

Abstract
Background and purpose:
Recently, some ligands targeting the sphingosine-1-phosphate receptor subtype 3 (S1P3) have become available. The characterization of these compounds was mainly based on one functional read-out system, although S1P3 receptors are known to activate different signal transduction pathways. Therefore, this study pharmacologically characterizes these compounds using different assays.
Experimental approach:
Using CHO-FlpIn cells expressing the human S1P3 receptor the potencies and maximal effects of S1P, FTY720-P, VPC23019, VPC23153 and VPC24191 were determined in three different assays [inhibition of cAMP accumulation, elevation of intracellular calcium concentrations ([Ca2+]i) and S1P3 receptor internalization].
Key results:
All compounds tested inhibited forskolin-induced cAMP accumulation, increased [Ca2+]i and induced S1P3 receptor internalization but with different potencies and maximal effects. S1P was the most potent compound in all assays followed by FTY720-P. The VPC compounds were generally less potent than S1P and FTY720-P. Regarding the maximal effects, all compounds except VPC23153, behaved as full agonists in the cAMP accumulation assay. In the calcium assay, FTY720-P, VPC23019 and VPC24191 displayed partial and VPC23153 weak partial agonist activity, relative to S1P. Interestingly, treatment with the Gi inactivator Pertussis toxin, did not affect S1P-induced [Ca2+]i elevations but inhibited those in response to the other compounds, by about 50%.
Conclusions and implications:
This study demonstrated differential response patterns at the S1P3 receptor for a range of ligands. These differences could indicate the presence of functional selectivity at this receptor as FTY720-P and the VPC compounds seemed to signal predominantly via Gi– whereas S1P activated Gi and Gq-coupled pathways.
Keywords: S1P3 receptor, ligand-directed signalling, G-protein-coupled receptor, functional selectivity, FTY720, VPC23019
Introduction
The sphingosine-1-phosphate (S1P) receptors, which bind the bioactive lipid S1P, comprise a group of five proteins (S1P1 to S1P5; nomenclature follows Alexander et al., 2008) which belongs to the family of G-protein-coupled receptors (Chun et al., 2002). One of the S1P receptor subtypes, the S1P3 receptor, plays a critical role in the control of cardiac rhythm (Forrest et al., 2004; Sanna et al., 2004) and lung epithelial barrier function (Gon et al., 2005). Although the precise role of the S1P3 receptor in the cardiac (Alewijnse et al., 2004) and pulmonary system still needs further elucidation, S1P3 receptors may represent potential new targets in cardiac diseases (Alewijnse and Peters, 2008) and disease states associated with a dys-regulated epithelial barrier function as in the adult respiratory distress syndrome (ARDS).
In recent years, several new S1P receptor ligands with some selectivity for the S1P3 receptor have become commercially available and these ligands could be useful tools to further elucidate the physiological role of S1P3 receptors in the cardiovascular and pulmonary system (Davis et al., 2005). Although S1P3 receptors are known to activate Gi as well as Gq and G12/13-mediated signal transduction pathways (Young and Van Brocklyn, 2006), the pharmacological characterization of these compounds was based mainly on a single functional read-out system (35S-GTPγS binding) (Clemens et al., 2003; 2004; Davis et al., 2005; Foss et al., 2007). The S1P ligands include VPC23019 [S1P1/S1P3 receptor antagonist, full agonist at S1P4 and partial agonist at S1P5 receptor (Davis et al., 2005; Foss et al., 2007)], VPC23153 [agonistic activity at S1P1, S1P3, S1P4 and S1P5 receptor (Clemens et al., 2004)], VPC24191 [agonistic activity at S1P1, S1P3, S1P4 and S1P5 receptor (Clemens et al., 2003)] and FTY720-P [full agonist S1P1, S1P4, S1P5 receptor, partial agonist S1P3 receptor (Albert et al., 2005)]. Importantly, it is known that the choice of the functional assay system can have implications for, for example, the observed efficacy and/or potency of a ligand and this has been demonstrated for the S1P5 receptor (Niedernberg et al., 2003). Assay-dependent variations in ligand behaviour can, for example, be the result of differences in the efficiency of the receptor to couple to and activate distinct subtypes of G-proteins (Kenakin, 2007). These variations in no instance change the actual rank order of activity of ligands and can therefore readily be explained by classic receptor theory which assumes that a single active state of the receptor controls all activation properties of this receptor (Kenakin, 2007). However, over the past 10 years, it has been observed in numerous cases that the actual rank order of activity of different ligands can change when studying different signal transduction pathways. These observations are incompatible with the classic receptor theory and can only be explained assuming that different ligands can induce different receptor activation states (Kenakin, 2007). This receptor active-state-based selectivity has alternatively been referred to as ligand-directed signalling, stimulus trafficking, biased agonism, conformational selectivity and functional selectivity (Hill, 2006; Kenakin, 2007; 2008; Urban et al., 2007).
Because of the existence of assay-based and/or active-state-based functional selectivity, it becomes of importance to characterize ligands pharmacologically, using different read-out systems. In the present study therefore, we, compared the pharmacological properties of the above mentioned ligands and the active metabolite of FTY720 on three human S1P3-receptor-mediated functional responses in transfected CHO cells expressing the human S1P3 receptor.
Methods
Molecular cloning, transfection and cell culture
An N-terminal HisG-tag was added to the S1P3 receptor via cloning into pcDNA3.1/HisA using BamHI&XhoI. A second cloning step was performed using HindIII&XhoI to clone the HisG-tagged S1P receptor into the expression vector pcDNA5/FRT/TO. CHO-FlpIn cells stably expressing HisG-tagged S1P3 receptors were constructed and cultured as described before (Jongsma et al., 2006). The HisG-tag had no influence on signal transduction induced via this receptor (data not shown). The cDNA of the S1P3 receptor obtained from the UMR cDNA Resource Center contained a point mutation (G962A which results in R321Q). This point mutation was removed by polymerase chain reaction (PCR) using the following primers (forward: TGCCTGGTCAGGGGGCGGGGGGCCCG, reverse: CGGGCCCCCCGCCCCCTGACCAGGCA). The S1P3 receptor coding DNA in the resulting plasmid as well as the stable cell line constructed with this plasmid have been confirmed by sequencing.
cAMP assay
The LANCE™ cAMP 384 kit was used to determine cAMP concentrations according to the manufacturer's protocol. CHO-FlpIn cells, after overnight serum-starvation, were detached from the surface using dissociation buffer. Cells were washed once with Hank's balanced salt solution (HBSS) and subsequently resuspended in stimulation buffer, containing HBSS with 0.05% bovine serum albumin (BSA) (fatty-acid free) and 5 mmol·L−1 4–(2–hydroxyethyl)piperazine–1–ethane sulfonic acid (HEPES). Stimulation mixtures consisted of stimulation buffer with 1 mmol·L−1 (final concentration 0.5 mmol·L−1) 3-isobutyl-1-methylxanthine, 6 µmol·L−1 (final concentration 3 µmol·L−1) forskolin and the indicated concentrations of the S1P receptor agonists. Cells were added to the stimulation mixtures 1:1 in a 384-well optiplate at 2500 cells·well−1 and stimulated for 5 min at room temperature in a total volume of 20 µL·well−1. The cAMP formed during stimulation was measured, according to the manufacturer's protocol, on a multiplate reader (Victor 2, Wallac, Perkin Elmer) 16–24 h after adding detection buffer and antibody mixture.
Intracellular calcium measurement
Intracellular calcium measurements were performed as previously described (Jongsma et al., 2006) with minor changes. CHO-FlpIn cells were plated in a black, clear bottom 96-well plate at 40 000 cells·well−1. After 1 day growth, cells were serum-starved overnight. Cells were then loaded for 1 h with basic buffer (HBSS containing 20 mmol·L−1 HEPES, 2.5 mmol·L−1 probenecid) containing 4 µmol·L−1 Fluo-4 AM ester and 0.42% v/v pluronic acid and incubated at 37°C. After loading, cells were washed twice with basic buffer and incubated at 37°C with basic buffer for 60 min. The fluorescence signal was measured at basal level, followed by ligand stimulation, Triton (5 v/v %) addition (resulting in Fmax) and 250 mmol·L−1 ethylene glycol tretraacetic acid addition (resulting in Fmin). The intracellular calcium concentration ([Ca2+]i) was calculated via the equation: [Ca2+]i= Kd*[(F−Fmin)/(Fmax−F)], Kd being the dissociation constant of the binding of Fluo-4 to calcium (345 nmol·L−1). The increase in [Ca2+]i upon ligand stimulation was calculated as the difference between the [Ca2+]i for the basal level and after adding a ligand.
Immunocytochemistry
Immunocytochemistry was performed as described previously (Jongsma et al., 2007). Briefly, after overnight serum starvation, cells were stimulated with the indicated ligand in serum-free medium for 30 min at 37 °C. The N-terminal HisG-tag added to the S1P3 receptor was detected using, first, an anti-HisG antibody followed by a goat-anti-mouse Alexa488 fluorescent antibody. The fluorescent signal measured on a microplate reader indicates the amount of receptors on the membrane.
Data analysis
Concentration response curves were analysed by fitting sigmoidal functions to the experimental data using Prism 4 (Graphpad Software, Inc., San Diego, CA, USA). Data are expressed as means ± SEM. One-way anova with a Dunnett's correction or Student's t-test was applied where appropriate. P < 0.05 was considered significant.
Materials
pcDNA3.1 containing the entire coding region of the human S1P3 receptor was purchased from UMR cDNA Resource Center (Rolla, MO, USA). Cell culture media, hygromycine B, Lipofectamine™ 2000, Optimem, HBSS, pOG44, pcDNA3.1/HisA, pcDNA5/FRT/TO and CHO-FlpIn cells, were obtained from Invitrogen (Breda, the Netherlands). Restriction enzymes (BamHI, XhoI and HindIII) were obtained from Fermentas Life Sciences (St. Leon-Rot, Germany). Pluronic acid and Fluo-4 AM were obtained from Molecular Probes (via Invitrogen). Foetal calf serum, enzyme-free cell dissociation buffer and penicillin/streptomycin were obtained from Gibco (via Invitrogen). LANCE™ cAMP 384 kit and white 384-well optiplates were obtained from Perkin Elmer (Zaventem, Belgium). S1P, (R)-phosphoric acid mono-[2-amino-2-(6-octyl-1H-benzoimiazol-2-yl) ethyl] ester (VPC23153), (R)-phosphoric acid mono-[2-amino-2-(3-octyl-phenylcarbamoyl)-ethyl] ester (VPC23019) and (S)-phosphoric acid mono-[2-amino-3-(4-octyl-phenylamino)-propyl] ester (VPC24191) were obtained from Avanti-Polar Lipids (via Instruchemie, Delfzijl, the Netherlands). Probenecid, EGTA, Triton X-100, 3-isobutyl-1-methylxanthine, Pertussis toxin (PTX), BSA (fatty-acid free) and activated charcoal were obtained from Sigma Aldrich (Zwijndrecht, the Netherlands). Black, clear bottom 96-well plates were obtained from Greiner Bio One (Alphen aan den Rijn, the Netherlands). 2-Amino-2-[2-(4-octylphenyl)ethyl]-1,3-propanediol mono(dihydrogen phosphate) ester (FTY720-P) was synthesized according to previously described methods (Albert et al., 2005).
Results
Ligand-induced inhibition of forskolin-induced cAMP accumulation
In mock-transfected CHO-FlpIn cells, S1P did not affect the forskolin-induced cAMP production (data not shown). In CHO-FlpIn cells expressing the S1P3 receptor, S1P at low concentrations concentration-dependently inhibited the forskolin-induced cAMP accumulation (pEC50 10.0 ± 0.2, n= 3) whereas at concentrations above 1 × 10−8 mol·L−1 this inhibitory effect was counteracted by a stimulatory effect (Figure 1A, Table 1). FTY720-P, VPC23019, VPC23153 and VPC24191 also inhibited the forskolin-induced cAMP accumulation in these cells but with a significantly lower potency than S1P (Figure 1A, Table 1). The maximal response induced by FTY720-P, VPC23019 and VPC24191 was similar to that of S1P. VPC23153 showed a significantly lower maximal response compared with S1P, that is, behaved as a partial agonist (Figure 1A, Table 1). Of note and in contrast to S1P, neither FTY720-P nor any of the VPC compounds exhibited a bell-shaped concentration-response curve.
Figure 1.
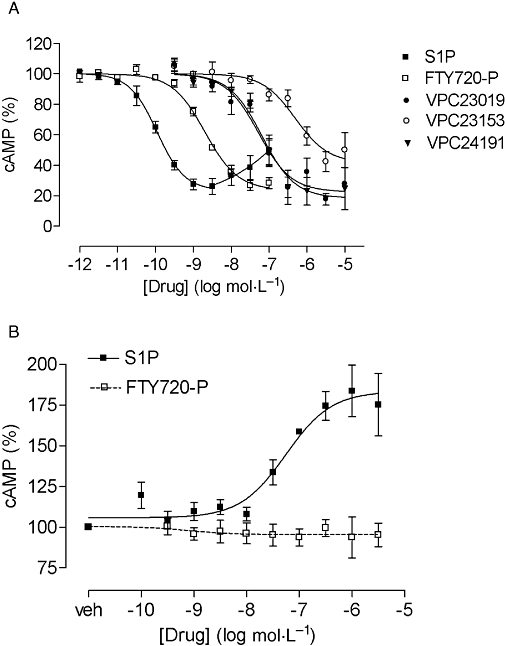
Effect of the S1P receptor ligands shown on the forskolin (3 µmol·L−1)-induced cAMP accumulation in CHO-FlpIn S1P3 cells in the absence (A) or presence of PTX (B). Cells were stimulated with increasing concentrations of the ligands for 5 min at room temperature. The effect of PTX was measured after overnight incubation with 100 ng·mL−1. The cAMP accumulation induced by forskolin (3 µmol·L−1) in the presence of vehicle is set at 100% in each experiment and data are expressed as % of this value. Values are means ± SEM (n= 3–4). PTX, Pertussis toxin; S1P, sphingosine-1-phosphate.
Table 1.
Potency and efficacy [maximal response (Emax) expressed as % of S1P response] for various ligands at the human S1P3 receptor determined in various assays (inhibition of forskolin-induced cAMP accumulation, elevations of [Ca2+]i, receptor internalization)
| Ligand | cAMP inhibition | [Ca2+]i | Internalization | ||||||
|---|---|---|---|---|---|---|---|---|---|
| pEC50 | Emax (% of S1P) | n | pEC50 | Emax (% of S1P) | n | pEC50 | Emax (% of SIP) | n | |
| S1P | 10.0 ± 0.2 | 100 | 3 | 8.4 ± 0.1 | 100 | 9 | 7.5 ± 0.1 | 100 | 8 |
| FTY720-P | 8.8 ± 0.1* | 101 ± 4 | 3 | 6.8 ± 0.1* | 51 ± 4* | 13 | 6.8 ± 0.1* | 109 ± 6 | 7 |
| VPC23019 | 7.2 ± 0.1* | 103 ± 7 | 4 | 6.1 ± 0.1* | 48 ± 4* | 5 | 5.7 ± 0.2* | 72 ± 10* | 4 |
| VPC23153 | 6.3 ± 0.1* | 76 ± 7* | 4 | 5.9 ± 0.1* | 25 ± 2* | 6 | 5.6 ± 0.4* | 52 ± 18* | 4 |
| VPC24191 | 7.2 ± 0.0* | 100 ± 15 | 4 | 5.9 ± 0.1* | 56 ± 10* | 6 | 6.0 ± 0.3* | 88 ± 5 | 4 |
The Emax of S1P in each assay was arbitrarily set to 100% (cAMP assay 100% = 90% inhibition of cAMP accumulation; calcium assay 100% = 1947 nmol·L−1 increase in [Ca2+]i; internalization assay 100% = 60% decrease in fluorescent signal).
P < 0.05, significantly different from corresponding values for S1P.
[Ca2+]i, intracellular calcium concentration; S1P, sphingosine-1-phosphate.
To further investigate the bell-shaped S1P concentration-response curve, cells were incubated with PTX to block Gi activation. Overnight incubation with PTX (100 ng·mL−1) abolished the inhibitory effect of S1P on the forskolin-induced cAMP formation and, therefore, only a concentration-dependent stimulation in cAMP accumulation by S1P was observed in these cells (Figure 1B). PTX treatment also abolished the inhibitory effect of FTY720-P (Figure 1B) but did not disclose any FTY720-P-induced stimulation of cAMP accumulation (Figure 1B).
Ligand-induced elevation of [Ca2+]i
In mock-transfected CHO-FlpIn cells, S1P only slightly increased [Ca2+]i (pEC50 6.9 ± 0.2, Emax= 198 ± 32 nmol·L−1, n= 4) whereas FTY720-P had no effect (data not shown). All compounds concentration-dependently increased [Ca2+]i in CHO-FlpIn S1P3 cells (Figure 2A,B). The potency of FTY720-P to increase [Ca2+]i was significantly lower than that of S1P and the maximal effect of FTY720-P was only 51 ± 4% of that of S1P (Figure 2B, Table 1). VPC23019, VPC23153 and VPC24191 also all raised [Ca2+]i but with significantly lower potencies and lower maximal effects compared with S1P (Figure 2B, Table 1).
Figure 2.
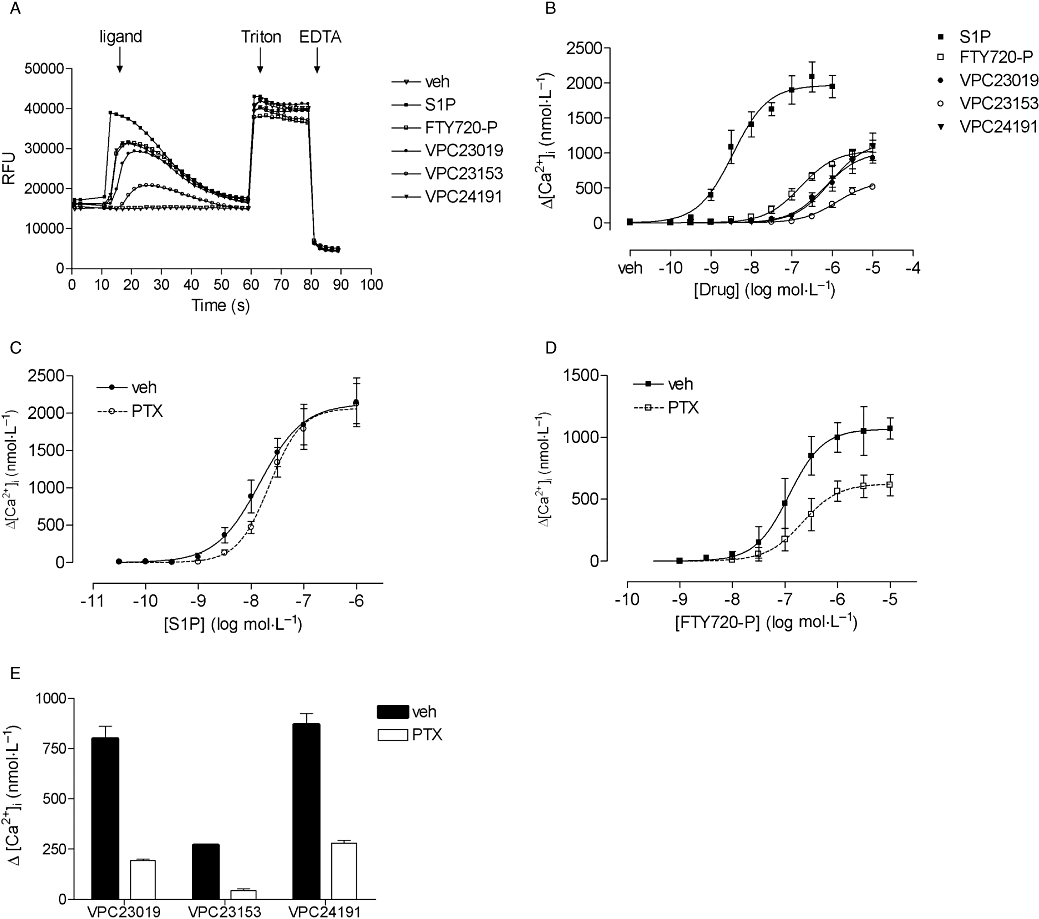
Effect of the S1P receptor ligands shown on the elevation of [Ca2+]i. Values are means ± SEM. (A) Representative tracings of vehicle (veh) or the ligands (1 µmol·L−1) in calcium experiments. The time of ligand addition, addition of 5% Triton and addition of 250 mmol·L−1 EGTA is indicated. (B) S1P, FTY720-P, VPC23019, VPC23153 and VPC24191-induced increases in [Ca2+]i in CHO-FlpIn S1P3 cells (n= 5–9). (C) Effect of overnight PTX (100 ng·mL−1) incubation on S1P-induced increases in [Ca2+]i (n= 4). (D) Effect of overnight PTX incubation on FTY720-P-induced increases in [Ca2+]i (n= 3). (E) Effect of overnight PTX incubation on 10 µmol·L−1 VPC23019, VPC23153 and VPC24191-induced increases in [Ca2+]i (n= 3). [Ca2+]i, intracellular calcium concentration; PTX, Pertussis toxin; S1P, sphingosine-1-phosphate.
Because both Gq-and Gi-activated pathways can mediate elevations in [Ca2+]i, the involvement of Gi was investigated by studying the effect of PTX treatment (100 ng·mL−1 overnight) on the calcium responses. The S1P-induced increase in [Ca2+]i in mock-transfected CHO-FlpIn cells was almost completely PTX-sensitive (data not shown). Interestingly, overnight incubation with PTX did not significantly affect the S1P-induced calcium responses in CHO-FlpIn S1P3 cells [pEC50 vehicle vs. PTX treated cells: 7.7 ± 0.2 vs. 7.5 ± 0.1, Emax (nmol·L−1) vehicle vs. PTX treated cells: 2475 ± 342 vs. 2537 ± 334, n= 4] although the concentration response curve seemed to be somewhat shifted to the right (Figure 2C). However, the FTY720-P-induced maximal responses but not the potency in these cells were significantly reduced [pEC50 vehicle vs. PTX treated cells: 6.8 ± 0.2 vs. 6.6 ± 0.2, Emax (nmol·L−1) vehicle vs. PTX treated cells: 1125 ± 124 vs. 661 ± 82, n= 3] (Figure 2D). The increase in [Ca2+]i induced by 10 µmol·L−1 of VPC23019, VPC23153 or VPC24191 was also significantly reduced upon PTX treatment (Figure 2E).
Ligand-induced receptor internalization
All tested compounds induced a concentration-dependent internalization of the HisG-tagged S1P3 receptor after 30 min stimulation represented by a decrease in membrane fluorescence (Figure 3, Table 1). The potency of FTY720-P as well as all three VPC compounds to induce internalization was significantly lower than that of S1P (Table 1). The maximal internalization induced was only significantly decreased for VPC23019 and VPC23153 compared with S1P, whereas the maximal effect of VPC24191 and FTY720-P was comparable to that of S1P (Figure 3,Table 1).
Figure 3.
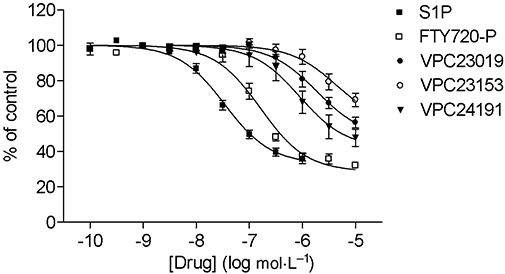
Concentration-dependent effect of indicated compounds on internalization of HisG-S1P3 receptors in CHO-FlpIn cells. Stimulations were carried out at 37°C for 30 min. Values are expressed as percentage of fluorescent signal measured for unstimulated cells which was set at 100% and are presented as means ± SEM (n= 7–8). S1P, sphingosine-1-phosphate.
Discussion
Recently, several new S1P ligands with some specificity for the S1P3 receptor have become commercially available but these compounds have not been intensively characterized yet. Because it is known that ligands can show assay-based selectivity and, as it has become evident during the last 10 years, even active state-based functional selectivity, it is important to characterize ligands pharmacologically using a range of assays. In this study we have, investigated the pharmacological properties of some S1P3 ligands using different signal transduction assays. Specifically, we determined the potency and efficacy [maximal response (Emax) expressed as % of S1P response] of all ligands with regard to their ability to inhibit forskolin-induced cAMP accumulation, to increase [Ca2+]i and to internalize the S1P3 receptor in CHO-FlpIn cells expressing the human S1P3 receptor.
In our experiments, all the tested ligands, to some extent, inhibited forskolin-induced cAMP accumulation, increased [Ca2+]i and induced S1P3 receptor internalization (Table 1). Depending on the assay used, in our studies, VPC23019 either behaved as a partial (calcium assay) or full S1P3 agonist (cAMP assay), rather than an antagonist of the human S1P3 receptor, as described by Davis et al. (2005). This difference in efficacy between our study and that by Davis et al. is most likely explained by differences in the cellular responses measured and/or the cellular system used. Such differences have been described before for the S1P5 receptor (Niedernberg et al., 2003) and could, for instance, reflect differences in receptor expression levels (Sato et al., 2007).
Comparing the potency of the different compounds tested in the cAMP accumulation versus the calcium assay, revealed that the potency in the cAMP assay is generally higher than that determined in the calcium assay except for VPC23153 which shows comparable potencies in both assays (Table 1). However, the rank order of potencies did not show remarkable differences between both assays.
In addition, the effect of all compounds on internalization of the S1P3 receptor has been investigated in this study. Although receptor occupancy is known to be an important factor in the induction of receptor regulatory processes, the potency and efficacy of internalization of the tested ligands were similar to the values observed in the calcium assay. The potencies determined in the calcium assay can thus be concluded to most closely resemble the affinity of the tested ligand and thus its receptor occupancy. Consequently, we may conclude that there was a receptor reserve for the inhibitory effect on cAMP accumulation.
When considering the S1P-mediated inhibition of the forskolin-induced cAMP production in more detail, it is worthwhile to mention that the decrease in forskolin-induced cAMP accumulation observed at low S1P concentrations was followed by an increase in cAMP accumulation at higher concentrations of S1P (<10 nmol·L−1). In contrast to S1P, FTY720-P did not increase cAMP accumulation at higher concentrations. Because, CHO cells have been reported to endogenously express S1P receptors (Holdsworth et al., 2005), the observed S1P effect could be due to stimulation of endogenous S1P receptors. However, as S1P did not induce an effect in mock transfected cells, a role for endogenously expressed receptors in the stimulatory cAMP effect of this compound is unlikely. The S1P-induced increase in cAMP accumulation was even more pronounced after PTX treatment indicating that, in this system, S1P activated not only Gi-coupled pathways but also led to an increase in cAMP accumulation probably, although we have no direct evidence for this, via the activation of Gs-proteins. Alternatively, the S1P-induced increases in cAMP accumulation may be explained by a Gq-mediated activation of calcium-dependent adenylyl cyclases. However, S1P3 receptor-mediated increases in cAMP accumulation in CHO cells stably expressing this receptor, have been described before and, in that study, were shown to be independent of calcium signalling (Kon et al., 1999). Similar to our observations the Gi-coupled muscarinic M2 cholinoceptor was also shown to increase cAMP accumulation at high ligand concentrations and high receptor density (Michal et al., 2001). These authors proposed that in systems with high receptor expression, Gi-protein activation becomes saturated at a certain point and that beyond this limit, any additionally activated receptors start coupling to Gs-proteins. Although, because of technical problems, we are not able to directly quantify the receptor density in our system, immunocytochemical measurements indicate that the S1P receptor expression in the CHO-FlpIn cells is rather high (Jongsma et al., 2007). The mechanism proposed by Michal et al. (2001) may thus also explain the effects observed in our study. In addition, similar observations have been described for the adenosine A1 receptor, for which the activation of Gs-proteins was both dependent on the receptor density (Cordeaux et al., 2000) and on the agonist used (Cordeaux et al., 2004). Because the S1P-induced increases in cAMP accumulation observed in our study are most likely to be observed only at a high receptor density, the physiological relevance of this finding is open to argument.
The observation in our study that only S1P and not FTY720-P induced an increase in cAMP accumulation may be a first indication for active state-based functional selectivity. Only the receptor conformation induced by S1P and not that induced by FTY720-P is able to activate Gs-proteins. An alternative explanation could be that the increases in cAMP accumulation are only observed for S1P because of the involvement of calcium dependent adenylyl cyclases. As became evident from our experiments, the potency and efficacy of S1P to induce increases in [Ca2+]i was significantly higher than that of FTY720-P which fits with the observation that S1P induced increases in cAMP accumulation, whereas at the concentrations tested no stimulatory effects were observed for FTY720-P. The differential effects of S1P and FTY720-P may thus be a first indication of active state-based functional selectivity but any definitive conclusions cannot be drawn based upon our data and we will thus address this point in future studies.
Besides the indication for functional selectivity in the experiments on cAMP accumulation, further evidence supporting this indication came from experiments studying the effects of the compounds on [Ca2+]i. In contrast to the S1P-induced increases in [Ca2+]i, the FTY720-P induced effects on calcium were, to an important extent, PTX-sensitive and thus mediated via the activation of Gi-proteins. The difference in PTX-sensitivity of the S1P versus the FTY720-P induced calcium response cannot be explained by effects on endogenous S1P receptors. Only S1P induces minor increases in [Ca2+]i in mock–transfected cells and these effects are almost completely Gi-mediated. We thus conclude that the active state induced by S1P probably activates both, Gq and Gi-mediated signalling pathways, whereas the state induced by FTY720-P probably prefers the activation of Gi-coupled pathways. Interestingly, similar conclusions can be drawn for the VPC compounds, which are structurally closely related to FTY720-P. However, it should be kept in mind that the S1P3-receptor expression in our study is relatively high and that the observed differences in the effects can probably also be the result of a high receptor reserve for the S1P-induced effect.
Shortly after completion of our study, a study by Sensken et al. (Sensken et al., 2008) also reported on functional selectivity at the S1P3 receptor. In line with our observations, Sensken and coworkers showed in their study that FTY720-P selectively activated Gi-coupled pathways via the S1P3 receptor whereas S1P activated both Gi and Gq-mediated pathways.
Interestingly, the existence of functional selectivity has also been described for the S1P1 receptor. S1P1 receptors, internalized upon stimulation by S1P or SEW2871, were recycled to the plasma membrane, while FTY720-P-induced S1P1 receptor internalization resulted in irreversible lysosomal degradation of these receptors (Oo et al., 2007). This difference in receptor fate has been related to polyubiquitinylation of the S1P1 receptor induced by FTY720-P, but not by S1P (Oo et al., 2007).
Overall, our study provides some evidence, in line with findings by Sensken et al. (Sensken et al., 2008), which may indicate the presence of functional selectivity at the S1P3 receptor. This conclusion is most importantly based upon the finding that the ligands tested in this study show differential response patterns at the S1P3 receptor. In addition, the signalling induced by FTY720-P and the VPC compounds seems to be predominantly Gi-mediated whereas for S1P both Gi-and Gq-coupled signalling was observed. Besides these important findings, this study for the first time extensively investigated and described the pharmacological properties of VPC23019, VPC23153 and VPC24191 at the human S1P3 receptor expressed in CHO-FlpIn cells. The above mentioned findings may be of major importance as FTY720, which is presently in clinical trials for the treatment of multiple sclerosis, has some cardiac side effects which are mediated by S1P3 receptors. At the moment it is not known whether these cardiac side effects are related to the fact that FTY720-P may preferentially activate Gi-mediated signalling and this issue awaits further investigation.
Glossary
Abbreviations:
- [Ca2+]i
intracellular calcium concentration
- HBSS
Hank's balanced salt solution
- PTX
Pertussis toxin
- S1P
sphingosine-1-phosphate
Conflict of interest
None.
References
- Albert R, Hinterding K, Brinkmann V, Guerini D, Muller-Hartwieg C, Knecht H, et al. Novel immunomodulator FTY720 is phosphorylated in rats and humans to form a single stereoisomer. Identification, chemical proof, and biological characterization of the biologically active species and its enantiomer. J Med Chem. 2005;48:5373–5377. doi: 10.1021/jm050242f. [DOI] [PubMed] [Google Scholar]
- Alewijnse AE, Peters SL. Sphingolipid signalling in the cardiovascular system: good, bad or both? Eur J Pharmacol. 2008;585:292–302. doi: 10.1016/j.ejphar.2008.02.089. [DOI] [PubMed] [Google Scholar]
- Alewijnse AE, Peters SL, Michel MC. Cardiovascular effects of sphingosine-1-phosphate and other sphingomyelin metabolites. Br J Pharmacol. 2004;143:666–684. doi: 10.1038/sj.bjp.0705934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 3rd edition. Br J Pharmacol. 2008;153(2):S1–S209. doi: 10.1038/sj.bjp.0707746. Suppl. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun J, Goetzl EJ, Hla T, Igarashi Y, Lynch KR, Moolenaar W, et al. International Union of Pharmacology. XXXIV. Lysophospholipid receptor nomenclature. Pharmacol Rev. 2002;54:265–269. doi: 10.1124/pr.54.2.265. [DOI] [PubMed] [Google Scholar]
- Clemens JJ, Davis MD, Lynch KR, Macdonald TL. Synthesis of para-alkyl aryl amide analogues of sphingosine-1-phosphate: discovery of potent S1P receptor agonists. Bioorg Med Chem Lett. 2003;13:3401–3404. doi: 10.1016/s0960-894x(03)00812-6. [DOI] [PubMed] [Google Scholar]
- Clemens JJ, Davis MD, Lynch KR, Macdonald TL. Synthesis of benzimidazole based analogues of sphingosine-1-phosphate: discovery of potent, subtype-selective S1P4 receptor agonists. Bioorg Med Chem Lett. 2004;14:4903–4906. doi: 10.1016/j.bmcl.2004.07.030. [DOI] [PubMed] [Google Scholar]
- Cordeaux Y, Briddon SJ, Megson AE, McDonnell J, Dickenson JM, Hill SJ. Influence of receptor number on functional responses elicited by agonists acting at the human adenosine A1 receptor: evidence for signaling pathway-dependent changes in agonist potency and relative intrinsic activity. Mol Pharmacol. 2000;58:1075–1084. doi: 10.1124/mol.58.5.1075. [DOI] [PubMed] [Google Scholar]
- Cordeaux Y, IJzerman AP, Hill SJ. Coupling of the human A1 adenosine receptor to different heterotrimeric G proteins: evidence for agonist-specific G protein activation. Br J Pharmacol. 2004;143:705–714. doi: 10.1038/sj.bjp.0705925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MD, Clemens JJ, Macdonald TL, Lynch KR. Sphingosine 1-phosphate analogs as receptor antagonists. J Biol Chem. 2005;280:9833–9841. doi: 10.1074/jbc.M412356200. [DOI] [PubMed] [Google Scholar]
- Forrest M, Sun SY, Hajdu R, Bergstrom J, Card D, Doherty G, et al. Immune cell regulation and cardiovascular effects of sphingosine 1-phosphatereceptor agonists in rodents are mediated via distinct receptor subtypes. J Pharmacol Exp Ther. 2004;309:758–768. doi: 10.1124/jpet.103.062828. [DOI] [PubMed] [Google Scholar]
- Foss FW, Jr, Snyder AH, Davis MD, Rouse M, Okusa MD, Lynch KR, et al. Synthesis and biological evaluation of gamma-aminophosphonates as potent, subtype-selective sphingosine 1-phosphate receptor agonists and antagonists. Bioorg Med Chem. 2007;15:663–677. doi: 10.1016/j.bmc.2006.10.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gon Y, Wood MR, Kiosses WB, Jo E, Sanna MG, Chun J, et al. S1P3 receptor-induced reorganization of epithelial tight junctions compromises lung barrier integrity and is potentiated by TNF. Proc Natl Acad Sci USA. 2005;102:9270–9275. doi: 10.1073/pnas.0501997102. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Hill SJ. G-protein-coupled receptors: past, present and future. Br J Pharmacol. 2006;147:S27–S37. doi: 10.1038/sj.bjp.0706455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holdsworth G, Slocombe P, Hutchinson G, Milligan G. Analysis of endogenous S1P and LPA receptor expression in CHO-K1 cells. Gene. 2005;350:59–63. doi: 10.1016/j.gene.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Jongsma M, Hendriks-Balk MC, Michel MC, Peters SL, Alewijnse AE. BML-241 fails to display selective antagonism at the sphingosine-1-phosphate receptor, S1P3. Br J Pharmacol. 2006;149:277–282. doi: 10.1038/sj.bjp.0706872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jongsma M, Florczyk UM, Hendriks-Balk MC, Michel MC, Peters SL, Alewijnse AE. Validation of a rapid, non-radioactive method to quantify internalisation of G-protein coupled receptors. Naunyn Schmiedebergs Arch Pharmacol. 2007;375:329–336. doi: 10.1007/s00210-007-0164-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T. Functional selectivity through protean and biased agonism: who steers the ship? Mol Pharmacol. 2007;72:1393–1401. doi: 10.1124/mol.107.040352. [DOI] [PubMed] [Google Scholar]
- Kenakin T. What systems can and can't do. Br J Pharmacol. 2008;153:841–843. doi: 10.1038/sj.bjp.0707677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kon J, Sato K, Watanabe T, Tomura H, Kuwabara A, Kimura T, et al. Comparison of intrinsic activities of the putative sphingosine-1-phosphate receptor subtypes to regulate several signalling pathways in their cDNA-transfected Chinese Hamster Ovary cells. J Biol Chem. 1999;274:23940–23947. doi: 10.1074/jbc.274.34.23940. [DOI] [PubMed] [Google Scholar]
- Michal P, Lysíková M, Tucek S. Dual effects of muscarinic M2 acetylcholine receptors on the synthesis of cyclic AMP in CHO cells: dependence on time, receptor density and receptor agonists. Br J Pharmacol. 2001;132:1217–1228. doi: 10.1038/sj.bjp.0703931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedernberg A, Tunaru S, Blaukat A, Harris B, Kostenis E. Comparative analysis of functional assays for characterization of agonist ligands at G protein-coupled receptors. J Biomol Screen. 2003;8:500–510. doi: 10.1177/1087057103257555. [DOI] [PubMed] [Google Scholar]
- Oo ML, Thangada S, Wu MT, Liu CH, Macdonald TL, Lynch KR, et al. Immunosuppressive and anti-angiogenic sphingosine 1-phosphate receptor-1 agonists induce ubiquitinylation and proteasomal degradation of the receptor. J Biol Chem. 2007;282:9082–9089. doi: 10.1074/jbc.M610318200. [DOI] [PubMed] [Google Scholar]
- Sanna MG, Liao J, Jo E, Alfonso C, Ahn MY, Peterson MS, et al. Sphingosine 1-phosphate (S1P) receptor subtypes S1P1 and S1P3, respectively, regulate lymphocyte recirculation and heart rate. J Biol Chem. 2004;279:13839–13848. doi: 10.1074/jbc.M311743200. [DOI] [PubMed] [Google Scholar]
- Sato M, Horinouchi T, Hutchinson DS, Evans BA, Summers RJ. Ligand-directed signaling at the β3-adrenoceptor produced by 3-(2-Ethylphenoxy)-1-[(1,S)-1,2,3,4-tetrahydronapth-1-ylamino]-2S-2-propanol oxalate (SR59230A) relative to receptor agonists. Mol Pharmacol. 2007;72:1359–1368. doi: 10.1124/mol.107.035337. [DOI] [PubMed] [Google Scholar]
- Sensken SC, Stäubert C, Keul P, Levkau B, Schöneberg T, Gräler MH. Selective activation of G alpha i mediated signalling of S1P3 by FTY720-phosphate. Cell Signal. 2008;20:1125–1133. doi: 10.1016/j.cellsig.2008.01.019. [DOI] [PubMed] [Google Scholar]
- Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, et al. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther. 2007;320:1–13. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- Young N, Van Brocklyn JR. Signal transduction of sphingosine-1-phosphate G protein-coupled receptors. Sci World J. 2006;6:946–966. doi: 10.1100/tsw.2006.182. [DOI] [PMC free article] [PubMed] [Google Scholar]