogen synthase kinase 3 impairs insulin action. Proc Natl Acad Sci USA. 1997;94:9660–9664. doi: 10.1073/pnas.94.18.9660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldar-Finkelman H, Schreyer SA, Shinohara MM, LeBoeuf RC, Krebs EG. Increased glycogen synthase kinase-3 activity in diabetes- and obesity-prone C57BL/6J mice. Diabetes. 1999;48:1662–1666. doi: 10.2337/diabetes.48.8.1662. [DOI] [PubMed] [Google Scholar]
- Embi N, Rylatt DB, Cohen P. Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur J Biochem. 1980;107:519–527. [PubMed] [Google Scholar]
- Engler TA, Henry JR, Malhotra S, Cunningham B, Furness K, Brozinick J, et al. Substituted 3-imidazo[1,2-a]pyridin-3-yl- 4-(1,2,3,4-tetrahydro-[1,4]diazepino-[6,7,1-hi]indol-7-yl)pyrrole-2,5-diones as highly selective and potent inhibitors of glycogen synthase kinase-3. J Med Chem. 2004;47:3934–3937. doi: 10.1021/jm049768a. [DOI] [PubMed] [Google Scholar]
- Engler TA, Malhotra S, Burkholder TP, Henry JR, Mendel D, Porter WJ, et al. The development of potent and selective bisarylmaleimide GSK3 inhibitors. Bioorg Med Chem Lett. 2005;15:899–903. doi: 10.1016/j.bmcl.2004.12.063. [DOI] [PubMed] [Google Scholar]
- Ferkey DM, Kimelman D. Glycogen synthase kinase-3 beta mutagenesis identifies a common binding domain for GBP and Axin. J Biol Chem. 2002;277:16147–16152. doi: 10.1074/jbc.M112363200. [DOI] [PubMed] [Google Scholar]
- Fischer PM. CDK versus GSK-3 inhibition: a purple haze no longer? Chem Biol. 2003;10:1144–1146. doi: 10.1016/j.chembiol.2003.12.009. [DOI] [PubMed] [Google Scholar]
- Fornoni A, Pileggi A, Molano RD, Sanabria NY, Tejada T, Gonzalez-Quintana J, et al. Inhibition of c-jun N terminal kinase (JNK) improves functional beta cell mass in human islets and leads to AKT and glycogen synthase kinase-3 (GSK-3) phosphorylation. Diabetologia. 2008;51:298–308. doi: 10.1007/s00125-007-0889-4. [DOI] [PubMed] [Google Scholar]
- Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem J. 2001;359:1–16. doi: 10.1042/0264-6021:3590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame S, Cohen P, Biondi RM. A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol Cell. 2001;7:1321–1327. doi: 10.1016/s1097-2765(01)00253-2. [DOI] [PubMed] [Google Scholar]
- Frame S, Zheleva D. Targeting glycogen synthase kinase-3 in insulin signalling. Expert Opin Ther Targets. 2006;10:429–444. doi: 10.1517/14728222.10.3.429. [DOI] [PubMed] [Google Scholar]
- Garrido JJ, Simón D, Varea O, Wandosell F. GSK3 alpha and GSK3 beta are necessary for axon formation. FEBS Lett. 2007;581:1579–1586. doi: 10.1016/j.febslet.2007.03.018. [DOI] [PubMed] [Google Scholar]
- Goñi-Oliver P, Lucas JJ, Avila J, Hernández F. N-terminal cleavage of GSK-3 by calpain: a new form of GSK-3 regulation. J Biol Chem. 2007;282:22406–22413. doi: 10.1074/jbc.M702793200. [DOI] [PubMed] [Google Scholar]
- Greene MW, Garofalo RS. Positive and negative regulatory role of insulin receptor substrate 1 and 2 (IRS-1 and IRS-2) serine/threonine phosphorylation. Biochemistry. 2002;41:7082–7091. doi: 10.1021/bi015992f. [DOI] [PubMed] [Google Scholar]
- ter Haar E, Coll JT, Austen DA, Hsiao HM, Swenson L, Jain J. Structure of GSK3beta reveals a primed phosphorylation mechanism. Nat Struct Biol. 2001;8:593–596. doi: 10.1038/89624. [DOI] [PubMed] [Google Scholar]
- Hagen T, Vidal-Puig A. Characterisation of the phosphorylation of beta-catenin at the GSK-3 priming site Ser45. Biochem Biophys Res Commun. 2002;294:324–328. doi: 10.1016/S0006-291X(02)00485-0. [DOI] [PubMed] [Google Scholar]
- Hagen T, Cross DA, Culbert AA, West A, Frame S, Morrice N, et al. FRAT1, a substrate-specific regulator of glycogen synthase kinase-3 activity, is a cellular substrate of protein kinase A. J Biol Chem. 2006;281:35021–35029. doi: 10.1074/jbc.M607003200. [DOI] [PubMed] [Google Scholar]
- Halse R, Bonavaud SM, Armstrong JL, McCormack JG, Yeaman SJ. Control of glycogen synthesis by glucose, glycogen, and insulin in cultured human muscle cells. Diabetes. 2001;50:720–726. doi: 10.2337/diabetes.50.4.720. [DOI] [PubMed] [Google Scholar]
- Hamann M, Alonso D, Martín-Aparicio E, Fuertes A, Pérez-Puerto MJ, Castro A, et al. Glycogen synthase kinase-3 (GSK-3) inhibitory activity and structure-activity relationship (SAR) studies of the manzamine alkaloids. Potential for Alzheimer's disease. J Nat Prod. 2007;70:1397–1405. doi: 10.1021/np060092r. [DOI] [PubMed] [Google Scholar]
- Han SI, Aramata S, Yasuda K, Kataoka K. MafA stability in pancreatic beta cells is regulated by glucose and is dependent on its constitutive phosphorylation at multiple sites by glycogen synthase kinase 3. Mol Cell Biol. 2007;27:6593–6605. doi: 10.1128/MCB.01573-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanashiro I, Roach PJ. Mutations of muscle glycogen synthase that disable activation by glucose 6-phosphate. Arch Biochem Biophys. 2002;397:286–292. doi: 10.1006/abbi.2001.2623. [DOI] [PubMed] [Google Scholar]
- Harwood AJ. Regulation of GSK-3: a cellular multiprocessor. Cell. 2001;105:821–824. doi: 10.1016/s0092-8674(01)00412-3. [DOI] [PubMed] [Google Scholar]
- Henriksen EJ, Dokken BB. Role of glycogen synthase kinase-3 in insulin resistance and type 2 diabetes. Curr Drug Targets. 2006;7:1435–1441. doi: 10.2174/1389450110607011435. [DOI] [PubMed] [Google Scholar]
- Henriksen EJ, Teachey MK. Short-term in vitro inhibition of glycogen synthase kinase 3 potentiates insulin signaling in type I skeletal muscle of Zucker Diabetic Fatty rats. Metabolism. 2007;56:931–938. doi: 10.1016/j.metabol.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksen EJ, Kinnick TR, Teachey MK, O'Keefe MP, Ring D, Johnson KW, et al. Modulation of muscle insulin resistance by selective inhibition of GSK-3 in Zucker diabetic fatty rats. Am J Physiol Endocrinol Metab. 2003;284:E892–E900. doi: 10.1152/ajpendo.00346.2002. [DOI] [PubMed] [Google Scholar]
- Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature. 2000;406:86–90. doi: 10.1038/35017574. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science. 1996;271:665–668. doi: 10.1126/science.271.5249.665. [DOI] [PubMed] [Google Scholar]
- Hughes K, Nikolakaki E, Plyte SE, Totty NF, Woodgett JR. Modulation of the glycogen synthase kinase-3 family by tyrosine phosphorylation. EMBO J. 1993;12:803–808. doi: 10.1002/j.1460-2075.1993.tb05715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilouz R, Kowalsman N, Eisenstein M, Eldar-Finkelman H. Identification of novel glycogen synthase kinase-3beta substrate-interacting residues suggests a common mechanism for substrate recognition. J Biol Chem. 2006;281:30621–30630. doi: 10.1074/jbc.M604633200. [DOI] [PubMed] [Google Scholar]
- Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Jope RS, Roh MS. Glycogen synthase kinase-3 (GSK3) in psychiatric diseases and therapeutic interventions. Curr Drug Targets. 2006;7:1421–1434. doi: 10.2174/1389450110607011421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaidanovich-Beilin O, Eldar-Finkelman H. Long-term treatment with novel glycogen synthase kinase-3 inhibitor improves glucose homeostasis in ob/ob mice: molecular characterization in liver and muscle. J Pharmacol Exp Ther. 2006;316:17–24. doi: 10.1124/jpet.105.090266. [DOI] [PubMed] [Google Scholar]
- Kim WY, Zhou FQ, Zhou J, Yokota Y, Wang YM, Yoshimura T, et al. Essential roles for GSK-3s and GSK-3-primed substrates in neurotrophin-induced and hippocampal axon growth. Neuron. 2006;52:981–996. doi: 10.1016/j.neuron.2006.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunick C, Lauenroth K, Leost M, Meijer L, Lemcke T. 1-Azakenpaullone is a selective inhibitor of glycogen synthase kinase-3 beta. Bioorg Med Chem Lett. 2004;14:413–416. doi: 10.1016/j.bmcl.2003.10.062. [DOI] [PubMed] [Google Scholar]
- Kunick C, Zeng Z, Gussio R, Zaharevitz D, Leost M, Totzke F, et al. Structure-aided optimization of kinase inhibitors derived from alsterpaullone. Chembiochem. 2005;6:541–549. doi: 10.1002/cbic.200400099. [DOI] [PubMed] [Google Scholar]
- Kuo GH, Prouty C, DeAngelis A, Shen L, O'Neill DJ, Shah C, et al. Synthesis and discovery of macrocyclic polyoxygenated bis-7-azaindolylmaleimides as a novel series of potent and highly selective glycogen synthase kinase-3beta inhibitors. J Med Chem. 2003;46:4021–4031. doi: 10.1021/jm030115o. [DOI] [PubMed] [Google Scholar]
- Leclerc S, Garnier M, Hoessel R, Marko D, Bibb JA, Snyder GL, et al. Indirubins inhibit glycogen synthase kinase-3 beta and CDK5/p25, two protein kinases involved in abnormal tau phosphorylation in Alzheimer's disease. A property common to most cyclin-dependent kinase inhibitors? J Biol Chem. 2001;276:251–260. doi: 10.1074/jbc.M002466200. [DOI] [PubMed] [Google Scholar]
- Lee HC, Tsai JN, Liao PY, Tsai WY, Lin KY, Chuang CC, et al. Glycogen synthase kinase 3 alpha and 3 beta have distinct functions during cardiogenesis of zebrafish embryo. BMC Dev Biol. 2007;7:93. doi: 10.1186/1471-213X-7-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Kim MS. The role of GSK3 in glucose homeostasis and the development of insulin resistance. Diabetes Res Clin Pract. 2007;77:49–57. doi: 10.1016/j.diabres.2007.01.033. [DOI] [PubMed] [Google Scholar]
- Leost M, Schultz C, Link A, Wu YZ, Biernat J, Mandelkow EM, et al. Paullones are potent inhibitors of glycogen synthase kinase-3beta and cyclin-dependent kinase 5/p25. Eur J Biochem. 2000;267:5983–5994. doi: 10.1046/j.1432-1327.2000.01673.x. [DOI] [PubMed] [Google Scholar]
- Liberman Z, Eldar-Finkelman H. Serine 332 phosphorylation of insulin receptor substrate-1 by glycogen synthase kinase-3 attenuates insulin signaling. J Biol Chem. 2005;280:4422–4428. doi: 10.1074/jbc.M410610200. [DOI] [PubMed] [Google Scholar]
- Lipina C, Huang X, Finlay D, McManus EJ, Alessi DR, Sutherland C. Analysis of hepatic gene transcription in mice expressing insulin-insensitive GSK3. Biochem J. 2005;15:633–639. doi: 10.1042/BJ20051046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Tanabe K, Bernal-Mizrachi E, Permutt MA. Mice with beta cell overexpression of glycogen synthase kinase-3beta have reduced beta cell mass and proliferation. Diabetologia. 2008;51:623–631. doi: 10.1007/s00125-007-0914-7. [DOI] [PubMed] [Google Scholar]
- Lochhead PA, Coghlan M, Rice SQ, Sutherland C. Inhibition of GSK-3 selectively reduces glucose-6-phosphatase and phosphatase and phosphoenolypyruvate carboxykinase gene expression. Diabetes. 2001;50:937–946. doi: 10.2337/diabetes.50.5.937. [DOI] [PubMed] [Google Scholar]
- Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- Logé C, Testard A, Thiéry V, Lozach O, Blairvacq M, Robert JM, et al. Novel 9-oxo-thiazolo[5,4-f]quinazoline-2-carbonitrile derivatives as dual cyclin-dependent kinase 1 (CDK1)/glycogen synthase kinase-3 (GSK-3) inhibitors: Synthesis, biological evaluation and molecular modeling studies. Eur J Med Chem. 2007;43:1469–1477. doi: 10.1016/j.ejmech.2007.09.020. [DOI] [PubMed] [Google Scholar]
- Luo J, Chen J, Deng ZL, Luo X, Song WX, Sharff KA, et al. Wnt signaling and human diseases: what are the therapeutic implications? Lab Invest. 2007;87:97–103. doi: 10.1038/labinvest.3700509. [DOI] [PubMed] [Google Scholar]
- Ma C, Wang J, Gao Y, Gao TW, Chen G, Bower KA, et al. The role of glycogen synthase kinase 3beta in the transformation of epidermal cells. Cancer Res. 2007;67:7756–7764. doi: 10.1158/0008-5472.CAN-06-4665. [DOI] [PubMed] [Google Scholar]
- MacAulay K, Hajduch E, Blair AS, Coghlan MP, Smith SA, Hundal HS. Use of lithium and SB-415286 to explore the role of glycogen synthase kinase-3 in the regulation of glucose transport and glycogen synthase. Eur J Biochem. 2003;270:3829–3838. doi: 10.1046/j.1432-1033.2003.03777.x. [DOI] [PubMed] [Google Scholar]
- MacAulay K, Blair AS, Hajduch E, Terashima T, Baba O, Sutherland C, et al. Constitutive activation of GSK3 down-regulates glycogen synthase abundance and glycogen deposition in rat skeletal muscle cells. J Biol Chem. 2005;280:9509–9518. doi: 10.1074/jbc.M411648200. [DOI] [PubMed] [Google Scholar]
- MacAulay K, Doble BW, Patel S, Hansotia T, Sinclair EM, Drucker DJ, et al. Glycogen synthase kinase 3alpha-specific regulation of murine hepatic glycogen metabolism. Cell Metab. 2007;6:329–337. doi: 10.1016/j.cmet.2007.08.013. [DOI] [PubMed] [Google Scholar]
- McManus EJ, Sakamoto K, Armit LJ, Ronaldson L, Shpiro N, Marquez R, et al. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 2005;24:1571–1583. doi: 10.1038/sj.emboj.7600633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markou T, Cullingford TE, Giraldo A, Weiss SC, Alsafi A, Fuller SJ, et al. Glycogen synthase kinases 3alpha and 3beta in cardiac myocytes: regulation and consequences of their inhibition. Cell Signal. 2008;20:206–218. doi: 10.1016/j.cellsig.2007.10.004. [DOI] [PubMed] [Google Scholar]
- Mazanetz MP, Fischer PM. Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases. Nat Rev Drug Discov. 2007;6:464–479. doi: 10.1038/nrd2111. [DOI] [PubMed] [Google Scholar]
- Meijer L, Thunnissen AM, White AW, Garnier M, Nikolic M, Tsai LH, et al. Inhibition of cyclin-dependent kinases, GSK-3beta and CK1 by hymenialdisine, a marine sponge constituent. Chem Biol. 2000;7:51–63. doi: 10.1016/s1074-5521(00)00063-6. [DOI] [PubMed] [Google Scholar]
- Meijer L, Skaltsounis AL, Magiatis P, Polychronopoulos P, Knockaert M, Leost M, et al. GSK-3-selective inhibitors derived from Tyrian purple indirubins. Chem Biol. 2003;10:1255–1266. doi: 10.1016/j.chembiol.2003.11.010. [DOI] [PubMed] [Google Scholar]
- Meijer L, Flajolet M, Greengard P. Pharmacological inhibitors of glycogen synthase kinase 3. Trends Pharmacol Sci. 2004;25:471–480. doi: 10.1016/j.tips.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Mettey Y, Gompel M, Thomas V, Garnier M, Leost M, Ceballos-Picot I, et al. Aloisines, a new family of CDK/GSK-3 inhibitors. SAR study, crystal structure in complex with CDK2, enzyme selectivity, and cellular effects. J Med Chem. 2003;46:222–236. doi: 10.1021/jm020319p. [DOI] [PubMed] [Google Scholar]
- Miyazaki Y, Maeda Y, Sato H, Nakano M, Mellor GW. Rational design of 4-amino-5,6-diaryl-furo[2,3-d]pyrimidines as potent glycogen synthase kinase-3 inhibitors. Bioorg Med Chem Lett. 2008;18:1967–1971. doi: 10.1016/j.bmcl.2008.01.113. [DOI] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Elluru R, Ratner N, Brady ST. Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J. 2002;21:281–293. doi: 10.1093/emboj/21.3.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai F, Ishiguro K, Sano Y, Fujita SC. Alternative splicing isoform of tau protein kinase l/glycogen synthase kinase 3beta. J Neurochem. 2002;81:1073–1083. doi: 10.1046/j.1471-4159.2002.00918.x. [DOI] [PubMed] [Google Scholar]
- Mussmann R, Geese M, Harder F, Kegel S, Andag U, Lomow A, et al. Inhibition of GSK3 promotes replication and survival of pancreatic beta cells. J Biol Chem. 2007;282:12030–12037. doi: 10.1074/jbc.M609637200. [DOI] [PubMed] [Google Scholar]
- Nielsen JN, Wojtaszewski JF. Regulation of glycogen synthase activity and phosphorylation by exercise. Proc Nutr Soc. 2004;63:233–237. doi: 10.1079/PNS2004348. [DOI] [PubMed] [Google Scholar]
- Nikoulina SE, Ciaraldi TP, Mudaliar S, Mohideen P, Carter L, Henry RR. Potential role of glycogen synthase kinase-3 in skeletal muscle insulin resistance of type 2 diabetes. Diabetes. 2000;49:263–271. doi: 10.2337/diabetes.49.2.263. [DOI] [PubMed] [Google Scholar]
- Nikoulina SE, Ciaraldi TP, Mudaliar S, Carter L, Johnson K, Henry RR. Inhibition of glycogen synthase kinase 3 improves insulin action and glucose metabolism in human skeletal muscle. Diabetes. 2002;51:2190–2198. doi: 10.2337/diabetes.51.7.2190. [DOI] [PubMed] [Google Scholar]
- O'Brien WT, Harper AD, Jové F, Woodgett JR, Maretto S, Piccolo S, et al. Glycogen synthase kinase-3beta haploinsufficiency mimics the behavioral and molecular effects of lithium. J Neurosci. 2004;24:6791–6798. doi: 10.1523/JNEUROSCI.4753-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega MA, Montoya ME, Zarranz B, Jaso A, Aldana I, Leclerc S, et al. Pyrazolo[3,4-b]quinoxalines. A new class of cyclin-dependent kinases inhibitors. Bioorg Med Chem. 2002;10:2177–2184. doi: 10.1016/s0968-0896(02)00069-x. [DOI] [PubMed] [Google Scholar]
- Patel S, Doble B, Woodgett JR. Glycogen synthase kinase-3 in insulin and Wnt signalling: a double-edged sword? Biochem Soc Trans. 2004;32:803–808. doi: 10.1042/BST0320803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce NJ, Arch JR, Clapham JC, Coghlan MP, Corcoran SL, Lister CA, et al. Development of glucose intolerance in male transgenic mice overexpressing human glycogen synthase kinase-3beta on a muscle-specific promoter. Metabolism. 2004;53:1322–1330. doi: 10.1016/j.metabol.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Peat AJ, Garrido D, Boucheron JA, Schweiker SL, Dickerson SH, Wilson JR, et al. Novel GSK-3 inhibitors with improved cellular activity. Bioorg Med Chem Lett. 2004;14:2127–2130. doi: 10.1016/j.bmcl.2004.02.037. [DOI] [PubMed] [Google Scholar]
- Phiel CJ, Klein PS. Molecular targets of lithium action. Annu Rev Pharmacol Toxicol. 2001;41:789–813. doi: 10.1146/annurev.pharmtox.41.1.789. [DOI] [PubMed] [Google Scholar]
- Phiel CJ, Wilson CA, Lee VM, Klein PS. GSK-3alpha regulates production of Alzheimer's disease amyloid-beta peptides. Nature. 2003;423:435–439. doi: 10.1038/nature01640. [DOI] [PubMed] [Google Scholar]
- Plotkin B, Kaidanovich O, Talior I, Eldar-Finkelman H. Insulin mimetic action of synthetic phosphorylated peptide inhibitors of glycogen synthase kinase-3. J Pharmacol Exp Ther. 2003;305:974–980. doi: 10.1124/jpet.102.047381. [DOI] [PubMed] [Google Scholar]
- Rao R, Hao CM, Redha R, Wasserman DH, McGuinness OP, Breyer MD. Glycogen synthase kinase 3 inhibition improves insulin-stimulated glucose metabolism but not hypertension in high-fat-fed C57BL/6J mice. Diabetologia. 2007;50:452–460. doi: 10.1007/s00125-006-0552-5. [DOI] [PubMed] [Google Scholar]
- Ribas J, Bettayeb K, Ferandin Y, Knockaert M, Garrofé-Ochoa X, Totzke F, et al. 7-Bromoindirubin-3′-oxime induces caspase-independent cell death. Oncogene. 2006;25:6304–6318. doi: 10.1038/sj.onc.1209648. [DOI] [PubMed] [Google Scholar]
- Ring DB, Johnson KW, Henriksen EJ, Nuss JM, Goff D, Kinnick TR. Selective glycogen synthase kinase 3 inhibitors potentiate insulin activation of glucose transport and utilization in vitro and in vivo. Diabetes. 2003;52:588–595. doi: 10.2337/diabetes.52.3.588. [DOI] [PubMed] [Google Scholar]
- Rossetti L. Normalization of insulin sensitivity with lithium in diabetic rats. Diabetes. 1989;38:648–652. doi: 10.2337/diab.38.5.648. [DOI] [PubMed] [Google Scholar]
- Rowe MK, Wiest C, Chuang DM. GSK-3 is a viable potential target for therapeutic intervention in bipolar disorder. Neurosci Biobehav Rev. 2007;31:920–931. doi: 10.1016/j.neubiorev.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P. Binding of GSK3beta to the APC-beta-catenin complex and regulation of complex assembly. Science. 1996;272:1023–1026. doi: 10.1126/science.272.5264.1023. [DOI] [PubMed] [Google Scholar]
- Ruel L, Bourouis M, Heitzler P, Pantesco V, Simpson P. Drosophila shaggy kinase and rat glycogen synthase kinase-3 have conserved activities and act downstream of Notch. Nature. 1993;362:557–560. doi: 10.1038/362557a0. [DOI] [PubMed] [Google Scholar]
- Ryves WJ, Harwood AJ. Lithium inhibits glycogen synthase kinase-3 by competition for magnesium. Biochem Biophys Res Commun. 2001;280:720–725. doi: 10.1006/bbrc.2000.4169. [DOI] [PubMed] [Google Scholar]
- Seidensticker MJ, Behrens J. Biochemical interactions in the wnt pathway. Biochim Biophys Acta. 2000;495:168–182. doi: 10.1016/s0167-4889(99)00158-5. [DOI] [PubMed] [Google Scholar]
- Semiz S, McNeill JH. Oral treatment with vanadium of Zucker fatty rats activates muscle glycogen synthesis and insulin-stimulated protein phosphatase-1 activity. Mol Cell Biochem. 2002;236:123–131. doi: 10.1023/a:1016116700632. [DOI] [PubMed] [Google Scholar]
- Shakoori A, Mai W, Miyashita K, Yasumoto K, Takahashi Y, Ooi A, et al. Inhibition of GSK-3? activity attenuates proliferation of human colon cancer cells in rodents. Cancer Sci. 2007;98:1388–1393. doi: 10.1111/j.1349-7006.2007.00545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Prouty C, Conway BR, Westover L, Xu JZ, Look RA, et al. Synthesis and biological evaluation of novel macrocyclic bis-7-azaindolylmaleimides as potent and highly selective glycogen synthase kinase-3 beta (GSK-3 beta) inhibitors. Bioorg Med Chem. 2004;12:1239–1255. doi: 10.1016/j.bmc.2003.09.047. [DOI] [PubMed] [Google Scholar]
- Shin D, Lee S, Heo Y, Lee W, Cho Y, Yong E, et al. Design and synthesis of 7-hydroxy-1H-benzoimidazole derivatives as novel inhibitors of glycogen synthase kinase-3. Bioorganic & Medicinal Chemistry Letters. 2007;17:5686–5689. doi: 10.1016/j.bmcl.2007.07.056. [DOI] [PubMed] [Google Scholar]
- Smith DG, Buffet M, Fenwick AE, Haigh D, Ife RJ, Saunders M, et al. 3-Anilino-4-arylmaleimides: potent and selective inhibitors of glycogen synthase kinase-3 (GSK-3) Bioorg Med Chem Lett. 2001;11:635–639. doi: 10.1016/s0960-894x(00)00721-6. [DOI] [PubMed] [Google Scholar]
- Soos TJ, Meijer L, Nelson PJ. CDK/GSK-3 inhibitors as a new approach for the treatment of proliferative renal diseases. Drug News Perspect. 2006;19:325–328. doi: 10.1358/dnp.2006.19.6.985939. [DOI] [PubMed] [Google Scholar]
- Srinivasan S, Ohsugi M, Liu Z, Fatrai S, Bernal-Mizrachi E, Permutt MA. Endoplasmic reticulum stress-induced apoptosis is partly mediated by reduced insulin signaling through phosphatidylinositol 3-kinase/Akt and increased glycogen synthase kinase-3beta in mouse insulinoma cells. Diabetes. 2005;54:968–975. doi: 10.2337/diabetes.54.4.968. [DOI] [PubMed] [Google Scholar]
- Stambolic V, Woodgett JR. Mitogen inactivation of glycogen synthase kinase-3 beta in intact cells via serine 9 phosphorylation. Biochem J. 1994;303:701–704. doi: 10.1042/bj3030701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoothoff WH, Cho JH, McDonald RP, Johnson GV. FRAT-2 preferentially increases glycogen synthase kinase 3 beta-mediated phosphorylation of primed sites, which results in enhanced tau phosphorylation. J Biol Chem. 2005;280:270–276. doi: 10.1074/jbc.M410061200. [DOI] [PubMed] [Google Scholar]
- Stukenbrock H, Mussmann R, Geese M, Ferandin Y, Lozach O, Lemcke T. 9-Cyano-1-azapaullone (Cazpaullone), a glycogen synthase kinase-3 (GSK-3) inhibitor activating pancreatic beta cell protection and replication. J Med Chem. 2008;51:196–207. doi: 10.1021/jm701582f. [DOI] [PubMed] [Google Scholar]
- Summers SA, Kao AW, Kohn AD, Backus GS, Roth RA, Pessin JE, et al. The role of glycogen synthase kinase 3beta in insulin-stimulated glucose metabolism. J Biol Chem. 1999;274:17934–17940. doi: 10.1074/jbc.274.25.17934. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Lanner C, Kim JH, Vilardo PG, Zhang H, Yang J, et al. Insulin control of glycogen metabolism in knockout mice lacking the muscle-specific protein phosphatase PP1G/RGL. Mol Cell Biol. 2001;21:2683–2694. doi: 10.1128/MCB.21.8.2683-2694.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe K, Liu Z, Patel S, Doble BW, Li L, Cras-Méneur C, et al. Genetic deficiency of glycogen synthase kinase-3beta corrects diabetes in mouse models of insulin resistance. PLoS Biol. 2008;6:e37. doi: 10.1371/journal.pbio.0060037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavares FX, Boucheron JA, Dickerson SH, Griffin RJ, Preugschat F, Thomson SA, et al. N-Phenyl-4-pyrazolo[1,5-b]pyridazin-3-ylpyrimidin-2-amines as potent and selective inhibitors of glycogen synthase kinase 3 with good cellular efficacy. J Med Chem. 2004;47:4716–4730. doi: 10.1021/jm040063i. [DOI] [PubMed] [Google Scholar]
- Testard A, Logé C, Léger B, Robert JM, Lozach O, Blairvacq M, et al. Thiazolo[5,4-f]quinazolin-9-ones, inhibitors of glycogen synthase kinase-3. Bioorg Med Chem Lett. 2006;16:3419–3423. doi: 10.1016/j.bmcl.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Thomas GM, Frame S, Goedert M, Nathke I, Polakis P, Cohen P. A GSK3-binding peptide from FRAT1 selectively inhibits the GSK3-catalysed phosphorylation of axin and beta-catenin. FEBS Lett. 1999;458:247–251. doi: 10.1016/s0014-5793(99)01161-8. [DOI] [PubMed] [Google Scholar]
- Thornton TM, Pedraza-Alva G, Deng B, Wood CD, Aronshtam A, Clements JL, et al. Phosphorylation by p38 MAPK as an alternative pathway for GSK3beta inactivation. Science. 2008;320:667–670. doi: 10.1126/science.1156037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tighe A, Ray-Sinha A, Staples OD, Taylor SS. GSK-3 inhibitors induce chromosome instability. BMC Cell Biol. 2007;14:8–34. doi: 10.1186/1471-2121-8-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twomey C, McCarthy JV. Presenilin-1 is an unprimed glycogen synthase kinase-3beta substrate. FEBS Lett. 2006;580:4015–4020. doi: 10.1016/j.febslet.2006.06.035. [DOI] [PubMed] [Google Scholar]
- Ueki K, Yamamoto-Honda R, Kaburagi Y, Yamauchi T, Tobe K, Burgering BM, et al. Potential role of protein kinase B in insulin-induced glucose transport, glycogen synthesis, and protein synthesis. J Biol Chem. 1998;237:5315–5322. doi: 10.1074/jbc.273.9.5315. [DOI] [PubMed] [Google Scholar]
- Wang QM, Fiol CJ, DePaoli-Roach AA, Roach PJ. Glycogen synthase kinase-3 beta is a dual specificity kinase differentially regulated by tyrosine and serine/threonine phosphorylation. J Biol Chem. 1994;269:14566–14574. [PubMed] [Google Scholar]
- Witherington J, Bordas V, Gaiba A, Garton NS, Naylor A, Rawlings AD, et al. 6-aryl-pyrazolo[3,4-b]pyridires: potent inhibitors of glycogen synthase kinase-3 (GSK-3) Bioorg Med Chem Lett. 2003;13:3055–3057. doi: 10.1016/s0960-894x(03)00645-0. [DOI] [PubMed] [Google Scholar]
- Woodgett JR. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 1990;9:2431–2438. doi: 10.1002/j.1460-2075.1990.tb07419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodgett JR. cDNA cloning and properties of glycogen synthase kinase-3. Methods Enzymol. 1991;200:564–577. doi: 10.1016/0076-6879(91)00172-s. [DOI] [PubMed] [Google Scholar]
- Zeng X, Tamai K, Doble B, Li S, Huang H, Habas R, et al. A dual-kinase mechanism for Wnt co-receptor phosphorylation and activation. Nature. 2005;438:873–877. doi: 10.1038/nature04185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng X, Huang H, Tamai K, Zhang X, Harada Y, Yokota C, et al. Initiation of Wnt signaling: control of Wnt coreceptor Lrp6 phosphorylation/activation via frizzled, dishevelled and axin functions. Development. 2008;135:367–375. doi: 10.1242/dev.013540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HC, White KB, Ye H, McComsey DF, Derian CK, Addo MF, et al. Macrocyclic bisindolylmaleimides as inhibitors of protein kinase C and glycogen synthase kinase-3. Bioorg Med Chem Lett. 2003;13:3049–3053. doi: 10.1016/s0960-894x(03)00644-9. [DOI] [PubMed] [Google Scholar]
- Zhang HC, Boñaga LV, Ye H, Derian CK, Damiano BP, Maryanoff BE. Novel bis(indolyl)maleimide pyridinophanes that are potent, selective inhibitors of glycogen synthase kinase-3. Bioorg Med Chem Lett. 2007;17:2863–2868. doi: 10.1016/j.bmcl.2007.02.059. [DOI] [PubMed] [Google Scholar]