

Abstract
Background and purpose:
Carisbamate is being developed for adjuvant treatment of partial onset epilepsy. Carisbamate produces anticonvulsant effects in primary generalized, complex partial and absence-type seizure models, and exhibits neuroprotective and antiepileptogenic properties in rodent epilepsy models. Phase IIb clinical trials of carisbamate demonstrated efficacy against partial onset seizures; however, its mechanisms of action remain unknown. Here, we report the effects of carisbamate on membrane properties, evoked and spontaneous synaptic transmission and induced epileptiform discharges in layer II-III neurones in piriform cortical brain slices.
Experimental approach:
Effects of carisbamate were investigated in rat piriform cortical neurones by using intracellular electrophysiological recordings.
Key results:
Carisbamate (50–400 µmol·L−1) reversibly decreased amplitude, duration and rise-time of evoked action potentials and inhibited repetitive firing, consistent with use-dependent Na+ channel block; 150–400 µmol·L−1 carisbamate reduced neuronal input resistance, without altering membrane potential. After microelectrode intracellular Cl− loading, carisbamate depolarized cells, an effect reversed by picrotoxin. Carisbamate (100–400 µmol·L−1) also selectively depressed lateral olfactory tract-afferent evoked excitatory synaptic transmission (opposed by picrotoxin), consistent with activation of a presynaptic Cl− conductance. Lidocaine (40–320 µmol·L−1) mimicked carisbamate, implying similar modes of action. Carisbamate (300–600 µmol·L−1) had no effect on spontaneous GABAA miniature inhibitory postsynaptic currents and at lower concentrations (50–200 µmol·L−1) inhibited Mg2+-free or 4-aminopyridine-induced seizure-like discharges.
Conclusions and implications:
Carisbamate blocked evoked action potentials use-dependently, consistent with a primary action on Na+ channels and increased Cl− conductances presynaptically and, under certain conditions, postsynaptically to selectively depress excitatory neurotransmission in piriform cortical layer Ia-afferent terminals.
Keywords: RWJ-333369, carisbamate, lidocaine, piriform cortical brain slices, intracellular recording
Introduction
Carisbamate (RWJ-333369; (S)-2-O-carbamoyl-1-O-chlorophenyl-ethanol) is a novel neuromodulator and antiepileptic drug (AED) currently under clinical development for adjunctive treatment of partial epilepsy (Rogawski, 2006). This AED is a monocarbamate with structural similarities to the AED felbamate, and both are comparably effective in standard rodent seizure models (Sofia et al., 1993; Bialer et al., 2006; White et al., 2006; Novak et al., 2007; Grabenstatter and Dudek, 2008). Carisbamate exhibits potent anticonvulsant effects in primary generalized, complex partial and absence-type models of epilepsy (Nehlig et al., 2005; White et al., 2006; Francois et al., 2008). At higher doses, carisbamate dramatically inhibits the development of spontaneous recurrent seizures and exhibits neuroprotective effects in the hippocampus, thalamus, amygdala and piriform cortical areas following lithium-pilocarpine-induced status epilepticus (Francois et al., 2005), suggesting additional antiepileptogenic or disease-modifying properties. A Phase IIb clinical trial with carisbamate demonstrated both efficacy and tolerability in patients with refractory partial onset seizures (Bialer et al., 2006), and evaluation of carisbamate in photosensitive epileptic patients has also shown significant dose-related suppression of paroxysmal electroencephalogram (EEG) responses to intermittent photic stimulation (Trenitéet al., 2007). Carisbamate therefore possesses powerful anticonvulsant and potential antiepileptogenic activities although the cellular mechanisms that underlie these effects currently remain unknown.
In the present work, we used rat piriform (olfactory) cortical slices (piriform cortex, PC; Whalley et al., 2005) to study the direct effects of carisbamate on intracellularly recorded neuronal membrane properties, evoked spike firing, excitatory synaptic transmission, induced epileptiform discharge activity and spontaneous miniature inhibitory postsynaptic currents (mIPSCs). The PC was chosen because it is susceptible to limbic epileptogenesis, leading to complex partial seizures in human epilepsy (Löscher and Ebert, 1996) and has been used in studies of epileptogenic processes and anticonvulsant actions (Libri et al., 1996; Löscher, 2002; Russo and Constanti, 2004). Here, we report three principal effects of carisbamate on PC neurones: use-dependent depression of evoked action potentials (consistent with a block of voltage-gated Na+ channels), layer-specific inhibition of evoked excitatory synaptic transmission and, at higher concentrations, a decrease in membrane input resistance and cell excitability, most likely mediated via activation of a presynaptic or postsynaptic Cl− conductance respectively but not via modulation of GABA release or reuptake. These data suggest a mechanism whereby block of action potentials and excitatory transmission contributes towards the observed anticonvulsant activity of this novel drug.
Methods
Preparation and maintenance of brain slices
Experiments were carried out in accordance with the UK Animals (Scientific Procedures) Act 1986, and every effort was made to minimize the number of animals used and their suffering. Rostro-caudal PC slices were prepared from male and female adult (P40-P80) Wistar rats, as previously described (Whalley et al., 2005). Animals were lightly anaesthetized with isoflurane (BDH, Loughborough, UK) and rapidly decapitated. The brain was removed and ∼450 µm thick slices cut along the axis of the lateral olfactory tract (LOT) in gassed (95% O2 : 5% CO2) ice-cold (4°C) Krebs solution [containing (mmol·L−1): NaCl 118; KCl 3; NaHCO3 25; MgCl2.6H2O 1; CaCl2 2.5 and D-glucose 11; pH 7.4] by using a Campden Vibroslice 752 mol·L−1 slicer. After stabilization in Krebs solution (32°C; minimum 30 min), slices were transferred to the recording chamber and superfused at ∼4 mL·min−1 with gassed Krebs solution (30–32°C). Mg2+-free Krebs solution was prepared by eliminating MgCl2.6H2O without replacement.
Electrophysiological techniques
Microelectrode recording
Intracellular recordings were made with glass microelectrodes filled with 4 mol·L−1 potassium acetate or 2 mol·L−1 potassium chloride (for intracellular Cl− loading); pH adjusted to 7.2 with glacial acetic acid; tip resistance 40–80 MΩ, connected to an Axoclamp 2A preamplifier (switching frequency 2–3 KHz, 30% duty cycle). To reveal Ca2+-mediated action potentials (Galvan et al., 1985a), some cells were recorded with 3 mol·L−1 caesium acetate-filled electrodes (tip resistance 30–50 MΩ) in the presence of 1 µmol·L−1 tetrodotoxin (TTX) to block fast Na+ spikes. Firing was assessed by positive current pulse injection (∼0.25–3 nA; 160 ms) and the resulting evoked action potentials recorded. Mean spike firing frequency was derived by dividing number of spike events by the duration of the depolarizing current pulse (+1.5 nA) applied through the recording microelectrode at resting membrane potential. First or second spike amplitudes were measured (from spike threshold to peak) under ‘bridge’ recording mode to avoid sampling limitations of the discontinuous sample-and-hold preamplifier. Spike durations were measured from threshold to return to baseline, while spike rate of rise was measured between 20% and 80% of spike amplitude, following a +1.5 nA, 160 ms injected current pulse. Membrane input resistance was calculated from ≤20 mV electrotonic potentials evoked at resting potential following injection of −0.5 nA, 160 ms current pulses through the recording microelectrode. Injected current and sampled voltage data were monitored on a storage oscilloscope and Gould RS3200 chart recorder (LDS Test and Measurement Ltd, Royston, UK) and captured via a Digidata 1200 interface (Axon CNS Molecular Devices, Sunnyvale, CA, USA) using pCLAMP8.1 software (Axon CNS Molecular Devices) that was also used for off-line analysis. Recordings were typically stable for 1–5 h. Data were collected from cells in layers II-III (LII-III) of the central PC region, identified as deep pyramidal neurones from their regular spike firing pattern in response to a long (∼200 ms) intracellular current pulse and absence of spike fractionation. PC interneurones show the same electrophysiological properties as deep cells in control conditions (Libri et al., 1994), therefore all recorded data were pooled for the purpose of the present analysis. Cells showing intrinsic membrane and evoked firing properties (spike fractionation) characteristic of superficial pyramidal neurones (Libri et al., 1994) were not recorded. The slow post-stimulus afterhyperpolarization (sAHP) was evoked by injection of a long (1.5 s) depolarizing current pulse, while maintaining the membrane potential at −70 mV (near firing threshold) by injecting steady positive holding current through the recording microelectrode.
Patch-clamp recording
PC brain slices (300 µm thickness), prepared as described above, were placed in a recording chamber at room temperature and superfused at 2–4 mL·min−1 with gassed Krebs solution (composition as described above). Whole-cell recordings were made from visually identified deep pyramidal neurones in LII-III of the PC by using an EPC-9 patch-clamp amplifier (HEKA Electronik, Lambrecht, Germany), controlled by Pulse software (HEKA Electronik, Lambrecht, Germany) with a Macintosh G4 computer. Patch electrodes were made from borosilicate glass (GC150-F10, Harvard Apparatus, Kent, UK) and, when filled with an intracellular solution containing (in mmol·L−1) CsCl 140, MgCl2 1, CaCl2 1, EGTA 10, MgATP 4, NaGTP 0.4 and HEPES 10, pH 7.3, had resistances of 3–7 MΩ. Series resistance was typically 3–7 MΩ and was monitored and compensated by 70–90% throughout. Data were sampled at 7 kHz and filtered at 2.33 kHz. Spontaneous mIPSCs were recorded in the presence of 1 µmol·L−1 TTX (Alomone, Jerusalem, Israel) and the non-NMDA (N-methyl-D-aspartate) glutamate receptor antagonist, 6-nitro-7-sulphamoylbenzo(f)quinoxaline-2-3-dione (NBQX, 5 µmol·L−1; Tocris, Bristol, UK). For measurement of mIPSC half-width, events were low-pass filtered at 200 Hz. Data were analysed by using Pulsefit (HEKA Electronik, Lambrecht, Germany), Axograph (Molecular Devices, Sunnyvale, CA, USA), Mini Analysis Program (Synaptosoft, Chapel Hill, NC, USA), Igor (Wavemetrics, Lake Oswego, OR, USA), Origin (Microcal, Northampton, MA, USA) and Excel (Microsoft, Redmond, WA, USA) software. Cumulative frequency plots were constructed for mIPSC inter-event intervals (bin width: 5 ms) or mIPSC amplitudes (bin width: 2 pA) and were analysed by Kolmogorov-Smirnov tests. In all cases, P < 0.05 was considered significant.
Recording of evoked synaptic potentials
Synaptic responses were evoked by using two bipolar stimulating electrodes, each composed of twisted 35 µm (15 µm inner core diameter, 10 µm insulator thickness, insulated except at tip) diameter nichrome wire, attached to a Digitimer DS2 (Digitimer Ltd., Welwyn Garden City, UK) isolated stimulus generator (stimuli: 5–20 V amplitude, 0.2 ms duration). One electrode was placed near the pial surface in PC layer Ia (LIa) to evoke excitatory afferent LOT depolarizing postsynaptic potentials (DPSPs) (Tseng and Haberly, 1988; Libri et al., 1994) and the other within the less opaque and deeper LII-III, to independently stimulate intrinsic excitatory LII-III fibres (Hasselmo and Bower, 1992). When eliciting DPSPs, cells were held at −90 mV membrane potential (i.e. ∼5 mV more negative than normal resting potential) by negative current injection, while varying stimulus strengths were applied and a range of DPSPs, from subthreshold to suprathreshold (eliciting an action potential) were produced. Stimulus strengths were adjusted to elicit reproducible DPSPs large enough to remain measurable after drug-induced suppression, yet small enough to elicit a subthreshold response of <40% peak suprathreshold amplitude. Stimulus strengths that produced small, subthreshold, measurable synaptic responses of identical amplitude for both LIa-afferent and intrinsic fibre stimulation from the recorded cell were used. Each stimulus was repeated five times, with a minimum interval of 30 s between stimuli. DPSP amplitudes were measured from resting potential to peak response. Repeats were performed in control, in the presence of drug and after washout (minimum 60 min) where possible. In recordings where DPSPs were evoked in the presence of picrotoxin, the membrane potential was maintained at −90 mV throughout by negative current injection to avoid spike firing. All synaptic potentials illustrated are composite averages of at least three consecutive traces unless otherwise indicated.
Paired-pulse facilitation
Assessment of a possible presynaptic site of action of carisbamate on excitatory neurotransmission was made by using a paired-pulse protocol (Whalley and Constanti, 2006), in which increases in early paired-pulse facilitation (PPF; a paired-pulse ratio, PPR > 1) of evoked DPSPs at facilitating synapses have been interpreted as indicative of presynaptic inhibition of transmission (Hasselmo and Bower, 1992). A conditioning stimulus (P1) was initially applied to the cell via a stimulating electrode followed by an identical stimulus at an interstimulus interval (ISI) of 25–200 ms (25 ms increments; P2) and the effect of time period variation upon second evoked potential amplitude observed from plots of PPR (P2/P1) versus ISI. Only short ISIs, yielding a facilitation of the second DPSP (PPF) were used and any significant differences between data sets calculated at the point of maximal facilitation (ISI = 25 ms). To avoid summation effects, any remaining P1 post-stimulus depolarization was subtracted from overall P2 DPSP amplitude.
Assessment of carisbamate on seizure-like activity induced in PC brain slices
The activity of carisbamate against spontaneous seizure-like discharges induced by 30 min exposure to Mg2+-free or 200 µmol·L−1 4-aminopyridine (4-AP)-containing Krebs medium (Libri et al., 1994) was also assessed. Effects on the initiation and manifestation of seizure-like activity were measured from changes to mean amplitude, duration and frequency of induced ictal discharges (paroxysmal depolarizing shifts: PDSs) over an epoch time of 15 min from digitized signals and chart records. These values were then used to calculate global averages ± SEM over the total number of cells recorded. PDS duration was measured from the time at which the voltage signal initially deflected from (and remained above) the baseline, until the point of return to the control membrane potential level. A PDS detection threshold was arbitrarily set at twice the peak level of the baseline noise. PDS amplitude was measured from baseline to peak, mid-PDS depolarized potential, after subtraction of superimposed spike amplitude. Inter-PDS interval was measured as the time between the onsets of individual PDSs.
Data analysis and statistical treatment of results
All data are expressed as means ± SEM. Statistical significance between normally distributed means was determined by using a two-tailed Student's paired t-test. Distribution-free, normalized data were tested for significance by using a non-parametric Mann–Whitney U-test. Normalized data are expressed as a percentage change of control measures. Reductions of measures are shown as negative percentages, increases as positive percentages. Use-dependence parameters [time constant for Na+ spike block onset (τ) and estimated maximal normalized steady-state block level (y0)] were calculated from mathematical curve-fitting to a single exponential decay function by using the Levenberg-Marquardt algorithm applied in Origin 6.0 (Microcal, Northampton, MA, USA). All curve-fitting routines used a nonlinear least-squares method. Statistical differences between parameters were determined by using a paired Student's t-test. Differences between data sets are reported at P < 0.1 but significance accepted at P < 0.05.
Preparation and application of drugs
Carisbamate (Johnson & Johnson Pharmaceutical Research & Development, L.L.C., Titusville, NJ, USA) was dissolved in dimethylsulphoxide (DMSO), with a final bath concentration of DMSO < 0.01%v/v. In control experiments, DMSO had no deleterious effects on cell properties at this concentration (n= 3). The stock solution of carisbamate was stored at 4°C, and appropriate stock volumes were added to the bathing solution to provide desired bath concentrations. Lidocaine hydrochloride (Astra Pharmaceuticals, London, UK), 4-AP, bicuculline methiodide, TTX and picrotoxin (all from Sigma-Aldrich Co. Ltd., Poole, UK) and NBQX (Tocris, Bristol, UK) were prepared as stock solutions in distilled water and stored at 4°C, then subsequently diluted in Krebs solution immediately before use. All measurements were taken before, during and (if possible) after bath drug applications (bath-exchange time ∼30 s), so each neurone served as its own control. Carisbamate was applied for a minimum of 20 min to achieve a steady state concentration unless stated otherwise. The results presented following are based on recordings obtained from a total of ∼150 cortical neurones; each PC slice was used for a single recording.
Results
Effects of carisbamate on membrane properties and evoked spike parameters
Bath application of carisbamate (50–400 µmol·L−1) produced clear effects on electrophysiological properties of pyramidal neurones. Cell firing properties were tested by injecting a brief (160 ms, +1.5 nA) depolarizing current pulse (Figure 1A). The number of action potentials evoked during the pulse was 6.8 ± 1.1, which decreased to 2.8 ± 0.2 in 400 µmol·L−1 carisbamate (∼−60%; P < 0.05, n= 10), indicating decreased cell excitability. Carisbamate induced a significant reduction in mean frequency of evoked action potentials at concentrations ≥100 µmol·L−1 to about 86% inhibition at 400 µmol·L−1 (P < 0.001, n= 11; Figure 1C i). Cell input resistance at resting membrane potential was minimally affected at ≤250 µmol·L−1 carisbamate, although at higher concentrations it was slowly, but significantly decreased, by about 35% at 400 µmol·L−1 (P < 0.01, n= 9; Figure 1B,C ii). There was no accompanying change in cell resting potential (−83.6 ± 0.9 mV in control vs. −83.2 ± 0.8 mV in carisbamate 50–400 µmol·L−1 after 20 min; n= 26, P > 0.5; Figure 1B). Carisbamate also caused a concentration-related progressive reduction in the amplitude, duration and rate of rise of evoked spikes (particularly the second and subsequent spikes; Figure 2) suggesting a possible use-dependent Na+ channel block. Significant effects on second spike amplitude were seen at carisbamate concentrations ≥300 µmol·L−1 (Figure 2B), whereas duration and rate of rise were affected at lower concentrations (≥50 µmol·L−1; Figure 2C,D). The most dramatic changes in first and second spike parameters occurred at carisbamate concentrations ≥300 µmol·L−1. All carisbamate effects were slowly but fully reversed after ∼50 min drug washout (not shown).
Figure 1.
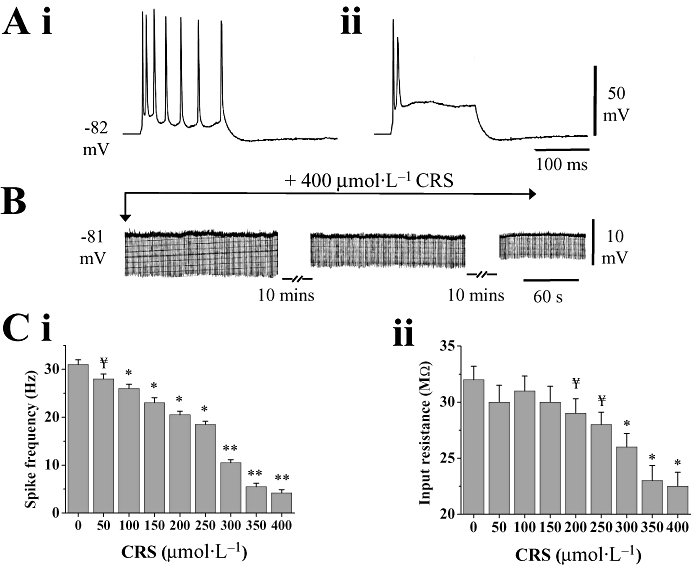
Effects of carisbamate (CRS) on rat piriform cortical (PC) neurones. (A) Representative evoked spike firing following application of a 160 ms, +1.5 nA current injection in (i) control and in (ii) 400 µmol·L−1 CRS (resting potential =−82 mV). (B) Slow decrease in membrane input resistance induced by CRS (400 µmol·L−1, 20 min) at −81 mV resting potential. Injected negative current pulses (−0.1 nA, 160 ms, 0.5 Hz) evoked electrotonic potentials throughout (downward deflections); CRS caused a time-dependent decrease in input resistance, but had minimal effect upon resting potential. (C) Histograms showing the effects of CRS (50–400 µmol·L−1) on (i) evoked mean spike firing frequency and (ii) neuronal input resistance. While CRS significantly decreased firing frequency at concentrations ≥100 µmol·L−1, it only significantly reduced input resistance at concentrations >250 µmol·L−1. Data shown are means ± SEM (n > 9) for all histograms. ¥ indicates a trend assessed by P < 0.1; * and ** indicate differences from control at P < 0.05 and P < 0.01 respectively. Significance accepted at P < 0.05.
Figure 2.
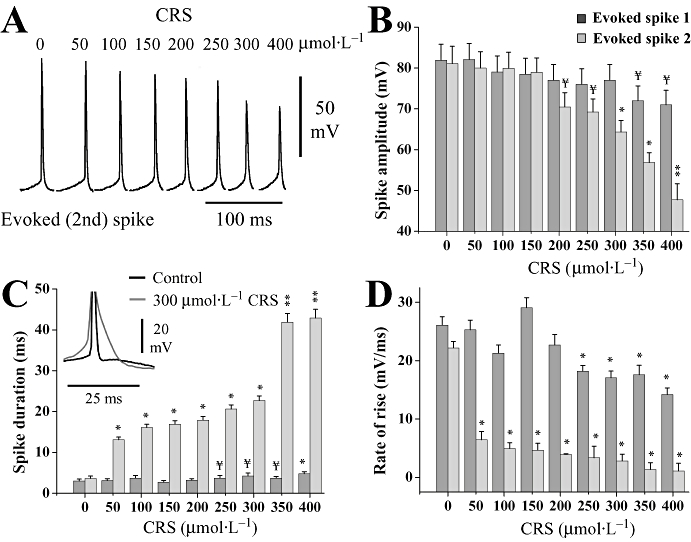
Effect of carisbamate (CRS) upon evoked spike parameters. (A) A depolarizing current pulse (+1.5 nA, 160 ms) was injected to evoke a train of action potentials from a neurone at −80 mV resting potential. Traces show the isolated second spike in each evoked spike train recorded in control and in CRS (50–400 µmol·L−1; 15 min). Note the progressive reduction in spike amplitude and increase in spike duration with increasing drug concentration, with the most pronounced effect at 300 and 400 µmol·L−1. (B) Histogram comparing the depressant effect of CRS (50–400 µmol·L−1) on evoked first and second spike amplitudes, measured from threshold to peak following injection of a +1.5 nA current pulse at resting membrane potential. (C,D) Histograms illustrating the effect of CRS on evoked first and second spike duration and rate of rise. Inset to (C) shows an overlay (centred around spike peaks) of second spikes (truncated) in control and 300 µmol·L−1 CRS. CRS had the most dramatic effect on spike properties at concentrations ≥300 µmol·L−1. Data values are means ± SEM (n= 6) for all histograms; ¥ indicates a trend assessed by P < 0.1; * and ** indicate differences from control at P < 0.05 and P < 0.01 respectively. Significance accepted at P < 0.05.
Comparison of the membrane effects of lidocaine with those of carisbamate
To further test the hypothesis that carisbamate blocks fast voltage-dependent Na+ channels, its basic membrane effects were compared with those of the prototypic use-dependent local anaesthetic lidocaine (Courtney, 1975; Hille, 1977). Like carisbamate, lidocaine (at ≥80 µmol·L−1) progressively reduced mean spike firing frequency (P < 0.05, n= 6; Figure 3A). A small, insignificant decrease in first spike amplitude, but more dramatic, concentration-dependent reduction in second spike amplitude at lidocaine concentrations ≥80 µmol·L−1 (Figure 3C; P < 0.05, n= 6) was also seen, in keeping with a use-dependent Na+ channel-blocking mechanism. As with carisbamate, lidocaine decreased both first and second spike rates of rise in a concentration-dependent manner (Figure 3D). Finally, lidocaine produced a more marked increase in second spike duration, at concentrations ≥40 µmol·L−1 (Figure 3E). Lidocaine effects were fully reversed after ∼45 min drug washout (not shown). Carisbamate and lidocaine therefore exerted similar effects on PC neurones, most likely via a use-dependent blocking mechanism. In a manner comparable to carisbamate, lidocaine (80–320 µmol·L−1; 15 min application) also reversibly decreased membrane input resistance (P < 0.05; n= 6; Figure 3B), without changing the membrane potential. Interestingly, lidocaine caused a similar decrease in input resistance at the highest concentration tested (∼−30%, 320 µmol·L−1) as that produced by carisbamate (∼−32%, 400 µmol·L−1; Figures 1C ii and 3b), suggesting the possible modulation of a conductance at resting membrane potential.
Figure 3.
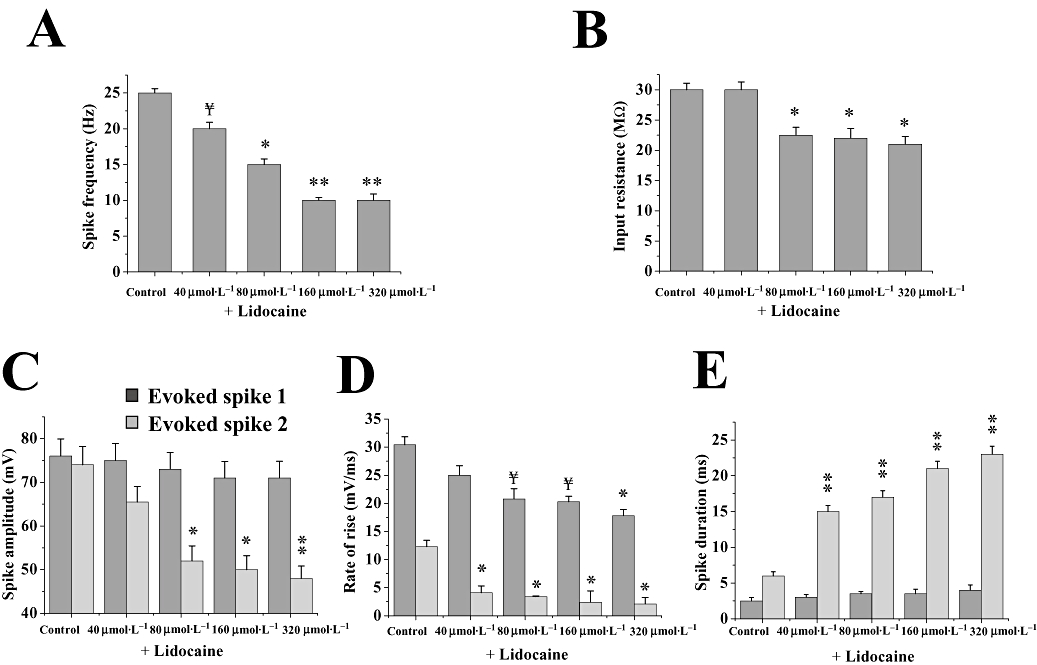
Effect of lidocaine upon evoked spike parameters. Histograms showing the effects of lidocaine (40–320 µmol·L−1; 15 min) on neuronal membrane properties and evoked spike parameters: (A) mean spike firing frequency (n= 5), (B) input resistance (n= 7), (C) first and second spike amplitude (n= 5), (D) first and second spike rate of rise (n= 5) and (E) first and second spike duration (n= 5). Data in A, C–E were all calculated following a +1.5 nA, 160 ms current pulse; evoked first spike and evoked second spike. All responses were elicited from resting potential (∼−83 mV). Note that the effects of carisbamate (see Figs 1 and 2) were comparable to those of lidocaine, on spike frequency, second spike amplitude, rate of rise and duration, suggesting a common mechanism of action; also, input resistance was significantly reduced by lidocaine at concentrations ≥80 µmol·L−1. ¥ indicates a trend assessed by P < 0.1; * and ** indicate differences from control at P < 0.05 and P < 0.01 respectively. Significance accepted at P < 0.05. Data shown are means ± SEM for all histograms.
Comparison of use-dependent blocking effects of carisbamate and lidocaine on evoked action potentials
A range of carisbamate concentrations (100–400 µmol·L−1; 15 min) was tested upon single action potential amplitudes evoked following injection of a train of brief, depolarizing current pulses (+4 nA; 1 ms) at 25, 50 and 100 Hz frequencies (15 pulses; 25, 100 Hz; Figure 4A). The progressive blocking effect of carisbamate (50 Hz stimulation; 400 µmol·L−1) is illustrated in Figure 4A ii (inset). A similar protocol was used to assess the use-dependent action potential blocking effect of lidocaine (40–160 µmol·L−1, 15 min) at 25, 50 and 100 Hz (Figure 4B); a sample trace of lidocaine's effect (50 Hz stimulation; 160 µmol·L−1) is shown in Figure 4B ii (inset). Carisbamate demonstrated a cumulative use-dependent blocking effect on evoked action potential amplitude at all stimulus frequencies investigated. Plots of peak action potential amplitude (normalized to control at t= 0 s) versus time (Figure 4A i, ii) were well fitted by first order exponential decays and showed that the decline in spike amplitude was increased at higher stimulation frequencies. Mean time constants (τ) for carisbamate block, calculated from the fitted curves of the maximum concentration used (400 µmol·L−1) across the frequencies tested, were: 25 Hz: 399 ± 59 ms; 50 Hz: 145 ± 19 ms and 100 Hz: 55 ± 6 ms, and the estimated final amplitude values attained (normalized; y0) were: 25 Hz: 0.42 ± 0.04; 50 Hz: 0.23 ± 0.04 and 100 Hz: 0.21 ± 0.03 (minimum n= 6 in each case) (cf. Wang et al., 2003). The blocking effect of carisbamate was thus clearly dependent upon both applied concentration and stimulation frequency.
Figure 4.
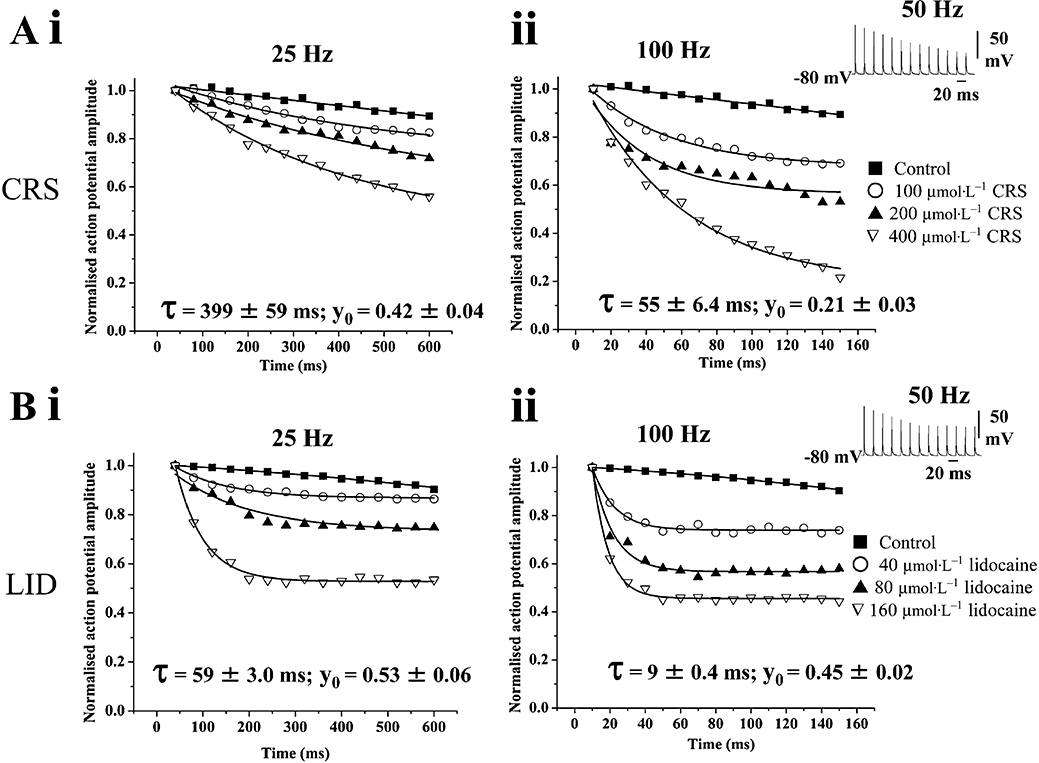
Use-dependent block of Na+ channels by carisbamate (CRS) and lidocaine (LID). Plots comparing the proposed use-dependent Na+ channel-blocking effects of CRS (100–400 µmol·L−1), with those of LID (40–160 µmol·L−1) on peak action potential amplitudes evoked by applying a train of brief depolarizing current injections (1 ms, +2.5 nA) at different stimulus frequencies. Results are plotted as mean action potential amplitude (normalized to control at t= 0) ± SEM versus time; n= 4 for all drug concentrations and frequencies (error bars were within width of points). (A) Concentration- and frequency-dependent effects of CRS at (i) 25 Hz and (ii) 100 Hz stimulus frequencies; inset shows a representative trace of the use-dependent effect of 400 µmol·L−1 CRS at 50 Hz stimulation; (B) shows concentration- and frequency-dependent effects of LID under comparable conditions to (A) at (i) 25 Hz and (ii) 100 Hz stimulation; inset shows use-dependent effect of LID (160 µmol·L−1) at 50 Hz stimulation. Note that for comparable final levels of spike suppression, the onset of block by CRS was notably slower than that produced by LID. Data for 400 µmol·L−1 CRS and 160 µmol·L−1 LID block at 25 and 100 Hz stimulus frequencies were fitted by single exponential functions using a least-squares method. The estimated time constants for block onset (τ) and plateau normalized peak values at the end of the stimulus train (y0) at the maximum concentrations used (derived from the best fits) are shown.
As anticipated, lidocaine also demonstrated similar use-dependent blocking effects (Figure 4B i, ii), although the mean time constants for block calculated from best fit curves at 160 µmol·L−1 concentration (25 Hz: 59 ± 3 ms; 50 Hz: 32 ± 3.4 ms and 100 Hz: 9 ± 0.4 ms) were ∼3–6-fold faster than those determined for carisbamate (P < 0.05 at each frequency, minimum n= 6 in each case). This indicated a relatively slower onset of action for carisbamate (i.e. a greater number of action potentials required to fire before achieving steady-state block y0) than lidocaine. However, a comparison of estimated y0 values at 160 µmol·L−1 lidocaine (25 Hz: 0.53 ± 0.06; 50 Hz: 0.44 ± 0.01 and 100 Hz: 0.45 ± 0.02) with carisbamate equivalents indicated a significantly greater maximal action potential-suppressing effect of carisbamate (P < 0.05 at each frequency) across the concentration and frequency ranges tested.
The effect of carisbamate on slow Ca2+-mediated action potentials and sAHP
We next tested whether carisbamate affected Ca2+-mediated conductances in PC neurones, and thereby influenced the generation of the slow post-stimulus Ca2+-dependent afterhyperpolarization (sAHP). By recording with Cs+-filled microelectrodes (to minimize outward K+ conductances) and in the presence of TTX (1 µmol·L−1; to block Na+ channels), Ca2+-mediated action potentials were revealed by applying brief depolarizing (30 ms; +1.5 nA) current pulses (Galvan et al., 1985a). These were typically followed by a slow depolarizing after potential (Figure 5A; **), probably due to a Ca2+-activated non-selective cation conductance (Kang et al., 1998). Carisbamate caused a small, but significant decrease in Ca2+ spike duration (−18 ± 6%; P < 0.05; 400 µmol·L−1, 15 min; n= 8), although the reduction in amplitude was not statistically significant (−9 ± 4%; P < 0.1, measured from baseline potential to spike peak).
Figure 5.
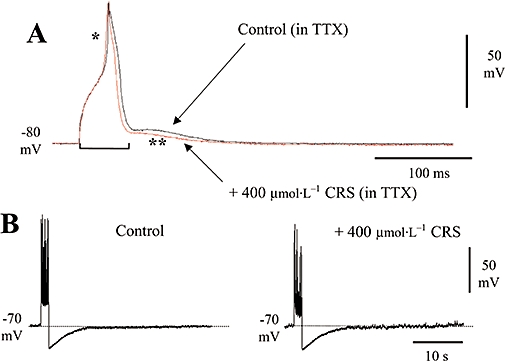
Assessment of the effects of carisbamate (CRS) upon Ca2+ conductances. (A) Superimposed slow action potentials evoked by a +1.5 nA, 30 ms depolarizing current pulse recorded by using a Cs acetate-filled microelectrode in the presence of 1 µmol·L−1 tetrodotoxin (TTX); a prolonged Ca2+ spike can be seen (*), followed by an afterdepolarization (**). In the presence of 400 µmol·L−1 CRS, Ca2+ spike duration but not amplitude were slightly but significantly reduced; (bar indicates where spike duration measurements were made; spike amplitude here was measured from baseline to peak). Holding membrane potential was −80 mV. (B) CRS (400 µmol·L−1) had no effect upon the slow, Ca2+-dependent afterhyperpolarization elicited by a +2 nA, 1.6 s current pulse (experiment conducted in the absence of TTX), although it did produce some reduction in spike amplitude and firing frequency during the stimulus train. Holding membrane potential was −70 mV.
The sAHP that follows a burst of action potentials is attributed to a Ca2+-dependent K+ conductance (Constanti and Sim, 1987). In neurones held close to firing threshold (∼−70 mV) by positive current injection through the recording microelectrode, sAHPs were evoked by using depolarizing current injection (+2 nA; 1.6 s) to produce a burst of action potentials (Figure 5B). As carisbamate may exert a Na+ channel-blocking effect and so reduce repetitive action potential firing during depolarizing stimuli (upon which the sAHP is partially dependent), quantitative measurements of sAHP amplitude were performed in 1 µmol·L−1 TTX to eliminate action potential effects. Application of carisbamate (400 µmol·L−1) in the presence of TTX had no effect upon sAHP amplitude (17.8 ± 1.2 mV in control vs. 16.6 ± 0.9 mV in carisbamate; P > 0.5; n= 7). In similar experiments performed in the absence of TTX, repetitive action potential firing and spike amplitude during depolarizing stimuli were reduced by carisbamate, likely due to Na+ channel block (see preceding) (control spikes = 24 ± 4, 400 µmol·L−1 carisbamate = 11 ± 3; P < 0.05, n= 7; ∼−54%), along with a reduction in peak amplitude of the last spike in the train (measured from −70 mV holding potential; 82 ± 6 mV control vs. 51.4 ± 5 mV in carisbamate; P < 0.05, n= 7; ∼−37%). The spike activation threshold (between ∼−60 and −65 mV) and sAHP amplitude, however, were not notably altered (Figure 5B). Taken together, these data suggest that carisbamate has only a weak interaction with voltage-dependent and/or voltage-independent Ca2+ conductances.
Effects of carisbamate on evoked excitatory synaptic transmission
Individual subthreshold and suprathreshold DPSPs were recorded from LII-III neurones following LOT (LIa)-afferent or intrinsic excitatory LII-III fibre stimulation in control and in carisbamate (25–400 µmol·L−1, 20 min). Subthreshold LIa-evoked DPSPs were significantly suppressed in a reversible, concentration-dependent manner (P < 0.05, n= 6 in each case; Figure 6). In marked contrast, no inhibition of intrinsic LII-III excitatory synaptic transmission, even at high carisbamate concentrations [e.g. +1 ± 6% (350 µmol·L−1 carisbamate); P > 0.5, n= 6; not shown] was seen. These data suggest a selectivity of carisbamate towards some central excitatory synapses over others. The log concentration–response relationship showing the percentage depression of LIa-afferent DPSP amplitude by carisbamate (averaged from n= 6 cells) is shown in Figure 6B; the estimated EC50 from the best-line fit to the curve was 118 µmol·L−1. Full reversal of the effects of carisbamate on transmission was observed after ∼70 min washout in control Krebs solution (cf. ∼50 min recovery time of input resistance and action potential parameters). Notably, carisbamate-induced inhibition of evoked synaptic transmission occurred at a lower threshold concentration (100 µmol·L−1), than postsynaptic membrane effects on input resistance (300 µmol·L−1) and was therefore unlikely to be directly related to changes in intrinsic membrane properties (Figure 1). However, the observed depression in spike amplitude may have also involved a postsynaptic component.
Figure 6.
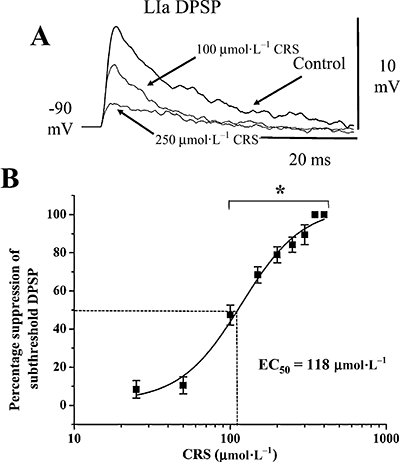
Effects of carisbamate (CRS) upon excitatory synaptic transmission. CRS (100–400 µmol·L−1) produced a selective, concentration-dependent suppression of depolarizing postsynaptic potentials (DPSPs) evoked by subthreshold lateral olfactory tract (layer Ia, LIa)-afferent fibre stimulation. (A) Superimposed subthreshold LIa DPSPs are elicited in control conditions and in CRS (100 and 250 µmol·L−1 as indicated). (B) Pooled log–concentration response curve showing the inhibitory effect of CRS upon LIa DPSP amplitude, expressed as percentage of control (data points are means ± SEM, n= 6; best-line fit gave EC50= 118 µmol·L−1; data points significantly different from control are indicated by *, P < 0.05).
To further test the proposed presynaptic action of carisbamate on synaptic transmission, a standard PPR analysis (Whalley and Constanti, 2006; see Methods) was conducted as presynaptically acting inhibitors of neurotransmitter release typically produce an increase in early PPF (Hasselmo and Bower, 1992). 200 µmol·L−1 carisbamate significantly increased PPF following LIa-afferent transmission (ISI = 25 ms, PPF = 1.27 ± 0.10 in control vs. 1.50 ± 0.14 in carisbamate, P < 0.05, n= 6; traces not shown) indicating a presynaptic aspect to the action of carisbamate. No significant increase in PPF was seen following intrinsic LII-LII fibre stimulation (PPF = 1.32 ± 0.21 in control vs. 1.37 ± 0.12 in carisbamate, P > 0.5, n= 6; traces not shown).
As a comparison, we also tested the effect of lidocaine in a similar manner. 160 µmol·L−1 lidocaine (15 min) produced a significant reversible depression of LIa fibre-evoked DPSPs (−40 ± 7% vs. control, P < 0.01; n= 7), but no significant effect on intrinsic LII-III excitatory synaptic transmission (−2 ± 5%; P > 0.5, n= 7; traces not shown), suggesting that the synaptic depressant effects of carisbamate and lidocaine were not merely due to a non-specific ‘local anaesthetic’ action at excitatory nerve terminals.
Effects of carisbamate on Mg2+-free medium or 4-AP-induced epileptiform discharges
The effects of carisbamate (50–200 µmol·L−1) upon seizure-like activity in PC slices were tested by using two established methods. The range of carisbamate concentrations used was chosen to avoid significant postsynaptic membrane effects on input resistance (Figure 1), a property that could have an important bearing on its efficacy to inhibit epileptiform discharges.
Thirty minutes pre-exposure to Mg2+-free Krebs medium (to remove Mg2+-induced blockade of NMDA-type glutamate receptors) induced spontaneous seizure-like activity (Figure 7A), comprising rhythmic PDSs (52 ± 6 mV) with superimposed spikes that lasted 31 ± 7 s and occurred at 1.5 ± 0.3 events·min−1 (n= 7). At concentrations up to 150 µmol·L−1, carisbamate application (15 min) had no notable effect on established Mg2+-free-induced epileptiform discharges (n= 6 experiments, not shown). However, 200 µmol·L−1 carisbamate reliably abolished PDS events (onset time for inhibition = 15 ± 3 min; n= 6) and returned the recorded cell to a quiescent state, with input resistance and resting potential returning to control values. This action was consistent with carisbamate causing a concentration-related disruption of synaptically generated and maintained seizure-like activity.
Figure 7.
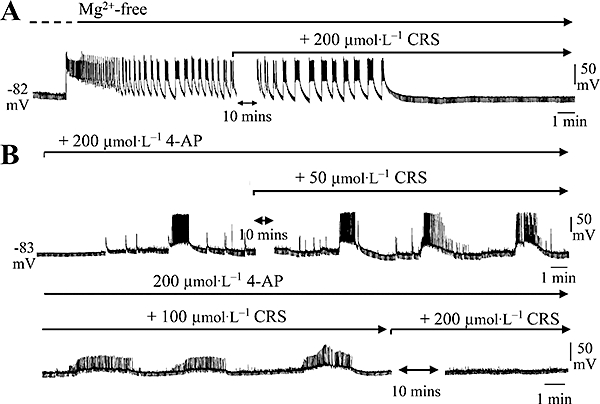
The effect of carisbamate (CRS) upon epileptiform activity. Chart records (chart speed 0.5 mm·s−1) showing the abolition by CRS of spontaneous epileptiform discharge activity recorded intracellularly in piriform cortex neurones from resting potential (∼−80 mV) during continued exposure to (A) Mg2+-free Krebs medium or (B) 200 µmol·L−1 4-aminopyridine (4-AP). Downward deflections are hyperpolarizing electrotonic potentials produced by −0.1 nA current stimuli (160 ms, 0.5 Hz), to monitor input resistance. Note that after blockade of Mg2+-free or 4-AP-induced discharges in CRS, the input resistance returned to the control level, measured prior to CRS application. Also, CRS modified 4-AP-induced discharge properties even at 50 µmol·L−1 concentration.
Application of the K+ channel blocker, 4-AP (200 µmol·L−1, 20 min), also induced similar epileptiform activity in recorded neurones (Figure 7B). PDS amplitude in 4-AP was 38 ± 7 mV with a duration of 64 ± 15 s and frequency of 0.25 ± 0.1 events·min−1 (n= 5). At 50 µmol·L−1, carisbamate had no obvious effect upon 4-AP-induced PDS incidence or amplitude (Figure 7B), although the frequency of superimposed spike firing was significantly reduced (4-AP: 2.6 ± 0.3 Hz vs. carisbamate: 2.1 ± 0.2 Hz, P < 0.05; n= 6). Increasing concentrations of carisbamate (≥100 µmol·L−1) produced more distinct anti-seizure effects such as a reduction in peak PDS amplitude (18 ± 5 mV in 100 µmol·L−1 carisbamate, P < 0.01, n= 6; ∼−50%; 9 ± 4 mV in 150 µmol·L−1 carisbamate, P < 0.01, n= 6; ∼−78%) and superimposed spike firing frequency (1.2 ± 0.2 and 0.75 ± 0.2 Hz in 100 and 150 µmol·L−1 carisbamate respectively, P < 0.01 in both cases). Interestingly, the ‘disrupted’ discharge duration was increased at higher carisbamate concentrations (140 ± 12 and 160 ± 21 s in 100 and 150 µmol·L−1 carisbamate, P < 0.01; Figure 7B). At 200 µmol·L−1 carisbamate, 4-AP-induced discharge activity was fully abolished, although some increased baseline noise did remain, most likely due to enhanced transmitter release from excitatory nerve terminals (Rogawski and Barker, 1983). Recovery of epileptiform discharges on removal of carisbamate while in 4-AP or Mg2+-free Krebs was not investigated.
CRS may decrease membrane input resistance via modulation of Cl− channels
While the actions of carisbamate suggest a use-dependent blockade of voltage-gated Na+ channels (see preceding), these effects were only manifested at membrane potentials beyond action potential firing threshold (∼−68 mV; Figure 1A). Therefore, this does not account for the observed carisbamate-induced membrane resistance decrease at resting membrane potential without an accompanying change in membrane potential (Figure 1B). Because the Cl− equilibrium potential (ECl) in PC neurones is around −70 mV (close to resting potential: ∼−83 mV, Whalley and Constanti, 2006), we considered whether the observed decrease in input resistance produced by carisbamate (Figure 1B) might involve activation of a Cl− conductance. Thus, recordings were made by using 2 mol·L−1 KCl-filled electrodes that shift ECl to a more depolarized level (Spain et al., 1987). An increase in control baseline noise was evident (Figure 8A*,C,D; cf. Figure 1B), likely to be due to GABAA-mediated spontaneous inhibitory postsynaptic potentials, arising from GABA release by inhibitory interneurone activity (Galvan et al., 1985b). Under these conditions, brief (150 s) applications of carisbamate (400 µmol·L−1) produced a rapid depolarization (38.1 ± 2.5 mV; n= 5) that reversed slowly on washout (Figure 8A), consistent with activation of a Cl− conductance. When the same protocol was applied by using lidocaine (200 µmol·L−1; Figure 8D), comparable effects were seen. The application of brief hyperpolarizing (160 ms; 1.5 nA) current injection through the recording microelectrode before and during carisbamate or lidocaine application revealed clear membrane resistance reductions (Figure 8B; cf. membrane resistance changes reported in Figure 1B,C ii). Subsequent application of the GABAA receptor/ion channel antagonist picrotoxin (100 µmol·L−1) in the continued presence of carisbamate fully reversed the depolarization induced by carisbamate (Figure 8C), consistent with a blockade of picrotoxin-sensitive Cl− channels under these conditions.
Figure 8.
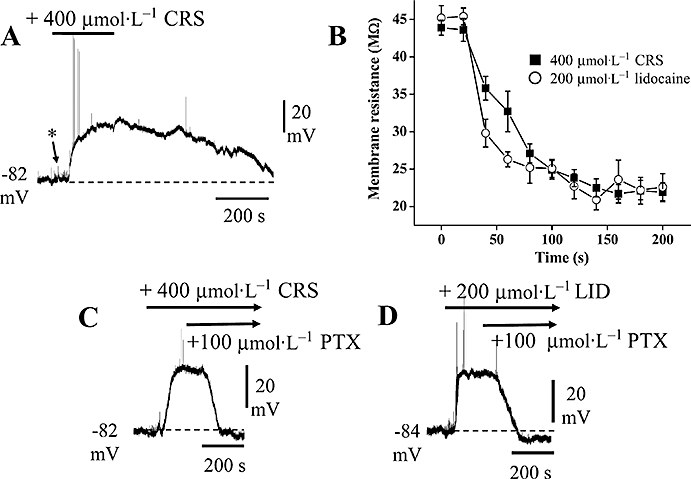
Effects of carisbamate (CRS) and lidocaine (LID) in Cl−-loaded cells. Chart records showing changes in membrane potential and input resistance produced by CRS and LID in cells recorded at normal resting potential, by using 2 mol·L−1 KCl-filled microelectrodes [* indicates increase in baseline ‘noise’ (cf. Figure 1B) probably due to spontaneous, depolarizing GABA release]. (A) A brief (150 s) application of CRS (400 µmol·L−1) produced a depolarization and slow recovery on washout; note the spontaneous firing induced at the onset of the depolarizing response (action potentials truncated by chart recorder). (B) Comparison of the input resistance changes produced by 400 µmol·L−1 CRS and 200 µmol·L−1 LID recorded in Cl−-loaded cells (as in a,c,d) (data points are means ± SEM, n= 4 for both CRS and LID). (C) CRS response recorded in a different neurone: application of picrotoxin (PTX; 100 µmol·L−1) in the continued presence of CRS reversed the CRS-induced depolarization. (D) Application of LID (200 µmol·L−1) in a different cell evoked a similar depolarization that was also reversed by applying PTX (in the presence of LID).
Lidocaine (200 µmol·L−1) was also tested after Cl− loading and produced a comparably rapid depolarization (32 ± 3.1 mV; n= 5; Figure 8B,D) that was reversed by subsequent application of picrotoxin, implying a similar mechanism of action to carisbamate. The membrane resistance change produced by lidocaine in Cl−-loaded cells was also similar (Figure 8B). For clarity, all measurements were made after intermittent returns of membrane potential to resting levels by injection of negative holding current. The input resistance decreased significantly by about 50% in either 400 µmol·L−1 carisbamate (P < 0.001, n= 4) or 200 µmol·L−1 lidocaine (P < 0.005, n= 4; 200 s applications in each case). We thus conclude that, at higher micromolar concentrations, carisbamate may also activate Cl− channels at potentials close to resting values in PC neurones, an effect that is mimicked by lidocaine.
Carisbamate may selectively depress evoked PC LIa-afferent synaptic transmission by activating a presynaptic Cl− conductance
In view of the above result and using conventional 2 mol·L−1 potassium acetate-filled microelectrodes, we investigated whether the selective effect of carisbamate on evoked LOT LIa fibre-evoked synaptic transmission at values close to the resting membrane potential could also be due to an increase in Cl− conductance in presynaptic nerve terminals. Subthreshold (i.e. not eliciting a superimposed action potential) LIa DPSPs were evoked in control conditions, then the effects of carisbamate (400 µmol·L−1) were examined; the effects of 100 µmol·L−1 picrotoxin in the continued presence of 400 µmol·L−1 carisbamate were then investigated (Figure 9A). The membrane potential was held at −90 mV by injection of hyperpolarizing current through the recording microelectrode. As previously shown (Figure 6), carisbamate (400 µmol·L−1; 15 min) produced near full suppression of the control DPSP response (−95.5 ± 1.8%; P < 0.001, n= 5). Addition of picrotoxin (100 µmol·L−1, 15 min) in the continued presence of carisbamate caused an initial small increase in DPSP amplitude (+9.1 ± 8.1%; measured as indicated by * in Figure 9A; P > 0.5, n= 4), followed by a prolonged increase in synaptic transmission. Interestingly, lidocaine (200 µmol·L−1; 15 min) similarly suppressed control DPSP amplitude (−58.9 ± 2.9%; P < 0.01, n= 4); picrotoxin (100 µmol·L−1, 15 min) caused a similar, small initial opposition of the lidocaine effect (DPSP suppression =−7.7 ± 4.8% of control; P > 0.5, n= 4; Figure 9B*), followed by a prolonged DPSP increase. We propose that picrotoxin initially opposes the effects of carisbamate and lidocaine in this system, potentially by occluding an effect on a presynaptic GABAA-like Cl− conductance. Prolonged DPSPs and depressed spikes may be due to a picrotoxin-induced removal of GABA-mediated inhibition and carisbamate/lidocaine effects on Na+ channels respectively.
Figure 9.
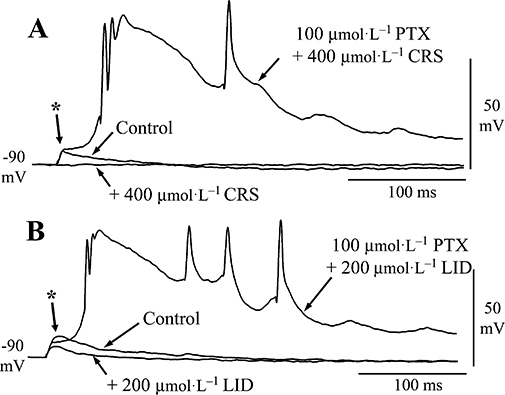
Effects of carisbamate (CRS) upon evoked depolarizing postsynaptic potentials (DPSPs). Synaptic potentials recorded by using potassium acetate-filled microelectrodes. (A) DPSPs evoked following layer Ia fibre stimulation (4 V, 0.2 ms) in control, CRS (400 µmol·L−1, 15 min) and CRS plus picrotoxin (PTX; 100 µmol·L−1). (B) Layer Ia DPSPs evoked in a different neurone (3 V, 0.2 ms) in control, lidocaine (LID; 200 µmol·L−1, 15 min) and LID plus PTX. Traces in PTX are single representative records; other traces are averages of at least three consecutive sweeps. Resting potential was maintained at −90 mV throughout by negative current injection.
Effect of carisbamate on action potential-independent mIPSCs
The experiments preceding show that carisbamate causes depolarization in Cl−-loaded cells and a depression of evoked (action potential-dependent) DPSPs at membrane potentials close to resting values. Moreover, the GABAA receptor antagonist picrotoxin opposed these effects. To investigate whether the effects of carisbamate were indirectly caused by modulation of ‘tonic’ GABA release and/or inhibition of GABA uptake mechanisms, we tested carisbamate effects on spontaneous inhibitory transmission in layer II-III neurones using patch-clamp recording. In order to avoid potential contaminating effects of carisbamate-induced Na+ channel block (see preceding) and to isolate presynaptic effects, action potential-independent mIPSCs were recorded in the presence of TTX plus NBQX. Recordings were conducted by using CsCl-filled pipettes (ECl−∼0 mV), to elicit inward GABAA currents at a holding potential of −70 mV (Figure 10A). Under these recording conditions, control mIPSCs had a mean amplitude of −37.7 ± 3.0 pA and mean frequency of 1.46 ± 0.44 Hz (n= 9 separate cells; Figure 10A,C i, ii). Application of 300 µmol·L−1 carisbamate (n= 6), 400 µmol·L−1 carisbamate (n= 3) or 600 µmol·L−1 carisbamate (n= 3) produced no significant effects upon mIPSC amplitude (−36.0 ± 2.2 pA; n= 12; P > 0.5; Figure 10A,C i) or frequency (1.72 ± 0.57 Hz; n= 12; P > 0.5; Figure 10A,C ii) when these data were grouped (or when examined for each concentration vs. control; data not shown). There was also no clear effect on mIPSC half-width (control = 12.1 ± 1.1 ms vs. 300 µmol·L−1 carisbamate = 11.9 ± 1.1 ms; n= 6; P > 0.5; from 551 events, n= 6 cells; Figure 10A,B). These data indicate a lack of effect of carisbamate on release or removal of GABA from synapses (for instance by uptake systems). Finally, we investigated the effects of carisbamate on holding currents measured under patch clamp, in order to determine potential activation of tonic GABA-mediated Cl− currents in the cerebral cortex (Drasbek and Jensen, 2005). Within these cells, there was no clear percentage change in holding current during carisbamate application (P > 0.5; n= 12; grouped data: 300–600 µmol·L−1 carisbamate; Figure 10D). Application of 10 µmol·L−1 bicuculline methiodide also failed to change normalized holding current (n= 4; P > 0.5; Figure 10D), suggesting little or no tonic activation of GABA receptors. However, application of the non-selective GABA transporter blocker nipecotic acid (500 µmol·L−1; Sigma-Aldrich, Gillingham, UK) was able to induce a readily detectable change in holding current in these neurones (n= 4; P < 0.05; Figure 10D). Taken together, the lack of effect of carisbamate in these experiments suggests that carisbamate does not modulate spontaneous, action potential-independent synaptic or extrasynaptic GABA release or reuptake at inhibitory synapses onto PC cells.
Figure 10.
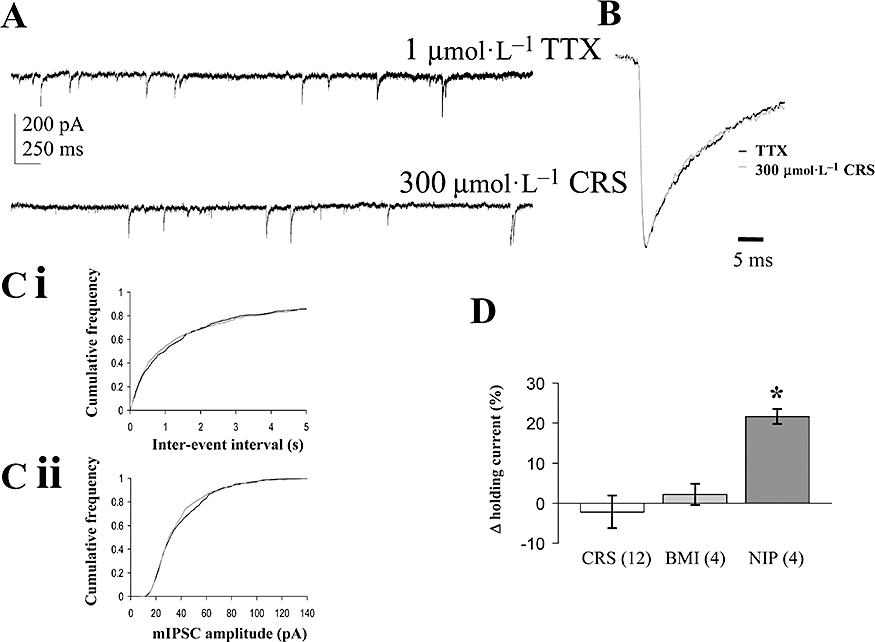
Effects of carisbamate (CRS) on spontaneous inhibitory synaptic transmission. (A) Examples of continuous traces showing GABAA miniature inhibitory postsynaptic currents (mIPSCs) recorded from layer II-III piriform cortical neurones under whole-cell patch clamp in 1 µmol·L−1 tetrodotoxin (TTX) plus 5 µmol·L−1 NBQX (6-nitro-7-sulphamoylbenzo(f)quinoxaline-2-3-dione) (control) and in the presence of CRS (300 µmol·L−1), 15 min application, holding potential −70 mV throughout. (B) Overlaid, normalized data showing averaged mIPSC event profile (TTX control: 492 events; CRS: 551 events); note that TTX control and CRS events had very similar half-widths. (C) Pooled cumulative frequency plot (n= 9) for effects of CRS on control for (i) mIPSC inter-event intervals and (ii) mIPSC amplitude (note lack of significant effects for both graphs: each replicant P > 0.5, Kolmogorov-Smirnov test). (D) Effects of CRS (300–600 µmol·L−1), bicuculline methiodide (BMI, 10 µmol·L−1) and nipecotic acid (NIP, 500 µmol·L−1) on holding current levels expressed as percentage change relative to control baseline level (Δ%; *P < 0.05); number of replicates (n) given in parentheses. For CRS, grouped data are presented based on experiments by using 300 µmol·L−1 (n= 6), 400 µmol·L−1 (n= 3) and 600 µmol·L−1 CRS (n= 3) respectively (as detailed in text).
Discussion
Carisbamate depressed evoked repetitive firing and excitatory synaptic transmission
We have shown that carisbamate (50–400 µmol·L−1) reversibly decreased the amplitude, duration and rate of rise of evoked action potentials and inhibited sustained repetitive firing of PC neurones, indicating decreased cell excitability. The nature of this block (greater effect on second vs. first evoked spike) is consistent with a use-dependent effect on fast voltage-gated Na+ channels. In support, the local anaesthetic lidocaine exerted similar effects to carisbamate on the spike parameters measured, although, in general, carisbamate was at least an order of magnitude less potent than lidocaine.
Inhibition of sustained repetitive firing is also produced by the established AEDs phenytoin, carbamazepine and lamotrigine (Willow et al., 1985; Lang et al., 1993) that act via partial fast Na+ channel blockade. On the basis of our data, we suggest a similar mechanism of action for carisbamate. In keeping with this proposal, recent evidence indicates that carisbamate, like some other Na+ channel-blocking AEDs (e.g. lamotrigine; Blackburn-Munro and Erichsen, 2005) may also be useful for treating neuropathic pain (Codd et al., 2008). Although the precise mechanism of Na+ channel blockade by carisbamate remains unknown, our observed concentration-dependent changes in spike parameters suggest a direct Na+ channel-mediated effect (Strichartz and Cohen 1978). In contrast, the lack of effect of carisbamate on the Ca2+-dependent sAHP (Constanti and Sim, 1987) suggests only minor action on neuronal Ca2+ conductances, despite the weak block of Ca2+ spikes revealed in Cs+-loaded cells and relatively small effect on Na+ spike firing frequency (∼17 Hz) exhibited during the long depolarizing stimulus (Figure 5B).
At higher concentrations (300–400 µmol·L−1), carisbamate also reversibly reduced neuronal input resistance without altering membrane potential, suggesting activation of a conductance around resting membrane potential. After intracellular Cl− loading, carisbamate induced a rapid depolarization and decreased input resistance that was reversed on washout, suggesting that carisbamate may also increase a Cl− conductance. Picrotoxin, a non-competitive GABAA receptor antagonist (Solís and Nicoll, 1992), opposed the carisbamate-induced depolarization in Cl−-loaded cells (Figure 8), suggesting that the activation of a GABAA-linked Cl− conductance may contribute to the mode of action of carisbamate. Interestingly, the dicarbamates, felbamate and meprobamate, potentiate GABA-evoked membrane currents in hippocampal neurones (Rho et al., 1997), and meprobamate-evoked Cl− currents are blocked by picrotoxin and bicuculline (a competitive GABAA receptor antagonist). Conversely, our experiments on action potential-independent mIPSCs indicate that carisbamate did not cause membrane depolarization indirectly via facilitation of presynaptic GABA release (Lei et al., 2007) or blockade of exogenous ‘tonic’ GABA uptake (Keros and Hablitz, 2005), because 300–600 µmol·L−1 carisbamate failed to affect spontaneous GABA-mediated mIPSC properties under whole-cell patch-clamp conditions. The apparent lack of holding current change produced by carisbamate in patch-clamp experiments may be due to possible differences between the Cl− reversal potentials in the whole cell and microelectrode (2 mol·L−1 KCl) configurations. Alternatively, whole-cell recording may lead to dialysis of an important intracellular mediator necessary for activation of a Cl− conductance that is preserved in ‘sharp’ microelectrode recording.
Interestingly, carisbamate, applied to cells held close to the resting membrane potential, reduced evoked excitatory synaptic transmission in LIa-afferent fibres (EC50= 118 µmol·L−1), but not in intrinsic LII-III fibres. These data suggest a potential selectivity towards some excitatory synapses over others, an observation that may be relevant for the central anticonvulsant profile of carisbamate, particularly in the treatment of limbic seizures involving the PC (Löscher and Ebert, 1996). The synaptic inhibitory effects of carisbamate were most likely presynaptically-mediated, as was confirmed by using a paired-pulse protocol (Whalley and Constanti, 2006) although whether this layer selectivity can also be demonstrated in other brain areas has yet to be established.
Based on our postsynaptic observations in Cl−-loaded cells, we suggest that the effects of carisbamate on action potential-dependent glutamate release at LIa terminals may be exerted via a Cl− conductance present on presynaptic terminals innervating pyramidal neurones. Interestingly, lidocaine also selectively depressed LIa DPSPs, and this effect was also opposed by picrotoxin, suggesting a similar presynaptic mechanism (rather than an action potential conduction block in LIa fibre terminals; cf. Scholfield and Harvey, 1975). Although the intracellular Cl− concentration in presynaptic terminals is unknown here, carisbamate actions will be dependent on the Cl− gradient at synapses onto pyramidal cells. Such gradients appear to be developmentally regulated in the CNS by specific transporters (Chavas and Marty, 2003) and will determine the relative contribution of effects on Cl− conductances to the mechanism of action of carisbamate used therapeutically. At present, we cannot rule out the possibility that carisbamate has differential effects on evoked versus spontaneous excitatory transmission in the PC.
Our experiments also do not allow a definitive identification of the proposed Cl− conductances affected by carisbamate (or lidocaine) or an assessment of their contribution towards the anticonvulsant effects of carisbamate at therapeutic plasma levels in vivo (∼20–40 µmol·L−1; see following). The possible carisbamate- (or lidocaine-) induced activation of other anion channel or possibly Cl− transporter types should therefore also be considered, for example, volume-sensitive Cl− channels (Inoue and Okada, 2007) or glycine-gated Cl− channels, which are present on adult hippocampal cells and are blocked by picrotoxin (Chattipakorn and McMahon, 2002; Kondratskaya et al., 2004). However, the latter may not be expressed in mature cortical neurones (Malosio et al., 1991), although glycine α2 and β receptor subunit expression has been detected in the adult PC. Whether these form functional Cl− channels is presently unknown.
Carisbamate produced use-dependent blocking effects on evoked spike amplitude
In our experiments, the use-dependent Na+ channel block exerted by carisbamate was evident as a cumulative depression of the action potential evoked by brief depolarizing stimulus trains applied at frequencies of 25, 50 and 100 Hz. With carisbamate, the time constant of use-dependence was notably longer (400 µmol·L−1, 100 Hz, τ=∼6 s) compared with lidocaine (160 µmol·L−1, 100 Hz, τ=∼1 s) to produce a comparable plateau level of block, suggesting a possible difference in the mechanisms of access and exit of both drugs from the Na+ channel-blocking site.
According to Hille (1977), use-dependent local anaesthetic block arises from preferential binding to open and inactivated Na+ channel states that predominate during repetitive firing and accumulates because drug dissociation occurs at a slower rate than the stimulation frequency (although see Feyh, 1993; Nau and Wang, 2004). On this basis, we suggest that repeated neuronal depolarization leads to progressive accumulation of Na+ channel states to which carisbamate (and lidocaine) preferentially bind, stabilizing the deactivated state. The slower onset of carisbamate block may reflect the relatively greater hydrophobicity of the carisbamate molecule compared with lidocaine which is more polar and so would be expected to show rapid use dependency (Liu et al., 1994). The possibility that carisbamate also binds to pre-open channel states to facilitate subsequent closing cannot be excluded (Liu et al., 1994). Our results are thus in accordance with those of Chernoff and Strichartz (1989) who showed that net positive charge on the local anaesthetic molecule is not necessarily required to produce use-dependent block of Na+ channels. It would be of interest to test whether carisbamate requires intracellular access for Na+ channel blockade, or whether its effects may be solely explained via the hydrophobic route (Hille, 1977).
Our experiments revealed a similarity between the membrane effects of the prototypical local anaesthetic lidocaine (Nau and Wang, 2004) and those of carisbamate on all spike parameters tested. Lidocaine (80–320 µmol·L−1) reduced evoked spike firing frequency and decreased first, and particularly, second spike amplitudes, at concentrations ≥80 µmol·L−1. It also decreased spike rates of rise and dramatically increased second spike duration at concentrations ≥40 µmol·L−1 (cf. Capek and Esplin, 1994; Schwarz and Puil, 1998). It is therefore likely that a common action on neuronal Na+-channels may contribute towards the in vivo anticonvulsant effects of both carisbamate (Nehlig et al., 2005; White et al., 2006) and low doses of lidocaine (DeToledo, 2000). Lidocaine also reversibly decreased neuronal input resistance at ≥80 µmol·L−1 without affecting membrane potential, as reported in thalamocortical neurones by Schwarz and Puil (1998) who suggested that an increased GABAA receptor-mediated Cl− conductance might be responsible. However, this action was not blocked by bicuculline and was therefore unlikely to be mediated by direct GABAA receptor activation (Schwarz and Puil, 1999). After intracellular Cl− loading, both carisbamate and lidocaine depolarized PC neurones, an effect opposed by co-application of 100 µmol·L−1 picrotoxin, suggesting a common underlying Cl− channel activation mechanism.
The effect of carisbamate on induced epileptiform discharges
In keeping with its proven anti-seizure efficacy in vivo (Nehlig et al., 2005), carisbamate (50–200 µmol·L−1) also modified epileptiform discharge activity induced by exposure to Mg2+-free Krebs medium or 4-AP (Libri et al., 1996). A similar effect of carisbamate on low Mg2+-induced discharges and repetitive firing in hippocampal neurones was also reported by Deshpande et al. (2008). Carisbamate was more effective in influencing 4-AP-induced discharges (threshold 50 µmol·L−1 carisbamate) that rely predominantly upon facilitation of presynaptic excitatory transmission (Perreault and Avoli, 1989), compared with the Mg2+-free-induced activity (threshold 200 µmol·L−1 carisbamate), which depends on enhanced NMDA receptor-mediated mechanisms. These reports are in accordance with our finding that lower concentrations of carisbamate were required to suppress LIa-afferent synaptic transmission than those required to produce pronounced postsynaptic effects. The concentrations of carisbamate found effective against seizure-like activity in vitro were comparable to the active free plasma levels of carisbamate observed clinically and in in vivo epilepsy models. A recent study in photosensitive epilepsy patients showed that a single 500 mg dose of carisbamate (=plasma Cmax of ∼48 µmol·L−1; Trenitéet al., 2007) reduced the EEG photoparoxysmal response while repeated administration of 200–500 mg total daily doses yielded maximal plasma concentrations of ∼17–130 µmol·L−1 (Yao et al., 2006). Moreover, in a study of kainate-induced epilepsy in rats (Grabenstatter and Dudek, 2008), a significant (∼50%) suppression of spontaneous motor seizures was achieved after a dose of 10 mg·kg−1 carisbamate, producing a blood plasma concentration of ∼5 µg·mL−1 (=∼20 µmol·L−1, measured after 4 h). Taken together with these values, our data suggest that carisbamate-induced inhibition of neuronal firing and/or action potential-dependent transmitter release operating via Cl− channel activation may be relevant only at higher plasma concentrations. However, while in vitro brain slices are an excellent model for the elucidation of drug mechanisms, the drug concentrations necessary to elicit measurable responses in vitro can often be difficult to relate to clinical or in vivo states due to compound lipophilicity and the cellular density of brain slice preparations (Becker and Liu, 2006).
Conclusions and general therapeutic implications
In conclusion, our study has revealed that carisbamate decreased the excitability of cortical neurones in vitro. We propose that the block of Na+ channels and excitatory transmission are the major mechanisms contributing to the anticonvulsant profile of carisbamate observed clinically and in vivo, whereas effects on Cl− channels may become relevant only at higher than clinically studied plasma concentrations, or potentially in patients whose neuronal intracellular Cl− levels are raised (e.g. see Ben-Ari, 2006). There is already some evidence that a Na+ channel-blocking property of carisbamate may not be its sole mechanism of anticonvulsant action, because in the 6 Hz seizure model of partial epilepsy (Barton et al., 2001), it showed anti-seizure activity even at high stimulus intensities where two recognized Na+ channel blockers (phenytoin and lamotrigine) proved ineffective. Also, carisbamate was protective in the amygdala kindling model of epilepsy where lamotrigine was inactive (White et al., 2006).
The Na+ channel-blocking effects of carisbamate may also be of potential therapeutic value in treating chronic neuropathic pain (see Kulig and Malawska, 2007), particularly if a relative selectivity for the TTX-resistant Na+ channel isoforms could be demonstrated (Hargus and Patel, 2007). Furthermore, block of Na+ channels can be neuroprotective following cerebral ischaemia, probably through inhibition of (excitotoxic) glutamate release and inhibition of persistent neuronal Na+ fluxes (Callaway, 2001). Carisbamate has already been shown to exert neuroprotective properties following status epilepticus (Francois et al., 2005). Finally, because the PC is an area particularly prone to epileptogenesis and excitotoxicity (Löscher and Ebert, 1996), our observed selective inhibition of PC LIa fibre-evoked neurotransmission would be a useful property for the management of limbic seizures that occur in temporal lobe epilepsy patients.
Acknowledgments
This work was supported by a grant from Johnson & Johnson Pharmaceutical Research & Development, L.L.C, Titusville, NJ, USA.
Glossary
Abbreviations:
- 4-AP
4-aminopyridine
- AED
antiepileptic drug
- DMSO
dimethylsulphoxide
- DPSP
depolarizing postsynaptic potential
- EEG
electroencephalogram
- ISI
interstimulus interval
- LIa
layer Ia
- LII-III
layers II-III
- LOT
lateral olfactory tract
- mIPSC
miniature inhibitory postsynaptic current
- NBQX
6-nitro-7-sulphamoylbenzo(f)quinoxaline-2-3-dione
- NMDA
N-methyl-D-aspartate
- PC
piriform cortex
- PDS
paroxysmal depolarizing shift
- PPF
paired-pulse facilitation
- PPR
paired-pulse ratio
- sAHP
slow afterhyperpolarization
- TTX
tetrodotoxin
Conflict of interest
None.
References
- Barton ME, Klein BD, Wolf HH, White HS. Pharmacological characterization of the 6 Hz psychomotor seizure model of partial epilepsy. Epilepsy Res. 2001;47:217–227. doi: 10.1016/s0920-1211(01)00302-3. [DOI] [PubMed] [Google Scholar]
- Becker S, Liu X. Evaluation of the utility of brain slice methods to study brain penetration. Drug Metab Dispos. 2006;34:855–861. doi: 10.1124/dmd.105.007914. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y. Seizures beget seizures: the quest for GABA as a key player. Crit Rev Neurobiol. 2006;18:135–144. doi: 10.1615/critrevneurobiol.v18.i1-2.140. [DOI] [PubMed] [Google Scholar]
- Bialer M, Johannessen SI, Kupferberg HJ, Levy RH, Perucca E, Tomson T. Progress report on new antiepileptic drugs: a summary of the Eigth Eilat Conference (EILAT VIII) Epilepsy Res. 2006;51:31–71. doi: 10.1016/j.eplepsyres.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Blackburn-Munro G, Erichsen HK. Antiepileptics and the treatment of neuropathic pain: evidence from animal models. Curr Pharm Des. 2005;11:2961–2976. doi: 10.2174/1381612054865000. [DOI] [PubMed] [Google Scholar]
- Callaway JK. Investigation of AM-36: a novel neuroprotective agent. Clin Exp Pharmacol Physiol. 2001;28:913–918. doi: 10.1046/j.1440-1681.2001.03547.x. [DOI] [PubMed] [Google Scholar]
- Capek R, Esplin B. Effects of lidocaine on hippocampal pyramidal cells: depression of repetitive firing. Neuroreport. 1994;5:681–684. doi: 10.1097/00001756-199402000-00005. [DOI] [PubMed] [Google Scholar]
- Chattipakorn SC, McMahon LL. Pharmacological characterization of glycine-gated chloride currents recorded in rat hippocampal slices. J Neurophysiol. 2002;87:1515–1525. doi: 10.1152/jn.00365.2001. [DOI] [PubMed] [Google Scholar]
- Chavas J, Marty A. Coexistence of excitatory and inhibitory GABA synapses in the cerebellar interneuron network. J Neurosci. 2003;23:2019–2031. doi: 10.1523/JNEUROSCI.23-06-02019.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernoff DM, Strichartz GR. Tonic and phasic block of neuronal sodium currents by 5-hydroxyhexano-2′,6′-xylide, a neutral lidocaine homologue. J Gen Physiol. 1989;93:1075–1090. doi: 10.1085/jgp.93.6.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codd EE, Martinez RP, Molino L, Rogers KE, Stone DJ, Tallarida RJ. Tramadol and several anticonvulsants synergize in attenuating nerve injury-induced allodynia. Pain. 2008;134:254–262. doi: 10.1016/j.pain.2007.04.019. [DOI] [PubMed] [Google Scholar]
- Constanti A, Sim JA. Calcium-dependent potassium conductance in guinea-pig olfactory cortex neurones in vitro. J Physiol. 1987;387:173–194. doi: 10.1113/jphysiol.1987.sp016569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney KR. Mechanism of frequency-dependent inhibition of sodium currents in frog myelinated nerve by the lidocaine derivative GEA 968. J Pharmacol Exp Ther. 1975;195:225–236. [PubMed] [Google Scholar]
- Deshpande LS, Nagarkatti N, Sombati S, Delorenzo RJ. The novel antiepileptic drug carisbamate (RWJ 333369) is effective in inhibiting spontaneous recurrent seizure discharges and blocking sustained repetitive firing in cultured hippocampal neurons. Epilepsy Res. 2008;79:158–165. doi: 10.1016/j.eplepsyres.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeToledo JC. Lidocaine and seizures. Ther Drug Monit. 2000;22:320–322. doi: 10.1097/00007691-200006000-00014. [DOI] [PubMed] [Google Scholar]
- Drasbek KR, Jensen K. THIP, a hypnotic and antinociceptive drug, enhances an extrasynaptic GABAA receptor-mediated conductance in mouse neocortex. Cereb Cortex. 2005;16:1134–1141. doi: 10.1093/cercor/bhj055. [DOI] [PubMed] [Google Scholar]
- Feyh LS. The chemistry and pharmacology of local anesthetics. CRNA. 1993;4:161–169. [PubMed] [Google Scholar]
- Francois J, Ferrandon A, Koning E, Nehlig A. A new drug RWJ 333369 protects limbic areas in the lithium-pilocarpine model (li-pilo) of epilepsy and delays or prevents the occurrence of spontaneous seizures. Epilepsia. 2005;46(Suppl.8):269–270. [Google Scholar]
- Francois J, Boehrer A, Nehlig A. Effects of Carisbamate (RWJ-333369) in two models of genetically determined generalized epilepsy, the GAERS and the audiogenic Wistar AS. Epilepsia. 2008;49:393–399. doi: 10.1111/j.1528-1167.2007.01278.x. [DOI] [PubMed] [Google Scholar]
- Galvan M, Constanti A, Franz P. Calcium-dependent action potentials in guinea-pig olfactory cortex neurones. Pflugers Arch. 1985a;404:252–258. doi: 10.1007/BF00581247. [DOI] [PubMed] [Google Scholar]
- Galvan M, Franz P, Constanti A. Spontaneous inhibitory postsynaptic potentials in guinea pig neocortex and olfactory cortex neurones. Neurosci Lett. 1985b;57:131–135. doi: 10.1016/0304-3940(85)90051-5. [DOI] [PubMed] [Google Scholar]
- Grabenstatter HL, Dudek FE. A new potential AED, carisbamate, substantially reduces spontaneous motor seizures in rats with kainate-induced epilepsy. Epilepsia. 2008;49:1787–1794. doi: 10.1111/j.1528-1167.2008.01657.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargus NJ, Patel MK. Voltage-gated Na+ channels in neuropathic pain. Expert Opin Investig Drugs. 2007;16:635–646. doi: 10.1517/13543784.16.5.635. [DOI] [PubMed] [Google Scholar]
- Hasselmo ME, Bower JM. Cholinergic suppression specific to intrinsic not afferent fiber synapses in rat piriform (olfactory) cortex. J Neurophysiol. 1992;67:1222–1229. doi: 10.1152/jn.1992.67.5.1222. [DOI] [PubMed] [Google Scholar]
- Hille B. Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. J Gen Physiol. 1977;69:497–515. doi: 10.1085/jgp.69.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue H, Okada Y. Roles of volume-sensitive chloride channel in excitotoxic neuronal injury. J Neurosci. 2007;27:1445–1455. doi: 10.1523/JNEUROSCI.4694-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Okada T, Ohmori H. A phenytoin-sensitive cationic current participates in generating the afterdepolarization and burst afterdischarge in rat neocortical pyramidal cells. Eur J Neurosci. 1998;10:1363–1375. doi: 10.1046/j.1460-9568.1998.00155.x. [DOI] [PubMed] [Google Scholar]
- Keros S, Hablitz JJ. Subtype-specific GABA transporter antagonists synergistically modulate phasic and tonic GABAA conductances in rat neocortex. J Neurophysiol. 2005;94:2073–2085. doi: 10.1152/jn.00520.2005. [DOI] [PubMed] [Google Scholar]
- Kondratskaya EL, Fisyunov AI, Chatterjee SS, Krishtal OA. Ginkgolide B preferentially blocks chloride channels formed by heteromeric glycine receptors in hippocampal pyramidal neurons of rat. Brain Res Bull. 2004;63:309–314. doi: 10.1016/j.brainresbull.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Kulig K, Malawska B. Carisbamate, a new carbamate for the treatment of epilepsy. IDrugs. 2007;10:720–727. [PubMed] [Google Scholar]
- Lang DG, Wang CM, Cooper BR. Lamotrigine, phenytoin and carbamazepine interactions on the sodium current present in N4TG1 mouse neuroblastoma cells. J Pharmacol Exp Ther. 1993;266:829–835. [PubMed] [Google Scholar]
- Lei S, Deng PY, Porter JE, Shin H. Adrenergic facilitation of GABAergic transmission in rat entorhinal cortex. J Neurophysiol. 2007;98:2868–2877. doi: 10.1152/jn.00679.2007. [DOI] [PubMed] [Google Scholar]
- Libri V, Constanti A, Calaminici M, Nisticó G. A comparison of the muscarinic response and morphological properties of identified cells in the guinea-pig olfactory cortex in vitro. Neuroscience. 1994;59:331–347. doi: 10.1016/0306-4522(94)90600-9. [DOI] [PubMed] [Google Scholar]
- Libri V, Constanti A, Zibetti M, Nisticó S. Effects of felbamate on muscarinic and metabotropic-glutamate agonist-mediated responses and magnesium-free or 4-aminopyridine-induced epileptiform activity in guinea pig olfactory cortex neurons in vitro. J Pharmacol Exp Ther. 1996;277:1759–1769. [PubMed] [Google Scholar]
- Liu L, Wendt DJ, Grant AO. Relationship between structure and sodium channel blockade by lidocaine and its amino-alkyl derivatives. J Cardiovasc Pharmacol. 1994;24:803–812. doi: 10.1097/00005344-199424050-00016. [DOI] [PubMed] [Google Scholar]
- Löscher W. Basic pharmacology of valproate: a review after 35 years of clinical use for the treatment of epilepsy. CNS Drugs. 2002;16:669–694. doi: 10.2165/00023210-200216100-00003. [DOI] [PubMed] [Google Scholar]
- Löscher W, Ebert U. The role of the piriform cortex in kindling. Prog Neurobiol. 1996;50:427–481. doi: 10.1016/s0301-0082(96)00036-6. [DOI] [PubMed] [Google Scholar]
- Malosio ML, Marquèze-Pouey B, Kuhse J, Betz H. Widespread expression of glycine receptor subunit mRNAs in the adult and developing rat brain. EMBO J. 1991;10:2401–2409. doi: 10.1002/j.1460-2075.1991.tb07779.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nau C, Wang GK. Interactions of local anesthetics with voltage-gated Na+ channels. J Membr Biol. 2004;201:1–8. doi: 10.1007/s00232-004-0702-y. [DOI] [PubMed] [Google Scholar]
- Nehlig A, Rigoulot M-A, Boehrer A. A new drug, RWJ 333369 displays potent antiepileptic properties in genetic models of absence and audiogenic epilepsy. Epilepsia. 2005;46:215. [Google Scholar]
- Novak GP, Kelley M, Zannikos P, Klein B. Carisbamate (RWJ-333369) Neurotherapeutics. 2007;4:106–109. doi: 10.1016/j.nurt.2006.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perreault P, Avoli M. Effects of low concentrations of 4-aminopyridine on CA1 pyramidal cells of the hippocampus. J Neurophysiol. 1989;61:953–970. doi: 10.1152/jn.1989.61.5.953. [DOI] [PubMed] [Google Scholar]
- Rho JM, Donevan SD, Rogawski MA. Barbiturate-like actions of the propanediol dicarbamates felbamate and meprobamate. J Pharmacol Exp Ther. 1997;280:1383–1391. [PubMed] [Google Scholar]
- Rogawski MA. Diverse mechanisms of antiepileptic drugs in the development pipeline. Epilepsy Res. 2006;69:273–294. doi: 10.1016/j.eplepsyres.2006.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogawski MA, Barker JL. Effects of 4-aminopyridine on calcium action potentials and calcium current under voltage clamp in spinal neurons. Brain Res. 1983;280:180–185. doi: 10.1016/0006-8993(83)91190-3. [DOI] [PubMed] [Google Scholar]
- Russo E, Constanti A. Topiramate hyperpolarizes and modulates the slow poststimulus AHP of rat olfactory cortical neurones in vitro. Br J Pharmacol. 2004;141:285–301. doi: 10.1038/sj.bjp.0705617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholfield CN, Harvey JA. Local anesthetics and barbiturates: effects on evoked potentials in isolated mammalian cortex. J Pharmacol Exp Ther. 1975;195:522–531. [PubMed] [Google Scholar]
- Schwarz SK, Puil E. Analgesic and sedative concentrations of lignocaine shunt tonic and burst firing in thalamocortical neurones. Br J Pharmacol. 1998;124:1633–1642. doi: 10.1038/sj.bjp.0702015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz SK, Puil E. Lidocaine produces a shunt in rat [correction of rats] thalamocortical neurons, unaffected by GABA(A) receptor blockade. Neurosci Lett. 1999;269:25–28. doi: 10.1016/s0304-3940(99)00415-2. [DOI] [PubMed] [Google Scholar]
- Sofia RD, Gordon R, Gels M, Diamantis W. Effects of felbamate and other anticonvulsant drugs in two models of status epilepticus in the rat. Res Commun Chem Pathol Pharmacol. 1993;79:335–341. [PubMed] [Google Scholar]
- Solís JM, Nicoll RA. Pharmacological characterization of GABAB-mediated responses in the CA1 region of the rat hippocampal slice. J Neurosci. 1992;12:3466–3472. doi: 10.1523/JNEUROSCI.12-09-03466.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spain WJ, Schwindt PC, Crill WE. Anomalous rectification in neurons from cat sensorimotor cortex in vitro. J Neurophysiol. 1987;57(5):1555–1576. doi: 10.1152/jn.1987.57.5.1555. [DOI] [PubMed] [Google Scholar]
- Strichartz G, Cohen I. Vmax as a measure of GNa in nerve and cardiac membranes. Biophys J. 1978;23:153–156. doi: 10.1016/S0006-3495(78)85440-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trenité DG, French JA, Hirsch E, Macher JP, Meyer BU, Grosse PA, et al. Evaluation of carisbamate, a novel antiepileptic drug, in photosensitive patients: an exploratory, placebo-controlled study. Epilepsy Res. 2007;74:193–200. doi: 10.1016/j.eplepsyres.2007.03.012. [DOI] [PubMed] [Google Scholar]
- Tseng GF, Haberly LB. Characterization of synaptically mediated fast and slow inhibitory processes in piriform cortex in an in vitro slice preparation. J Neurophysiol. 1988;59:1352–1376. doi: 10.1152/jn.1988.59.5.1352. [DOI] [PubMed] [Google Scholar]
- Wang GK, Russell C, Wang SY. State-dependent block of wild-type and inactivation-deficient Na+ channels by flecainide. J Gen Physiol. 2003;122:365–374. doi: 10.1085/jgp.200308857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalley BJ, Constanti A. Developmental changes in presynaptic muscarinic modulation of excitatory and inhibitory neurotransmission in rat piriform cortex in vitro: relevance to epileptiform bursting susceptibility. Neuroscience. 2006;140:939–956. doi: 10.1016/j.neuroscience.2006.02.046. [DOI] [PubMed] [Google Scholar]
- Whalley BJ, Postlethwaite M, Constanti A. Further characterization of muscarinic agonist-induced epileptiform bursting activity in immature rat piriform cortex, in vitro. Neuroscience. 2005;134:549–566. doi: 10.1016/j.neuroscience.2005.04.018. [DOI] [PubMed] [Google Scholar]
- White HS, Srivastava A, Klein B, Zhao B, Choi YM, Gordon R, et al. The novel investigational neuromodulator RWJ 333369 displays a broad-spectrum anticonvulsant profile in rodent seizure and epilepsy models. Epilepsia. 2006;47(Suppl. 4):200. Abst. [Google Scholar]
- Willow M, Gonoi T, Catterall WA. Voltage clamp analysis of the inhibitory actions of diphenylhydantoin and carbamazepine on voltage-sensitive sodium channels in neuroblastoma cells. Mol Pharmacol. 1985;27:549–558. [PubMed] [Google Scholar]
- Yao C, Doose DR, Novak G, Bialer M. Pharmacokinetics of the new antiepileptic and CNS drug RWJ-333369 following single and multiple dosing to humans. Epilepsia. 2006;11:1822–1829. doi: 10.1111/j.1528-1167.2006.00814.x. [DOI] [PubMed] [Google Scholar]