

Abstract
Background and purpose:
Statins and fibrates can produce mild to life-threatening skeletal muscle damage. Resting chloride channel conductance (gCl), carried by the ClC-1 channel, is reduced in muscles of rats chronically treated with fluvastatin, atorvastatin or fenofibrate, along with increased resting cytosolic calcium in statin-treated rats. A high gCl, controlled by the Ca2+-dependent protein kinase C (PKC), maintains sarcolemma electrical stability and its reduction alters muscle function. Here, we investigated how statins and fenofibrate impaired gCl.
Experimental approach:
In rats treated with fluvastatin, atorvastatin or fenofibrate, we examined the involvement of PKC in gCl reduction by the two intracellular microelectrodes technique and ClC-1 mRNA level by quantitative real time-polymerase chain reaction. Direct drug effects were tested by patch clamp analysis on human ClC-1 channels expressed in human embryonic kidney (HEK) 293 cells.
Key results:
Chelerythrine, a PKC inhibitor, applied in vitro on muscle dissected from atorvastatin-treated rats fully restored gCl, suggesting the involvement of this enzyme in statin action. Chelerythrine partially restored gCl in muscles from fluvastatin-treated rats but not in those from fenofibrate-treated rats, implying additional mechanisms for gCl impairment. Accordingly, a decrease of ClC-1 channel mRNA was found in both fluvastatin-and fenofibrate-treated rat muscles. Fenofibric acid, the in vivo metabolite of fenofibrate, but not fluvastatin, rapidly reduced chloride currents in HEK 293 cells.
Conclusions and implications:
Our data suggest multiple mechanisms underlie the effect of statins and fenofibrate on ClC-1 channel conductance. While statins promote Ca2+-mediated PKC activation, fenofibrate directly inhibits ClC-1 channels and both fluvastatin and fenofibrate impair expression of mRNA for ClC-1.
Keywords: hypolipidemic drugs, skeletal muscle, chloride channel, cell signaling, gene expression
Introduction
Statins (HMG CoA reductase inhibitors) are widely used to reduce plasma cholesterol levels, and several clinical trials have established the benefit of these drugs in the prevention of coronary heart disease (Davidson and Robinson, 2007). Recent findings have also shown that statins may have cholesterol-independent beneficial effects during inflammatory syndromes, Alzheimer's disease and cancer, that may expand their clinical utility (Weitz-Schmidt, 2002; Stuve et al., 2003; Demierre et al., 2005). However, statin therapy can be severely limited by adverse effects involving many tissues, especially skeletal muscle. Cerivastatin, marketed in the United States in 1998, was subsequently withdrawn because of a high rate of rhabdomyolysis-induced deaths associated with the drug. Although rhabdomyolysis is very uncommon, clinical use of these medications is more frequently associated with a variety of less severe adverse effects on muscle including myalgia, cramps and weakness that arise during long-term use (Thompson et al., 2003). However, the mechanism determining muscle damage is still not clear, although some investigators have implicated certain consequences of inhibiting cholesterol biosynthesis, such as depletion of ubiquinone or of small GTP-binding proteins involved in myocyte maintenance (Vaughan and Gotto, 2004).
Fibrates are another important class of lipid-lowering drugs useful for the management of dyslipidemia. By acting on the peroxisome proliferator-activated receptor (PPAR)-α, mainly expressed in tissues that metabolize fatty acids, these drugs lower serum triglyceride levels and raise high-density lipoprotein-cholesterol (Cannon, 2008). As with statins, fibrate monotherapy is associated with a risk of myopathy (Davidson et al., 2007) and this risk is enhanced when these agents are given together. However, whereas gemfibrozil can increase plasma concentration of the commonly prescribed statins, fenofibrate has less influence on the pharmacokinetics of statins (Cannon, 2008).
We have previously found that lipophilic statins can affect skeletal muscle function by reducing the resting chloride channel conductance (gCl) in rats, through a mechanism independent of their ability to lower endogenous cholesterol (Pierno et al., 2006). We have also demonstrated that chronic treatment with clofibrate reduces resting gCl (Conte Camerino et al., 1984). A large gCl, carried by the ClC-1 chloride channel, is important for muscle function as it stabilizes resting membrane potential (RMP) and helps to repolarize the membrane after action potentials (Aromataris and Rychkov, 2006). Loss of function mutations in ClC-1 cause myotonia congenita, an inherited condition characterized by delayed skeletal muscle relaxation after voluntary contraction (Koch et al., 1992; Jentsch et al., 2005). Resting gCl strictly depends on ClC-1 chloride channel expression and regulation. A Ca2+-and phospholipid-dependent protein kinase C (PKC) controls ClC-1 channel activity and activation of PKC closes the channel (Rosenbohm et al., 1999), reducing gCl (Bryant and Conte Camerino, 1991; De Luca et al., 1994). In fast-twitch muscles, the chloride channel is maintained in a dephosphorylated state allowing high resting gCl to preserve muscle membrane potential stability. In contrast, slow-twitch, anti-gravity muscles, such as soleus muscle, are characterized by a lower gCl, due to both a lower expression of the ClC-1 channel and different regulation by PKC (Pierno et al., 2007). Indeed, the use of pharmacological tools, such as chelerythrine, a PKC inhibitor, suggests that a higher basal activity of this enzyme in soleus muscle is responsible for the phosphorylation and closure of a fraction of ClC-1 channels at rest. We previously demonstrated that statins also affect the excitation–contraction coupling mechanism (Pierno et al., 1999a). Accordingly, recent studies confirmed that statins either chronically administered or applied acutely in vitro produce a mitochondria-mediated increase of resting cytosolic calcium in intact muscle cells (Sirvent et al., 2005; Liantonio et al., 2007).
To address the myotoxic potential of statins and fibrates, we investigated the molecular mechanisms by which these drugs reduce resting gCl. We treated rats chronically with fluvastatin (5 mg·kg−1and 20 mg·kg−1), atorvastatin (10 mg·kg−1) or fenofibrate (60 mg·kg−1) and then analysed the effects of drug treatments on the signalling pathways related to PKC-mediated gCl regulation and on ClC-1 gene expression. We also evaluated the direct effects of drugs by measuring macroscopic gCl in native skeletal muscle fibres or chloride current from human ClC-1 channels expressed in human embryonic kidney (HEK) 293 cells. Our experiments have demonstrated different mechanisms responsible for drug-induced reduction of gCl. The activation of Ca2+-dependent PKC was involved in statin-induced gCl decline, whereas fenofibrate was able to reduce gCl by a direct action on ClC-1. A concomitant decrease in ClC-1 mRNA expression occurred in both fluvastatin-and fenofibrate-treated animals and probably contributed to the reduced gCl.
Methods
Animal care and treatments
All experiments were conducted in accordance with the Italian Guidelines for the use of laboratory animals, which conforms with the European Community Directive published in 1986 (86/609/EEC). Adult male Wistar rats (Charles River Laboratories, Italy) were used for chronic and acute studies. The animals, weighing 300–350 g at the beginning of the treatment, were housed individually in rat appropriate cages in an environmentally controlled room and fed with 30 g·day−1 of a commercial rodent chow (Charles River, 4RF21) in addition to tap water ad libitum. Five groups of five rats each were chronically treated with 20 mg·kg−1 of fluvastatin, 5 mg·kg−1 of fluvastatin, 10 mg·kg−1 of atorvastatin, 60 mg·kg−1 of fenofibrate and with the vehicle (carboxy-methylcellulose, CMC) used to dissolve the drugs. Fluvastatin (Lescol, Novartis), atorvastatin (Torvast, Pfizer) and fenofibrate (Lipsin, Caber) dissolved in CMC (0.5%) were administered to the different groups of animals orally via an esophageal cannula, once a day, for 2 months (Pierno et al., 2006). During treatment, all rats were weighed weekly and vital parameters (general health conditions, water and food consumption) were recorded. Rats treated with the highest dose of fluvastatin (20 mg·kg−1) exhibited a slight decrease in body weight gain, compared with control and all other treated animals. This effect, related to a lower food intake (25 g·day−1 vs. 30 g·day−1), was probably due to a non-muscle effect. Skeletal muscle performance was evaluated daily by testing each rat for the righting reflex (the ability of the rat to straighten itself on four legs when turned on its back) which, when abnormal, is a sign of myotonia. In all the animals, the righting reflex evaluated during the entire treatment period was normal. Drug and channel nomenclature conforms to the British Journal of Pharmacology Guide to Receptors and Channels (Alexander et al., 2008).
Electromyographic recordings
Electromyographic (EMG) technique was used to evaluate the presence of muscular abnormalities due to drug treatment or myotonic-like signs (Heller et al., 1982). EMG traces were recorded in all rats in each experimental condition at the beginning and every 2 weeks until the end of treatment by using the Biopack MP100 acquisition system (EMG 100C, Biopack Systems Inc., Santa Barbara, CA, USA). EMG activity was measured by two thin concentric needle electrodes inserted in the gastrocnemius muscle of rats anaesthetized with pentobarbital (60 mg·kg−1, i.p., supplemented if required). Insertional activity was initiated by slight movement of the electrode within the muscle and the presence of spontaneous activity (not due to the hind-limb movement) was evaluated. EMG activity was monitored continuously over a 3-to 4-min period starting from the insertion of the electrodes. A qualitative estimate of myotonic-like activity was based on various parameters such as the voltage excursion, the frequency and the duration of the burst. EMG activity was also recorded in three rats acutely injected with anthracene-9-carboxylic acid (9-AC) (5 mg·kg−1 i.p.) (Sigma Aldrich, Italy) a well-known blocker of ClC-1 channel conductance and, because of this, a myotonia-inducer (Kwieciński et al., 1988). We used 9-AC as a reference compound for observing myotonia discharges on EMG recordings, with the aim of comparing the effects of long-term treatments with statins or fenofibrate.
Recordings of skeletal muscle gCl by two-intracellular microelectrode technique: in vivo and in vitro studies
The extensor digitorum longus (EDL) and soleus muscles were dissected from treated and control animals under urethane anesthesia (1.2 g·kg−1 i.p.). The preparations were immediately placed in a 25 mL muscle bath, maintained at 30°C and perfused with normal or chloride-free physiological solution (gassed with 95% O2 and 5% CO2; pH = 7.2–7.3) (Bryant and Conte Camerino, 1991). The normal physiological solution had the following composition (in mmol·L−1): NaCl 148, KCl 4.5, CaCl2 2.0, MgCl2 1.0, NaHCO3 12.0, NaH2PO4 0.44 and glucose 5.55. The chloride-free solution was prepared by equimolar replacement of methylsulphate salts for NaCl and KCl and nitrate salts for CaCl2 and MgCl2. The contralateral EDL muscles were also surgically removed and frozen in liquid nitrogen until mRNA analysis.
As previously detailed (Bryant and Conte Camerino, 1991; De Luca et al., 1994; Pierno et al., 2007), by means of standard two-intracellular-microelectrode technique, RMP, cable parameters, component conductances and excitability characteristics of treated and untreated muscle fibres were measured in current-clamp mode. Cable parameters, in both normal and chloride-free solutions, were calculated from the electrotonic potential elicited by square-wave hyperpolarizing current pulse (100 ms duration) injected by the current electrode. The membrane voltage responses were monitored by the voltage electrode at two distances from the current electrode. The current pulse generation, the acquisition of the voltage records and the calculation of the fibre constants were performed in real time under computer control as described elsewhere (Bryant and Conte Camerino, 1991). From the experimentally determined values of input resistance, space and time constant, and from an assumed myoplasmic resistivity, of 125 Ω·cm, fibre diameter, membrane resistance (rm) and membrane capacitance were calculated (Bryant and Conte Camerino, 1991). The reciprocal of rm from each fibre in normal physiological solution was assumed to be gm and the same parameter measured in chloride-free solution was considered to be the potassium conductance (gK). The mean gCl was estimated as the mean gm minus the mean gK (Bryant and Conte Camerino, 1991).
For in vitro studies atorvastatin calcium salt (Synfine Laboratories, Richmond Hill, Canada), fluvastatin sodium salt (Calbiochem, Milan, Italy), simvastatin (Tocris Bioscience, Bristol, UK) and fenofibric acid, the in vivo metabolite of fenofibrate (Sigma Aldrich, Milan, Italy), were dissolved in dimethyl sulphoxide and tested on isolated EDL muscle dissected from control rats. Fenofibric acid was obtained by hydrolysis of fenofibrate. Resting gCl was measured before and 30 min after addition of increasing concentrations of the different hypolipidemic drugs. The concentration-response curves obtained were well fitted with a first-order binding function:
where IC50 (µmol·L−1) is the half-maximum inhibitory concentration, h is the slope factor and min is the minimal gCl.
Chelerythrine (Tocris Bioscience) 1 µmol·L−1 was applied in vitro on EDL muscles dissected from control and chronically treated rats. To study if chelerythrine prevented the reduction of gCl induced acutely by hypolipidemic drugs, we pre-incubated EDL control muscles with chelerythrine for 1 h and then measured gCl 30 min after hypolipidemic drug addition.
Real-time quantitative polymerase chain reaction (PCR)
For each muscle sample, total RNA was isolated by using Trizol reagent. Total RNA (3 µg) was used for reverse transcription. Synthesis of cDNA was performed by using random hexamers (annealed 10 min, 25°C) and Superscript II reverse transcriptase (Invitrogen-Life Technologies, Carlsbad, CA, USA) incubated at 42°C for 50 min. We assayed expression of ClC-1 mRNA using a pre-designed TaqMan assay (Assay ID Rn00565736_ml; Applied Biosystems, Foster City, CA, USA) and a custom assay for β-actin (primer and probes sequences available on request). Triplicate reactions were carried out in parallel for each individual muscle sample. The results were compared with a gene-specific standard curve and normalized to expression of the housekeeping gene β-actin in the same samples. The template used for determining standard curves consisted of plasmid DNA containing the expected target sequence evaluated by Spectrophotometer (ND-1000 NanoDrop, Wilmington, DE, USA) (Rogers et al., 2002; Lundquist et al., 2005).
Whole-cell patch-clamp recordings of hClC-1 chloride currents
Full-length hClC-1 constructs were subcloned into the mammalian expression vector pRc/CMV as previously described (Rosenbohm et al., 1999). The HEK 293 cells used in the study were from stocks maintained in this laboratory. They were cultured in standard Dulbecco's modified Eagle's medium with penicillin/streptomycin (DMEM+P/S) before and after transfection. The cells were passaged in a 100 mm dish about 24 h before transfection. Cell density for transfection was about 60–80%. The HEK 293 cells were cotransfected with 4 µg of plasmid DNA encoding the chloride channel and lower amount of plasmid DNA encoding the CD8 receptor, using the calcium phosphate co-precipitation method (Desaphy et al., 2001). About 15 h after transfection (full confluence was reached), the cells were washed with DMEM+P/S, divided in small 60 mm dishes and maintained in DMEM+P/S until patch-clamp (36–72 h after transfection). The cells were washed several times with recording solution before patch-clamping.
For patch-clamp recording, successfully transfected cells were identified using Dynal microbeads coated with anti-CD8 antibody (Dynal AS, Oslo, Norway). Whole-cell ClC-1 chloride currents were recorded at room temperature (20–22°C) using an Axopatch 200B amplifier (Axon Instruments, Union City, CA, USA). Voltage clamp protocols and data acquisition were performed with PClamp 9.2 software (Axon Instruments) through a 12-bit A-D/D-A interface (Digidata 1322A, Axon Instruments). The external solution contained (mmol·L−1): 140 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 5 HEPES and 5 glucose, and the pH was set to 7.4 with NaOH. The pipette solution contained (mmol·L−1): 130 CsCl, 2 MgCl2, 5 EGTA and 10 HEPES, and the pH was adjusted to 7.4 with CsOH. With such solutions, pipettes made from Corning 7052 glass (Garner Glass, Claremont, CA, USA) had a resistance of 1–3 MΩ. Currents were low-passed filtered at 5 kHz (−3 dB) by the amplifier and digitized at 10–20 kHz. Series resistance and whole-cell capacitance were almost fully compensated for by the amplifier circuitry and no run-down was observed during the experiments.
Statistical analyses
Data are expressed as mean ± SEM. The estimates for SEM and number of fibres (n) of gCl were obtained from the variance of gm and gK, assuming no covariance, and from the number of fibres sampled for gm and gK respectively (Bryant and Conte Camerino, 1991). Analysis of variance (anova) was used to evaluate statistical differences between treated and control groups followed by Bonferroni's t-test or Tukey's post-test (only for real-time PCR). Comparison of means from each treated rat and the related control group was evaluated by unpaired Student's t-test.
Results
To determine the cellular and molecular mechanisms responsible for diminished muscle membrane gCl, we used several ex vivo and in vitro approaches. We will first present the results of the in vivo studies followed by our findings from in vitro and molecular experiments.
In vivo studies
Effects of drug treatment on EMG activity of gastrocnemius muscle
The gastrocnemius muscle of control rats showed normal electrical activity characterized by rapid and brief bursts occurring only during voluntary movements and followed by long periods of rest during muscle inactivity. We recorded slight spontaneous electrical activity in one out of 10 animals treated with fluvastatin at both doses, in two out of 10 animals treated with atorvastatin and in two out of 10 rats treated with fenofibrate. Figure 1 illustrates a representative EMG trace recorded from an atorvastatin-treated rat in which brief high-frequency repetitive discharges, lasting 500 ms, appeared after muscle movement. In the fenofibrate-treated rat, we also observed high-frequency repetitive discharges that persisted for 4 s, at most. However, these EMG abnormalities found in statin and fenofibrate-treated rats appeared only after 7–8 weeks of treatment. In comparison, as previously documented (Heller et al., 1982; Kwieciński et al., 1988), animals acutely treated with 9-AC exhibited the expected myotonic-like discharges with spontaneous high-frequency electrical activity recorded starting 1 h after the injection, and lasting several seconds (<5 s) after voluntary movement (Figure 1). These recordings were more dramatic than those observed in animals treated with hypolipidemic drugs.
Figure 1.
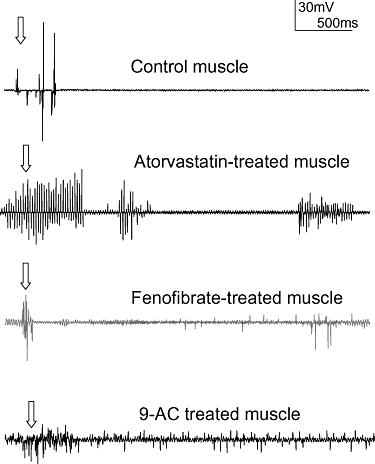
Example of EMG activity recorded from the gastrocnemius muscle of control rats and rats treated with atorvastatin (10 mg·kg−1) or fenofibrate (60 mg·kg−1) anaesthetized with pentobarbital. In the control animal, repetitive discharges occurred with needle insertion (normal insertional activity indicated by the arrow) and were followed by a long period of muscle inactivity. Repetitive discharges with variable frequency and amplitude were apparent and persisted for a few ms in a atorvastatin-treated rat. Additional bursts could be elicited by minimal movement of the needle in situ. This effect was observed in two of 10 treated rats. In fenofibrate-treated rats spontaneous discharges lasting a few seconds appeared after rat movement. This effect was observed in two of 10 treated rats. In comparison, the EMG activity of rats treated with 9-AC showed higher activity than in atorvastatin and fenofibrate-treated rats, lasting several seconds. This effect was observed in three out of three animals. Calibration bar, 30 mV, 500 ms. 9-AC, anthracene-9-carboxylic acid; EMG, electromyographic.
Ex vivo studies
Effects of statins and fibrate chronic treatment on muscle gCl: involvement of PKC
As previously demonstrated (Pierno et al., 2006), gCl was reduced by 20–35% in EDL muscles of rats chronically treated with the different drugs, compared with the gCl in muscles from control animals (Figure 2). In contrast, gCl measured in soleus muscle was unchanged (control 1511 ± 178 µS·cm−2, n = 27; 20 mg·kg−1 fluvastatin 1832 ± 99 µS·cm−2, n = 25; 10 mg·kg−1 atorvastatin 1441 ± 83 µS·cm−2, n = 29). We analysed the involvement of PKC in the reduction of gCl by using the PKC inhibitor chelerythrine. As observed previously (Pierno et al., 2007), the in vitro application of chelerythrine to EDL muscle of rats treated with the vehicle induced a slight increase of gCl (Figure 2) because of the high proportion of ClC-1 channels in a dephosphorylated state. When chelerythrine (1 µmol·L−1) was applied to EDL muscle dissected from rats treated with 5 mg·kg−1 or 20 mg·kg−1 fluvastatin, gCl was partially restored towards the control value, increasing by 13% and 15%, with respect to the value measured before the application of the PKC inhibitor (Figure 2). Chelerythrine significantly increased gCl of muscles from atorvastatin-treated rat by 40%, reaching the value measured in control rats. In contrast, chelerythrine was unable to antagonize the reduction of gCl induced by fenofibrate treatment (Figure 2).
Figure 2.
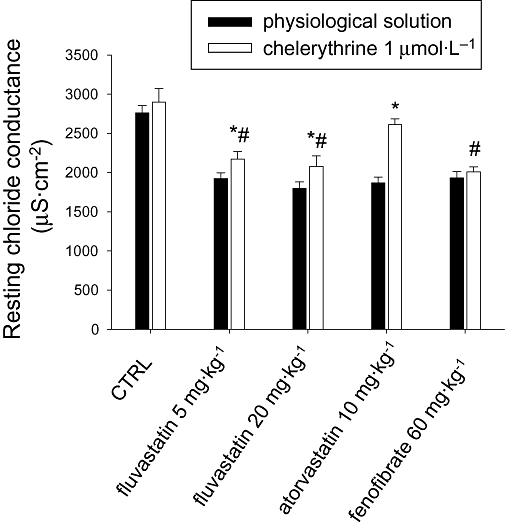
Effects of chelerythrine application in vitro on the chloride channel conductance (gCl) of extensor digitorum longus muscle of fluvastatin (5 mg·kg−1 or 20 mg·kg−1), atorvastatin (10 mg·kg−1) and fenofibrate (60 mg·kg−1)-treated and control (CTRL) rats. Each bar represents the mean ± SEM of gCl measured in muscle fibres in the presence of chelerythrine (1 µmol·L−1) with respect to the value recorded in the absence of the compound (physiological solution). The means have been calculated from 25–35 fibres of 3–5 rats. *Significantly different with respect to the value measured in the absence of chelerythrine (by Student's t-test) (P < 0.05 or less). #Significantly different with respect to control value measured in physiological solution (by Student's t-test) (P < 0.001).
Effects of chronic treatment with fluvastatin and fenofibrate on ClC-1 mRNA expression
Because the reduction of gCl in fluvastatin-treated rats was not completely attributable to an increase of PKC activity, as observed for atorvastatin treatment, we examined the effects of fluvastatin on ClC-1 gene expression. We also investigated ClC-1 mRNA levels in fenofibrate-treated animals in which PKC is not involved in reducing gCl. The results obtained by real-time quantitative (RT)-PCR analysis demonstrated a reduction of ClC-1 mRNA in EDL muscle of fluvastatin-treated rats, even at the lower dose (5 mg·kg−1) (Figure 3A). Also fenofibrate-treated rats showed lower expression of ClC-1 mRNA than control rats (Figure 3A). The relationship between ClC-1 mRNA levels and gCl is illustrated in Figure 3B. In particular, to better evaluate the individual response to drug treatment, in this figure we plotted the gCl measured only in the contra-lateral EDL muscles of the same fluvastatin (5 mg·kg−1) or fenofibrate-treated animals in which the ClC-1 mRNA amount had been determined. In these animals, gCl was significantly reduced by 16% and 20%, respectively, slightly different from the data in Figure 2 probably because of variability among individual animals encountered with in vivo pharmacological treatments. The degree of reduction in ClC-1 mRNA expression is greater than the respective decrease in gCl and below we discuss potential explanations for this phenomenon.
Figure 3.
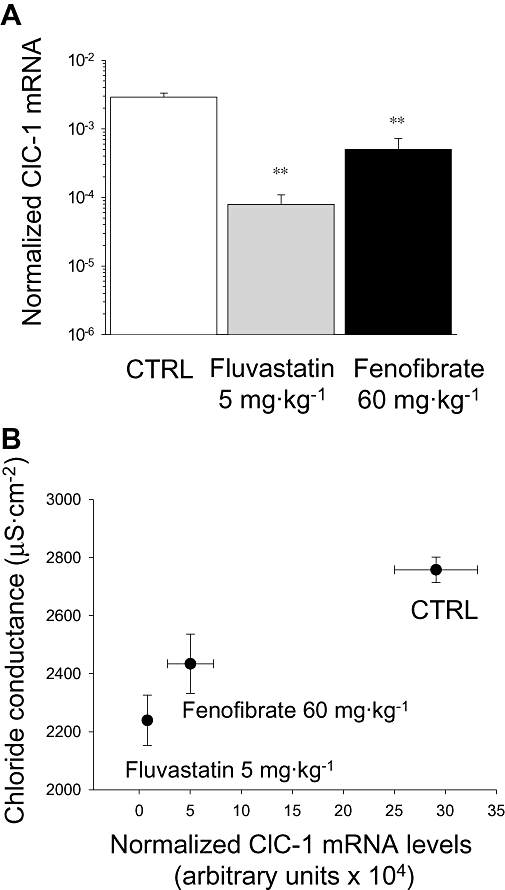
Effects of fluvastatin and fenofibrate treatment on muscle ClC-1 mRNA expression. (A) Normalized ClC-1 mRNA level measured by real time-polymerase chain reaction in extensor digitorum longus (EDL) muscle of 3–4 fluvastatin (5 mg·kg−1) and fenofibrate (60 mg·kg−1)-treated rats with respect to control rats. The level of ClC-1 were normalized to β-actin which was constant in all muscle preparations. **Significantly different compared with control (CTRL) (P < 0.05, one-way anova test followed by Tukey's post-test). (B) Relationship between ClC-1 mRNA expression and resting gCl measured in the contralateral EDL muscles of the same animals. gCl, chloride channel conductance.
In vitro studies
Effects of in vitro application of fluvastatin, atorvastatin and fenofibric acid on muscle gCl
When tested in vitro on EDL muscle fibres, fluvastatin reduced gCl in a concentration-dependent manner, with a 30% reduction at the higher concentration tested (50 µmol·L−1, Figure 4A). Atorvastatin tested at the same concentrations was slightly less effective in reducing gCl (50 µmol·L−1, 2183 ± 97 µS·cm−2, n = 13; control, 2736 ± 45 µS·cm−2, n = 24). However, from the concentration-response curve, similar values of IC50 (half-maximal inhibiting concentration) were calculated (fluvastatin: IC50 = 9.2 ± 3.3 µmol·L−1; h = 1.33 ± 0.6; minimum = 0.645 ± 0.06. atorvastatin: IC50 = 8.6 ± 7.2 µmol·L−1; h = 0.73 ± 0.4; minimum = 0.746 ± 0.07). Chelerythrine completely prevented the reduction of gCl induced by either fluvastatin (Figure 4B) or atorvastatin (2183 ± 97 µS·cm−2, n = 13 and 2982 ± 61 µS·cm−2, n = 20, in the absence and in the presence of chelerythrine respectively). Fenofibric acid, the metabolite of fenofibrate responsible for the in vivo drug effect, concentration-dependently reduced gCl (100 µmol·L−1, 2430 ± 79 µS·cm−2, n = 23; 500 µmol·L−1, 1964 ± 121 µS·cm−2, n = 28; no drug, 2745 ± 80 µS·cm−2, n = 23). As observed for in vivo treatment, chelerythrine did not prevent the reduction of gCl induced by the in vitro application of fenofibric acid (data not shown).
Figure 4.
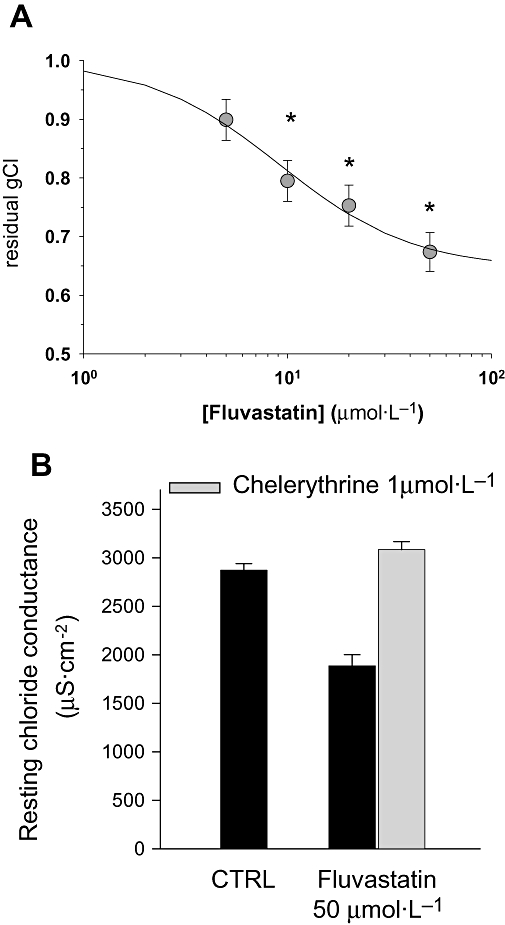
Effects of acute application of fluvastatin (A) on resting chloride channel conductance (gCl) measured in untreated rat extensor digitorum longus muscle. The dose-response relationship was constructed by calculating the residual gCl at different concentrations of fluvastatin. Each data point was calculated as the mean ± SEM of gCl from 27 to 60 fibres of 5–7 rats. *Significantly different with respect to control (P < 0.05 or less) by Student's t-test. In (B) is shown the effect of chelerythrine (1 µmol·L−1) application in muscle bath containing 50 µmol·L−1 of fluvastatin. The protein kinase C inhibitor completely prevented the reduction of gCl induced by the statin by shifting gCl towards the control (CTRL) value.
Atorvastatin at the higher concentration tested (50 µmol·L−1) did not modify resting gCl of soleus muscle, being 1540 ± 65 µS·cm−2, n = 17 and 1552 ± 47 µS·cm−2, n = 21; before and after addition of the compound. In contrast, the in vitro application of fenofibric acid (500 µmol·L−1) to soleus muscle markedly reduced gCl from 1478 ± 74 µS·cm−2, n = 16 of control to 908 ± 52 µS·cm−2, n = 24, respectively, strongly corroborating the results obtained in vivo and giving additional support to the hypothesis of the direct block of the channel with minor involvement of PKC.
Effects of statins and fenofibrate on human ClC-1 channel expressed in HEK 293 cells
To confirm the possible direct effect of drugs on the chloride channels responsible for the gCl, fenofibric acid, fluvastatin and simvastatin (500 µmol·L−1) were tested in vitro in HEK cells expressing human hClC-1 channels. We used the more lipophilic simvastatin as a reference compound. Whole-cell chloride currents were recorded before and 5 min after drug application. Neither fluvastatin (Figure 5A,B) nor simvastatin (not shown) had any effect on chloride current amplitude. In contrast, as observed for the structurally related 2-p-(chlorophenoxy)-propionic acid (CPP) derivatives (Aromataris et al., 1999; Liantonio et al., 2003), fenofibric acid caused a rapid and marked decrease of inward hClC-1 currents and acceleration of current deactivation (Figure 5C,D). These data strongly suggest that, like the CPP derivatives, fenofibric acid is capable of interfering with ClC-1 gating by interacting with an intracellular binding site (Pusch et al., 2000).
Figure 5.
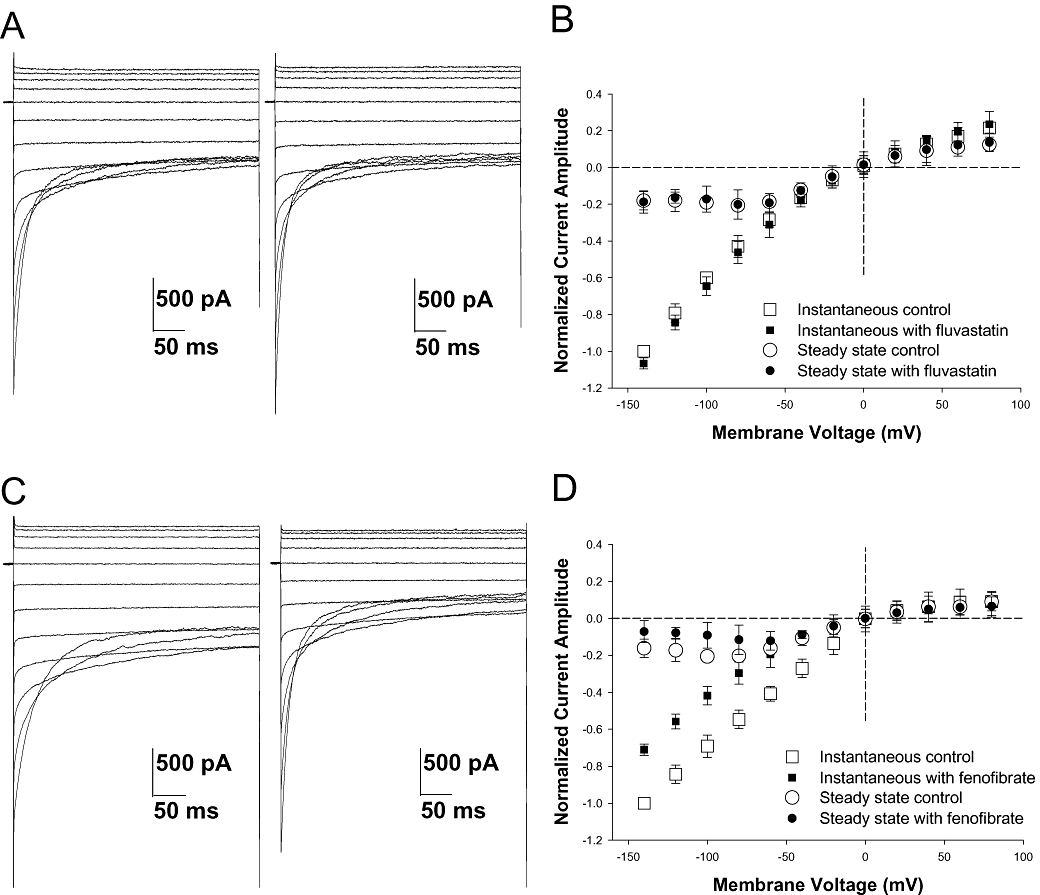
Effects of in vitro application of fluvastatin (500 µmol·L−1) and fenofibric acid (500 µmol·L−1) on heterologously expressed hClC-1 channel. Whole-cell patch-clamp chloride currents were elicited from a holding potential of 0 mV by 400 ms test pulses ranging from −140 to +80 mV in 20-mV increments. Current was recorded in the absence (A, C left panels), then presence of 500 µmol·L−1 fluvastatin (a, right panel) or 500 µmol·L−1 fenofibric acid (C, right panel). Normalized current-voltage relationships were obtained for instantaneous currents, measured at the beginning of the test pulse, and steady state currents, measured at the end of test pulse, in control and in presence of 500 µmol·L−1 fluvastatin (B) or 500 µmol·L−1 fenofibric acid (D). Each data point is the mean ± SEM from four cells (fluvastatin) or five cells (fenofibrate). In each cell, the current amplitude was normalized with respect to current value measured at −140 mV in control condition. Fluvastatin had no effect, whereas fenofibric acid significantly reduced inward chloride currents.
Discussion
The finding that hypolipidemic drugs, such as fenofibrate, clofibrate and lipophilic statins, reduced in vivo the resting chloride conductance (gCl) in rat skeletal muscle has been already established (Conte Camerino et al., 1984; Pierno et al., 1995; Pierno et al., 2006). We previously showed that only simvastatin, but not pravastatin, was able to affect gCl, an effect we attributed to the greater lipophilic properties of the former. Nevertheless, statins of more recent generation, such as fluvastatin and atorvastatin, were more potent than simvastatin in reducing gCl, independently of lipophilic status. Here, we attempted to clarify the molecular mechanisms by which statins and fibrate inhibit gCl.
We first investigated the effects of the hypolipidemic drugs on the metabolic pathway responsible for the regulation of ClC-1 channels, the main determinant of sarcolemmal gCl, which involves the calcium-dependent PKC. The high gCl typical of fast-twitch muscles is sustained by a low basal activity of PKC that maintains ClC-1 in a dephosphorylated, active state (De Luca et al., 1994; Pierno et al., 2007). In vitro PKC activation by phorbol esters concentration-dependently reduced the gCl, while PKC inhibition by chelerythrine produced little or no effect. We reported here a new finding: the reduction of skeletal muscle gCl induced by statins is mediated by the activation of the PKC pathway. When chelerythrine was applied in vitro to skeletal muscle from fluvastatin and atorvastatin-treated rats, the gCl was restored towards that of control, suggesting that these drugs, by activating PKC, were likely to have increased phosphorylation and closure of the ClC-1 chloride channel. We argue that this is a rapid effect specific to muscle because acute in vitro application of statins to muscle fibres reduced the gCl and this reduction was totally prevented by prior incubation with chelerythrine, whereas no effect of statins was found on heterologously expressed ClC-1 channels. This hypothesis is further supported by the observation that statin treatment had no effect, either in vivo or when applied in vitro, on the gCl in the slow-twitch soleus muscle, a muscle type characterized by a higher basal PKC activity, than that found in the fast-twitch muscle (Pierno et al., 2007) and that in contrast, fenofibrate markedly reduced gCl in this muscle. One possible mechanism for PKC activation by statins may be elevated intracellular Ca2+ concentrations. Indeed, we have demonstrated that in vivo administration as well as acute in vitro application of either fluvastatin or atorvastatin increased resting intracellular Ca2+ concentration in EDL muscle fibres, through a mechanism that involves the sequential release from mitochondria and then sarcoplasmic reticulum (Liantonio et al., 2007). Thus, the modification of calcium handling mechanisms induced by these drugs may account for a rapid, calcium-dependent, PKC activation. However, other mechanisms may contribute to in vivo effects, that is, increased expression of PKC through PPAR activation (Martin et al., 2001; Inoue et al., 2002).
Although PKC activation contributes to the action of both statins, chelerythrine did not fully reverse the reduction of gCl induced by fluvastatin, suggesting that additional events might contribute to the reduction of gCl. Also chelerythrine had no effect on the gCl reduction induced by fenofibrate. This is in accord with the lack of change in cytosolic calcium and mechanical threshold for contraction after fenofibrate treatment (Pierno et al., 2006; Liantonio et al., 2007). Thus, fenofibrate acts through a different mechanism. Patch clamp studies showed that fenofibric acid directly reduced the ClC-1 chloride current. The correlation between chemical structure and blocking properties between fenofibric acid and clofibric acid (Liantonio et al., 2003) lead us to envisage a direct effect of fenofibrate in blocking this channel. In contrast, neither fluvastatin nor simvastatin modifies ClC-1 currents after incubation not exceeding 5 min, again corroborating the key role of PKC in statin-induced gCl modification.
Because the reduction of gCl in fluvastatin-treated animals was not completely antagonized by chelerythrine and was not due to a direct block of the channel, a modification of ClC-1 expression was hypothesized. Statins and fibrates can modify the expression of various genes through activation of PPAR (Martin et al., 2001) or GTP-ase protein inhibition (Habib et al., 2007). Data obtained by quantitative RT-PCR demonstrated a significant reduction of ClC-1 mRNA in fluvastatin as well as in fenofibrate-treated rats with respect to controls. Although the reduction of ClC-1 mRNA is proportionally larger than the decrease of gCl, similar apparent discrepancies have been described in other conditions. For instance, in heterozygous ADR+/− mice, in which one ClC allele is disrupted, the mRNA for ClC-1 channels was reduced to 50% of that measured in wild-type mice (Chen et al., 1997) but gCl was normal, suggesting that post-translational modulation can compensate to some extent for a reduction in ClC-1 mRNA level, at least in pathogenic conditions. A similar mechanism may account for the disproportional changes in ClC-1 mRNA expression and gCl in the drug-treated rats. The measurement of gCl gives a direct assessment of protein function that plays a crucial role in controlling muscle excitability and, secondarily, muscle contractility. Our results demonstrate that gCl is reduced in drug-treated rats and that this effect is, at least partly, related to reduced expression of ClC-1 mRNA. More direct effects on protein function may also contribute to the reduction of gCl, in a manner specific to each drug.
In conclusion, the reduction of gCl we measured in statin and fenofibrate chronically treated rats was not greater than 30%, even at doses much higher than those clinically used. It is unlikely that myotonia-like phenomena will occur in this situation, because myotonia is normally characterized by stronger reduction of gCl (more than 50% of the normal value) (Pusch, 2002). Accordingly, the EMG activity recorded on fluvastatin, atorvastatin and fenofibrate-treated animals showed signs of altered excitability characterized by brief bursts of AP following normal movement-induced electrical activity, which were different from those seen during 9-AC acutely induced myotonia but closer to those recorded in humans during muscle cramping or fasciculation (Costa et al., 2005; Serrao et al., 2007). Taking into account the already observed lack of histological damage (Pierno et al., 2006), it is feasible that the reduction of gCl observed can be responsible for the mild adverse effects of hypolipidemic drugs, such as myalgia and cramps. The EMG alteration was observed only after 7–8 weeks of treatment but we can not exclude occurrence at earlier stages, as these phenomena may be episodic in nature and their frequency most probably increased with duration of treatment. Our data provide a molecular link between the reduction of gCl induced by statins and fenofibrate and the pathogenesis of skeletal muscle side effects. Figure 6 and Figure S1 summarizes all the results obtained in this study and hypothesizes the sequence of events involved in the mechanism determining muscle ClC-1 channel impairment. It should be considered that physiopathological conditions in which there is an increase of PKC activity, such as exercise (Rose et al., 2004; Ort et al., 2007) or aging (De Luca et al., 1994; Pierno et al., 2007), statin therapy should be continuously monitored. Higher rates of adverse side effects occur in aged patients, in which gCl can be affected due to development of sarcopenia (Pierno et al., 1999b). The use of compounds able to reduce muscle PKC activity should be considered to prevent skeletal muscle side effects that may arise during hypolipidemic drug therapy.
Figure 6.
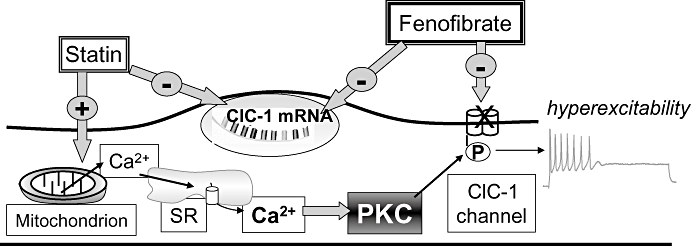
Diagram of the mechanisms by which statins and fenofibrate affect resting chloride channel conductance (gCl), carried by ClC-1 channels in skeletal muscle. It is known that protein kinase C (PKC) activation is responsible for the phosphorylation and closure of ClC-1 channel producing a reduction of gCl together with an increase of sarcolemma excitability (De Luca et al., 1994; Pierno et al., 2007). As described in the text, statins affect ClC-1 channel regulation by increasing PKC activity via calcium-release from mitochondria and sarcoplasmic reticulum (SR) (Liantonio et al., 2007). Fluvastatin also reduced ClC-1 mRNA expression. Fenofibrate acted by directly blocking ClC-1 channels and by reducing its expression.
Acknowledgments
This work was supported by Italian ‘Ministero dell'Istruzione, dell'Università e della Ricerca’ (FIRB RBAU015E9T) to D. C. C. We are grateful to Dr G. Fracchiolla for the preparation of fenofibric acid from fenofibrate (commercially obtained from Sigma Aldrich, Italy) and Dr C. Digennaro for technical assistance. We also thanks Dr M. Pusch for preliminary experiments and helpful suggestions.
Glossary
Abbreviations:
- 9-AC
anthracene-9-carboxylic acid
- CPP
2-p-(chlorophenoxy)-propionic acid
- EDL
extensor digitorum longus
- EMG
electromyographic
- gCl
chloride channel conductance
- gK
potassium conductance
- HEK
human embryonic kidney
- PKC
protein kinase C
- rm
membrane resistance
- RMP
resting membrane potential
Conflict of interest
The authors state no conflict of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. The mechanisms by which fluvastatin and fenofibrate affect resting chloride conductance, sustained by CIC-1 channels in rat skeletal muscle (fast-twitch-phenotype) fibres.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. (3rd edn.) 2008;153(2) Suppl.:S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aromataris EC, Astill DS, Rychkov GY, Bryant SH, Bretag AH, Roberts ML. Modulation of the gating of ClC-1 by S-(-) 2-(4-chlorophenoxy) propionic acid. Br J Pharmacol. 1999;126:1375–1382. doi: 10.1038/sj.bjp.0702459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aromataris EC, Rychkov GY. ClC-1 chloride channel: matching its properties to a role in skeletal muscle. Clin Exp Pharmacol Physiol. 2006;33:1118–1123. doi: 10.1111/j.1440-1681.2006.04502.x. [DOI] [PubMed] [Google Scholar]
- Bryant SH, Conte Camerino D. Chloride channel regulation in the skeletal muscle of normal and myotonic goats. Pflügers Arch. 1991;417:605–610. doi: 10.1007/BF00372958. [DOI] [PubMed] [Google Scholar]
- Cannon CP. Combination therapy in the management of mixed dyslipidaemia. J Intern Med. 2008;263:353–365. doi: 10.1111/j.1365-2796.2008.01933.x. [DOI] [PubMed] [Google Scholar]
- Chen MF, Niggeweg R, Iaizzo PA, Lehmann-Horn F, Jockusch H. Chloride conductance in mouse muscle is subject to post-transcriptional compensation of the functional Cl-channel 1 gene dosage. J Physiol. 1997;504:75–81. doi: 10.1111/j.1469-7793.1997.075bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conte Camerino D, Tortorella V, Ferranini E, Bryant SH. The toxic effects of clofibrate and its metabolite on mammalian skeletal muscle: an electrophysiological study. Arch Toxicol. 1984;7:482–484. doi: 10.1007/978-3-642-69132-4_101. Suppl. [DOI] [PubMed] [Google Scholar]
- Costa J, Graça P, Evangelista T, De Carvalho M. Pain and calf hypertrophy associated with spontaneous repetitive discharges treated with botulinum toxin. Clin Neurophysiol. 2005;116:2847–2852. doi: 10.1016/j.clinph.2005.07.016. [DOI] [PubMed] [Google Scholar]
- Davidson MH, Armani A, McKenney JM, Jacobson TA. Safety considerations with fibrate therapy. Am J Cardiol. 2007;99:3C–18C. doi: 10.1016/j.amjcard.2006.11.016. [DOI] [PubMed] [Google Scholar]
- Davidson MH, Robinson JG. Safety of aggressive lipid management. J Am Coll Cardiol. 2007;49:1753–1762. doi: 10.1016/j.jacc.2007.01.067. [DOI] [PubMed] [Google Scholar]
- De Luca A, Tricarico D, Pierno S, Conte Camerino D. Aging and chloride channel regulation in rat fast-twitch muscle fibers. Pflugers Arch. 1994;427:80–85. doi: 10.1007/BF00585945. [DOI] [PubMed] [Google Scholar]
- Demierre MF, Higgins PD, Gruber SB, Hawk E, Lippman SM. Statins and cancer prevention. Nat Rev Cancer. 2005;5:930–942. doi: 10.1038/nrc1751. [DOI] [PubMed] [Google Scholar]
- Desaphy J-F, De Luca A, Tortorella P, De Vito D, George AL, Jr, Conte Camerino D. Gating of myotonic Na channel mutants defines the response to mexiletine and a potent derivative. Neurology. 2001;57:1849–1857. doi: 10.1212/wnl.57.10.1849. [DOI] [PubMed] [Google Scholar]
- Habib A, Shamseddeen I, Nasrallah MS, Antoun TA, Nemer G, Bertoglio J, et al. Modulation of COX-2 expression by statins in human monocytic cells. FASEB J. 2007;21:1665–1674. doi: 10.1096/fj.06-6766com. [DOI] [PubMed] [Google Scholar]
- Heller AH, Eicher EM, Hallett M, Sidman RL. Myotonia, a new inherited muscle disease in mice. J Neurosci. 1982;2:924–933. doi: 10.1523/JNEUROSCI.02-07-00924.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue I, Itoh F, Aoyagi S, Tazawa S, Kusama H, Akahane M, et al. Fibrate and statin synergistically increase the transcriptional activities of PPARalpha/RXRalpha and decrease the transactivation of NFkappaB. Biochem Biophys Res Commun. 2002;290:131–139. doi: 10.1006/bbrc.2001.6141. [DOI] [PubMed] [Google Scholar]
- Jentsch TJ, Poët M, Fuhrmann JC, Zdebik AA. Physiological functions of CLC Cl-channels gleaned from human genetic disease and mouse models. Annu Rev Physiol. 2005;67:779–807. doi: 10.1146/annurev.physiol.67.032003.153245. [DOI] [PubMed] [Google Scholar]
- Koch MC, Steinmeyer K, Lorenz C, Ricker K, Wolf F, Otto M, et al. The skeletal muscle chloride channel in dominant and recessive human myotonia. Science. 1992;257:797–800. doi: 10.1126/science.1379744. [DOI] [PubMed] [Google Scholar]
- Kwieciński H, Lehmann-Horn F, Rüdel R. Drug-induced myotonia in human intercostal muscle. Muscle Nerve. 1988;11:576–581. doi: 10.1002/mus.880110609. [DOI] [PubMed] [Google Scholar]
- Liantonio A, De Luca A, Pierno S, Didonna MP, Loiodice F, Fracchiolla G, et al. Structural requisites of 2-(p-chlorophenoxy)-propionic acid analogues for activity on native rat skeletal muscle chloride conductance and on heterologously expressed CLC-1. Br J Pharmacol. 2003;139:1255–1264. doi: 10.1038/sj.bjp.0705364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liantonio A, Giannuzzi V, Cippone V, Pierno S, Conte Camerino D. Fluvastatin and atorvastatin affect calcium homeostasis of rat skeletal muscle by impairing the sarcoplasmic reticulum/mitochondria Ca2+-release system in vivo and in vitro. J Pharmacol Exp Ther. 2007;321:626–634. doi: 10.1124/jpet.106.118331. [DOI] [PubMed] [Google Scholar]
- Lundquist AL, Manderfield LJ, Vanoye CG, Rogers CS, Donahue BS, Chang PA, et al. Expression of multiple KCNE genes in human heart may enable variable modulation of IKs. J Mol Cell Cardiol. 2005;38:277–287. doi: 10.1016/j.yjmcc.2004.11.012. [DOI] [PubMed] [Google Scholar]
- Martin G, Duez H, Blanquart C, Berezowski V, Poulain P, Fruchart JC, et al. Statin-induced inhibition of the Rho-signaling pathway activates PPARalpha and induces HDL apoA-I. J Clin Invest. 2001;107:1423–1432. doi: 10.1172/JCI10852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ort T, Gerwien R, Lindborg KA, Diehl CJ, Lemieux AM, Eisen A, et al. Alterations in soleus muscle gene expression associated with a metabolic endpoint following exercise training by lean and obese Zucker rats. Physiol Genomics. 2007;29:302–311. doi: 10.1152/physiolgenomics.00257.2006. [DOI] [PubMed] [Google Scholar]
- Pierno S, De Luca A, Tricarico D, Roselli A, Natuzzi F, Ferrannini E, et al. Potential risk of myopathy by HMG-CoA reductase inhibitors: a comparison of pravastatin and simvastatin effects on membrane electrical properties of rat skeletal muscle fibres. J Pharmacol Exp Ther. 1995;275:1490–1496. [PubMed] [Google Scholar]
- Pierno S, De Luca A, Liantonio A, Camerino C, Conte Camerino D. Effects of HMG-CoA reductase inhibitors on excitation-contraction coupling of rat skeletal muscle. Eur J Pharmacol. 1999a;364:43–48. doi: 10.1016/s0014-2999(98)00817-6. [DOI] [PubMed] [Google Scholar]
- Pierno S, De Luca A, Beck CL, George AL, Jr, Conte Camerino D. Aging-associated down-regulation of ClC-1 expression in skeletal muscle: phenotypic-independent relation to the decrease of chloride conductance. FEBS Lett. 1999b;449:12–16. doi: 10.1016/s0014-5793(99)00202-1. [DOI] [PubMed] [Google Scholar]
- Pierno S, Desaphy J-F, Liantonio A, De Luca A, Zarrilli A, Mastrofrancesco L, et al. Disuse of rat muscle in vivo reduces protein kinase C activity controlling the sarcolemma chloride conductance. J Physiol. 2007;584:983–995. doi: 10.1113/jphysiol.2007.141358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierno S, Didonna MP, Cippone V, De Luca A, Pisoni M, Frigeri A, et al. Effects of chronic treatment with statins and fenofibrate on rat skeletal muscle: a biochemical, histological and electrophysiological study. Br J Pharmacol. 2006;149:909–919. doi: 10.1038/sj.bjp.0706917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch M. Myotonia caused by mutations in the muscle chloride channel gene ClCN1. Hum Mutat. 2002;19:423–434. doi: 10.1002/humu.10063. [DOI] [PubMed] [Google Scholar]
- Pusch M, Liantonio A, Bertorello L, Accardi A, De Luca A, Pierno S, et al. Pharmacological characterization of chloride channels belonging to the ClC family by the use of chiral clofibric acid derivatives. Mol Pharmacol. 2000;58:498–507. doi: 10.1124/mol.58.3.498. [DOI] [PubMed] [Google Scholar]
- Rogers CS, Vanoye CG, Sullenger BA, George AL., Jr Functional repair of a mutant chloride channel using a trans-splicing ribozyme. J Clin Invest. 2002;110:1783–1789. doi: 10.1172/JCI200216481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose AJ, Michell BJ, Kemp BE, Hargreaves M. Effect of exercise on protein kinase C activity and localization in human skeletal muscle. J Physiol. 2004;561:861–870. doi: 10.1113/jphysiol.2004.075549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbohm A, Rudel R, Fahlke C. Regulation of the human skeletal muscle chloride channel hClC-1 by protein kinase C. J Physiol. 1999;514:677–685. doi: 10.1111/j.1469-7793.1999.677ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrao M, Arendt-Nielsen L, Ge H-Y, Pierelli F, Sandrini G, Farina D. Experimental muscle pain decreases the frequency threshold of electrically elicited muscle cramps. Exp Brain Res. 2007;182:301–308. doi: 10.1007/s00221-007-0985-1. [DOI] [PubMed] [Google Scholar]
- Sirvent P, Bordenave S, Vermaelen M, Roels B, Vassort G, Mercier J, et al. Simvastatin induces impairment in skeletal muscle while heart is protected. Biochem Biophys Res Commun. 2005;338:1426–1434. doi: 10.1016/j.bbrc.2005.10.108. [DOI] [PubMed] [Google Scholar]
- Stuve O, Youssef S, Steinman L, Zamvil SS. Statins as potential therapeutic agents in neuroinflammatory disorders. Curr Opin Neurol. 2003;16:393–401. doi: 10.1097/01.wco.0000073942.19076.d1. [DOI] [PubMed] [Google Scholar]
- Thompson PD, Clarkson P, Karas RH. Statin-associated myopathy. JAMA. 2003;289:1681–1690. doi: 10.1001/jama.289.13.1681. [DOI] [PubMed] [Google Scholar]
- Vaughan CJ, Gotto AM., Jr Update on statins: 2003. Circulation. 2004;110:886–892. doi: 10.1161/01.CIR.0000139312.10076.BA. [DOI] [PubMed] [Google Scholar]
- Weitz-Schmidt G. Statins as anti-inflammatory agents. Trends Pharmacol Sci. 2002;23:482–486. doi: 10.1016/s0165-6147(02)02077-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.